

Introduction
The frequency of follow-up visits in hypertrophic cardiomyopathy (HCM) patients is mainly determined by their symptoms, age and severity of disease. Clinical visits should focus on sudden cardiac death (SCD) and embolic risk-assessment, changes in symptoms, cardiac rhythm, left ventricular outflow tract obstruction (LVOTO) and left ventricular (LV) systolic function1.
Patients with HCM phenotype
In asymptomatic patients, a clinical examination with 12-lead electrocardiogram (ECG) and transthoracic echocardiography should be preferably performed on a yearly basis or every two years at most1. 24-hour Holter monitoring is recommended every 1–2 years or every 6 months in patients with left atrial (LA) dilation of more than 45 mm in order to detect asymptomatic atrial fibrillation that would warrant oral anticoagulation therapy1. The rationale for this specific threshold is that it has been observed that the risk of thromboembolism rises exponentially with LA diameter above 45–50 mm2.
Indication for implanted cardioverter-defibrillator (ICD), based on the HCM risk-score, should be re-evaluated in each follow-up visit3, as well as indication for oral anticoagulation.
Symptomatic patients with LVOTO need closer follow-up every 3–6 months in order to evaluate response to medical treatment and to plan eventual septal reduction therapies. Once an invasive reduction therapy is performed, a clinical follow-up including ECG, transthoracic echocardiogram and ambulatory Holter monitoring should be performed at 3 and then at 6–12 months1. Additionally, cardiac magnetic resonance imaging (MRI) evaluation could be considered every 2–3 years1, especially when clinical progression is detected or to decide ICD indication in borderline and doubtful cases, given the prognostic value of late gadolinium enhancement (LGE)4. Also a cardiopulmonary exercise test is a valuable tool to objectively assess functional capacity once heart failure (HR) is present. In a large study with HCM patients who underwent cardiopulmonary exercise test, peak oxygen consumption, ventilatory efficiency and ventilatory anaerobic threshold were found to be predictors of death and heart transplant5, so this tool can be useful in providing prognostic information.
Patients with non-obstructive HCM also present symptoms due to diastolic dysfunction or microvascular angina, and have an earlier onset of HF and a more rapid progression to advanced HF and adverse outcome as compared to patients with LVOTO6. The average time between diagnosis and onset of HF is reported to be 11 years, but once HF is present only a mean period of 4 years is necessary until death or heart transplantation (Figure 1).
Figure 1. Time lines for the three different hypertrophic cardiomyopathy phenotypes.

Patient ages at intervals describe clinical evolution. FU, follow-up. Reproduced with permission from Melacini P et al. Eur Heart J. 2010; 31: 2111–2123.6
Mutation carriers without a phenotype
Although data regarding follow-up in mutation carriers without a phenotype are scarce, a recent study from Rotterdam has shown that during a mean follow-up period of 7 years 16% of genotype-positive individuals develop left ventricular hypertrophy (LVH) consistent with HCM diagnosis with survival at 5/10 years of 99%/95%7. Other recent studies also suggest that clinical course in most unaffected mutation carriers is benign1,8. Clinical screening with ECG and echocardiography in first degree relatives of HCM patients can start during childhood or up to 10–12 years9, as clinically important events in asymptomatic children are very rare before puberty8.
The recommended intervals for follow-up are different in European and American HCM guidelines. The former do not establish a specific interval but recommend a long-term evaluation in normal healthy mutation carriers1. The American College of Cardiology Foundation/American Heart Association (ACCF/AHA) recommends screening at least every 5 years above the age of 21 and every 12-18 months from age 12 to 18-2110. Moreover, a position statement from the European Society of Cardiology (ESC) working group on myocardial and pericardial diseases established two possible scenarios in the follow-up of mutation carriers without HCM phenotype: causal mutation identified or not in the probands with HCM phenotype. In the first case, phenotype negative relatives who are proved to be non-carriers do not require further cardiac evaluation and can be discharged. On the other hand, mutation carriers could benefit from an initial cardiac MRI apart from ECG and echocardiogram in order to assess early signs of HCM and a yearly follow-up is recommended between the age of 10 and 20, and then every 1 to 3 years9.
In the second scenario, when a causal genetic variant has not been found in the proband, a repeated cardiac evaluation with ECG and echocardiography is recommended every 3–5 years before 10 years of age, every 1–2 years between 10 and 20 years of age and every 2–5 years thereafter in first degree relatives9.
The age at which genetic studies should be performed remains controversial. The American Society of Human Genetics (ASHG) published a statement on genetic testing in children and adolescents in 2015 that encouraged parents to defer predictive or pre-dispositional testing for adult-onset conditions such as HCM until at least adolescence, unless there is a specific clinical intervention during childhood11. It was argued that due to the complexity of genetic information, this should be offered when the patient can really understand it. Likewise, the European Society of Genetics recommends delaying the time of genetic studies in children as the genetic diagnosis can add anxiety, and promotes overprotection12. On the other hand, some studies have observed that mutation carriers of HCM causing mutations do not have a worse health-related quality of life or more anxiety as compared to a representative sample of children from the general population13. Despite these recommendations it is not uncommon that predictive genetic testing is offered to minors and even the ESC HCM guidelines suggest that it could be considered in children over 10 years-old. The arguments of those who favor early genetic testing is that it allows orientation of children sport activities and removes uncertainty to parents. Although there is no current consensus among cardiologists about performing genetic testing in asymptomatic children without signs of HCM, the child’s well-being and interests should always be taken into consideration.
Advanced echocardiographic characterization with tissue Doppler and strain imaging is useful in mutation carriers without an overt HCM phenotype. Tissue Doppler imaging (TDI) techniques have shown decreased systolic and diastolic velocities independently of left ventricular hypertrophy14 and could be used to plan a closer follow-up in those patients with lower TDI velocities. Likewise, strain analysis has shown a delay in untwist and unstrain rates in mutation carriers without HCM phenotype15. Regarding cardiac MRI, it can be useful in genotype-positive subjects with borderline ventricular thicknesses by echocardiographic analysis or when acoustic window is suboptimal. In early stages of disease, it may show structural abnormalities such as crypts, papillary muscle anomalies or septo-apical bundles, as well as focal LGE16.
General lifestyle considerations for patients with hypertrophic cardiomyopathy
Most HCM patients are able to carry out normal activities without restriction, but during follow-up visits the cardiologist or general practitioner should spend some time explaining certain general indications that need to be taken into account in daily life.
Sport
One of the patients’ main concerns is exercise and sport. Patients with HCM should generally avoid competitive sports and focus on recreational activities. Current algorithms to predict the risk of SCD in to HCM are to be applied in the general population17, but there is no evidence that they can be used in athletes. Thus, both the ESC and the AHA restrict intense competitive sports in HCM patients1,10. On the other hand, asymptomatic HCM patients with mild LV hypertrophy below 20 mm, no ventricular arrhythmia at exercise treadmill test or ECG monitoring and no family history of SCD may participate in competitive low-intensity sports such as golf or bowling17.
The recently published RESET-HCM trial observed that HCM patients with moderate-intensity training presented a statistically significant increase in exercise capacity at 16 weeks measured with peak oxygen consumption compared with usual activity18. Hence, it seems reasonable to recommend low to moderate intensity exercise in HCM patients in order to maintain a healthy lifestyle, but dissuade them from practicing high intensity sports until more data is available in this matter. Regarding definite mutation carriers with no HCM phenotype, the 2015 AHA/ACC eligibility and disqualification recommendations for athletes state that participation in competitive athletics is reasonable in the absence of family history of SCD related to HCM17. As for the ESC HCM guidelines, sports activities are allowed taking into account the underlying mutation and the type of sport activity1.
Occupation and life insurance
The vast majority of patients with HCM will be able to continue with their job1. Some exceptions are military services, pilots, law enforcement or firefighting. For instance, in the case of the US military, HCM is considered disqualifying. If newly developed once enlisted, each case is independently assessed19. Each country has its own legislation regarding this issue and it is important to discuss the potential professional implications during genetic counselling in HCM relatives. Life insurance depends again on the rules that apply in different countries. In any case, HCM patients might find mores difficulties trying to find life insurances or mortgages.
Prevention of infective endocarditis
Current European and American guidelines recommend good oral hygiene in HCM patients but not antibiotic prophylaxis in HCM patients undergoing dental procedures20,21.
In the past, infective endocarditis (IE) antibiotic prophylaxis was recommended for all HCM patients before invasive procedures22. However, in 2007, the AHA revised the recommendations and retired antibiotic prophylaxis for HCM patients due to an apparently significant morbidity associated with IE antibiotic prophylaxis therapy, and a lack of evidence supporting efficacy of antibiotic prophylaxis in IE prevention21. A recent study has observed that previous dental procedures and streptococcal infections are higher in IE in HCM patients compared to those with antibiotic prophylaxis indication, suggesting that these patients could benefit from prophylaxis23. Other issues regarding lifestyle are described in Table 1, based on the ESC guidelines1.
Table 1. Other lifestyle considerations for patients with hypertrophic cardiomyopathy.
Reproduced and adapted from 2014 HCM ESC guidelines (Elliott et al. Eur Heart J. 2014;35 (39):2733–2779).1
| Topic | General guidance |
|---|---|
| Diet, alcohol and weight | Patients should be encouraged to maintain a healthy body mass index Large meals can precipitate chest pain, particularly in patients with LVOTO. Smaller, more frequent meals may be helpful Avoid dehydration and excess alcohol, particularly in patients with LVOTO Constipation is a frequent side-effect of verapamil/disopyramide and should be managed with diet and if necessary aperients |
| Smoking | There are no data that show an interaction between tobacco smoking and HCM, but patients should be provided with general advice on the health risks associated with smoking and, when available, information on smoking cessation |
| Sexual activity | Patients should be given the opportunity to discuss their concerns about sexual activity. Anxiety and depression following a diagnosis are frequent and some patients may express guilt or fear about their genetic diagnosis and the risk of transmission to offspring Patients should be counselled on the potential effect of their medication on sexual performance In general, patients should avoid phosphodiesterase type 5 (PDE5) inhibitors, particularly when they have LVOTO |
| Medication | Patients should be provided with information about their medication, including potential side-effects and interactions with prescribed medications, over-the-counter remedies and other complementary therapies Where possible, peripheral vasodilators should be avoided in patients, particularly when they have LVOTO |
| Vaccination | In the absence of contraindications, symptomatic patients should be advised to have yearly influenza vaccination |
| Driving | Most patients should be eligible for an ordinary driving licence and can continue driving unless they experience distracting or disabling symptoms Advice on driving licences for heavy goods or passenger-carrying vehicles should be in line with local legislation For further advice on driving with ICD see European Heart Rhythm Association guidelines and local rules. |
| Holidays and travel insurance | |
| Education/schooling | Teachers and other carers should be provided with advice and written information relevant to the care of children with HCM In the absence of symptoms and risk factors, children should be allowed to perform low to moderate level aerobic physical activity, in accordance with advice from their cardiologist. Provision should be made for children with learning difficulties and other special needs |
Reproduction and contraception
Reproduction
In most cases women with HCM tolerate pregnancy fairly well1. However, a recent study with data from the ESC initiated Registry of Pregnancy and Cardiac disease (ROPAC) has observed that pregnancy may not be as benign as previously believed, as major cardiovascular events were present in 23% of cases. There was no maternal mortality, but 15% presented HF and 12% arrhythmic events24. A pooled analysis with 408 cases has observed that HCM mortality during pregnancy is 0.5%, and the 2 reported deaths corresponded to high risk patients: one very symptomatic with LVOTO and a 30 mm interventricular septum and the other with a strong family history of SCD and evidence of ventricular tachycardia (VT) before death25. Thus, risk assessment before pregnancy should include a detailed medical history to confirm the patients’ functional status and a thorough physical examination to detect signs of HF, which are important risk factors for further complications in pregnant women with HCM.
Follow-up of pregnant HCM women should include an echocardiogram each trimester or earlier if new symptoms occur. As previously stated, symptomatic patients before pregnancy present a higher risk of complications and should be assessed regularly.
Beta blockers should be continued if they were used before pregnancy and have a solid clinical indication1, and can be initiated during pregnancy if required. In any case, monitoring of fetal growth is advised when using these drugs. Acebutolol, pindolol and sotalol present a FDA pregnancy category B and frequently used drugs such as carvedilol, metoprolol or bisoprolol a pregnancy category C, as there are no well-controlled studies in humans but potential benefits may warrant its use despite potential risks. Only atenolol is categorized as D due to an apparently increased risk of intrauterine growth retardation26.
Calcium channel blockers such as verapamil can also be used during pregnancy with an FDA Class C recommendation. Amiodarone, on the other hand, increases the risk of fetal thyroid toxicity, neurological adverse effects and growth retardation27. Thus, it should be avoided and only used if strictly necessary.
Planned vaginal delivery is the preferred choice in most of the cases. However, caesarian section should be considered in patients with severe LVOTO or severely symptomatic HF1. Epidural and spinal anaesthesia must be administered with caution in women with severe LVOTO to avoid vasodilatation and hypotension, thus single-shot spinal anaesthesia should be avoided. Oxytocin should also be given cautiously avoiding hypotension, tachycardia and arrhythmia. Attention should be paid to volemia, avoiding not only blood loss and preload decreases but also fluid overload after delivery with high risk of pulmonary oedema28.
Contraception
Oral contraceptives are preferred to barrier methods in order to prevent unintended pregnancies, but estrogen-based pills should be avoided in patients with an increased thrombotic risk, such as women older than 35 years of age, smokers, previous history of atrial fibrillation, venous thromboembolism or HF. On the other hand, progesterone-only contraceptives increase liquid retention and are worse tolerated in women with HF. A safe alternative is the levonogestrel-releasing intra uterine device (IUD)1. In the event of pregnancy termination, prostaglandin F should not be administered as increases pulmonary artery pressure. Prostaglandin E1 or E2 are safer alternatives1.
Reproductive counselling
Most HCM cases present an autosomal dominant inheritance, so there is a 50% chance for the offspring to carry the cardiomyopathy-causing mutation. As penetrance is not usually complete, the parents-to-be need to know that their future child may inherit a predisposition for HCM, but not necessarily will develop a HCM phenotype. Furthermore, if the child develops HCM, expressivity is variable even between members of the same family, so information regarding a specific clinical phenotype or prognosis cannot be accurately provided. With this information, parents may consider a natural conception or different methods to avoid the inheritance of the disease-causing mutation. These include adoption, gamete donation (oocyte or sperm), prenatal diagnosis techniques or preimplantation genetic diagnosis (PGD) which are subject to the legislation of each country, as well as to the internal regulations of each health system.
The two routinely used techniques for prenatal diagnosis are chorionic villus sampling or amniocentesis. In both cases, if the foetus is affected the parents have the option to terminate pregnancy if permitted in their residing country. PGD combines in vitro fertilization (IVF) and genetic analysis to test an embryo for the specific familial mutation before implantation. Unaffected embryos are selected and transferred to the mother’s uterus. During reproductive counselling, parents should be advised that there are approximately 0.5% of false negative results29, which implies that the disease-causing mutation will be transmitted to the offspring.
Furthermore, each country is subject to specific laws on assisted reproduction that the doctor must know to offer reproductive advice to patients.
Prognosis
General prognosis of HCM patients
According to its relatively high prevalence (1/500 in general population, the highest for an inherited heart disease) and the low global incidence of complications, the general prognosis of a HCM patient is generally good and it is commonly regarded as a benign disease30. Thus, two thirds of patients with HCM present a normal life span without significant morbidity30. A recent series reviewed HCM mortality rates and described a 0.7%/year HCM-related mortality rate and a 1.1%/year rate for HCM-unrelated causes of death31. Most of the herein commented complications arise in patients at high risk of SCD or HF, many of these with severe LVOTO.
SCD prevention in HCM patients
As previously presented in a specific chapter, SCD risk stratification in HCM patients is evolving and remains challenging. The HCM-risk score adopted by ESC HCM Guidelines in 20141 may still be improved in the future to maximize its accuracy. Some interesting points in this field are commented on hereafter.
Prognostic factors in HCM
Age
Age has historically been warranted as a modulator of the outcomes in HCM patients. Notably, clinically stable patients who achieve the age of ≥60 years experience a subsequent clinical course with a particularly low SCD event rate (0.2%/year)32. A younger age at diagnosis has been thought to portend a reduced life expectancy and limited effective treatment options in the literature. Two contemporary papers have shed some light in the current scenario with updated management options. In both, idiopathic HCM in children exhibited a good 5-year survival of 94–97% for those diagnosed after 1 year of age and similar to middle-aged adult HCM patients33,34 which is reduced down to 82% for those diagnosed before 1 year of age33.
Considering HCM phenocopies in children and adolescents <18 years, similar outcomes were pointed out for those with neuromuscular disorders (5-year survival of 98%). On the other hand, children with HCM associated with an error of metabolism or malformation syndrome manifest the disease at a younger age, and had low 5-year survival rates (42% and 74%, respectively)33.
Actually, among HCM patients from 7 to 30 years old, the HCM-related mortality was as low as 0.54%/year, similar to that found in patients aged 30-59 and ≥60. These findings were explained by high rate of nonfatal HCM events aborted with contemporary treatment interventions in the pediatric population (>2-fold than that of older patients), which finally yielded a low incidence of SCD and HF HCM deaths of 0.39%/year and 0.17%/year, respectively34.
Finally, it has been repeatedly stressed that the ESC risk score model proved highly ineffective for identifying patients at the greatest SCD risk34. Endorsed by a current meta-analyses, among pediatric HCM population only 4 ‘major’ risk factors that have been shown to be statistically associated with increased risk of death in at least 2 studies (previous adverse cardiac event, non-sustained ventricular tachycardia (NSVT), syncope and LVH)35.
Apical aneurysms
Apical aneurysms are found in 5–8% of HCM patients, who exhibit a poorer prognosis36,37. Cardiac MRI emerges as the most useful technique for its diagnosis (40% can be missed with other imaging methods) and ventricular arrhythmia substrate analyses (LGE in the aneurysm rim)36. It has been suggested that apical aneurysms should be considered a new risk factor for SCD on the basis of a high rate of HCM-related deaths combined with life-saving aborted disease-related events (6.4–10.1%/year, more than 3-fold greater than the 2.0%/year event rate in 1,847 HCM patients without aneurysms, p < 0.001)36,37. Moreover, apical aneurysms also increase morbidity and could prompt physicians to initiate anticoagulation therapy due to an incidence of stroke of 1.1%–4.0%/year36,37. Anticoagulation due to clot identification in 15% of these patients prevented embolic complications in 4 years of follow-up and radiofrequency ablation of monomorphic ventricular tachycardia yielded a high success rate36.
Non-sustained ventricular tachycardia (NSVT)
NSVT are registered during holter monitoring in 20-30% of HCM patients1 and were included in the current ESC proposal of HCM risk stratification as ≥3 consecutive ventricular beats at ≥120 BPM lasting <30 seconds. Although it was acknowledged that no evidence supported a particular frequency, duration or rate in risk modulation, more recent publications on extended monitoring, confirmed that NSVT was independently associated with ICD-treated ventricular arrhythmias, but only when their rate was >200 beats per minute, when they lasted >7 beats, and when repetitive (adjusted hazard ratios 6–15, p < 0.05)38. Apart from resting NSVT, ventricular arrhythmias may also be exercise-induced. In this case, they appear to be closely related to myocardial fibrosis39. The promising results from the London cohort linking exercise induced-arrhythmias and a poor prognosis40 have not yet inspired more papers either to confirm or to argue their conclusions. In that series 27/1380 patients exhibited NSVT or ventricular fibrillation (VF) during exercise and those arrhythmias were associated to more severe hypertrophy, larger left atria, male sex and death or cardiac event (3.73-fold increase in risk of SD/ICD discharge), the latter also in the multivariate analysis (hazard ratio 3.14, 95% CI [1.29–7.61])40.
Myocardial fibrosis
With respect to prognosis in any cardiomyopathy the presence and magnitude of myocardial fibrosis (or LGE in cardiac MRI) has been a trending topic in the past 10 years. Particularly in HCM patients, it appears clear that the percentage of patients with LGE and its quantity (grams and percentage of LV myocardial mass with LGE) correlates with systolic impairment and HF (end-stage HCM)41. Regarding the arrhythmic risk, in a recent metaanalysis with pooled data the amount of LGE remained independently associated with SCD risk (adjusted hazard ratio 1.4 for every 10% increase in LGE of LV mass, and adjusted hazard ratio of 1.6 for 15% LGE)42. Based on these data, it may be reasonable to consider that patients with HCM and extensive LGE (≥15–20% left ventricular myocardium) present an increased risk, regardless of other high-risk features, with implications on management strategies including ICD implantation. Extensive data can be found in the chapter regarding cardiac imaging.
The blunted perfusion reserve of HCM patients (related to microvascular dysfunction and small vessel disease) precedes the development of myocardial fibrosis and systolic dysfunction by years43, so it could be an early marker of poor prognosis. Thus, early identification of microvascular flow abnormalities with PET studies represents an opportunity for pharmacological prevention of disease progression44 since once fibrosis has been established in HCM patients, little can be done from a curative therapeutic approach.
Genotype
Genetics may also have an influence in the general HCM prognosis. Patients with sarcomeric protein variants (nearly 44% of HCM patients) are characterized by younger age and higher prevalence of family history of HCM, family history of SCD, asymmetric septal hypertrophy, greater maximum LV wall thickness and an increased incidence of cardiovascular death (all p values<0.02)45. Besides the mutated gene, the literature underlines that also the number of genetic hits may be relevant. Thus being a triple mutation carrier, although rare (0.8%), conferred a remarkably increased risk of end-stage progression (14-fold increase) and ventricular arrhythmias (100% were deemed at high risk of SCD and were ICD carriers)46. Although the presence of mutations have been associated with worse prognosis that the absence of a positive genetic test, recent data have demonstrated that individual mutations currently do not allow to establish prognosis in the majority of patients47.
Diastolic dysfuntion
Diastolic dysfunction promotes adverse remodeling over time in HCM patients. It has often (but not always) been related to HF development. This observation is based on several studies reporting a 100% of impaired LV filling pattern in non-obstructive HCM patients with HF (43–50% with additional left ventricular ejection fraction (LVEF) <50% and 40-100% with restrictive pattern)48,49. Strikingly, the most adverse LV filling pattern alteration, a restrictive pattern, was depicted in 5.9% of HCM patients at initial evaluation and in 9.2% during follow-up50. Among these patients HCM behaved aggressively with a 6-fold increase in risk of developing end-stage HCM, a 0.95%/year of SCD or appropriate ICD interventions, and a 3.2%/year of HCM-related death or heart transplantation50. Thus, the restrictive filling pattern was a strong and independent marker of increased risk compared to patients without restrictive filling (hazard ratio for SCD 3.5, and for a wider endpoint also including heart transplantation, resuscitated cardiac arrest, and appropriate ICD intervention 5.1, p < 0.05 both)50,51.
Finally, not only the LV restrictive filling pattern but also the RV restrictive physiology appears to have significant predictive value in HCM, regardless of the presence of other detrimental risk factors52.
Left ventricular ejection fraction
Systolic dysfunction is regarded as a clear marker of adverse prognosis in HCM patients (see Heart failure in the complications section below).
Autonomic dysfunction
Autonomic dysfunction in HCM patients precedes systolic dysfunction and so may play a role in its genesis, can be depicted by nuclear medicine studies assessing the pre-synaptic level and could represent a future therapeutic target to improve outcomes44.
Left ventricular outflow tract obstruction
A number of studies have proved a significant association of the LVOT maximal gradient with an increased risk of SCD, as cited in the current guidelines30. However, less evidence supports the role of a provoked LVOT gradient and it appears that, in keeping with the same rationale, provoked LVOT gradients <30 mmHg, 30–90 mmHg and >90 mmHg could identify HCM patients of low-, intermediate- and high-SCD risk53.
Pulmonary hypertension
Pulmonary artery systolic pressure (PASP) over 36 mmHg is one of the features of end-stage HCM40,54. It is present in nearly 40% of HCM patients being its predictor factors older age, female sex, AF and class II-IV NYHA class54. More interestingly, it was identified as the only independent predictor of all-cause mortality except in patients with obstructive HCM who underwent septal reduction therapy (in non-obstructive HCM hazard ratio 1.59 per 10 mmHg PASP increase; in obstructive HCM without septal reduction therapy hazard ratio 1.15 per 10 mmHg PASP, both p < 0.05)54.
ECG repolarization
Inconstantly, exercise-induced microvolt T-wave alternans has been associated to ventricular arrhythmia susceptibility (as a subrogate marker of an increased risk in HCM patients)55,56.
Biomarkers
Several plasmatic biomarkers have been analyzed aiming to predict prognosis in HCM patients. A high hsCRP concentration associated to more adverse events (>3.0 mg/L versus <1.0 mg/L: adjusted hazard ratios for individual adverse events 4–11-fold, p < 0.05) and it was replicated when considered as a continuous variable (adjusted hazard ratios for individual adverse events 1.06–1,20, p < 0.05)57. NT-proBNP concentration independently predicted all-cause mortality or cardiac transplantation being a serum concentration of ≥135 pmol/L associated with an annual event rate of 6.1%58,59. Although NT-proBNP has repeatedly been identified as a significant independent predictor of HF and transplant-related deaths there is not total agreement in the literature concerning its accuracy to predict SCD or appropriate ICD shock51,58,59.
Several metalloproteinases (MMPs) are increased in the serum of HCM patients, such as MMP-9 and MMP-3, respectively associated with LGE and increased arrhythmic risk60. Since aldosterone is known to promote MMP expression and is elevated in HCM patients it has been repeatedly identified as a potential therapeutic target44. Also Copeptin (the stable C-terminal part of pro-arginine vasopressin, also termed antidiuretic hormone) remained as an independent predictor of HF and adverse cardiac events in multivariable logistic regression analysis on a small HCM cohort followed during 24 months58.
Scarce yet interesting evidence supports the role of the soluble suppression of tumourigenicity (sST2) in HCM risk stratification due to the presence of higher levels in HCM patients with NSVT61. Increased transforming growth factor-beta levels, however, identified patients with higher risk of developing postoperative AF after myectomy but not major adverse cardiac events62. Interleukin-6, tumour necrosis-alpha and soluble Fas ligand have been associated to fibrosis in HCM patients, and so indirectly linked to HCM prognosis44.
Finally, regarding the attractive role of microRNAs as key modulators of gene expression, the circulating levels of miR-29a were found to be up-regulated in HCM patients, correlating with both myocardial hypertrophy and fibrosis, and indirectly suggesting a potential prognosis value for this microRNA63.
Clinical complications of HCM
Heart failure
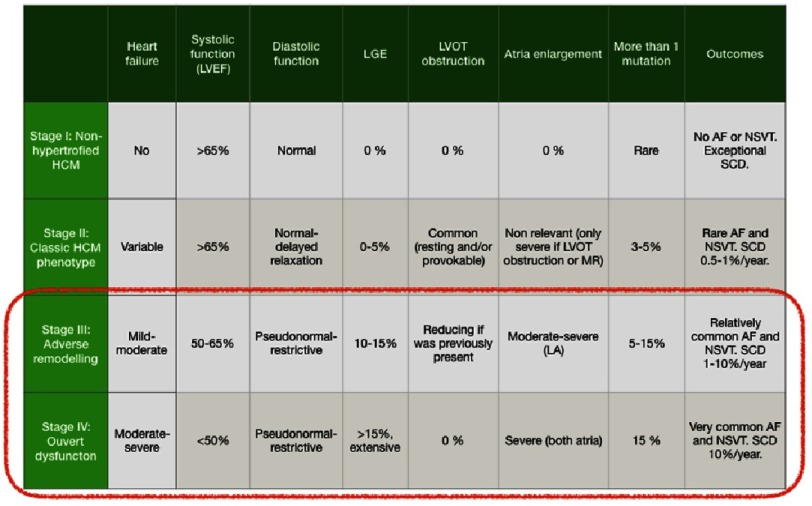
A life-long remodelling process takes place within the myocardium of HCM patients which leads to clinical progression and may culminate in the so-called end-stage or burnt-out phase in 5–7% of patients, at any age, and with a 10-fold increase in SCD estimates (Table 2)41. Four stages of disease have been proposed to define the natural history of the disease (i) ‘Non-hypertrophic HCM’, characterized by the absence of LVH in mutation carriers (the occurrence of live-threating ventricular arrhythmias is exceptional); (ii) ‘Classic HCM phenotype’, defined as fully developed hypertrophy with hyperdynamic left ventricle, often with LVOTO but without significant fibrosis; (iii) ‘Adverse remodelling’ (15% of HCM patients), where already unfavourable structural modifications have occurred (extensive LV fibrosis and LVEF in the low normal range of 50–65%), with preserved clinical and hemodynamic balance; (iv) ‘Overt dysfunction’ (5–7% of HCM patients), characterized by severe functional deterioration of the LV (defined as overt systolic dysfunction and/or restrictive pathophysiology), extreme LV fibrosis and atrial dilatation, haemodynamic decompensation and adverse outcome41,49. Treatment with HF drugs, aggressive management of AF and prevention of SCD in stages III and IV should be taken into account and the promising role of ranolazine could also open new doors to improve outcomes at this point41.
Table 2. Clinical characteristics of different HCM phenotypes.
Reproduced and adapted from Olivotto I, et al. Circ Heart Fail. 2012;5(4):535–46.41
|
Atrial fibrillation
An important proportion of HCM patients develop any kind of new onset atrial fibrillation (AF), namely 17–22% of patients during 9–22 year follow-up period, 2%/year65,65. The timing and frequency of paroxysmal AF episodes are unpredictable in HCM patients, with an average 2-year interval between the first and second symptomatic episode, but progressing to permanent AF uncommonly (26%)31. AF has been generally associated with increased morbidity and mortality in HCM patients. However, two recent studies showed opposite conclusions at this point: one yielded a 2-fold increased risk of cardiovascular death at 10 years in HCM patients with AF64 whereas the other pointed out that with the current strategies AF is not a major contributor to HF or a cause of arrhythmic sudden death, assuming that AF usually appears to represent a secondary event, a disease marker, or largely an innocent bystander to clinical events, rather than a primary cause31. Notably, the recruitment and follow-up periods were significantly different in both studies, from 1986–2008 the former64 and 2004-2014 the latter31 which may have influenced the treatment strategies employed. In the most contemporary series, no differences regarding AF development were found in all cause or HCM-related mortality (0.7%/year), and progression of HF symptoms from class I/II to III/IV at 5 years (approximately 5%, and without differences when considering paroxysmal or permanent AF)31.
The occurrence of new onset AF and the presence of permanent versus paroxysmal AF was associated with female sex, age, left atrial diameter, and NYHA class, whereas the new onset was additionally associated to hypertension and vascular disease and the progression to permanent AF to LVEF, larger LV end-diastolic cavity size and less common resting LVOTO or myectomy31,64. On multivariable analysis, the only independent predictors for development of AF were younger age at HCM diagnosis, larger LA, and lower LVEF31.
Regarding the most frequently used treatments, oral drugs were unsuccessful to maintain64 sinus rhythm and to restore it, same as Maze procedures31. Regarding catheter ablation for AF treatment, long-term outcomes was worse in patients with apical HCM, as compared to controls, but was similar to patients with septal HCM (50% free from AF/atrial tachycardia with a 44.7 ±30.8 months) and LA diameter was an independent predictor for recurrence66.
Stroke.
Stroke is considered the most disabling consequence of AF in HCM patients, and under the currently available management it presents with a wide range in 6–28% of these patients being 11% of them fatal strokes31,64,67. Given that HCM patients with AF usually would not fulfill anticoagulation criteria by CHADs scoring, anticoagulation is always recommended, irrespective to CHADs score system1,31,68. Although little evidence supports the use of direct oral anticoagulants, initial evidence supports that at least equals anti-vitamin K management with a higher treatment satisfaction among patients69.
Infective endocarditis.
The incidence of IE among HCM patients has been described to be 18 to 28 times higher than in the general population being the LVOTO and enlarged LA its reported risk factors70. The two largest retrospective cohorts (N = 30 and N = 34) pointed out the non-determinant presence of LVOTO and similar rates for surgical intervention (37–42%) but differed in the 1-year mortality (7% versus 42%), aortic and mitral valve involvement (similar versus predominantly mitral), and embolic complications (33% versus 18%).23,71 Although since 2009 antibiotic prophylaxis is no longer recommended by the ESC in HCM patients20, the very low incidence of prophylactic complication and the high risk of complications in an HCM patient with IE still generate debate and warrants reconsideration of the balance of risks/benefits in this scenario. Thus, some dedicated clinics currently recommend IE antibiotic prophylaxis.
References
- 1.Elliott PM, Anastasakis A, Borger MA, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J. 2014;35(39):2733–2779. doi: 10.1093/eurheartj/ehu284. [DOI] [PubMed] [Google Scholar]
- 2.Guttmann OP, Pavlou M, O’Mahony C, Monserrat L, Anastasakis A, Rapezzi C, et al. Prediction of thrombo-embolic risk in patients with hypertrophic cardiomyopathy (HCM Risk-CVA) Eur J Heart Fail. 2015;17(8):837–45. doi: 10.1002/ejhf.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Mahony C, Jichi F, Ommen SR, Christiaans I, Arbustini E, Garcia-Pavia P, et al. An International External Validation Study of the 2014 European Society of Cardiology Guideline on Sudden Cardiac Death Prevention in Hypertrophic Cardiomyopathy (Evidence from HCM) Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.117.030437. [DOI] [PubMed] [Google Scholar]
- 4.Chan RH, Maron BJ, Olivotto I, Pencina MJ, Assenza GE, Haas T, et al. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation. 2014;130(6):484–95. doi: 10.1161/CIRCULATIONAHA.113.007094. [DOI] [PubMed] [Google Scholar]
- 5.Coats CJ, Rantell K, Bartnik A, Patel A, Mist B, McKenna WJ, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Hear Fail. 2015;8(6):1022–31. doi: 10.1161/CIRCHEARTFAILURE.114.002248. [DOI] [PubMed] [Google Scholar]
- 6.Melacini P, Basso C, Angelini A, Calore C, Bobbo F, Tokajuk B, et al. Clinicopathological profiles of progressive heart failure in hypertrophic cardiomyopathy. Eur Heart J. 2010;31(17):2111–23. doi: 10.1093/eurheartj/ehq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Velzen HG, Schinkel AFL, Baart SJ, Oldenburg RA, Frohn-Mulder IME, van Slegtenhorst MA, et al. Outcomes of contemporary family screening in hypertrophic cardiomyopathy. Circ Genomic Precis Med. 2018;11(4) doi: 10.1161/CIRCGEN.117.001896. [DOI] [PubMed] [Google Scholar]
- 8.Jensen MK, Havndrup O, Christiansen M, Andersen PS, Diness B, Axelsson A, et al. Penetrance of hypertrophic cardiomyopathy in children and adolescents: A 12-year follow-up study of clinical screening and predictive genetic testing. Circulation. 2013;127(1):48–54. doi: 10.1161/CIRCULATIONAHA.111.090514. [DOI] [PubMed] [Google Scholar]
- 9.Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, et al. Genetic counselling and testing in cardiomyopathies: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. European Heart Journal. 2010;31:2715–28. doi: 10.1093/eurheartj/ehq271. [DOI] [PubMed] [Google Scholar]
- 10.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: Executive summary: A report of the American College of cardiology foundation/American heart association task force on practice guidelines. Circulation. 2011;124:2761–96. doi: 10.1161/CIR.0b013e318223e230. [DOI] [PubMed] [Google Scholar]
- 11.Botkin JR, Belmont JW, Berg JS, Berkman BE. ASHG POSITION STATEMENT points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am J Hum Genet. 2015;97:6–21. doi: 10.1016/j.ajhg.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker F, Van El CG, Ibarreta D, Zika E, Hogarth S, Borry P, et al. Genetic testing and common disorders in a public health framework: How to assess relevance and possibilities. Eur J Hum Genet. 2011;19(SUPPL 1) doi: 10.1038/ejhg.2010.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smets EM a, Stam MMH, Meulenkamp TM, van Langen IM, Wilde A a M, Wiegman A, et al. Health-related quality of life of children with a positive carrier status for inherited cardiovascular diseases. Am J Med Genet A. 2008;146A(6):700–7. doi: 10.1002/ajmg.a.32218. [DOI] [PubMed] [Google Scholar]
- 14.Nagueh SF, Bachinski LL, Meyer D, Hill R, Zoghbi WA, Tam JW, et al. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation. 2001;104(2):128–30. doi: 10.1161/01.cir.104.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kauer F, Van Dalen BM, Michels M, Schinkel AFL, Vletter WB, Van Slegtenhorst M, et al. Delayed and decreased LV untwist and unstrain rate in mutation carriers for hypertrophic cardiomyopathy. Eur Heart J Cardiovasc Imaging. 2017;18(4):383–9. doi: 10.1093/ehjci/jew213. [DOI] [PubMed] [Google Scholar]
- 16.Dominguez F, González-López E, Padron-Barthe L, Cavero MA, Garcia-Pavia P. Role of echocardiography in the diagnosis and management of hypertrophic cardiomyopathy. Heart. 2018;104(3):261–73. doi: 10.1136/heartjnl-2016-310559. [DOI] [PubMed] [Google Scholar]
- 17.Maron BJ, Udelson JE, Bonow RO, Nishimura RA, Ackerman MJ, Estes NAM, et al. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: task force 3: hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and other cardiomyopathies, and myocarditis: a scientif. J Am Coll Cardiol. 2015;66(21):2362–71. doi: 10.1016/j.jacc.2015.09.035. [DOI] [PubMed] [Google Scholar]
- 18.Saberi S, Wheeler M, Bragg-Gresham J, Hornsby W, Agarwal PP, Attili A, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy. JAMA. 2017;317(13):1349. doi: 10.1001/jama.2017.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naidu SS. Hypertrophic cardiomyopathy: foreword by Bernard Gersh and historical context by Eugene Braunwald. 2015 doi: 10.1007/978-1-4471-4956-9. [DOI] [Google Scholar]
- 20.Habib G, Lancellotti P, Antunes MJ, Bongiorni MG, Casalta J-P, Del Zotti F, Dulgheru R, El Khoury G, Erba PA, Iung B, Miro JM, Mulder BJ, Plonska-Gosciniak E, Price S, Roos-Hesselink J, Snygg-Martin U, Thuny F, Tornos Mas P, Vilacosta I, Zamorano JL, ESC Scientific Document Group 2015 ESC Guidelines for the management of infective endocarditis. European Heart Journal. 2015 ehv319. [Google Scholar]
- 21.Wilson W, Taubert KA, Gewitz M, Lockhart PB, Baddour LM, Levison M, Bolger A, Cabell CH, Takahashi M, Baltimore RS, Newburger JW, Strom BL, Tani LY, Gerber M, Bonow RO, Pallasch T, Shulman ST, Rowley AH, Burns JC, Ferrieri P, Gardner T, Goff D, Durack DT. Prevention of Infective Endocarditis: Guidelines From the American Heart Association: A Guideline From the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116(15):1736–1754. doi: 10.1161/CIRCULATIONAHA.106.183095. [DOI] [PubMed] [Google Scholar]
- 22.Dajani AS1, Taubert KA, Wilson W, Bolger AF, Bayer A, Ferrieri P, Gewitz MH, Shulman ST, Nouri S, Newburger JW, Hutto C, Pallasch TJ, Gage TW, Levison ME, Peter G, Zuccaro Jr G. Prevention of bacterial endocarditis: recommendations by the American Heart Association. Clin Infect Dis. 1997;25(6):1448–58. doi: 10.1086/516156. [DOI] [PubMed] [Google Scholar]
- 23.Dominguez F, Ramos A, Bouza E, Muñoz P, Valerio MC, Fariñas MC, de Berrazueta JR, Zarauza J, Pericás Pulido JM, Paré JC, de Alarcón A, Sousa D, Rodriguez Bailón I, Montejo-Baranda M, Noureddine M, García Vázquez E, Garcia-Pavia P. Infective endocarditis in hypertrophic cardiomyopathy. Med (United States) 2016;95(26) doi: 10.1097/MD.0000000000004008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goland S, Van Hagen IM, Elbaz-Greener G, Elkayam U, Shotan A, Merz WM, et al. Pregnancy in women with hypertrophic cardiomyopathy: Data from the European Society of Cardiology initiated Registry of Pregnancy and Cardiac disease (ROPAC) Eur Heart J. 2017;38(35):2683–90. doi: 10.1093/eurheartj/ehx189. [DOI] [PubMed] [Google Scholar]
- 25.Schinkel AFL. Pregnancy in women with hypertrophic cardiomyopathy. Cardiol Rev. 2014;22(5):217–222. doi: 10.1097/CRD.0000000000000010. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka K, Tanaka H, Kamiya C, Katsuragi S, Sawada M, Tsuritani M, et al. Beta-Blockers and Fetal Growth Restriction in Pregnant Women With Cardiovascular Disease. Circ J. 2016;80(10):2221–6. doi: 10.1253/circj.CJ-15-0617. [DOI] [PubMed] [Google Scholar]
- 27.European Society of Gynecology (ESG) Association for European Paediatric Cardiology (AEPC) German Society for Gender Medicine (DGesGM) Regitz-Zagrosek V, Blomstrom Lundqvist C, Borghi C, et al. ESC Guidelines on the management of cardiovascular diseases during pregnancy: the Task Force on the Management of Cardiovascular Diseases during Pregnancy of the European Society of Cardiology (ESC) Eur Heart J. 2011;32(24):3147–97. doi: 10.1093/eurheartj/ehr218. [DOI] [PubMed] [Google Scholar]
- 28.Pieper PG, Walker F. Pregnancy in women with hypertrophic cardiomyopathy. Netherlands Heart Journal. 2013;21(1):14–18. doi: 10.1007/s12471-012-0358-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dreesen J, Destouni A, Kourlaba G, Degn B, Mette WC, Carvalho F, et al. Evaluation of PCR-based preimplantation genetic diagnosis applied to monogenic diseases: A collaborative ESHRE PGD consortium study. Eur J Hum Genet. 2014;22(8):1012–8. doi: 10.1038/ejhg.2013.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121(7):749–770. doi: 10.1161/CIRCRESAHA.117.311059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowin EJ, Hausvater A, Link MS, Abt P, Gionfriddo W, Wang W, Rastegar H, Estes NAM, Maron MS, Maron BJ. Clinical profile and consequences of atrial fibrillation in hypertrophic cardiomyopathy. Circulation. 2017;136(25):2420–2436. doi: 10.1161/CIRCULATIONAHA.117.029267. [DOI] [PubMed] [Google Scholar]
- 32.Maron BJ, Rowin EJ, Casey SA, Haas TS, Chan RH, Udelson JE, Garberich RF, Lesser JR, Ap-pelbaum E, Manning WJ, Maron MS. Risk stratification and outcome of patients with hypertrophic cardiomyopathy ≥60 years of age. Circulation. 2013;127:585–593. doi: 10.1161/CIRCULATIONAHA.112.136085. [DOI] [PubMed] [Google Scholar]
- 33.Wilkinson JD, Landy DC, Colan SD, Towbin JA, Sleeper LA, Orav EJ, Cox GF, Canter CE, Hsu DT, Webber SA, Lipshultz SE. The pediatric cardiomyopathy registry and heart failure: key results from the first 15 years. Heart Fail Clin. 2010;6(4):401–13. doi: 10.1016/j.hfc.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maron BJ, Rowin EJ, Casey SA, Lesser JR, Garberich RF, McGriff DM, Maron MS. Hyper-trophic cardiomyopathy in children, adolescents, and young adults associated with low cardiovascular mortality with contemporary management strategies. Circulation. 2016;133(1):62–73. doi: 10.1161/CIRCULATIONAHA.115.017633. [DOI] [PubMed] [Google Scholar]
- 35.Norrish G, Cantarutti N, Pissaridou E, Ridout DA, Limongelli G, Elliott PM, Kaski JP. Risk factors for sudden cardiac death in childhood hypertrophic cardiomyopathy: A systematic review and meta-analysis. Eur J Prev Cardiol. 2017;24(11):1220–1230. doi: 10.1177/2047487317702519. [DOI] [PubMed] [Google Scholar]
- 36.Rowin EJ, Maron BJ, Haas TS, Garberich RF, Wang W, Link MS, Maron MS. Hypertrophic cardiomyopathy with left ventricular apical aneurysm: Implications for risk stratification and management. J Am Coll Cardiol. 2017;69(7):761–773. doi: 10.1016/j.jacc.2016.11.063. [DOI] [PubMed] [Google Scholar]
- 37.Ichida M, Nishimura Y, Kario K. Clinical significance of left ventricular apical aneurysms in hypertrophic cardiomyopathy patients: the role of diagnostic electrocardiography. J Cardiol. 2014;64(4):265–72. doi: 10.1016/j.jjcc.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of non-sustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopa-thy. Circ Arrhythm Electrophysiol. 2017;10(3) doi: 10.1161/CIRCEP.116.004604. [DOI] [PubMed] [Google Scholar]
- 39.van Rijsingen IA, Bekkers SC, Schalla S, Hermans-van Ast JF, Snoep G, Alzand BS, Arens YH, van den Wijngaard A, Crijns HJ, Pinto YM. Exercise related ventricular arrhythmias are related to cardiac fibrosis in hypertrophic cardiomyopathy mutation carriers. Neth Heart J. 2011;19(4):168–174. doi: 10.1007/s12471-011-0090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gimeno JR, Tomé-Esteban M, Lofiego C, Hurtado J, Pantazis A, Mist B, Lambiase P, McKenna WJ, Elliott PM. Exercise-induced ventricular arrhythmias and risk of sudden car-diac death in patients with hypertrophic cardiomyopathy. Eur Heart J. 2009;30(21):2599–605. doi: 10.1093/eurheartj/ehp327. [DOI] [PubMed] [Google Scholar]
- 41.Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Patterns of disease progression in hypertrophic cardiomyopathy: an individualized approach to clinical staging. Circ Heart Fail. 2012;5(4):535–46. doi: 10.1161/CIRCHEARTFAILURE.112.967026. [DOI] [PubMed] [Google Scholar]
- 42.Weng Z, Yao J, Chan RH, He J, Yang X, Zhou Y, He Y. Prognostic value of LGE-CMR in HCM: A meta-analysis. JACC Cardiovasc Imaging. 2016;9(12):1392–1402. doi: 10.1016/j.jcmg.2016.02.031. [DOI] [PubMed] [Google Scholar]
- 43.Cecchi F, Sgalambro A, Baldi M, Sotgia B, Antoniucci D, Camici PG, Sciagrà R, Olivotto I. Microvascular dysfunction, myocardial ischemia, and progression to heart failure in patients with hypertrophic cardiomyopathy. J Cardiovasc Transl Res. 2009;2(4):452–61. doi: 10.1007/s12265-009-9142-5. [DOI] [PubMed] [Google Scholar]
- 44.Olivotto I, d’Amati G, Basso C, Van Rossum A, Patten M, Emdin M, Pinto Y, Tomberli B, Camici PG, Michels M. Defining phenotypes and disease progression in sarcomeric cardio-myopathies: contemporary role of clinical investigations. Cardiovasc Res. 2015;105(4):409–23. doi: 10.1093/cvr/cvv024. [DOI] [PubMed] [Google Scholar]
- 45.Lopes LR, Syrris P, Guttmann OP, O’Mahony C, Tang HC, Dalageorgou C, Jenkins S, Hubank M, Monserrat L, McKenna WJ, Plagnol V, Elliott PM. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardio-myopathy. Heart. 2015;101(4):294–301. doi: 10.1136/heartjnl-2014-306387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, Torricelli F, Yeates L, Cecchi F, Ackerman MJ, Olivotto I. Clinical features and outcome of hypertrophic cardiomyopathy as-sociated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010;55(14):1444–53. doi: 10.1016/j.jacc.2009.11.062. [DOI] [PubMed] [Google Scholar]
- 47.Restrepo-Cordoba MA, Campuzano O, Ripoll-Vera T, Cobo-Marcos M, Mademont-Soler I, Gámez JM, Dominguez F, Gonzalez-Lopez E, Padron-Barthe L, Lara-Pezzi E, Alonso-Pulpon L, Brugada R, Garcia-Pavia P. Usefulness of genetic testing in hypertrophic cardiomyopathy. An analysis using real-world data. J Cardiovasc Trans Res. 2017;10:35–46. doi: 10.1007/s12265-017-9730-8. [DOI] [PubMed] [Google Scholar]
- 48.Rowin EJ, Maron BJ, Kiernan MS, Casey SA, Feldman DS, Hryniewicz KM, Chan RH, Harris KM, Udelson JE, DeNofrio D, Roberts WC, Maron MS. Advanced heart failure with pre-served systolic function in nonobstructive hypertrophic cardiomyopathy: under-recognized subset of candidates for heart transplant. Circ Heart Fail. 2014;7(6):967–75. doi: 10.1161/CIRCHEARTFAILURE.114.001435. [DOI] [PubMed] [Google Scholar]
- 49.Pasqualucci D, Fornaro A, Castelli G, Rossi A, Arretini A, Chiriatti C, Targetti M, Girolami F, Corda M, Orrù P, Matta G, Stefàno P, Cecchi F, Porcu M, Olivotto I. Clinical spectrum, Therapeutic options, and outcome of advanced heart failure in hypertrophic cardiomyopathy. Circ Heart Fail. 2015;8(6):1014–21. doi: 10.1161/CIRCHEARTFAILURE.114.001843. [DOI] [PubMed] [Google Scholar]
- 50.Biagini E, Spirito P, Rocchi G, Ferlito M, Rosmini S, Lai F, Lorenzini M, Terzi F, Bacchi-Reggiani L, Boriani G, Branzi A, Boni L, Rapezzi C. Prognostic implications of the Doppler restrictive filling pattern in hypertrophic cardiomyopathy. Am J Cardiol. 2009;104(12):1727–31. doi: 10.1016/j.amjcard.2009.07.057. [DOI] [PubMed] [Google Scholar]
- 51.D’Amato R, Tomberli B, Castelli G, Spoladore R, Girolami F, Fornaro A, Caldini A, Torricelli F, Camici P, Gensini GF, Cecchi F, Olivotto I. Prognostic value of N-terminal pro-brain natriuretic Peptide in outpatients with hypertrophic cardiomyopathy. Am J Cardiol. 2013;112(8):1190–6. doi: 10.1016/j.amjcard.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 52.Pagourelias ED, Efthimiadis GK, Parcharidou DG, Gossios TD, Kamperidis V, Karoulas T, Karvounis H, Styliadis IH. Prognostic value of right ventricular diastolic function indices in hypertrophic cardiomyopathy. Eur J Echocardiogr. 2011;12(11):809–17. doi: 10.1093/ejechocard/jer126. [DOI] [PubMed] [Google Scholar]
- 53.Lu DY, Hailesealassie B, Ventoulis I, Liu H, Liang HY, Nowbar A, Pozios I, Canepa M, Cress-well K, Luo HC, Abraham MR, Abraham TP. Impact of peak provoked left ventricular out-flow tract gradients on clinical outcomes in hypertrophic cardiomyopathy. Int J Cardiol. 2017;243:290–295. doi: 10.1016/j.ijcard.2017.04.039. [DOI] [PubMed] [Google Scholar]
- 54.Ong KC, Geske JB, Hebl VB, Nishimura RA, Schaff HV, Ackerman MJ, Klarich KW, Siontis KC, Coutinho T, Dearani JA, Ommen SR, Gersh BJ. Pulmonary hypertension is associated with worse survival in hypertrophic cardiomyopathy. Eur Heart J Cardiovasc Imaging. 2016;(6):604–10. doi: 10.1093/ehjci/jew024. [DOI] [PubMed] [Google Scholar]
- 55.Puntmann VO, Yap YG, McKenna W, Camm J. T-wave alternans and left ventricular wall thickness in predicting arrhythmic risk in patients with hypertrophic cardiomyopathy. Circ J. 2010;74(6):1197–204. doi: 10.1253/circj.cj-09-1003. [DOI] [PubMed] [Google Scholar]
- 56.Trzos E, Kasprzak JD, Krzemińska-Pakuła M, Rechciński T, Wierzbowska-Drabik K, Uznańska B, Śmiałowski A, Rudziński T, Kurpesa M. The prevalence and the prognostic value of mi-crovolt T-wave alternans in patients with hypertrophic cardiomyopathy. Ann Noninvasive Electrocardiol. 2011;16(3):276–86. doi: 10.1111/j.1542-474X.2011.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu L, Zou Y, Wang Y, Luo X, Sun K, Wang H, Jia L, Liu Y, Zou J, Yuan Z, Hui R, Kang L, Song L, Wang J. Prognostic significance of plasma high-sensitivity C-reactive protein in patients with hypertrophic cardiomyopathy. J Am Heart Assoc. 2017;6(2) doi: 10.1161/JAHA.116.004529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coats CJ, Gallagher MJ, Foley M, O’Mahony C, Critoph C, Gimeno J, Dawnay A, McKenna WJ, Elliott PM. Relation between serum N-terminal pro-brain natriuretic peptide and prognosis in patients with hypertrophic cardiomyopathy. Eur Heart J. 2013;34(32):2529–37. doi: 10.1093/eurheartj/eht070. [DOI] [PubMed] [Google Scholar]
- 59.Sahin I, Gungor B, Ozkaynak B, Uzun F, Küçük SH, Avci II, Ozal E, Ayça B, Cetın S, Okuyan E, Dinckal MH. Higher copeptin levels are associated with worse outcome in patients with hypertrophic cardiomyopathy. Clin Cardiol. 2017;40(1):32–37. doi: 10.1002/clc.22602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zachariah JP, Colan SD, Lang P, Triedman JK, Alexander ME, Walsh EP, Berul CI, Cecchin F. Circulating matrix metalloproteinases in adolescents with hypertrophic cardiomyopathy and ventricular arrhythmia. Circ Heart Fail. 2012;5(4):462–6. doi: 10.1161/CIRCHEARTFAILURE.111.966200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gawor M, Śpiewak M, Janas J, Kozuch K, Wróbel A, Mazurkiewicz Ł, Baranowski R, Mar-czak M, Grzybowski J. The usefulness of sST2 and galectin-3 as novel biomarkers for better risk stratification in hypertrophic cardiomyopathy. Kardiol Pol. 2017;75(10):997–1004. doi: 10.5603/KP.a2017.0118. [DOI] [PubMed] [Google Scholar]
- 62.Guo Y, Wu X, Zheng X, Lu J, Wang S, Huang X. Usefulness of preoperative transforming growth factor-beta to predict new onset atrial fibrillation after surgical ventricular septal myectomy in patients with obstructive hypertrophic cardiomyopathy. Am J Cardiol. 2017;120(1):118–123. doi: 10.1016/j.amjcard.2017.03.252. [DOI] [PubMed] [Google Scholar]
- 63.Roncarati R, Viviani Anselmi C, Losi MA, Papa L, Cavarretta E, Da Costa Martins P, Contaldi C, Saccani Jotti G, Franzone A, Galastri L, Latronico MV, Imbriaco M, Esposito G, DeWindt L, Betocchi S, Condorelli G. Circulating miR-29a, among other up-regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardio-myopathy. J Am Coll Cardiol. 2014;63:920–927. doi: 10.1016/j.jacc.2013.09.041. [DOI] [PubMed] [Google Scholar]
- 64.Guttmann OP, Pavlou M, O’Mahony C, Monserrat L, Anastasakis A, Rapezzi C, Biagini E, Gimeno JR, Limongelli G, Garcia-Pavia P, McKenna WJ, Omar RZ, Elliott PM, Hypertrophic Cardiomyopathy Outcomes Investigators Predictors of atrial fibrillation in hypertrophic cardiomyopathy. Heart. 2017;103(9):672–678. doi: 10.1136/heartjnl-2016-309672. [DOI] [PubMed] [Google Scholar]
- 65.Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001;104:2517–2524. doi: 10.1161/hc4601.097997. [DOI] [PubMed] [Google Scholar]
- 66.Roh SY, Kim DH, Ahn J, Lee KN, Lee DI, Shim J, Choi JI, Park SW, Kim YH. Long-term out-come of catheter ablation for atrial fibrillation in patients with apical hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol. 2016;27(7):788–95. doi: 10.1111/jce.12985. [DOI] [PubMed] [Google Scholar]
- 67.Camm CF, Camm AJ. Atrial fibrillation and anticoagulation in hypertrophic cardiomyopathy. Arrhythm Electrophysiol Rev. 2017;6(2):63–68. doi: 10.15420/aer.2017.4.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, Castella M, Diener HC, Heidbuchel H, Hendriks J, Hindricks G, Manolis AS, Oldgren J, Popescu BA, Schotten U, Van Putte B, Vardas P, ESC Scientific Document Group 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37(38):2893–2962. doi: 10.1093/eurheartj/ehw210. [DOI] [PubMed] [Google Scholar]
- 69.Dominguez F, Climent V, Zorio E, Ripoll-Vera T, Salazar-Mendiguchía J, García Pinilla JM, Urbano-Moral JA, Fernández-Fernández X, Lopez-Cuenca D, Ajo-Ferrer R, Sanz-Sanchez J, Gomez-Perez Y, López-Garrido MA, Barriales-Villa R, Gimeno JR, Garcia-Pavia P. Direct oral anticoagulants in patients with hypertrophic cardiomyopathy and atrial fibrillation. Int J Cardiol. 2017;248:232–238. doi: 10.1016/j.ijcard.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 70.Spirito P, Rapezzi C, Bellone P, Betocchi S, Autore C, Conte MR, et al. Infective endocarditis in hypertrophic cardiomyopathy: Prevalence, incidence, and indications for antibiotic prophylaxis. Circulation. 1999;99(16):2132–7. doi: 10.1161/01.cir.99.16.2132. [DOI] [PubMed] [Google Scholar]
- 71.Sims JR, Anavekar NS, Bhatia S, O’Horo JC, Geske JB, Chandrasekaran K, et al. Clinical, radiographic, and microbiologic features of infective endocarditis in patients with hypertrophic cardiomyopathy. American Journal of Cardiology. 2017 doi: 10.1016/j.amjcard.2017.11.010. [DOI] [PubMed] [Google Scholar]