

A growing body of evidence indicates that endotoxemia is in close association with alcoholic liver disease (ALD). Endotoxins stimulate different cell in liver releasing cytokines, chemokines and reactive oxygen species (ROS) by a toll-like receptor-4 (TLR-4)-mediated mechanisms. Intestinal microflora is the source of circulating endotoxins, and the gut barrier dysfunction leading to elevated intestinal permeability is considered the main cause of endotoxemia in ALD. Understanding the mechanism of ethanol-induced gut barrier disruption is an active area of investigation. Evidence indicates that intestinal microflora, metabolism of ethanol and acetaldehyde-induced cell signaling are involved in the ethanol-induced intestinal barrier dysfunction. Recent advances in alcoholic endotoxemia, the mechanism of epithelial barrier disruption and the factors that prevent alcoholic endotoxemia are discussed in this article. The current understanding of these issues is illustrated in Fig. 1.
Fig. 1:
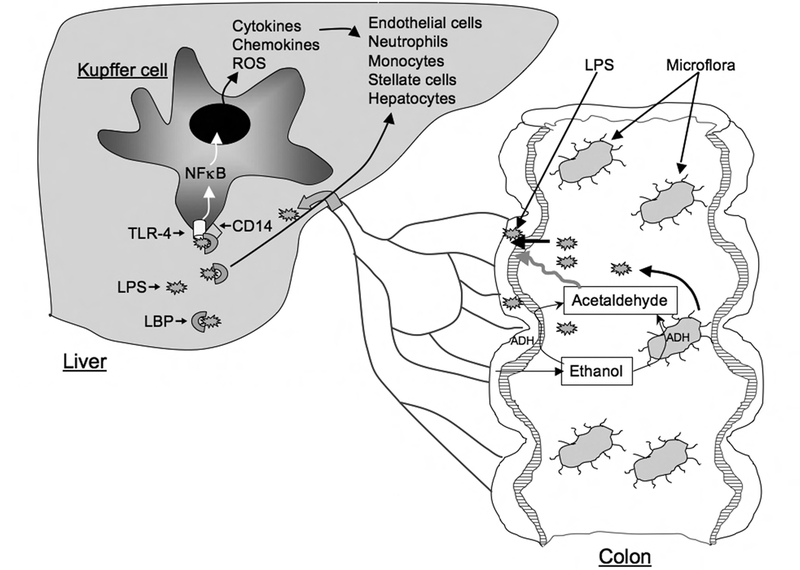
Diagrammatic representation of ethanol-induced intestinal permeability and endotoxemia. Ethanol in colonic lumen is metabolized to acetaldehyde by microflora and mucosa. Acetaldehyde disrupts epithelial barrier function and allows diffusion of bacterial lipopolysaccharide (LPS) into portal circulation. In the liver, LPS activates kupffer cells via LPB/CD14/TLR-4-dependent mechanism
Endotoxemia in ALD
Endotoxins are lipopolysaccharides (LPS) derived from the cell wall of Gram-negative bacteria. Dead bacteria and the LPS shed from the cell wall of viable organisms contribute to the circulating endotoxins. Normally, Kupffer cells in the liver detoxify endotoxins by phagocytosis. When the flux of endotoxins overwhelms the phagocytotic capacity of Kupffer cells, the endotoxins spill into the systemic circulation. Endotoxemia is conventionally believed as a condition when plasma endotoxin level rises to a greater than 2.5 EU (endotoxin unit)•m-1. Endotoxemia in ALD was first recognized by the detection of antibodies against E. Coli in the plasma of patients with ALD (1). Numerous subsequent studies demonstrated that plasma endotoxin level in patients with ALD is several-fold greater than those in healthy subjects (2–7) or in patients with non-alcoholic cirrhosis (4). Endotoxemia was recorded in alcoholics who showed only minimal symptoms of ALD (7), and therefore, endotoxemia appears to occur early during the pathogenesis of this disease (7).
The plasma endotoxin level in normal subjects ranges 0.3–10.4 pg/ml (2–8), while endotoxin level in ALD-patients ranges 8.5–206 pg/ml (2–8). Despite the wide variability in endotoxin values, the plasma endotoxin levels in patients with ALD were always found to be 5–20 folds greater than those in normal subjects. Although one study indicated the existence of a positive correlation between the degree of endotoxemia and the severity of disease (3), other studies failed to demonstrate such correlation (7).
Endotoxemia in ALD was confirmed in experimental models of alcoholic liver injury. Plasma endotoxin levels were elevated by acute (9, 10) or chronic (9, 11–13) ethanol administration, and the levels correlated well with the development of liver injury. The elevated plasma endotoxin level by acute ethanol administration indicated that endotoxemia occurs during the early stage of liver damage. Although there exists no single animal model that depicts different stages of ALD, there is a striking agreement between plasma endotoxin levels in patients with ALD and ethanol-fed animals. There is no evidence of species-dependent differences in alcoholic endotoxemia, however, a gender-dependent difference was observed (12, 14).
Endotoxin-mediated liver injury
The attenuation of alcohol-induced endotoxemia and liver damage by antibiotics indicated that endotoxin plays a crucial role in alcoholic liver damage (15). An emerging body of evidence indicates that LPS and ethanol synergistically affect the liver cells. LPS by itself failed to mimic ethanol-induced steatosis or hepatitis, however ethanol and LPS together effectively induced liver damage. Ethanol feeding sensitizes liver for LPS-induced cellular injury in experimental animals (16–18) and exacerbates LPS-induced cytokine release in liver (19). The mechanism of synergism between ethanol and LPS may involve multiple factors such as down regulation of IL-10-mediated protection (18), NADPH oxidase-dependent production of ROS (16) and adrenergic stimulation (17).
Cellular targets of LPS in liver include Kupffer cells (20), sinusoidal endothelial cells (20), stellate cells (21), neutrophils and hepatocytes (22). Inactivation of Kupffer cells prevents ethanol-induced liver injury in rats (23). LPS stimulates sinusoidal endothelial cells to release cytokines and chemokines (20). In stellate cells, LPS pretreatment enhances ethanol-induced collagen secretion (21). LPS binding protein (LBP) presents LPS to CD14, a 55 kDa glycoprotein (24). CD14 specifically binds to LPS and interacts with TLR-4 (25). Chronic ethanol feeding enhances the expression of LBP (11) and CD14 (26) and the ethanol-induced liver injury is absent in LBP (27), CD14 (26) and TLR-4 (28) knockout mice. TLR-4-mediated stimulation of different liver cells leads to secretion of a variety of proinflammatory factors such as cytokines (29), chemokines (30) and ROS (31).
Mechanism of alcoholic endotoxemia
Intestinal microflora is the source of circulating endotoxins. Normally, endotoxin absorption is impeded by the mucosal barrier function. Three major factors may contribute to alcoholic endotoxemia: 1) delayed endotoxin clearance from the circulation, 2) ethanol-induced bacterial over growth, and 3) dysfunctional gut barrier leading to elevated endotoxin absorption.
Evidence indicates that clearance of LPS from the circulation is delayed by ethanol feeding (32). Ethanol impairs the phagocytic function of Kupffer cells and attenuates the endotoxin uptake by these cells (33). The endotoxins that escape Kupffer cells spill into the circulation and contribute to endotoxemia. The numbers of both aerobic and anaerobic bacteria were found to be high in the jejunal aspirates of alcoholics compared to that in normal subjects (34). Ethanol has been shown to delay the gastrointestinal motility (35), and delayed gastrointestinal transit is known to increase bacterial growth in the lumen. Therefore, it is possible that delayed gastrointestinal motility is responsible for bacterial overgrowth in alcoholics. Although delayed clearance and bacterial over growth are potential contributors, enhanced intestinal permeability to endotoxins appears to be the primary cause of alcoholic endotoxemia.
Elevated gut permeability in ALD
Alcohol ingestion was shown to increase the intestinal permeability to macromolecules in both normal subjects and alcoholics (13, 34, 36), suggesting that disruption of intestinal epithelial barrier function plays an important role in causing endotoxemia in alcoholics. It is not clear whether ethanol-induced increase in permeability to macromolecules is caused by gastroduodenal barrier disruption or due to intestinal barrier disruption. This is an important issue as the intestinal microflora are confined to colon and the distal ileum. One study suggested that acute ethanol may increase the gastroduodenal permeability without altering the intestinal permeability, while chronic ethanol may elevate the intestinal permeability, without altering the gastroduodenal permeability (37).
Another important question is whether ethanol-induced intestinal permeability is a transient or a persistent response. Studies by Bjarnason etal (38) demonstrated that elevated intestinal permeability to EDTA exists in alcoholics after 3 days of abstaining from alcohol, while it persisted even after two weeks of sobriety in alcoholics with liver cirrhosis. Therefore, it appears that ethanol effect on intestinal barrier function is a transient effect in normal subjects as well as alcoholics without cirrhosis, whereas the ethanol effect is likely to persist much longer in alcoholics with the liver disease. As intestinal epithelial cells are renewed every 48–72 hours, it is likely that chronic ethanol feeding causes a delay in cell migration and renewal.
Increased intestinal permeability to macromolecules was also observed in animal models of alcoholic liver damage (39). Gastrointestinal permeability to macromolecular markers and LPS was elevated by both acute and chronic ethanol administration in experimental animals (9, 35, 36, 40, 41). It is likely that barrier disruption occurs much before liver injury, which is confirmed by a recent study (42). Ethanol-induced increase in gastrointestinal permeability was associated with endotoxemia and liver damage (9, 36, 39). Thus, experimental models of alcoholic liver damage are beneficial in understanding the pathogenesis of ALD.
Intestinal barrier disruption by ethanol and acetaldehyde
One of the important questions that have been addressed for a decade is whether ethanol directly disrupts the intestinal barrier function. All of the in vitro studies indicated that intestinal barrier disruption requires an ethanol concentration greater than 1%. Increases in paracellular permeability in rat jejunal everted sacs (43) and Caco-2 cell monolayers (44, 45) required an exposure to at least 2% ethanol. Ethanol concentration of 1.2% and 5% were used to disrupt the barrier function in IEC-6 (46) and MDCK (47) cell monolayers, respectively. Such a high concentration of ethanol is not expected to exist in the distal intestine of alcoholics. An early study by Halsted etal (48) demonstrated that 5% ethanol could be measured in the jejunal contents after alcohol consumption, while only 0.2–0.25% ethanol was detectable in the ileal and colonic lumen. Therefore, the results obtained in studies using ethanol concentrations greater than 0.25% may be relevant to barrier dysfunction in gastroduodenal and jejunal mucosa.
A significant body of evidence indicates that acetaldehyde plays a crucial role in the disruption of intestinal epithelial barrier function (41). Acetaldehyde level has been measured as high as 0.4 mM in saliva of alcoholics (49). The concentration of acetaldehyde in rat colonic lumen after ethanol administration was found to be in millimolar concentration (50). Acetaldehyde at 0.1–0.6 mM concentration disrupts the barrier function in Caco-2 cell monolayers (13, 45). A similar acetaldehyde-induced barrier disruption was demonstrated in human colonic mucosa (51). Furthermore, in vitro incubation of rat colonic strips mounted to Ussing chambers showed that ethanol up to 18 mM does not affect the barrier function (41). However, acetaldehyde (50–160 μM) dose-dependently increased the paracellular permeability, thus demonstrating that ethanol metabolism into acetaldehyde in the colonic lumen is required for the barrier disruption. The cellular mechanism involved in the acetaldehyde-induced barrier dysfunction involves the disruption of epithelial tight junctions (TJs) and adherens junctions (AJs) (13, 52–57).
Role of intestinal microflora in ethanol metabolism and barrier disruption
In addition to being the source of circulating endotoxins, intestinal microflora plays an important role in ethanol metabolism. Although intestinal epithelial cells express alcohol dehydrogenase (ADH), bacterial ADH seems to play a predominant role in the generation and accumulation of acetaldehyde in the intestinal lumen (58). The capacity of microflora and intestinal mucosa to further metabolize acetaldehyde into acetate by aldehyde dehydrogenase (ALDH) is low, thus resulting in acetaldehyde accumulation in the colonic lumen. A recent study (41) demonstrated that antibiotics partially reduced the colonic acetaldehyde. Therefore, intestinal microflora seem to play an important role in the production of acetaldehyde in the intestinal lumen. Ethanol-induced gastrointestinal permeability was attenuated by the pretreatment of rats with antibiotics for 12 days, which was also associated with the attenuation of ethanol-induced endotoxemia.
Acetaldehyde-induced disruption of TJs and AJs
TJs are the specialized intercellular junctional complexes that form a barrier to the diffusion of macromolecules across the epithelial monolayers (59). TJs are assembled by the organization of a variety of specific proteins such as occludin, claudins and zonula occludens. AJ is another intercellular junctional complex that lie beneath the TJ. AJs do not form a physical barrier to macromolecular diffusion. However, AJs indirectly regulate the integrity of TJs and therefore control the barrier function of the epithelium. AJ is organized by the interaction between E-cadherin and β-catenin, interaction between β-catenin and α-catenin and binding of α-catenin to the actin cytoskeleton. Both TJ and AJ are regulated by intracellular signal transduction
Disruption of TJs and AJs by ethanol and acetaldehyde was demonstrated in Caco-2 cell monolayers by immunofluorescence microscopy (44, 52–57). Acetaldehyde induced a redistribution of occludin and ZO-1 from the intercellular junctions (52, 54–56), and dissociates these proteins from the actin cytoskeleton (57). Acetaldehyde also caused a redistribution of E-cadherin and β-catenin from the intercellular junctions indicating the disruption of AJ (52, 55). A careful time course analysis of redistribution of E-cadherin and β-catenin indicated that acetaldehyde disrupts AJs as early as 10 min, while the distribution of occludin and ZO-1 was unaffected. This observation suggested that acetaldehyde alters the AJ integrity first, which in turn may trigger the disruption of TJs (55). Acetaldehyde-induced disruption of TJs and AJs was further validated in human colonic mucosa (51). Acetaldehyde induced disruption of TJs and AJs in human colonic mucosal biopsies, as indicated by redistribution of TJ and AJ proteins from the intercellular junctions and dissociation of these proteins from the actin cytoskeleton.
Mechanism of acetaldehyde-induced barrier dysfunction
Signaling elements such as protein kinases (60–64) and protein phosphatases (63) regulate the integrity of TJs in different epithelia. Acetaldehyde-induced disruption of TJs and AJs was associated with a rapid increase in the tyrosine-phosphorylation of ZO-1, E-cadherin and β-catenin, and the paracellular permeability was attenuated by tyrosine kinase inhibitors (52). Acetaldehyde inhibits protein tyrosine phosphatase (PTPase) activity, a likely cause of increased tyrosine-phosphorylation. Acetaldehyde inhibits PTPase activity in cell-free fractions, indicating that it directly interacts with the PTPases. Acetaldehyde not only inhibits the PTPase activity, but also disrupts its interaction with E-cadherin (55).
The current knowledge supports a model in which inhibition and dissociation of PTP1B from E-cadherin leads to tyrosine-phosphorylation of E-cadherin and β-catenin and loss of interaction between these two proteins (Fig. 2). Mass spectrometric analysis demonstrated that acetaldehyde increases phosphorylation of β-catenin on Y331, Y333, Y654 and Y670. In vitro protein-protein interaction studies using recombinant proteins demonstrated that tyrosine-phosphorylation of β-catenin reduces its interaction with E-cadherin, but tyrosine-phosphorylation of E-cadherin had no effect on this interaction (Fig. 2). On the other hand, tyrosine-phosphorylation of E-cadherin resulted in the loss of its interaction with PTP1B. Therefore, acetaldehyde disrupts interaction between E-cadherin and β-catenin by a phosphorylation-dependent mechanism.
Fig. 2: Diagrammatic model showing acetaldehyde-induced loss of interaction between PTP1B-E-cadherin-β-catenin.
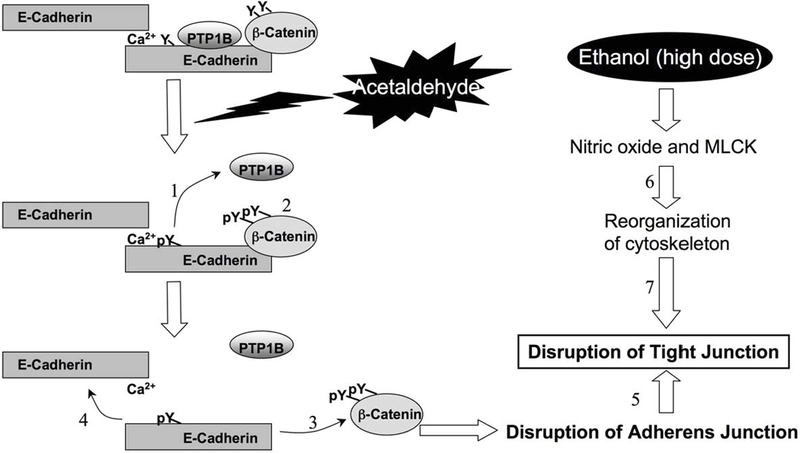
Cadherin-based cell-cell adhesion is mediated by interaction of E-cadherin with β-catenin. PTP1B binds to intracellular domain of E-cadherin and dephosphorylates β-catenin on Tyr residues. Treatment of cell monolayers with acetaldehyde induces inhibition and dissociation of PTP1B from E-cadherin (1), which results in increased Tyr-phosphorylation of β-catenin and E-cadherin (2). Tyr-phosphorylation of β-catenin results in the loss of its interaction with E-cadherin (3), and loss of interaction between E-cadherin and β-catenin leads to a loss of homophilic interaction between extracellular domains of E-cadherin and disruption of adherens junction(4). Disrption fof adherens junctions leads to disruption of tight junctions (5). Ethanol at high doses alters cytoskeletal structure by nitric oxide and MLCK-dependent mechanism (6), which in turn disrupts tight junctions (7).
Disruption of AJs is the likely signal that leads to disruption TJs. Although the precise mechanism involved in AJ-mediated TJ disruption is unknown, recent studies demonstrated that acetaldehyde induces tyrosine-phosphorylation of ZO-1 and threonine-dephosphorylation of occludin. The functions of occludin dephosphorylation and ZO-1 phosphorylation are unclear. However, such changes in phosphorylation status of occludin and ZO-1 have been consistently observed during the disruption of TJs and the barrier function by other mediators.
Controlling alcohol-induced intestinal permeability and endotoxemia
Several factors that ameliorate ethanol/acetaldehyde-induced barrier dysfunction have been identified. Understanding the mechanisms involved in the actions of these protective factors may be beneficial in the development of therapeutic strategies for ALD.
EGF, secreted in saliva and other gastrointestinal secretions (65), is known to protect the gastrointestinal mucosa from various insults (66, 67). A recent study demonstrated that EGF prevents acetaldehyde-induced TJ disruption (56) by preventing the reorganization of actin cytoskeleton and dissociation of TJ and AJ proteins from the actin cytoskeleton. EGF-mediated prevention of acetaldehyde effects on TJs involves the activation of PLCγ (57). Knockdown of PLCγ1 attenuates EGF-mediated protection of TJs from acetaldehyde. EGF induced membrane translocation of PKCβI and PKCε by a PLCγ-dependent mechanism (57). Prevention of PKCβI and PKCε translocation by interfering peptides also attenuated EGF-mediated protection of TJs. This study indicated that PLCγ-mediated activation of PKCβI and PKCε may be involved in stabilization of actin cytoskeleton.
Other mechanisms may be involved in barrier dysfunction by high dose of ethanol. Ethanol activates myosin light chain kinase (44) and induces nitric oxide generation (42), which alters the actin and microtubule cytoskeletal structures. Therefore, more than one mechanism may synergistically attenuate barrier function.
Glutamine is an essential nutrient for intestinal epithelial cell growth and differentiation (66). Pretreatment of Caco-2 cell monolayers with L-glutamine attenuated acetaldehyde-induced permeability to inulin and LPS (56). This protective effect of glutamine was associated with a prevention of acetaldehyde-induced redistribution of occludin, ZO-1, E-cadherin and β-catenin from the intercellular junctions. Interestingly enough, the protective effect of L-glutamine was mediated by the transactivation of EGF receptor. Glutamine rapidly activates EGF receptor and a selective inhibitor of EGF receptor kinase attenuated the glutamine effect on TJs.
Other studies showed that feeding oat bran or zinc attenuates ethanol-induced liver injury. Zinc supplementation attenuates ethanol-induced increase in plasma ALT and liver pathology (40), which was associated with a reduction of ethanol-induced intestinal permeability and endotoxemia. Similarly, oat bran supplementation prevents ethanol-induced liver injury (36). Once again, oat bran supplementation attenuates ethanol-induced intestinal permeability and endotoxemia.
Another potential factor that may ameliorate alcoholic liver injury is probiotic. Probiotics attenuate ethanol-induced liver injury in rats (68, 69). Probiotics are well known to reduce the growth of harmful bacteria (70), thus it is likely that probiotics prevent intestinal barrier disruption and endotoxemia in alcoholics. Additionally, Lactobacillus rhamnosus GG was shown to effectively metabolize acetaldehyde into acetate (71). Therefore, by reducing the acetaldehyde-producing commensal bacteria and by metabolizing acetaldehyde probiotics may alleviate colonic acetaldehyde level. However, further studies are necessary to confirm this possibility.
Summary and perspectives
In summary, the evidence is clear that alcohol consumption leads to increased intestinal permeability and endotoxemia, which is involved in the pathogenesis of ALD. Intestinal microflora and generation of acetaldehyde in the colonic lumen play a crucial role in the alcoholic intestinal permeability and endotoxemia. Evidence indicates that intestinal microflora is not only the source of circulating endotoxin, but it also plays a role in generation and accumulation of acetaldehyde in colonic lumen and subsequent influence on epithelial barrier dysfunction. The major mechanism of acetaldehyde-induced barrier dysfunction involves disruption of AJs and TJs by a phosphorylation-dependent mechanism. Gastrointestinal mucosal protective factors such as EGF, glutamine, zinc, oat bran and probioitcs prevent ethanol/acetaldehyde-induced intestinal permeability, endotoxemia and liver damage. It is therefore, important to delineate the mechanisms involved in the ethanol-induced intestinal epithelial barrier disruption, endotoxemia and endotoxin-mediated liver cell injury to understand the pathogenesis of ALD and in designing the preventive and therapeutic strategies for the treatment of ALD.
Supplementary Material
ACKNOWLEDGEMENTS
Preparation of this chapter was supported by National Institute of Health grants R01-DK55532 and R01-AA12307.
Abbreviations:
- ALD
alcoholic liver disease
- ROS
reactive oxygen species
- TLR
toll-like receptor
- EU
endotoxin unit
- LPS
lipopolysaccharide
- LBP
LPS binding protein
- TJ
tight junction
- AJ
adherens junction
- ADH
alcohol dehydrogenase
- ALDH
aldehyde dehydrogenase
- ZO-1
zonula occludens-1
- PTP1B
protein tyrosine phosphatase-1B
- PLCγ
phospholipase C-γ
- PKC
protein kinase-C
- EGF
epidermal growth factor
REFERENCES
- 1.Staun-Olsen P, Bjorneboe M, Prytz H, Thomsen AC, Orskov F. Escherichia coli antibodies in alcoholic liver disease. Correlation to alcohol consumption, alcoholic hepatitis, and serum IgA. Scand J Gastroenterol 1983;18:889–896. [DOI] [PubMed] [Google Scholar]
- 2.Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol 1987;4:8–14. [DOI] [PubMed] [Google Scholar]
- 3.Fujimoto M, Uemura M, Nakatani Y, Tsujita S, Hoppo K, Tamagawa T, Kitano H, et al. Plasma endotoxin and serum cytokine levels in patients with alcoholic hepatitis: relation to severity of liver disturbance. Alcohol Clin Exp Res 2000;24:48S–54S. [PubMed] [Google Scholar]
- 4.Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol 1991;12:162–169. [DOI] [PubMed] [Google Scholar]
- 5.Hanck C, Rossol S, Bocker U, Tokus M, Singer MV. Presence of plasma endotoxin is correlated with tumour necrosis factor receptor levels and disease activity in alcoholic cirrhosis. Alcohol Alcohol 1998;33:606–608. [DOI] [PubMed] [Google Scholar]
- 6.Parlesak A, Schafer C, Schutz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol 2000;32:742–747. [DOI] [PubMed] [Google Scholar]
- 7.Schafer C, Parlesak A, Schutt C, Bode JC, Bode C. Concentrations of lipopolysaccharide-binding protein, bactericidal/permeability-increasing protein, soluble CD14 and plasma lipids in relation to endotoxaemia in patients with alcoholic liver disease. Alcohol Alcohol 2002;37:81–86. [DOI] [PubMed] [Google Scholar]
- 8.Schafer C, Greiner B, Landig J, Feil E, Schutz ET, Bode JC, Bode C. Decreased endotoxin-binding capacity of whole blood in patients with alcoholic liver disease. J Hepatol 1997;26:567–573. [DOI] [PubMed] [Google Scholar]
- 9.Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology 2000;32:1008–1017. [DOI] [PubMed] [Google Scholar]
- 10.Nanji AA, Jokelainen K, Fotouhinia M, Rahemtulla A, Thomas P, Tipoe GL, Su GL, et al. Increased severity of alcoholic liver injury in female rats: role of oxidative stress, endotoxin, and chemokines. Am J Physiol Gastrointest Liver Physiol 2001;281:G1348–1356. [DOI] [PubMed] [Google Scholar]
- 11.Jokelainen K, Reinke LA, Nanji AA. Nf-kappab activation is associated with free radical generation and endotoxemia and precedes pathological liver injury in experimental alcoholic liver disease. Cytokine 2001;16:36–39. [DOI] [PubMed] [Google Scholar]
- 12.Nanji AA, Su GL, Laposata M, French SW. Pathogenesis of alcoholic liver disease--recent advances. Alcohol Clin Exp Res 2002;26:731–736. [PubMed] [Google Scholar]
- 13.Rao RK, Seth A, Sheth P. Recent Advances in Alcoholic Liver Disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 2004;286:G881–884. [DOI] [PubMed] [Google Scholar]
- 14.Schenker S Medical consequences of alcohol abuse: is gender a factor? Alcohol Clin Exp Res 1997;21:179–181. [DOI] [PubMed] [Google Scholar]
- 15.Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 1995;108:218–224. [DOI] [PubMed] [Google Scholar]
- 16.Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J Leukoc Biol 2006;79:1348–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.von Montfort C, Beier JI, Guo L, Kaiser JP, Arteel GE. Contribution of the sympathetic hormone epinephrine to the sensitizing effect of ethanol on LPS-induced liver damage in mice. Am J Physiol Gastrointest Liver Physiol 2008;294:G1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hill DB, Barve S, Joshi-Barve S, McClain C. Increased monocyte nuclear factor-kappaB activation and tumor necrosis factor production in alcoholic hepatitis. J Lab Clin Med 2000;135:387–395. [DOI] [PubMed] [Google Scholar]
- 19.Pennington HL, Hall PM, Wilce PA, Worrall S. Ethanol feeding enhances inflammatory cytokine expression in lipopolysaccharide-induced hepatitis. J Gastroenterol Hepatol 1997;12:305–313. [DOI] [PubMed] [Google Scholar]
- 20.Duryee MJ, Klassen LW, Freeman TL, Willis MS, Tuma DJ, Thiele GM. Lipopolysaccharide is a cofactor for malondialdehyde-acetaldehyde adduct-mediated cytokine/chemokine release by rat sinusoidal liver endothelial and Kupffer cells. Alcohol Clin Exp Res 2004;28:1931–1938. [DOI] [PubMed] [Google Scholar]
- 21.Quiroz SC, Bucio L, Souza V, Hernandez E, Gonzalez E, Gomez-Quiroz L, Kershenobich D, et al. Effect of endotoxin pretreatment on hepatic stellate cell response to ethanol and acetaldehyde. J Gastroenterol Hepatol 2001;16:1267–1273. [DOI] [PubMed] [Google Scholar]
- 22.Hoek JB, Pastorino JG. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol 2002;27:63–68. [DOI] [PubMed] [Google Scholar]
- 23.Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology 1994;20:453–460. [PubMed] [Google Scholar]
- 24.Haziot A, Chen S, Ferrero E, Low MG, Silber R, Goyert SM. The monocyte differentiation antigen, CD14, is anchored to the cell membrane by a phosphatidylinositol linkage. J Immunol 1988;141:547–552. [PubMed] [Google Scholar]
- 25.da Silva Correia J, Soldau K, Christen U, Tobias PS, Ulevitch RJ. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. transfer from CD14 to TLR4 and MD-2. J Biol Chem 2001;276:21129–21135. [DOI] [PubMed] [Google Scholar]
- 26.Yin M, Bradford BU, Wheeler MD, Uesugi T, Froh M, Goyert SM, Thurman RG. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol 2001;166:4737–4742. [DOI] [PubMed] [Google Scholar]
- 27.Uesugi T, Froh M, Arteel GE, Bradford BU, Wheeler MD, Gabele E, Isayama F, et al. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol 2002;168:2963–2969. [DOI] [PubMed] [Google Scholar]
- 28.Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001;34:101–108. [DOI] [PubMed] [Google Scholar]
- 29.McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 2004;287:G497–502. [DOI] [PubMed] [Google Scholar]
- 30.Bautista AP. Impact of alcohol on the ability of Kupffer cells to produce chemokines and its role in alcoholic liver disease. J Gastroenterol Hepatol 2000;15:349–356. [DOI] [PubMed] [Google Scholar]
- 31.Albano E Oxidative mechanisms in the pathogenesis of alcoholic liver disease. Mol Aspects Med 2008;29:9–16. [DOI] [PubMed] [Google Scholar]
- 32.Jarvelainen HA, Fang C, Ingelman-Sundberg M, Lindros KO. Effect of chronic coadministration of endotoxin and ethanol on rat liver pathology and proinflammatory and anti-inflammatory cytokines. Hepatology 1999;29:1503–1510. [DOI] [PubMed] [Google Scholar]
- 33.Fukui H, Kitano H, Okamoto Y, Kikuchi E, Matsumoto M, Kikukawa M, Morimura M, et al. Interaction of Kupffer cells to splenic macrophages and hepatocytes in endotoxin clearance: effect of alcohol. J Gastroenterol Hepatol 1995;10 Suppl 1:S31–34. [DOI] [PubMed] [Google Scholar]
- 34.Bode JC, Bode C, Heidelbach R, Durr HK, Martini GA. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology 1984;31:30–34. [PubMed] [Google Scholar]
- 35.Bode C, Bode JC. Effect of alcohol consumption on the gut. Best Pract Res Clin Gastroenterol 2003;17:575–592. [DOI] [PubMed] [Google Scholar]
- 36.Keshavarzian A, Choudhary S, Holmes EW, Yong S, Banan A, Jakate S, Fields JZ. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J Pharmacol Exp Ther 2001;299:442–448. [PubMed] [Google Scholar]
- 37.Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol 1999;94:200–207. [DOI] [PubMed] [Google Scholar]
- 38.Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet 1984;1:179–182. [DOI] [PubMed] [Google Scholar]
- 39.Purohit V, Bode JC, Bode C, Brenner DA, Choudhry MA, Hamilton F, Kang YJ, et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol 2008;42:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ. Preservation of intestinal structural integrity by zinc is independent of metallothionein in alcohol-intoxicated mice. Am J Pathol 2004;164:1959–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferrier L, Berard F, Debrauwer L, Chabo C, Langella P, Bueno L, Fioramonti J. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol 2006;168:1148–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keshavarzian A, Farhadi A, Forsyth CB, Rangan J, Jakate S, Shaikh M, Banan A, et al. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol 2009;50:538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carter EA, Harmatz PR, Udall JN, Walker WA. Barrier defense function of the small intestine: effect of ethanol and acute burn trauma. Adv Exp Med Biol 1987;216A:829–833. [DOI] [PubMed] [Google Scholar]
- 44.Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N. Ethanol modulation of intestinal epithelial tight junction barrier. Am J Physiol 1999;276:G965–974. [DOI] [PubMed] [Google Scholar]
- 45.Rao RK. Acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. Alcohol Clin Exp Res 1998;22:1724–1730. [PubMed] [Google Scholar]
- 46.Kishikawa H, Miura S, Nishida J, Nakano M, Hirano E, Sudo N, Morishita T, et al. Ethanol-induced CXC-chemokine synthesis and barrier dysfunction in intestinal epithelial cells. Alcohol Clin Exp Res 2005;29:2116–2122. [DOI] [PubMed] [Google Scholar]
- 47.Diebel LN, Liberati DM, Dulchavsky SA, Diglio CA, Brown WJ. Ethanol impairs intestinal barrier defense by modulation of immunoglobulin A transport. Surgery 2002;132:573–581; discussion 581. [DOI] [PubMed] [Google Scholar]
- 48.Halsted CH, Robles EA, Mezey E. Distribution of ethanol in the human gastrointestinal tract. Am J Clin Nutr 1973;26:831–834. [DOI] [PubMed] [Google Scholar]
- 49.Homann N, Tillonen J, Meurman JH, Rintamaki H, Lindqvist C, Rautio M, Jousimies-Somer H, et al. Increased salivary acetaldehyde levels in heavy drinkers and smokers: a microbiological approach to oral cavity cancer. Carcinogenesis 2000;21:663–668. [DOI] [PubMed] [Google Scholar]
- 50.Koivisto T, Salaspuro M. Aldehyde dehydrogenases of the rat colon: comparison with other tissues of the alimentary tract and the liver. Alcohol Clin Exp Res 1996;20:551–555. [DOI] [PubMed] [Google Scholar]
- 51.Basuroy S, Sheth P, Mansbach CM, Rao RK. Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: protection by EGF and L-glutamine. Am J Physiol Gastrointest Liver Physiol 2005;289:G367–375. [DOI] [PubMed] [Google Scholar]
- 52.Atkinson KJ, Rao RK. Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. Am J Physiol Gastrointest Liver Physiol 2001;280:G1280–1288. [DOI] [PubMed] [Google Scholar]
- 53.Rao RK. Acetaldehyde-induced barrier disruption and paracellular permeability in Caco-2 cell monolayer. Methods Mol Biol 2008;447:171–183. [DOI] [PubMed] [Google Scholar]
- 54.Seth A, Basuroy S, Sheth P, Rao RK. L-Glutamine ameliorates acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. Am J Physiol Gastrointest Liver Physiol 2004;287:G510–517. [DOI] [PubMed] [Google Scholar]
- 55.Sheth P, Seth A, Atkinson KJ, Gheyi T, Kale G, Giorgianni F, Desiderio DM, et al. Acetaldehyde dissociates the PTP1B-E-cadherin-beta-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem J 2007;402:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sheth P, Seth A, Thangavel M, Basuroy S, Rao RK. Epidermal growth factor prevents acetaldehyde-induced paracellular permeability in Caco-2 cell monolayer. Alcohol Clin Exp Res 2004;28:797–804. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki T, Seth A, Rao R. Role of phospholipase Cgamma-induced activation of protein kinase Cepsilon (PKCepsilon) and PKCbetaI in epidermal growth factor-mediated protection of tight junctions from acetaldehyde in Caco-2 cell monolayers. J Biol Chem 2008;283:3574–3583. [DOI] [PubMed] [Google Scholar]
- 58.Salaspuro M Bacteriocolonic pathway for ethanol oxidation: characteristics and implications. Ann Med 1996;28:195–200. [DOI] [PubMed] [Google Scholar]
- 59.Anderson JM, Van Itallie CM. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol 1995;269:G467–475. [DOI] [PubMed] [Google Scholar]
- 60.Basuroy S, Sheth P, Kuppuswamy D, Balasubramanian S, Ray RM, Rao RK. Expression of kinase-inactive c-Src delays oxidative stress-induced disassembly and accelerates calcium-mediated reassembly of tight junctions in the Caco-2 cell monolayer. J Biol Chem 2003;278:11916–11924. [DOI] [PubMed] [Google Scholar]
- 61.Rao RK, Basuroy S, Rao VU, Karnaky KJ Jr, Gupta A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J 2002;368:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rao RK, Li L, Baker RD, Baker SS, Gupta A. Glutathione oxidation and PTPase inhibition by hydrogen peroxide in Caco-2 cell monolayer. Am J Physiol Gastrointest Liver Physiol 2000;279:G332–340. [DOI] [PubMed] [Google Scholar]
- 63.Seth A, Sheth P, Elias BC, Rao R. Protein phosphatases 2A and 1 interact with occludin and negatively regulate the assembly of tight junctions in the CACO-2 cell monolayer. J Biol Chem 2007;282:11487–11498. [DOI] [PubMed] [Google Scholar]
- 64.Sheth P, Basuroy S, Li C, Naren AP, Rao RK. Role of phosphatidylinositol 3-kinase in oxidative stress-induced disruption of tight junctions. J Biol Chem 2003;278:49239–49245. [DOI] [PubMed] [Google Scholar]
- 65.Rao RK. Biologically active peptides in the gastrointestinal lumen. Life Sci 1991;48:1685–1704. [DOI] [PubMed] [Google Scholar]
- 66.Rao R, Baker RD, Baker SS. Inhibition of oxidant-induced barrier disruption and protein tyrosine phosphorylation in Caco-2 cell monolayers by epidermal growth factor. Biochem Pharmacol 1999;57:685–695. [DOI] [PubMed] [Google Scholar]
- 67.Basuroy S, Seth A, Elias B, Naren AP, Rao R. MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem J 2006;393:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nanji AA, Khettry U, Sadrzadeh SM. Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver (disease). Proc Soc Exp Biol Med 1994;205:243–247. [DOI] [PubMed] [Google Scholar]
- 69.Forsyth CB, Farhadi A, Jakate SM, Tang Y, Shaikh M, Keshavarzian A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol 2009;43:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nardone G, Rocco A. Probiotics: a potential target for the prevention and treatment of steatohepatitis. J Clin Gastroenterol 2004;38:S121–122. [DOI] [PubMed] [Google Scholar]
- 71.Nosova T, Jousimies-Somer H, Jokelainen K, Heine R, Salaspuro M. Acetaldehyde production and metabolism by human indigenous and probiotic Lactobacillus and Bifidobacterium strains. Alcohol Alcohol 2000;35:561–568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.