

Abstract
Introduction:
Lumacaftor-ivacaftor is indicated for treatment of cystic fibrosis (CF) in patients homozygous for the Phe-508del cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations. In clinical trials, treated patients showed improved pulmonary function, reduced pulmonary exacerbations, and other benefits. This article reviews safety of this therapy.
Areas covered:
Safety findings in ivacaftor, lumacaftor and combined therapy trials, and reported subsequently through post-approval evaluation, were accessed by PubMed and Google searches using key words “VX-770”, “ivacaftor”, “VX-809”, and “lumacaftor”. Transaminitis was seen in ivacaftor and combination trials. Non-congenital cataracts were seen in pre-clinical animal studies and in children taking ivacaftor and combined therapy. Dyspnea occurs in some patients taking lumacaftor and combined therapy and usually resolves without stopping treatment. Lumacaftor is a strong inducer of CYP3A while ivacaftor is a CYP3A sensitive substrate. Combination therapy can decrease systemic exposure of medications that are substrates of CYP3A, decreasing therapeutic effect. Co-administration of lumacaftor-ivacaftor with sensitive CYP3A substrates or CYP3A substrates with narrow therapeutic index is not recommended.
Expert opinion:
Lumacaftor-ivacaftor therapy may be associated with ocular and hepatic side effects. Specific recommendations for monitoring are available. Dyspnea occurs, especially during initiation of treatment. Potential drug interactions should be evaluated in patients taking combination therapy. The risk benefit ratio of lumacaftor-ivacaftor favors therapy.
Keywords: cystic fibrosis, lumacaftor, ivacaftor, cataract, transaminitis
1. Introduction:
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The CFTR protein transports ions at the luminal epithelial cell surface1,2. Abnormal or absent CFTR reduces epithelial chloride and bicarbonate transport. This causes viscous secretions dysfunction of multiple organs. Most CF patients have severe exocrine pancreatic insufficiency. Progressive lung disease, characterized by bronchiectasis and decline in forced expiratory volume in one second (FEV1), leads to premature mortality in the majority of affected individuals3,4. CF affects over 70,000 people worldwide3. Incidence varies with race and ethnicity. In many countries, diagnosis is in infancy through newborn screening. Otherwise, most cases are diagnosed by 2 years of age, but presentation in and after the second decade of life is well characterized4.
Quantitative pilocarpine iontophoresis sweat testing is the most reliable test for CF diagnosis. Sweat chloride ≥ 60 mmol/L with a positive newborn screening test and/or clinical manifestations of the disease confirms a CF diagnosis, though some patients with CF have intermediate (30–50 mmol/L) sweat chloride values5. Manifestations vary depending upon CFTR gene mutations, modifier genes, environmental factors, and age. Approximately 70% of patients worldwide have at least 1 copy of Phe-508del; almost 50% of US patients are homozygotes. This mutation causes defects in processing and trafficking of protein, leading to its degradation before reaching the cell surface1,2.
In CF, poor growth and low body mass index (BMI) are strongly associated with worse pulmonary function and increased mortality6–8. Pulmonary exacerbations, characterized by increase in cough, sputum production, weight loss, fatigue or lung function decline, are associated with more rapid decline in pulmonary function over time.
The management of CF is complex and time consuming, but advances in therapies have markedly increased survival over the past 3 decades. Until recently, therapies have been targeted towards end-organ manifestations of CF. Ivacaftor, a CFTR potentiator that improves chloride transport in class III and residual function mutations, was the first therapy approved that improves function of the mutant CFTR protein. Lumacaftor, a CFTR corrector, improves processing and conformational stability of Phe-508del CFTR, allowing mature protein to reach the surface. In vitro, lumacaftor acts early on biogenesis of CFTR by limiting defective folding and improving interactions between membrane-spanning and nucleotide binding proteins9,10.
Ivacaftor or lumacaftor monotherapy did not show effective restoration of chloride transport in homozygous Phe-508del cystic fibrosis in vivo or efficacy in phase II trials11–13. However, combination lumacaftor and ivacaftor therapy is efficacious for the management of CF in Phe-508del homozygotes and has received approval by a number of regulatory agencies since 2015. Initially approved for ages 12 and older, the label has recently been extended to 6–11 year olds, and a trial is underway in 2–5 year olds 14,15. Because Phe-508del homozygosity is the most common genotype causing CF, understanding the safety profile of combination therapy is essential.
2. Mechanism of action, pharmacokinetics and pharmacodynamics
2.1. Mechanism of action
The Phe-508del mutation results in CFTR misfolding which causes alterations in the cellular processing and trafficking. Premature protein degradation in the endoplasmic reticulum (ER) results in reduced functional protein reaching the cell surface. A small amount of Phe-508del-CFTR reaches the surface, but displays defective chloride ion channel activity. Lumacaftor, previously called VX-809, is a corrector that modulates conformation of specific region within the CFTR protein, enhancing protein folding, assembly, stability and transport to the cell surface9,10. It was discovered through screening of compounds that increase Phe-508del-CFTR mediated chloride transport. HEK-293 cells expressing Phe-508del-CFTR and treated with VX-809, showed a six fold increase in Phe-508del-CFTR exiting the ER consequently reaching cell surface, when compared to vehicle alone9. Human bronchial epithelial cells (HBE) from patients homozygous for Phe-508del mutation showed improvement in the chloride transport across the membrane and increase airway surface height when exposed to VX-809. At the most effective concentrations, VX-809 restores Phe-508del-CFTR function to approximately 15% of normal9. Enhancement of CFTR mediated chloride transport is achieved by addition of ivacaftor, which increases the probability of channel opening of activated CFTR and airway surface liquid height9.
2.2. Chemistry and Formulation
Lumacaftor is 3-[6-({[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-3-methylpyridin-2-yl]benzoic acid; ivacaftor is a N-(2,4-di-tert- butyl-5-hydroxyphenyl)-1,4-dihydro-4-oxoquinoline-3-carboxamide. The molecular formulas and molecular weights for lumacaftor and ivacaftor are C24H18F2N2O5/ 452.41 and C24H28N2O3/392.49, respectively. Lumacaftor-ivacaftor is available as a pink, oval-shaped, film-coated tablet for oral administration which contains 200 mg of lumacaftor and 125 mg of ivacaftor, dosed as two tablets twice daily for patients aged 12 years and older, and 100 mg lumacaftor and 125 mg ivacaftor, two tablets twice daily for patients 6–11 years old14.
2.3. Pharmacokinetics and Metabolism
During twice-daily dosing, steady-state plasma concentrations of lumacaftor and ivacaftor in healthy subjects were generally reached after approximately 7 days of treatment, The exposure (AUC) of lumacaftor is approximately 2-fold higher in healthy adult volunteers compared to exposure in patients with CF. The exposure of ivacaftor is similar between healthy adult volunteers and patients with CF. The steady-state exposure of ivacaftor is lower than that of Day 1 due to the CYP3A induction effect of lumacaftor. After a single oral dose of lumacaftor-ivacaftor was ingested with fat-containing foods, exposure increased to approximately two-times higher for lumacaftor and three-times higher for ivacaftor, respectively. Both compounds reach peak plasma concentrations at 4 hours14.
Lumacaftor and ivacaftor are both approximately 99% bound to plasma proteins. The half-life of lumacaftor, when consumed by itself, is approximately 26 hours with an apparent clearance estimated to be 2.38 L/hr in patients with CF. However, when ingested combined with ivacaftor, the half-life is approximately 9 hours in healthy subjects with an estimated clearance of a 25.1 L/hr in CF patients. After oral administration of 200 mg of lumacaftor every 24 hours for 28 days to patients with CF in a fed state the mean (±SD) for apparent volume of distribution was 86.0 (69.8L). Following twice daily dosing, steady-state plasma concentrations were reached approximately at 7 days. Lumacaftor and ivacaftor are both primarily excreted in the feces, with 50% of lumacaftor excreted unchanged and 87% of ivacaftor eliminated after metabolic conversion14.
2.4. Pharmacodynamics
Pharmacodynamics of lumacaftor-ivacaftor were assessed by sweat chloride, a measure of CFTR function; EKG QT interval was a measure of short term safety. The effect of multiple doses of lumacaftor-ivacaftor 600mg daily combined with 250 mg twice daily as well 1000mg daily combined with 450mg twice daily respectively, revealed no significant changes on QT interval analysis, although a decrease up to 8 beats per minute from baseline heart rate was noted14.
In a phase II double blind, placebo controlled trial of ivacaftor monotherapy in Phe-508del homozygous patients, the treatment group showed a small but statistically significant decrease in sweat chloride levels at day 15 that was sustained over 16 weeks11. In phase II trial in Phe-508del homozygotes, lumacaftor monotherapy was associated with a dose-dependent reduction in sweat chloride levels within 7 days of dosing that was sustained during treatment period and rapidly reversed following discontinuation of therapy12.
A phase II study of combined lumacaftor-ivacaftor was conducted using 3 cohorts13,16. Cohort 1 and 3 included Phe-508del homozygotes, while cohort 2 included homozygous and heterozygous individuals. Subjects initially allocated to received lumacaftor monotherapy or placebo for 14 days, followed by additional 7 days of combined lumacaftor and ivacaftor therapy or placebo, for a total of 21 days of therapy. The greatest reduction in sweat chloride occurred in subjects receiving lumacaftor combined with the highest dose of ivacaftor, 250 mg every 12 hours, a statistically significant change of - 12.6 mmol/L p< 0.002 when compared with those receiving placebo. In cohort 2, subjects received placebo or lumacaftor at varying doses 200 mg, 400mg or 600 mg once daily for a total of 28 days followed by addition of 250 mg every 12 hours of ivacaftor or placebo for another 28 days. A decrease in mean sweat chloride levels during the first 28 days in all active treatment groups, mean sweat chloride concentration decreased over the entire 56 day trial for those Phe-508del homozygotes who received lumacaftor combined with ivacaftor. In the last cohort, total change in sweat chloride over the entire study was −10.3 mmol/L (p= 0.004), when compared to placebo13.
The final cohort of the phase II study was comprised of subjects heterozygous for Phe-508del, who did not have a second mutation responsive to ivacaftor15. Lumacaftor 400 mg/ivacaftor 250 mg q12h or placebo were administered for 56 days. Sweat chloride decreased in the lumacaftor/ivacaftor group at day 14 (−9.4 mmol/L) decreased further at day 56(−11.8 mmol/L). Patients in the placebo group had no significant change in sweat chloride at any time point (−1.8 and −0.8 mmol/L at day 14 and day 56).
While the pivotal phase III studies in patients at least 12 years of age, conducted over 24 weeks with an open label follow-on through week 96 weeks did not measure sweat chloride 17,18, an open label phase III trial in patients 6–11 years of age revealed a significant decrease in sweat chloride level, with return to baseline 2 weeks following discontinuation of therapy19. Similar sweat chloride reductions were seen in a placebo controlled trial in this age group20.
3. Clinical applications, including key efficacy data
Key efficacy data have been noted in previous reviews21–23. In the phase II studies, neither ivacaftor nor lumacaftor monotherapy showed evidence of efficacy in Phe-508del homozygotes, including pulmonary function, weight, or CFQ-R respiratory sub-score in homozygous Phe-508del11,12.
In cohort 1 of the phase II combined therapy study, FEV1 did not change significantly during lumacaftor monotherapy or during combined therapy with either ivacaftor dose, compared to placebo group. Cohorts 2 and 3 showed no significant change in percent predicted FEV1 (ppFEV1) during monotherapy, but had a dose-dependent trend toward decline in ppFEV1 at higher doses. Mean absolute ppFEV1 improved significantly during combination therapy with lumacaftor 600 mg daily and 400 mg every 12 hours combined with ivacaftor 250 mg compared to placebo. There was improvement of ≥ 5% in ppFEV1 in half of patients receiving 600 mg daily and 400 mg every 12 hours of lumacaftor, while a similar change increase was seen in only in 13% of patients in the placebo group13. Overall, this trial revealed a modest improvement in lung function in treatment groups and provided data for dosing of combined therapy for phase III trials in Phe-508del homozygotes. Therapy with lumacaftor-ivacaftor in patients heterozygous for Phe-508del did not result in meaningful improvement in ppFEV1 or BMI16, and heterozygotes were excluded from phase III studies.
Two phase III clinical trials were conducted simultaneously in patients homozygous for Phe-508del, older than 12 years of age and with FEV1 < 90% predicted17. Patients were randomly assigned to 3 different groups in a 1:1:1 ratio to receive lumacaftor 600 mg daily combined with ivacaftor 250 mg every 12 hours, lumacaftor 400 mg with ivacaftor 250 mg both given at 12 hour intervals or placebo for 24 weeks. The average age of enrolled subjects was 25 years and the mean FEV1 at enrollment was 61% predicted. FEV1 improvements from baseline were seen by day 15 and sustained through week 24 in treatment groups. The difference between the treated group combined and the placebo group was 2.6 to 4.0 percentage points (P=<0.001) in mean absolute change in ppFEV1 from baseline to week 24. In each study the fraction of patients showing improvement in ppFEV1 of 5% or more was greater in the combination group than in placebo. Clinically meaningful reductions in the rates of pulmonary exacerbations of 30% and 39% for were seen in the treated groups, as was an increase in mean BMI and improvement in the CFQ-R respiratory subscore17.
In a 96-week open label extension study through 120 total weeks of therapy, patients who transitioned from placebo to lumacaftor 400 mg combined with ivacaftor 250 mg every 12 hours had improvement in ppFEV1 that was sustained through extension week 7218. Similar findings were seen at extension week 96, however, the number of subjects who continued therapy for 96 weeks was small. An increase from baseline BMI continued throughout study in both lumacaftor dosing groups. The CFQ-R respiratory domain score increase from baseline continued through week 96. Importantly, the annual rate of pulmonary exacerbation through 120 weeks remained lower in the lumacaftor-ivacaftor group18.
Two phase III trials of lumacaftor 200 mg and ivacaftor 250 mg BID were conducted in children 6–11 years old19,20. The primary endpoint was safety. An open label study showed no statistically significant increase in ppFEV1, but showed improvement in lung clearance index (LCI), indicating reduction of ventilation inhomogeneity, a sensitive marker of small airways disease19. A larger placebo controlled study showed a significant increase in ppFEV1 compared to placebo, though on a similar scale to that in the open-label study, as well as improvement in LCI20.
4. Safety Evaluation
4.1. Safety in clinical studies
The safety of ivacaftor has previously been reviewed24. Key efficacy and safety data for lumacaftor, ivacaftor and combined therapy is presented in the Table. During the phase II trial of lumacaftor monotherapy, incidence of adverse effects (AEs) was similar between all treated groups; with respiratory events being the most common complaint, cough occurring in 46% of individuals, ultimately leading to discontinuation of therapy in one patient from each dosing group12.
Table.
Key efficacy and safety data in lumacaftor-ivacaftor trials for Phe-508del homozygous cystic fibrosis patients.
| Author (ref #) | Phase | Treatment | Duration | Mean absolute change in pp FEV1 vs placebo or baseline | Adverse effects |
|---|---|---|---|---|---|
| Flume (11) | II | IVA 150 mg q 12h | 16 week vs placebo 24 week open label extension |
+ 1.7 Not sustained during OLE |
Cough, nausea, rash and dermatitis similar in active and placebo Pulmonary exacerbation lower in active arm |
| Clancy (12) | II | LUM 25, 50, 100, 200 mg qd Or placebo |
28 days | 25 mg: −2.46 50 mg: −2.15 100 mg: + 0.3 200 mg: + 0.47 |
Cough, fatigue, pulmonary exacerbation, sinus congestion. musculoskeletal discomfort |
| Boyle (13) | II |
Cohor1: LUM 200 mg qd, followed by LUM 200 mg qd with IVA 150 or 250 mg q 12 h Or placebo Cohort 2: LUM 200, 400, 600 mg qd followed by LUM with IVA 250 mg q 12 h Or placebo Cohort 3: LUM 400 mg qd, followed by LUM 400 mg with IVA 250 mg q 12 h Or Placebo |
Cohort 1: 14 days of monotherapy followed by 7 days of combination therapy Cohorts 2 and 3: 28 days of monotherapy followed by 28 days of combination therapy |
Cohort 1: NS Cohort 2: LUM 600 mg qd and IVA +6.2 LUM 400 mg q 12 h and IVA + 6.1 Cohort 3: LUM 400 mg and IVA + 6.1 |
Cough Chest tightness and dyspnea higher during monotherapy with higher doses of LUM Pulmonary exacerbations |
| Wainwright (17) | III | LUM 600mg qd or 400 mg q 12 h with IVA 250 mg q 12 hrs Or placebo |
24 weeks vs placebo | +2.6 – 4.0 | Dyspnea, chest tightness Pulmonary exacerbations Elevation of CK, AST, ALT Hemoptysis Bronchospasms Hypertension |
| Konstan (18) | III | LUM 600 qd or 400 mg q 12 h with IVA 250 mg q 12 h | 96 weeks open label extension | +0.5 week 72, +0.5 week 96 in previously treated subjects |
Dyspnea Chest tightness Pulmonary exacerbation Hemoptysis Distal intestinal obstruction syndrome Elevation of AST and ALT Hypertension |
| Milla (19) | III | LUM 200 mg and IVA 250 mg q 12 h Open label 6–11 year olds |
24 weeks | + 2.5 (NS) | Elevation of AST, ALT Rash |
| Ratjen (20) | III | LUM 200 mg and IVA 250 mg q 12 h | 24 weeks | + 2.4 | Cough, increased sputum, nasal congestion Elevation of AST, ALT |
Abbreviations: LUM, lumacator; IVA, ivacaftor; CK, creatine phosphokinase; AST, aspartate aminotransferase; ALT, alanine aminotransferase
In combined therapy phase II trials, overall AEs were evenly distributed between treatment and placebo groups. Chest tightness/discomfort, indicated as respiration abnormal, occurred more frequently during lumacaftor monotherapy, commonly occurring soon after initiation of therapy, and more frequent in the 400 mg BID group. Cases of dyspnea or chest tightness did not increase during combination therapy, suggesting increase occurrence to be related to treatment with lumacaftor as single agent13.
Serious events occurred in 7 of 97 participants during monotherapy. These included pulmonary exacerbation and hypersensitivity reaction, dyspnea, pneumonia and chest tightness in treated patients. Only 1 serious event was reported in the placebo group. In cohorts 2 and 3, the most common AEs reported were cough, pulmonary exacerbation and headache; dyspnea and chest tightness were rare. 8 serious AEs occurred in 6 out of the 90 participants receiving combination therapy, including 5 pulmonary exacerbations, 1 episode hemoptysis, 1 dyspnea and 1 elevation in creatinine phosphokinase. No patients discontinued treatment after AEs that started during this phase of the study. Subjects who had resolution of symptoms within 2–3 weeks13. Phase III trials overall revealed similar proportion of AEs across lumacaftor-ivacaftor and placebo groups. In all groups pulmonary exacerbations were the most common serious AE.
Among patients receiving combination therapy, AEs leading to discontinuation of therapy were elevation of creatine kinase levels, hemoptysis, bronchospasm, dyspnea, pulmonary exacerbation and skin rash, with respiratory events being most common complaint. Elevation of liver enzymes occurred in placebo and treatment groups, with no serious events reported in the placebo group. Marked increases in liver enzymes, increase in bilirubin levels and encephalopathy occurred in treated patients with pre-existing liver disease. Liver function returned to baseline in 6 out of the 7 patients with discontinuation of therapy13.
The rate of respiratory events, primarily dyspnea and abnormal respiration, occurred during initiation of combination therapy in phase III trials and was higher during the extension study in patients who transitioned from placebo to lumacaftor-ivacaftor. Subjects with lower ppFEV1 had an increased frequency of these AEs17.
Lumacaftor-ivacaftor can decrease effectiveness of hormonal contraceptives; the incidence of menstrual abnormalities (dysmenorrhea, amenorrhea and irregular periods) was 27% higher in females taking hormonal contraceptives with combination therapy compared to placebo14.
Lumacaftor is a strong inducer of CYP3A while ivacaftor is a CYP3A sensitive substrate. Combination therapy can decrease systemic exposure of medications which are substrates of CYP3A, decreasing therapeutic effect. On the other hand, co-administration of lumacaftor-ivacaftor with sensitive CYP3A substrates or CYP3A substrates with narrow therapeutic index is also not recommended; these include benzodiazepines (midazolam, triazolam) and immunosuppressants (tacrolimus, sirolimus, cyclosporine and everolimus)14. Systolic blood pressure is increased in patients taking lumacaftor-ivacaftor combination therapy; blood pressure should be monitored in patients on treatment14,15,17,18.
Although there is significant decrease in the rate of pulmonary exacerbations after treatment with lumacaftor-ivacaftor, there are no data regarding microbiologic changes. Ivacaftor monotherapy has been linked to reduction in Pseudomonas aeruginosa culture positivity, decreased odds of detection of mucoid strains of Pseudomonas aeruginosa, and of Aspergillus fumigatus25.
4.2. Post marketing data
Lumacaftor-ivacaftor is currently approved by the US Food and Drug Administration for the treatment of CF patients aged 6 through 11 who are homozygous for Phe-508del. The safety profile observed was similar to that during prior trials, except for a higher rate of transaminase elevations in these younger patients19,20. Recommendations for co-administration of lumacaftor-ivacaftor and strong CYP3A inhibitors have been modified to starting at lower than established dose, according to patient’s age, for the first week of treatment to allow for the steady-state induction effect of lumacaftor prior to increasing to the usual recommended daily dose26.
4.3. Use in Special Populations
Lumacaftor-ivacaftor should be prescribed with caution in patients with advanced liver disease, this combination can potentially worsen liver function including hepatic encephalopathy; patients should be monitored closely, with hepatic function panel assessment prior to initiation of therapy, every 3 months during the first year after initiation of therapy and annually thereafter. In patients with liver disease, specific adjustments in dose are recommended based on scores calculated using the Child-Pugh classification system, which categorizes liver disease as mild, moderate or severe based on serum bilirubin and albumin, prothrombin time, ascites and hepatic encephalopathy14.
Cataracts have been reported in children and adolescents receiving ivacaftor as monotherapy and when combined with lumacaftor. Therefore, is recommended that baseline and periodic eye examinations be performed in children who are prescribed these agents14.
Lumacaftor-ivacaftor has not been studied in patients with CF who have undergone organ transplantation. However, is not recommended due to potential drug interactions, particularly with calcineurin inhibitors14.
There are limited data regarding lumacaftor or ivacaftor in human milk. Both lumacaftor and ivacaftor are secreted into the milk of lactating rats, but due differences in lactation physiology, animal lactation may not be reliable to predict levels in human milk14.
5. Conclusion
Lumacaftor and ivacaftor, given in combination, results in improved pulmonary function as measured by increased ppFEV1, nutritional status, and a validated quality of life measurement. A decrease in the pulmonary exacerbation frequency is also noted. The side effect frequency is well characterized and can be monitored in routine clinical practice. Overall, there is a positive risk benefit ratio.
6. Expert Opinion
The successful application of potentiator and corrector drugs that target specific molecular defects in the CFTR protein has ushered in a new age in cystic fibrosis therapy. Ivacaftor monotherapy, while highly efficacious, is indicated for only a minority of CF patients with gating and residual function mutations, most of which are very rare. Combination therapy with lumacaftor and ivacaftor has more modest efficacy in improving FEV1. Phase II studies showed limited efficacy in patients heterozygous for Phe-508del, and the phase III studies were completed only in homozygotes. In addition to improving FEV1, benefits of therapy seen in placebo-controlled and open label studies include increased BMI, improved respiratory symptoms as indicated by CFQ-R respiratory sub-score, and decreased frequency of pulmonary exacerbations. The reduction in pulmonary exacerbations is especially important, since increased exacerbation frequency is associated with rate of pulmonary function decline. Furthermore, two year data comparing FEV1 in patients given lumacaftor-ivacaftor therapy continuously to propensity score matched Phe-508del homozygotes shows a significant reduction in the rate of FEV1 decline. If this effect is sustained over years of therapy, the ultimate difference in lung function between treated and untreated patients would be substantial. Long term follow-up data using registries will be needed to evaluate this potential benefit.
The safety profile of lumacaftor-ivacaftor is well characterized and generally favorable. Clinical trials have demonstrated multi-organ adverse events in both placebo and treatment groups, including respiratory abnormalities associated with initiation of treatment. This chest tightness and dyspnea are generally transient, and do not require termination of therapy in most treated patients. Importantly, these symptoms primarily occur at initiation of therapy. Patients who have difficulty adhering to treatment, or who must go off treatment for administration of a drug that is needed to treat a complication, may be at risk for more frequent symptoms when treatment is stopped and restarted.
Hepatic impairment can be serious in patients with underlying liver disease, and regular monitoring of hepatic function tests is necessary for all patients who start combination therapy. Cataracts have been reported in children and adolescents receiving ivacaftor as mono- and combined therapy; baseline and periodic eye examinations are recommended in children. Lumacaftor a strong inducer of CYP3A may decrease the therapeutic effect for medications that are CYP3A substrates. In the CF population, the most widely used concomitant drugs of concern include the azole antifungal agents (i.e., voriconazole and itraconazole). Oral contraceptive use is associated with menstrual abnormalities, and alternative contraceptive agents or dose changes may be needed to reduce this side effect and provide adequate contraception.
The limited efficacy of lumacaftor-ivacaftor may be related to the interaction between these agents. In vitro data has shown that rescue of Phe-508 del CFTR function is markedly decreased in cultures that have been chronically treated with lumacaftor and ivacaftor, translating in decreased chloride secretion. 27,28. On the other hand, limited follow-up data suggests ongoing benefits of therapy on FEV1 and BMI.
It is important to note that populations treated with lumacaftor-ivacaftor in clinical trials were a highly-treated population, using multiple maintenance therapies in accordance with the standard of care for cystic fibrosis. Patients starting therapy should be advised that their usual treatment should also be continued, as should regular monitoring. In patients with significant bronchiectasis, even complete correction of CFTR would not be expected to reverse longstanding structural changes in the lung. Whether introduction of CFTR modulation in infants and children will markedly reduce or eliminate bronchiectasis, or otherwise change the clinical course of CF, is unknown.
Alternative and additional agents are currently being studied in order to develop therapies with more correction of abnormal CFTR and therefore better efficacy. Tezacaftor is another CFTR corrector that, combined with ivacaftor, has recently shown efficacy in phase III studies for Phe-508 homozygotes and some heterozygotes29.So-called “next generation” therapies, given in combination with tezacaftor and ivacaftor, have shown evidence of increased efficacy, compared to active treatment with tezacaftor and ivacaftor alone, in phase II studies30.
Ultimately, the goal of CFTR modulation is to provide enough CFTR function to eliminate decline in FEV1 and multiple complications of cystic fibrosis. The degree of correction that would be meaningful has been described based on “milder” CFTR gene mutations associated with pancreatic sufficiency and lower sweat chloride levels. The efficacy of lumacaftor-ivacaftor provides good evidence for study of further agents that may have more pronounced effects on clinically important outcomes.
Drug Summary
| Drug name: lumacaftor-ivacaftor |
|---|
| Phase: launched |
| Indication: cystic fibrosis mediated by homozygosity for the Phe-508del CFTR mutation |
| Pharmacology/ mechanism of action: potentiates CFTR through a non-PK A dependent pathway, improving the probability of an open ion channel |
| Route of administration: oral |
Chemical structure of ivacaftor:
|
Chemical structure of lumacaftor: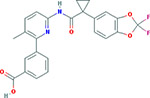 Essential pre-clinical study: Van Goor F, Hadida S, Groothenhuis PDJ, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M and Negulescu PA. Correction of the Phe-508del-CFTR protein processing defect in vitro by investigational drug VX-809. Proceedings of the National Academy of Sciences of the United States of America 2009; 108:18843–18848. Pivotal trials: Wainwright CE, Elborn JS, Ramsey BW et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. New England Journal of Medicine 2015, 2015;373:220–31. |
Acknowledgments
Funding
This paper has not been funded.
Footnotes
Declaration of interest
S McColley is a consultant speaker for Vertex Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as: * of interest ** of considerable interest
- 1.Ikuma M, Weslh M. Regulation of CFTR Cl- channel gating by ATP binding and hydrolysis. Proc Natl Acad. 2000; 97: 8675–8680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005; 352:1992–2001 [DOI] [PubMed] [Google Scholar]
- 3.A clinician’s guide to cystic fibrosis. CFTR Science https://www.cftrscience.com/?q=epidemiology [Accessed 6 August 2017]
- 4.Cystic Fibrosis Foundation Patient Data Registry Report. https://www.cff.org/Our-Research/CF-Patient-Registry/2015-Patient-Registry-Annual-Data-Report.pdf [Accessed 6 August 2017] [Google Scholar]
- 5.Farrell PM, White TB, Ren CL, et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. The Journal of pediatrics. 2017; 181S:S4–S15.e1 [DOI] [PubMed] [Google Scholar]
- 6.Konstan MW, Butler SM, Wohl ME, et al. Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. The Journal of pediatrics. June 2003;142(6):624–630. [DOI] [PubMed] [Google Scholar]
- 7.Yen EH, Quinton H, Borowitz D. Better nutritional status in early childhood is associated with improved clinical outcomes and survival in patients with cystic fibrosis. J Pediatr 2013;162:530–535 e531. [DOI] [PubMed] [Google Scholar]
- 8.McColley SA, Schechter MS, Morgan WJ, Pasta DJ, Craib ML, Konstan MW. Risk factors for mortality before age 18 years in cystic fibrosis. Pediatr Pulmonol. 2017;909–915. [DOI] [PubMed] [Google Scholar]
- 9.Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the Phe-508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren HY, Grove DE, De La Rosa O, et al. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Molecular biology of the cell 2013;24:3016–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flume PA, Liou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the Phe-508del-CFTR mutation. Chest 2012;142:718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clancy JP, Rowe SM, Accurso FJ et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012; 67: 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respiratory medicine 2014;2:527–538. [DOI] [PubMed] [Google Scholar]
- 14.Vertex Drug Information. http://pi.vrtx.com/files/uspi_lumacaftor_ivacaftor.pdf [accessed 6 August 2017]
- 15.ClinicalTrials.gov. A service of the National ClinicalTrials.gov. A service of the National Institutes of Health. http://www.clinicaltrials.gov/ct2/results?term=CFTR&Search=Search [Accessed 24 May 2017] Institutes of Health. http://www.clinicaltrials.gov/ct2/results?term=CFTR&Search=Search [Accessed 6 August 2017]
- 16.Rowe SM, McColley SA, Rietschel E, et al. Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for Phe-508del-CFTR. Annals of the American Thoracic Society 2017;14:213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.*.Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220–231.This is the report of two phase III placebo-controlled clinical trials of lumacaftor-ivacaftor therapy in Phe-508del homozugous cystic fibrosis. [DOI] [PubMed] [Google Scholar]
- 18.*.Konstan MW, McKone EF, Moss RB et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the Phe-508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respiratory medicine 2017;5(:107–118.This phase III follow-on study evaluates long term safety and efficacy of lumacaftor-ivacaftor therapy in Phe-508del homozygous cystic fibrosis. [DOI] [PubMed] [Google Scholar]
- 19.*.Milla CE, Ratjen F, Marigowda G et al. Lumacaftor/Ivacaftor in Patients Aged 6–11 Years with Cystic Fibrosis and Homozygous for Phe-508del-CFTR. AJRCCM 2017;195:912–920. This is the first published trial of lumacaftor-ivacaftor therapy in children aged 6–11 years old with Phe-508 del homozygous cystic fibrosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ratjen F, Hug C, Marigowda G et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR : a randomised, placebo-controlled phase 3 trial. Lancet Respiratory Medicine 2017; 5:557–567 [DOI] [PubMed] [Google Scholar]
- 21.Deeks ED. Lumacaftor/Ivacaftor: A Review in Cystic Fibrosis. Drugs 2016;76:1191–1201. [DOI] [PubMed] [Google Scholar]
- 22.McColley SA. Combination lumacaftor and ivacaftor therapy for cystic fibrosis. Expert opinion on orphan drugs. doi: 10.1517/21678707.2016.1133282 [DOI] [Google Scholar]
- 23.Kuk K, Taylor-Cousar JL. Lumacaftor and ivacaftor in the management of patients with cystic fibrosis: current evidence and future prospects. Therapeutic advances in respiratory disease 2015;9:313–326. [DOI] [PubMed] [Google Scholar]
- 24.McColley SA. A safety evaluation of ivacaftor for the treatment of cystic fibrosis, Expert Opinion on Drug Safety, 2016. DOI: 10.1517/14740338.2016.116566 [DOI] [PubMed]
- 25.Heltshe SL, Mayer-Hamblett N, Rowe S Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor et al. Clin Infect Dis 2015; 60:703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. https://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges/index.cfm?event=searchdetail.page&DrugNameID=234.
- 27.Cholon DM, Quinney NL, Fulcher ML, et al. Potentitor ivacaftor abrogates pharmachological correction of ΔF508 CFTR in cystic fibrosis. Sci Transl Med 2014; 6: 246ra96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veit G, Avramescu RG, Perdomo D, et al. Some gating potentiators, including VX-770, diminish ΔF508 CFTR functional expression. Sci Transl Med 2014; 6: 246ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vertex press release. http://investors.vrtx.com/releasedetail.cfm?ReleaseID=1019156 [accessed 7Aug2017]
- 30.Vertex press release. http://investors.vrtx.com/releasedetail.cfm?ReleaseID=995364 [accessed 7 Aug 2–17]