

Abstract
The passive monitoring of biological environments by soft materials has a variety of nanobiotechnology applications; however, invoking distinct transitions in geometric, mechanical or optical properties remains a prevalent design challenge. We demonstrate here that close-packed nanoparticle—hydrogel composites can progress through a substantial shift in such properties by the use of a chemical-to-physical cross-link transition mediated by the catalytic activity of different proteases. Catalytic cleavage of the original hydrogel network structure initiates the self-assembled formation of a secondary, physically cross-linked network, causing a 1200% increase in storage modulus. Furthermore, this unique mechanism can be fabricated as a 3D photonic crystal with broad (∼240 nm), visible responses to the targeted enzymes. Moreover, the material provided threshold responses, requiring a certain extent of proteolytic activity before the transition occurred. This allowed for the fabrication of Boolean logic gates (OR and AND), which responded to a specific assortment of proteases. Ultimately, this mechanism enables the design of stimuli-responsive hydrogels, which can proceed through a secondary network formation, after an energetic barrier has been breached. Protease responsive hydrogel nanocomposites, described here, could offer avenues in degradation-stiffening and collapsing materials for a variety of biomaterial applications.
Keywords: hydrogel, nanoparticle, photonic crystal, self-assembly, protease
Graphical Abstract
Intelligent soft matter systems have garnered a growing interest due to their ability to autonomously and dynamically respond to environmental stimuli.1 Particularly, hydrophilic systems (e.g., hydrogels) can have tailored responses in applications such as synthetic extracellular matrices,2 smart tissue cultures,3 autonomous sensors,4 injectable biomaterials,5 and drug delivery vesicles.6 A majority of these reported systems rely on the modulation of the forces that dictate the swelling equilibrium of a polymer network.
Initial reports of such systems primarily responded to simple, nonspecific chemical and physical stimuli such as changes in pH,7,8organic solvents,9 or temperature.10,11 The field has grown to develop more sophisticated systems with recognition properties for nanobiotechnology applications. This includes hydrogels with recognition sites such as boronic acids for the sensing of sugars12–14 and peptides specifically phosphorylated by kinases.15 These systems rely on changes in the number of immobile, polymer-bound charges, creating a Donnan potential, and therefore swell.
Another reported approach exploits mechanisms that alter the cross-link density of a hydrogel, in which the degradation of the network causes a swelling event. Hydrogels with specific response to proteases have been reported, including the use of matrix metalloproteinases2,16–19 and chymotrypsin, 20 which cleave peptide-based crosslinks. More recently, soft matter systems have been described that rearrange upon exposure to a stimulus.21–23 Enzymatic activity causes the formation of hydrophobic pendant groups on polymer chains triggering supramolecular complexes born from hydrophobic interactions. These systems can proceed through both sol—gel22 or gel—sol23 transitions depending on design as well as respond to proteolytic enzymes through changes in swelling.21 Ultimately, biospecific, stimuli-responsive hydrogels reported thus far only proceed through mechanisms involving dissolution of chemical bonds or mechanical degradation and swelling.
In this manuscript, we demonstrate that hydrogel—nanoparticle composites can respond to proteolytic degradation with an increase in stiffness through the formation of physical cross-links, contrary to previously reported systems, which normally mechanically degrade and swell. The nanocomposite consists of a colloidal array of 240 nm silica nanoparticles (NPs) distributed within a polyethylene glycol(PEG)-peptide hydrogel as seen in Figure 1. The silica nanoparticle size of 240 nm was chosen to provide hydrogel composites with a tunable color range of blue to red, dependent on hydrogel precursor concentration. The hydrogel itself was fabricated utilizing a step-growth thiol—ene polymerization between 4-arm norbornene-PEG (PEG4NB) and a dicysteine capped peptide.17 The peptide, CY↓KC,is hydrolyzed by chymotrypsin at the carboxyl side of the tyrosine residue. The negatively charged silica NPs used are also at a sufficiently high concentrations (>54%) to form a regular periodic structure within the hydrogel, acting as a photonic bandgap which can reflect visible wavelengths of light.12 The spacing of the silica NPs dictates the wavelength reflected, where a more swollen hydrogel and thus larger periodic nanoparticle spacing reflects larger wavelengths.
Figure 1.
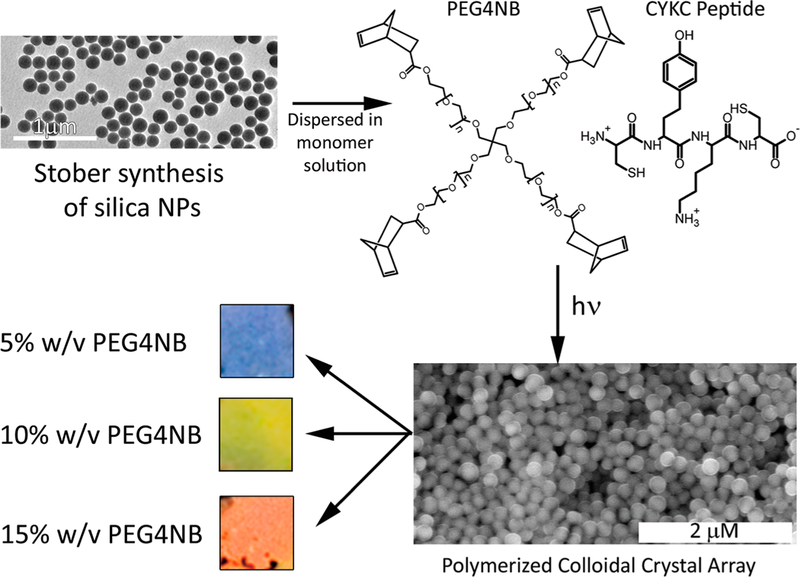
Schematic depicting the synthesis of the nanoparticle—hydrogel composites. Approximately 240 nm silica NPs were fabricated utilizing a modified Stober method. The resulting NPs were dispersed in a 1:1 -ene/thiol solution of PEG4NB and CYKC (cysteine tyrosine lysine cysteine) oligopeptide and exposed to UV radiation, initiating the thiol—ene polymerization and producing the nanocomposite. Increasing concentrations of the PEGNB and CYKC resulted in more swollen and therefore more red-shifted gels.
RESULTS AND DISCUSSION
The mechanism that dictates this unique collapse and stiffening response originates from pendant network chains adsorbing to the surface of the silica NPs. It is well-known that polymers can adsorb to the surface of nanoparticles and such phenomena have been exploited for use in wet adhesives24 and synthesis of physically cross-linked hydrogels.25 However, if the polymers are heavily cross-linked, such as in a covalent network, their entropy will be hindered preventing adsorption from occurring. Accordingly, it is hypothesized that if the cross-links in such a system are cleaved, the polymer chains would gain enough mobility for adsorption and thus formation of a tighter, secondary, physical network (Figure 2). Complete dissolution of the hydrogel does not occur due to the resulting crowding effects originating from the increased polymer volume fraction within the collapsing hydrogel, which prevent any further action by the chymotrypsin, leaving the original covalent network partially intact. The remaining covalent network provides linkages between pendant network chains adsorbed to the silica NP surface.
Figure 2.
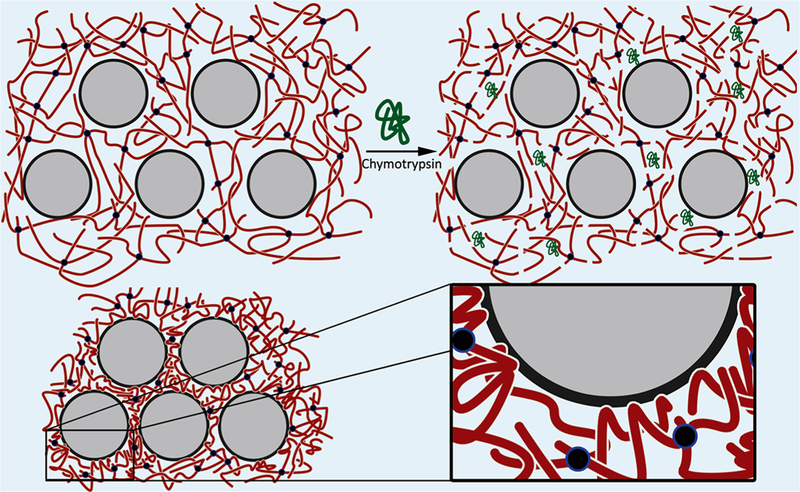
Depiction of the enzyme induced chemical-to-physical cross-link transition. When the nanocomposite is exposed to chymotrypsin, the CYKC peptides are catalytically cleaved. This imparts pendant network chains with enough mobility to physically adsorb onto the surface of the silica NPs causing the hydrogel to collapse.
The interaction producing this physical cross-linking event is likely mediated by the positive charged N-terminal amine that is produced by the catalytic cleavage of the peptide cross-linker. These generated positive charges can associate with the surface of the negatively charged silica, effectively causing the silica nanoparticles to aggregate with the polymer acting as a spacer. What remains is a hydrogel consisting of both a fraction of the original covalent network and physical interactions between the now positively charged polymer and the silica nanoparticles. Two observational experiments were performed to verify this phenomenon. First, collapsed and physically cross-linked hydrogels were exposed to alkaline conditions, which can cleave the original network’s ester bonds. The exposure degraded the hydrogel rapidly, indicating that the original network remaining partially intact is necessary for the “chemical” to “physical” transition to occur. The second observation was made after exposing both the original covalent gels and the collapsed physically cross-linked gels to the hydrogen fluoride. The hydrogen fluoride reacts with and destroys the silica nano-particles. Exposure of the hydrogen fluoride to the original covalent gels results in loss of structural color, due to destruction of the silica and what remains is a transparent hydrogel. Conversely, exposure of hydrogen fluoride to the collapsed physically cross-linked gels results in destruction of the hydrogel. This indicates that the hydrogel consists of physical cross-links between the silica nanoparticles.
This chemical-to-physical cross-link transition produces collapsing responses across a wide range of chymotrypsin concentrations. Exposure to chymotrypsin solutions ranging from 20 to 4000 nM initiated an ∼30–70% decrease in volume compared to the control case which increased 30% in volume (Figure 3). The swelling of the control case results from equilibrium swelling of the polymer network over time. Interestingly, at concentrations of 40 000 nM or higher, this mechanism did not occur and the hydrogel was eroded away by the enzymatic activity. At concentrations of chymotrypsin exceeding 40 000 nM, the peptide cross-links are likely cleaved faster than the network chains adsorbing to the silica NPs, allowing for diffusion of the polymer and NPs away from the bulk gel.
Figure 3.
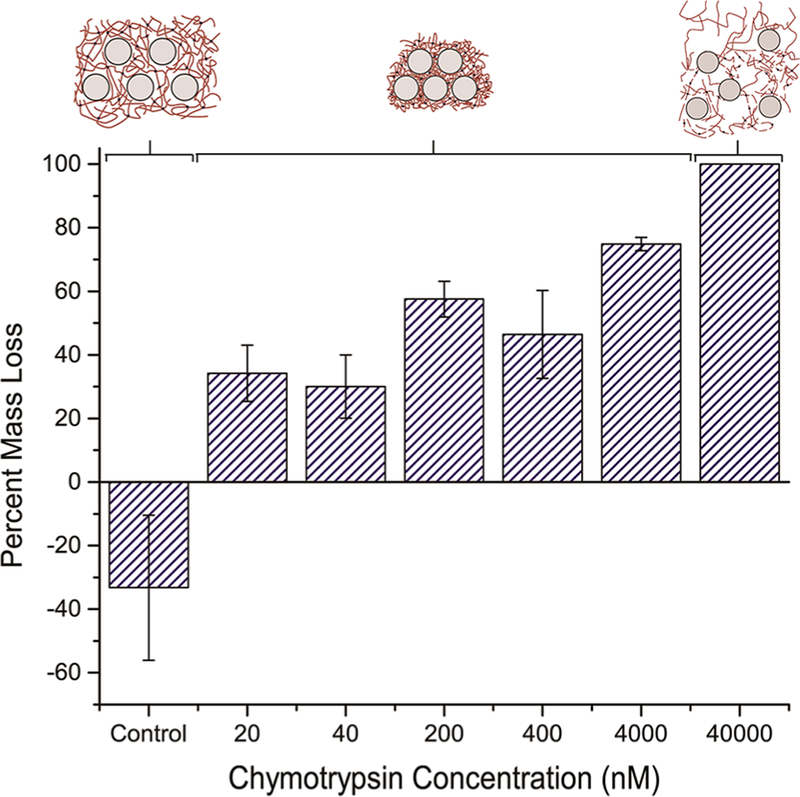
Exposure to chymotrypsin concentrations ranging from 20 to 4000 nM resulted in an∼40% mass loss. At higher concentrations, the proteolytic breakdown of the nanocomposite occurs faster than the physical adsorption mechanism, causing larger mass losses due to erosion of the hydrogel.
To further investigate the changes to material properties after this collapsing transition, rheology was performed to obtain the shear storage modulus, G′, and shear loss modulus, G″. The nanocomposites before and after chymotrypsin exposure were subjected to shear strain from 0 to 20%. G′ of the nano-composite increased from ∼70 to ∼850 Pa, a 1200% increase, after exposure to chymotrypsin (Figure 4). As the hydrogel collapses, several factors contribute to the increase in storage modulus including formation of physical cross-links and an increase in the polymer volume fraction within the hydrogel.26 This type of significant collapse and stiffening of a hydrogel system has only been reported for nonspecific stimuli such as temperature.10,11 In temperature responsive LCST hydrogels, the polymer chains have decreased solubility above their lower critical solution temperature, causing an increase in polymer volume fraction. There have also been reports of hydrogels that form secondary cross-links in response to a chemical stimulus,27 but the original cross-links reduce the mobility of the network chains, which hinders secondary cross-link formation and prevents large changes in polymer volume fraction. Alternatively, in the demonstrated nanocomposite, the chemically cross-linked network is partially degraded by chymotrypsin, which increases polymer chain mobility allowing for an increase in the formation of secondary physical cross-links. Furthermore, the distributed silica NPs act as localization sites for physical cross-link formation, producing a more compact structure than interchain cross-links.
Figure 4.
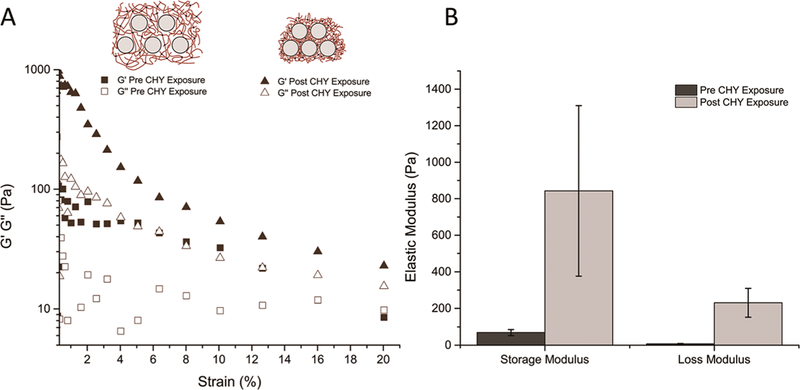
(A) Representative case of the storage and loss modulus under constant strain of the hydrogel composite before and after chymotrypsin exposure. (B) Before exposure to chymotrypsin, the nanocomposite gels had a storage modulus of ∼70 Pa, and post exposure, the storage modulus was ∼850 Pa (n = 3). The data for each case can found in Supporting Information Figure 4.
This mechanism of collapse and stiffening upon enzymatic degradation was further investigated utilizing fluorescence microscopy. The nanocomposite gels were exposed to a4000 nM chymotrypsin solution containing fluorescamine. As the peptide cross-links are cleaved, the fluorescamine reacts with generated N-terminal amines, producing a fluorescent signal that can be visualized using a 365 nm excitation, 450 nm emission microscope filter (Figure 5). The surrounding media did not increase in fluorescence, indicating that the hydrogel is indeed collapsing without the release or erosion of any polymeric mass. Conversely, the fluorescent response of the hydrogel increased linearly with time as the peptide cross-links are digested by chymotrypsin.
Figure 5.
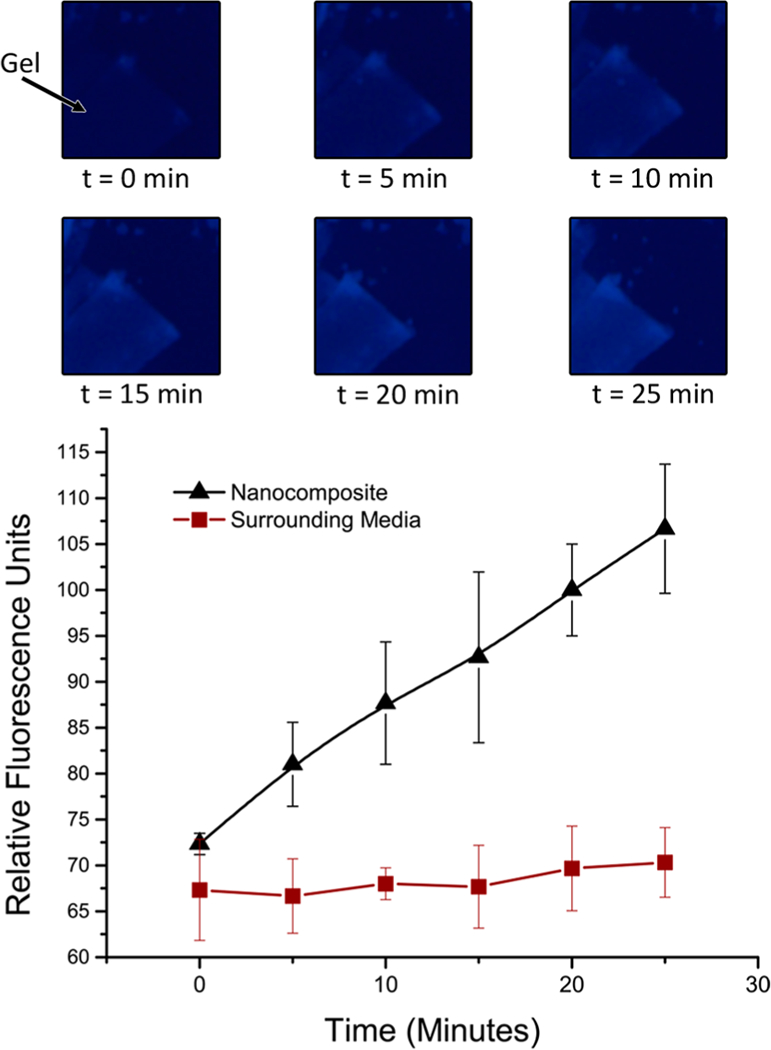
Exposure to fluorescamine during degradation by a 4000 nM chymotrypsin caused the hydrogel to fluoresce over time as the fluorescamine binds to generated N-terminal amines. The increase in fluorescence was linear, with no significant increase in the surrounding media, indicating the hydrogel was collapsing as opposed to eroding.
The collapsing mechanism is also evident based on the shift in photonic properties of the hydrogel nanocomposite. As previously mentioned, the nanocomposite has a photonic bandgap due to the periodic distribution of the NPs within the hydrogel. Periodic structures consisting of dielectrics will prohibit certain energies of electromagnetic radiation from propagating through the structure. The energies or wavelengths prohibited are dictated by both the indices of refraction and spacing of the dielectrics. In this case the dielectrics are the silica NPs and the water swollen network of the hydrogel. Several characterization methods were attempted to determine the periodic structure of the hydrogel nanocomposite, including the use of Small-angle X-ray Scattering (SAXS), laser scanning confocal microscopy and by wet Atomic Force Microscopy (AFM). Unfortunately, the nanoparticle periodicity was too large for the SAXS instrument range, too small to be resolved by confocal microscopy, and too rough for AFM.
The photonic bandgaps properties are dictated by the filling factor, or the ratio of the volume of silica nanoparticles to the volume of the polymer network. The swelling or collapsing of the polymer network changes the filling factor and thus the reflected color. The as-synthesized hydrogel reflects red wavelengths of light at a peak reflectance of 665 nm. When the nanocomposite collapses, such as in the presence of chymotrypsin, the filling factor increases, and the reflected wavelength blueshifts. Further evidence for the change in filling factor is the volumetric measurements presented in Figure 3, demonstrating a decrease in hydrogel volume.
Reflectance measurements, seen in Figure 6A, exhibit the broadband blueshift after exposure to chymotrypsin and the subsequent collapse mechanism occurs. The peak reflectance blueshifted ∼240 nm from 665 to 428 nm at a chymotrypsin concentration of 20 nM. This is the largest photonic response to a protein reported, especially given the analyte concentration, with similar systems giving responses of only a 15 nm shift to a 9 μM concentration,3 80 nm shift to a16000U/mLconcentration,15 anda100nmshifttoa 40 μM concentration.28
Figure 6.
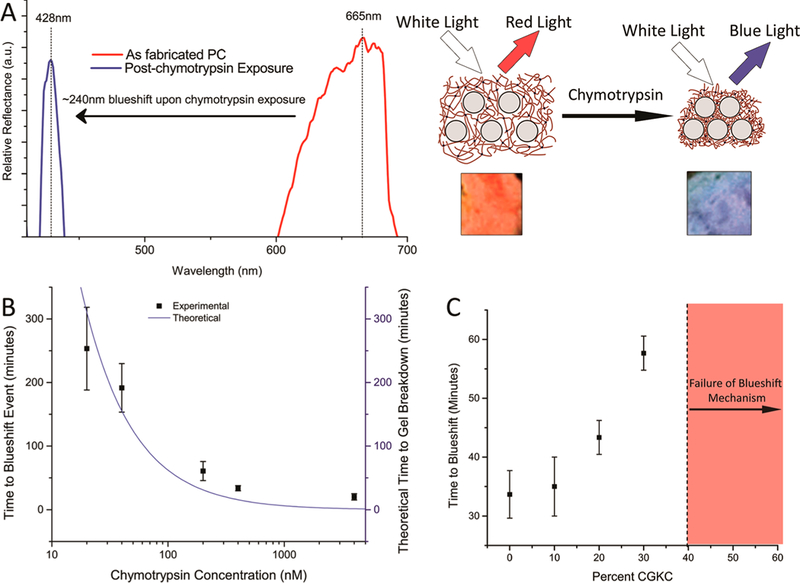
(A) As synthesized nanocomposite gels were initially red in color, giving a peak reflectance at ∼665 nm. Once exposed to chymotrypsin, the system collapsed due to the formation of physical cross-links and would reflect blue wavelength of light with a peak at ∼428 nm. This is a 240 nm blueshift, the largest response reported from a photonic crystal responding to a protein. (B) The blueshifting event results in the same color despite the concentration, but the time to blueshift increases with decreasing concentration. The time to blueshift followed a theoretical model of enzymatic degradation of a hydrogel. (C) Increasing mole fractions of the nondegradable CGKC peptide increased the time to blueshift at a chymotrypsin concentration of 400 nM until a mole fraction of 0.4, in which case the mechanism fails.
The broad spectral shift (∼240 nm) resulting from specific response to chymotrypsin is consistent for solution concentrations between 20 and 4000 nM (Figure 6B). The lowest concentration which produced a response was 20 nM. These concentrations are significantly lower than physiologic concentration of chymotrypsin which is hundreds of micrograms per milliliter of gastrointestinal fluid;29 however, the response to chymotrypsin simply serves as a model to demonstrate the properties of the investigated nanocomposite.
The blueshift response to chymotrypsin occurs abruptly, as the system transitions rapidly from the as-synthesized “red” state to the collapsed “blue” state. Increasing chymotrypsin concentrations leads to a decrease in time to blue shift while maintaining the same ultimate photonic response. This is in contrast to other photonic crystal sensors that report concentration dependent photonic behavior.12,14,28 The time in minutes to the collapsing event follows a theoretical relationship defined by eq 1:
| (1) |
where xc is the critical fraction of cross-links that must be cleaved for the collapsing mechanism to occur, So is the substrate concentration in micromolar, kcat is the protease turnover rate, and E is the protease concentration. This equation is adapted from Lutolf et al.19 Deviations between the theoretical and experimental values are likely due to unpredicted defects in the hydrogel network such as chain entanglements or network pendant groups. Under this consideration, the theoretical calculations of tb were reasonable as compared with the experimental data exhibited in Figure 6B. The value of kcat for chymotrypsin, toward the hydrogel specifically, was determined utilizing an adapted method in which the generation of fluorescent markers in response to peptide lysis events is measured.30The system followed zeroth order enzyme kinetics due to So >> Km (Michaelis enzyme constant, Supporting Information Figure 3) within the hydrogel, which has been described in other peptide based hydrogels.17,19
To determine xc, nanocomposites were synthesized with two different peptides, CYKC, which is sensitive to chymotrypsin, and CGKC, which is not chymotrypsin cleavable. Different ratios of CGKC:CYKC were utilized and the resulting nanocomposite was evaluated for time to blueshift, tb after exposure to a 400 nM chymotrypsin solution. Increasing the ratio caused an exponential increase in the time to the blueshift event and asymptotically approached a ratio of 0.4asseenin Figure 6C. Above ratios of0.3CGKC:CYKC, the collapse mechanism fails as an insufficient number of cross-links are cleaved to impart the pendant network chains with the mobility necessary for NP adsorption to occur. xc was evaluated to be 0.7, representing the mole fraction of peptide cross-links that must be cleaved.
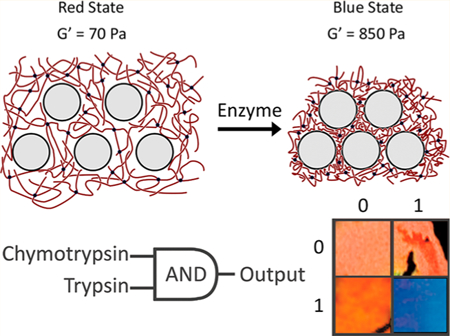
The discrete nature of the collapsing mechanism, after xc peptide cross-links are cleaved, causing a shift in color from red to blue with no transitional color is analogous to a digital output after sufficient input analog voltage. These discrete responses were exploited to fabricate Boolean logic gates (AND/OR) which can respond to different sets of proteases dictated by specific truth tables. Synthesis of an OR gate required the use of a peptide that could be specifically cleaved by both chymotrypsin as well as trypsin. The sequence CYKC was utilized as it will be cleaved by chymotrypsin at the carboxyl side of the tyrosine residue and by trypsin at the carboxyl side of the lysine residue. The presence of either protease at a concentration of 40 nM invoked the collapsing and subsequent blueshift of the nanocomposite (Figure 7). In contrast, to produce an AND gate, the threshold of requiring digestion of at least 0.7 of the peptide cross-links was employed by incorporating two peptides, CGGYC for chymotrypsin and CGKC for trypsin, ata1:1 ratio. When this system was exposed to just one protease at a concentration of 40 nM, only 0.5 of the peptide cross-links within the hydrogel will be cleaved, which does not breach the 0.7 threshold required for the collapsing mechanism to occur. However, when both proteases are present, the 0.7 threshold can be met, and the hydrogel will collapse, stiffen, and blueshift. The digital photographs in Figure 7 display the truth tables that can be devised by exposing the hydrogel to different combinations of chymotrypsin and trypsin. Discrepancies in the photographed color are a product of aberrations in the hydrogel network structure likely originating from the fabrication process. Molecular computations performed passively by the hydrogel would have applications in drug delivery and tissue engineering, in which the hydrogel would only response in very specific environments containing a combination of stimuli.
Figure 7.
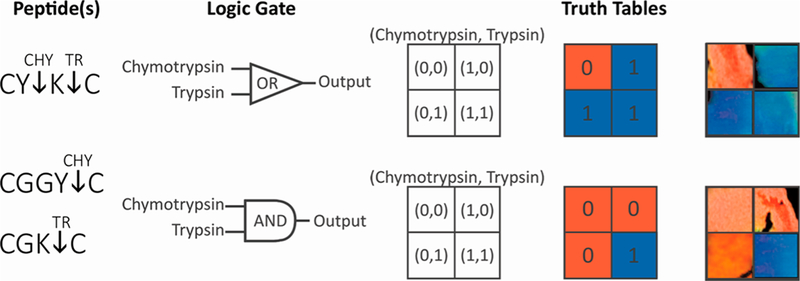
Hydrogels were synthesized containing either CYKC or a 1:1 ratio of CGGYC and CGKC. The CYKC peptide can be cleaved by either chymotrypsin or trypsin, producing an OR gate. In the latter hydrogel, the CGGYC is cleaved by chymotrypsin and the CGKC is cleaved by trypsin; therefore, both enzymes must be present for a blueshift response to occur.
CONCLUSIONS
By incorporating silica NPs at a high density within a degradable nanoparticle hydrogel composite, we were able to fabricate soft materials with a unique and counterintuitive response to proteolytic stimuli. The demonstrated hydrogels would collapse in response to diverse biological stimuli, causing a 1200% increase in elastic storage and loss modulus. The collapsing mechanism also invoked a blueshift in the optical properties of the hydrogel. This blueshift response was the highest reported for a photonic crystal toward a proteogenic stimulus, to our knowledge. The mechanism is consistent with various proteases and only requires a different peptide cross-linker that is specific toward the protease of interest. The hydrogels additionally exhibited abrupt and discrete responses, allowing for the installation of Boolean logic gates (AND, OR) in the hydrogel nanocomposite. Ultimately, this mechanism opens a new path in designing stimuli-responsive hydrogels, which can proceed through a secondary network formation, after an energetic barrier has been breached. Enzyme responsive hydrogel nanocomposites, described here, could offer avenues in degradation-stiffening and collapsing materials for a variety of bio-material applications.
METHODS
Materials.
All materials were purchased from Sigma-Aldrich unless specified otherwise.
Nanoparticle Synthesis.
Silica nanoparticles were synthesized utilizing a modified Stober method. An initial solution of 6 mL of tetraethoxy orthosilicate was prepared in 70 mL of ethanol. To this solution were added 25 mL of deionized water and 1.75 mL of ammonium hydroxide under 200 rpm stirring. The reaction was allowed to proceed for 2 h. The ammonium hydroxide can be changed to modulate particle size. Sulfonation of the particles was performed by first diluting the freshly synthesized NPs in an equal volume of deionized water. Six milliliters of (3-(trihydroxylsilyl)-1-propane-sulfonic acid (Gelest) was titrated to a pH of ∼6 and added to the stirring suspension. The reaction was refluxed at 80 LC for 6 h. The silica NPs were washed through centrifugation and resuspension in deionized water. This proce-dure was repeated 5 more times, and the particles were stored on a mixed bed ion-exchange resin (BioRad) until use.
Synthesis of PEG4NB.
Synthesis of the monomer was performed using Steglich esterification and adapted from Aimetti et al.17 Briefly, 2 g of 4-arm polyethylene glycol (MW ∼ 10 000, Creative PegWorks), 24 mg of dimethylaminopyridine, and 164 μL of pyridine were dissolved in 4 mL of dichloromethane. The solution was blanketed with nitrogen and allowed to stir at 300 rpm under nitrogen. A reaction was then prepared of 333 mg of 5-carboxylic acid-2-norbornene and 247 mg of dicyclo-hexylcarbodiimide in 2 mL of dichloromethane. The reaction proceeded for 30 min. During this time, white crystals of urea derivatives formed. The solution was passed through a 0.2 μm PTFE filter (Whatman) to remove the crystals. The filtered solution containing the activated 5-carboxylic acid-2-nobor-nene was then added to the PEG solution. The final reaction was then allowed to proceed for 24 h under nitrogen.
Fabrication of Hydrogel Silica NP Composite.
A 100 μL aqueous hydrogel precursor solution was prepared consisting of 0.1% (w/v) Igracure, 15 mg of PEG4NB, and 1.5 mg of peptide, whether the CYKC, CGGYC, or CGKC (Genscript). Then, 300 mL of the prepared silica NPs was centrifuged and decanted. The resulting pellet was then resuspended in the hydrogel precursor solution and centrifuged again at 14 000 rpm for 2 min. The remaining precursor supernatant was discarded and the resulting pellet was extracted and sandwich cast between two glass coverslips (Fisher Scientific). The sandwich cast pellet was then exposed to 3.5 mW/cm2, 365 nm radiation for 20 min, allowing the thiol ene polymerization to proceed. The resulting hydrogel film was then allowed to equilibrate in distilled water for 1 h.
Characterization.
Freeze-fracture scanning electron microscopy (Hitachi SU-70) and transmission electron microscopy (JEOL 2100F) were utilized to image the cross-section of the nanocomposite and the bare silica NPs, respectively. The diameter and zeta potential of the silica NPs were measured utilizing dynamic light scattering (Zetasizer Nano ZS90). Nuclear magnetic resonance of the synthesized PEG4NB was performed in deuterated chloroform using a Bruker AV-400 MHz. Mechanical measurements were taken using parallel plate rheology (Rheometrics RDA III) with a linear strain increase from 0 to 20%. Reflectance measurements of the nanocomposite were performed using a PerkinElmer UV—vis. The nanocomposites were immersed in a glass-slide holder filled with distilled water.
Enzyme Response Studies.
Chymotrypsin and trypsin solution were prepared at concentrations ranging from 1 ng to 100 μg/mL in 10 mM calcium chloride and 100 mM Tris-HCl solution, and titrated to a pH of 8. The nanocomposites were then immersed in these static solutions and observed until the collapsing event occurred. This protocol involved examining the hydrogels every 5 min after application of the enzyme solution until a visible color change had occurred. Since the blueshift is abrupt and there is no transitional color between the red initial state and blue digested state, the visible color change was defined as the moment the entire hydrogel could be observed as blue in color. This protocol was verified using reflectivity measurements shown in Figure 6A. Changes in mass were recorded by blotting the nanocomposites with a Kimwipe and measuring the mass using a Sartorius microbalance.
Fluorescence Measurements.
Fluorescamine was dissolved in acetone at a concentration of 40 mM. This solution was then diluted to a concentration of 5 mM using a 10 mM calcium chloride and 100 mM HEPES solution. A 4000 nM solution of chymotrypsin was prepared using the 5 mM fluorescamine solution. The hydrogel nanocomposites were then exposed to the fluorescamine—chymotrypsin solution. The hydrogels were then imaged using a fluorescent microscope using a DAPI filter (ex, 365; em, 450). RGB values extracted from the images were used to calculate the relative fluorescence units, which is an accepted method for fluorescence and colorimetric mea-surements.31
Supplementary Material
Acknowledgment.
We would like to acknowledge Joe White for his assistance with rheology. We would also like to acknowledge M. Ibrahim, G. Ngatha, W. Hwang, M. Vural and A. Behrens for their critical commentary.
Footnotes
Conflict of Interest: The authors declare no competing financial interest.
Supporting Information Available: NMR of the PEG4NB; FTIR of the gel—sol transition; DLS, TEM, and SEM of the silica nanoparticles; enzyme kinetics of chymotrypsin exposed to free CYKC peptide. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b01514.
REFERENCES AND NOTES
- 1.Stuart MAC; Huck WTS; Genzer J; Muller M; Ober C; Stamm M; Sukhorukov GB; Szleifer I; Tsukruk VV; Urban M; et al. Emerging Applications of Stimuli-Responsive Polymer Materails. Nat. Mater 2010, 9, 101–113. [DOI] [PubMed] [Google Scholar]
- 2.Fairbanks BD; Schwartz MP; Halevi AE; Nuttelman CR; Bowman C; Anseth KS A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photo-polymerization. Adv. Mater 2009, 21, 5005–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kilian K. a; Lai LMH; Magenau A; Cartland S; Böcking T; Di Girolamo N; Gal M; Gaus K; Gooding JJ Smart Tissue Culture: In Situ Monitoring of the Activity of Pro-tease Enzymes Secreted from Live Cells Using Nanostructured Photonic Crystals. Nano Lett 2009, 9, 2021–2025. [DOI] [PubMed] [Google Scholar]
- 4.Orosco MM; Pacholski C; Miskelly GM; Sailor MJ Protein-Coated Porous-Silicon Photonic Crystals for Amplified Optical Detection of Protease Activity. Adv. Mater 2006, 18, 1393–1396. [Google Scholar]
- 5.Purcell BP; Lobb D; Charati MB; Dorsey SM; Wade RJ; Zellars KN; Doviak H; Pettaway S; Logdon CB; Shuman J. a; et al. Injectable and Bioresponsive Hydrogels for on-Demand Matrix Metalloproteinase Inhibition. Nat. Mater 2014, 13, 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peppas NA Hydrogels and Drug Delivery. Curr. Opin. Colloid Interface Sci 1997, 2, 531–537. [Google Scholar]
- 7.Lowman AM; Morishita M; Kajita M; Nagai T; Peppas NA Oral Delivery of Insulin Using pH-Responsive Com-plexation Gels. J. Pharm. Sci 1999, 88, 933–937. [DOI] [PubMed] [Google Scholar]
- 8.Lee K; Asher SA Photonic Crystal Chemical Sensors: pH and Ionic Strength. J. Am. Chem. Soc 2000, 122, 9534–9537. [Google Scholar]
- 9.Xu X; Goponenko AV; Asher S. a. Polymerized Poly-HEMA Photonic Crystals: pH and Ethanol Sensor Materials. J Am. Chem. Soc 2008, 130, 3113–3119. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida T; Aoyagi T; Kokufuta E; Okano T Newly Designed Hydrogel with Both Sensitive Thermoresponse and Biodegradability. J. Polym. Sci., Part A: Polym. Chem 2003, 41, 779–787. [Google Scholar]
- 11.Yoshida R; Uchida K; Kaneko Y; Sakai K; Kikuchi A; Sakurai Y; Okano T Comb-Type Grafted Hydrogels with Rapid Deswelling Response to Temperature Changes. Nature 1995, 374, 240–242. [Google Scholar]
- 12.Holtz JH; Asher SA Polymerized Colloidal Crystal Hydrogel Films as Intelligent Chemical Sensing Materials. Nature 1997, 389, 829–832. [DOI] [PubMed] [Google Scholar]
- 13.Asher S. a; Alexeev VL; Goponenko AV; Sharma AC; Lednev IK; Wilcox CS; Finegold DN Photonic Crystal Carbohydrate Sensors: Low Ionic Strength Sugar Sensing. J Am. Chem. Soc 2003, 125, 3322–3329. [DOI] [PubMed] [Google Scholar]
- 14.Ayyub OB; Sekowski JW; Yang TI; Zhang X; Briber RM; Kofinas P Color Changing Block Copolymer Films for Chemical Sensing of Simple Sugars. Biosens. Bioelectron 2011, 28, 349–354. [DOI] [PubMed] [Google Scholar]
- 15.MacConaghy K; Geary C Photonic Crystal Kinase Bio-sensor. J. Am. Chem. Soc 2014, 136, 6896–6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson SB; Lin C; Kuntzler DV; Anseth KS Biomaterials The Performance of Human Mesenchymal Stem Cells Encapsulated in Cell-Degradable Polymer-Peptide Hydrogels. Biomaterials 2011, 32, 3564–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aimetti AA; Machen AJ; Anseth KS Biomaterials Poly (Ethylene Glycol) Hydrogels Formed by Thiol-Ene Photo-polymerization for Enzyme-Responsive Protein Delivery. Biomaterials 2009, 30, 6048–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mann B; Gobin A; Tsai A; Schmedlen R; West J Smooth Muscle Cell Growth in Photopolymerized Hydrogels with Cell Adhesive and Proteolytically Degradable Domains: Synthetic ECM Analogs for Tissue Engineering. Biomaterials 2001, 22, 3045–3051. [DOI] [PubMed] [Google Scholar]
- 19.Lutolf M Synthetic Matrix Metalloproteinase-Sensitive Hydrogels for the Conduction of Tissue Regeneration: Engineering Cell-Invasion Characteristics. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 5413–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plunkett KN; Berkowski KL; Moore JS Chymotrypsin Responsive Hydrogel: Application of a Disulfide Exchange Protocol for the Preparation of Methacrylamide Contain-ing Peptides Chymotrypsin Responsive Hydrogel: Applica-tion of a Disulfide Exchange Protocol for the Preparation of Methacrylamide. Biomacromolecules 2005, 6, 632–636. [DOI] [PubMed] [Google Scholar]
- 21.Thornton P; Mart R; Ulijn R Enzyme-Responsive Polymer Hydrogel Particles for Controlled Release. Adv. Mater 2007, 19, 1252–1256. [Google Scholar]
- 22.Toledano S; Williams R Enzyme-Triggered Self-Assembly of Peptide Hydrogels via Reversed Hydrolysis. J. Am. Chem. Soc 2006, 128, 1070–1071. [DOI] [PubMed] [Google Scholar]
- 23.Ikeda M; Tanida T; Yoshii T; Kurotani K; Onogi S; Urayama K; Hamachi I Installing Logic-Gate Responses to a Variety of Biological Substances in Supramolecular Hydrogel-Enzyme Hybrids. Nat. Chem 2014, 6, 511–518. [DOI] [PubMed] [Google Scholar]
- 24.Rose S; Prevoteau A; Elzière P; Hourdet D; Marcellan A; Leibler L Nanoparticle Solutions as Adhesives for Gels and Biological Tissues. Nature 2014, 505, 382–385. [DOI] [PubMed] [Google Scholar]
- 25.Schexnailder P; Schmidt G Nanocomposite Polymer Hydrogels. Colloid Polym. Sci 2009, 287, 1–11. [Google Scholar]
- 26.Anseth K; Bowman C; Brannon-Peppas L Mechanical Properties of Hydrogels and Their Experimental Determi-nation. Biomaterials 1996, 17, 1647–1657. [DOI] [PubMed] [Google Scholar]
- 27.Kimble KW; Walker JP; Finegold DN; Asher S. a. Progress toward the Development of a Point-of-Care Photonic Crystal Ammonia Sensor. Anal. Bioanal. Chem 2006, 385, 678–685. [DOI] [PubMed] [Google Scholar]
- 28.Orosco M; Pacholski C; Sailor M Real-Time Monitoring of Enzyme Activity in a Mesoporous Silicon Double Layer. Nat. Nanotechnol 2009, 4, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borgström B; Dahlqvist A Studies of Intestinal Digestion and Absorption in the Human. J. Clin. Invest 1957, 36, 1521–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lauer-Fields JL Hydrolysis of Triple-Helical Collagen Peptide Models by Matrix Metalloproteinases. J. Biol. Chem 2000, 275, 13282–13290. [DOI] [PubMed] [Google Scholar]
- 31.Yetisen AK; Martinez-Hurtado JL; Garcia-Melendrez A; da Cruz Vasconcellos F; Lowe CR A Smartphone Algo-rithm with Inter-Phone Repeatability for the Analysis of Colorimetric Tests. Sens. Actuators, B 2014, 196, 156–160. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.