

Abstract
Aim.
The purpose of this article is to provide an overview of current insights into the neurodevelopmental and psychiatric manifestations of 22q11.2 deletion syndrome (22q11DS) in children and adolescents.
Recent findings.
The pediatric neuropsychiatric expression of 22q11DS is characterized by high variability, both inter-individual and intra-individual (different expressions over the lifespan). Besides varying levels of intellectual disability, the prevalence of autism spectrum disorders, attention deficit disorders, anxiety disorders, and psychotic disorders in young individuals with 22q11DS is significantly higher than in the general population, or in individuals with idiopathic intellectual disability. Possible explanations for this observed phenotypic variability will be discussed, including genetic pleiotropy, gene-environment interactions, the age-dependency of phenotypes, but also the impact of assessment and ascertainment bias as well as the limitations of our current diagnostic classification system.
Implications.
The implications inferred by these observations mentioned above bear direct relevance to both scientists and clinicians. Observations regarding the neuropsychiatric manifestations in individuals with 22q11DS exemplify the need for a dimensional approach to neuropsychiatric assessment, in addition to our current categorical diagnostic classification system. The potential usefulness of 22q11DS as a genetic model to study the early phases of schizophrenia as well as the phenomenon of neuropsychiatric pleiotropy observed in many CNV’s will be delineated. From a clinical perspective, the importance of regular neuropsychiatric evaluations with attention to symptoms not always captured in diagnostic categories and of maintaining equilibrium between individual difficulties and competencies and environmental demands will be discussed.
Keywords: 22q11DS, psychiatry, pleiotropy, pediatric psychiatry, clinical implications, schizophrenia
1. INTRODUCTION
Ever since the first reports of psychotic disorders in individuals with the 22q11.2 deletion (22q11DS) were published, now some 25 years ago, there has been increasing interest in this remarkable association[Bassett and others 1998; Murphy and others 1999; Pulver and others 1994; Shprintzen and others 1992]. These initial findings have been replicated in several studies, confirming an approximately 25-fold increased risk of developing schizophrenia in patients with 22q11DS compared to a ~1% lifetime risk in the general population[McGrath and others 2008]. Understandably, this observed association continues to receive much attention from both clinicians and investigators. From a clinical perspective the increased risk mandates careful monitoring of patients, in particular during adolescence and early adulthood, when the risk of psychotic development is highest. From a research perspective the association represents the strongest known genetic risk for schizophrenia conferred by a single genetic variant[Van and others 2017]. In addition, the phenotypic expression of schizophrenia in 22q11DS has been described as indiscernible from schizophrenia in the general population[Bassett and others 2003; Chow and others 2006]. These observations have spurred the research community to examine 22q11DS as a unique genetically homogeneous model for schizophrenia {Gur, 2017 #5081}, and, in the words of Thomas Insel: initiate prospective studies of this population that will provide “important insights into the trajectory from risk to disorder”[Insel 2010].
While the emphasis on schizophrenia and associated psychotic disorders (commonly referred to as “schizophrenia spectrum”) in 22q11DS is clearly justified[McDonald-McGinn and others 2015], a potential downside of a highly specific focus may be that the occurrence of other neuropsychiatric phenotypes in individuals with this genetic disorder can be easily overlooked. Multiple independent studies indicate significantly increased rates of a number of psychiatric and other neurodevelopmental disorders (including anxiety, autism spectrum, attention deficit disorders) in addition to schizophrenia in individuals with 22q11DS [Antshel and others 2006; Jolin and others 2009; Niklasson and others 2009; Schneider and others 2014a; Vorstman and others 2006]. This article will review the current knowledge of these phenotypes in 22q11DS, with a focus on childhood and adolescence. In addition, potential pitfalls regarding these findings will be examined, including the effect of ascertainment bias and possible limitations of categorical diagnostic classifications. Furthermore, both research and clinical care implications of the neuropsychiatric phenotypes in 22q11DS will be discussed.
2. NEURODEVELOPMENTAL DISORDERS IN CHILDHOOD AND ADOLESCENCE
2.1.1. Overview of the neuropsychiatric and cognitive phenotype
The neurodevelopmental and psychiatric phenotype of children and adolescents with 22q11DS is highly diverse. From infancy onward, delayed and/or impaired speech and language development are frequently observed [Swillen and others 1999]. Moreover, intellectual functioning in the borderline range (FSIQ between 70–85) is most common, followed by mild intellectual disability (FSIQ 55–70). More severe levels of intellectual disability are uncommon in children, but more frequently observed in adults with 22q11DS[De Smedt and others 2007; Evers and others 2009; Swillen and others 1999] suggesting that cognitive abilities may not be stable in all individuals with 22q11DS. Indeed, a recent longitudinal study found that, overall, individuals with 22q11DS showed a modest but significant decline in IQ between the ages of 8 and 24[Vorstman and others 2015]. Notably, in those who developed a psychotic disorder, the decline, most pronounced in verbal IQ, was significantly steeper compared to those without a psychotic disorder. These findings provide evidence that cognitive decline might be a useful early clinical marker of psychotic disorders in people with 22q11DS[Vorstman and others 2015].
The international brain and behavior consortium (IBBC) on 22q11DS has provided the most comprehensive overview of psychopathology in individuals with 22q11DS to date [Schneider and others 2014a]. This first study from this multicenter consortium reported on the psychopathology of 1401 individuals with 22q11DS, recruited across multiple sites, aged 6–68 (mean = 18,78, SD = 10,66) [Gur and others 2017; Schneider and others 2014a]. Figure 1 presents a summary of the findings across three different age groups between 6 and 25 years. Developmental disorders such as attention deficit hyperactivity disorder (ADHD) and autism spectrum disorder (ASD) are reported more frequently in the younger age groups as compared to the older groups (see also: [Antshel and others 2006; Niklasson and others 2009; Vorstman and others 2006]). Disruptive disorders are relatively less frequent overall, and their prevalence declines with increasing age. Mood disorders are frequently observed, and notably their prevalence increases significantly over time, particularly for major depressive disorder. Anxiety disorders are frequently reported across all age groups, but are especially prevalent in children and adolescents (see also: [Jolin and others 2009]). The prevalence of schizophrenia spectrum disorders increases significantly over time, with prevalence rates of 24% in emerging adults and about 41% in individuals over 25 years old (see also: [Bassett and others 2005; Murphy and others 1999]).
Figure 1:
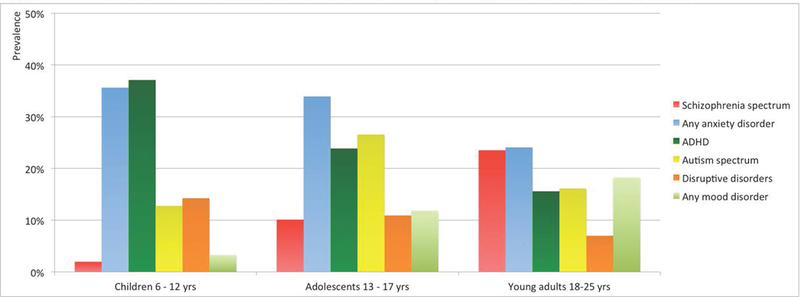
Prevalence of psychiatric disorders in 22q11DS from childhood to young adulthood.
2.1.2. The specificity of the psychiatric and neurodevelopmental profile in 22q11DS
Observations from epidemiological studies indicate that the overall rate of psychopathology is increased in youth with idiopathic intellectual impairment [Einfeld and others 2011]. One particularly salient question therefore is to what extent the prevalence of psychiatric disorders in 22q11DS deviates from what is reported in unselected cohorts with intellectual impairment. Table 1 lists the prevalence rates of developmental and psychiatric disorders in individuals with 22q11DS (6 through 17 years) compared to observations in individuals with idiopathic intellectual impairment (n=641)[Emerson and Hatton 2007] and the general population.
Table 1:
Prevalence rates of psychopathology in youth with 22q11DS compared to youth with idiopathic intellectual impairment and the general population.
| Youth with 22q11DSa
|
Youth with idiopathic intellectual impairmentb | Youth in general populationc | |
|---|---|---|---|
| ADHD | 32.1% | 8.3% | 5%I |
| Disruptive disordersd | 13.0% | 20.5% | 6%II |
| ASD | 21.5% | 8.0% | 1 – 1.5%III, IV |
| Any mood disorder | 7.0% | 1.4% | 10%II, V |
| Any anxiety disorder | 34.9% | 11.4% | 12%VI |
| Any psychotic disorder, including schizophrenia | 5.5% | not reported | << 1% VII, VIII, IX |
Data from [Schneider and others 2014a] (805 individuals, note that percentages deviate from figure 1 since we collapsed data obtained in the age range 6 to 17 years, most individuals assessed at a level of mild intellectual disability)
Data from [Emerson and Hatton 2007] (641 individuals, age range 5 to 16 years, intellectual impairment based on parental/teacher report, most individuals estimated at a level of mild intellectual disability, the numbers represent point prevalence (i.e. symptoms present during the month – half year preceding the assessment)
Data from cohorts including both children and adolescents were used to obtain these estimates; exact age ranges vary between the different studies.
Oppositional defiant disorder, conduct disorder.
Autism and others 2009
Developmental Disabilities Monitoring Network Surveillance Year Principal and others 2014
It is important to note that prevalence rates reported in table 1 are all to some extent influenced by variable degrees of ascertainment and/or assessment biases and should therefore be compared with caution. For instance, individuals in the idiopathic intellectual impairment group were not recruited through a clinical site and intellectual impairment was established based on parental / teacher report. In contrast, many studies contributing to the overview of 22q11DS findings were conducted at a clinical site (see discussion below) and in many individuals IQ was obtained through formal testing. Rates reported in the general population are not specific for youth.
Notwithstanding these precautions, some preliminary insights can be gained from table 1. First, from this young age onwards, psychotic disorders occur in the 22q11DS population (between age 13 and 17 years the rate is already 10%). Many individuals fall into the diagnostic category “psychotic disorder not otherwise specified”, likely due to the young age at assessment. Second, the prevalence of other developmental disorders (ASD and ADHD) and mood/anxiety disorders is increased in 22q11DS youth over what is observed in a population with idiopathic intellectual impairment. Third, the rate of disruptive disorders in people with 22q11DS is well below the rate in the idiopathic intellectual impairment population. These findings suggest that the 22q11.2 deletion has a specific impact on the behavioral phenotype and therefore that the risk of psychopathology cannot be solely explained as an unspecific consequence of intellectual impairment (Figure 2, scenario A).
Figure 2:
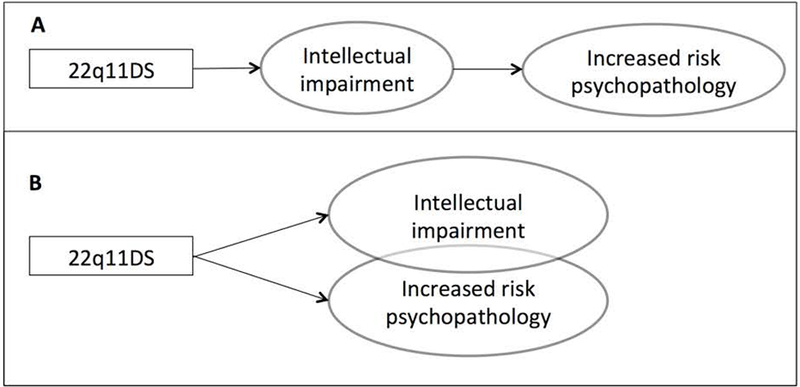
Two possible associations between intellectual impairment and the increased risk for psychopathology in 22q11DS.
Further support for the proposition that the observed increased rates of psychopathology are specific to 22q11DS (Figure 2, scenario B), as opposed to a non-specific effect of broad intellectual impairment, is provided by studies that show no correlation between cognitive level and the risk for psychiatric disorders in individuals with 22q11DS [Evers and others 2014; Niarchou and others 2014]. Accordingly, higher rates of psychopathology are reported in 22q11DS individuals compared to IQ-matched controls[Jansen and others 2007]. Inversely, the lower than expected prevalence rates of both disruptive disorders (albeit higher than in the general population, see Table 1) and substance use disorders in 22q11DS (Vingerhoets et al. under review), compared to both general and idiopathic intellectually impaired populations [Carroll Chapman and Wu 2012; Swerts and others 2016] provide further evidence for a specific genetic effect. These findings suggest that scenario B (Figure 2) is likely - although it does not exclude some effect by the mechanism of scenario A altogether. Furthermore, findings suggest that 22q11DS increases the risk of some psychiatric disorders, but not of others. Indeed, similar to observations in cohorts with other structural pathogenic variants, emerging evidence indicates that specific profiles of psychopathology can be distinguished when comparing 22q11DS to idiopathic, unselected populations[Bruining and others 2014; Smith and others 2012].
2.2. Understanding the high degree of phenotypic variability
2.2.1. Neuropsychiatric pleiotropy
The psychiatric phenotypic expression of 22q11DS is highly variable. Many different psychiatric disorders are associated with this CNV, phenotypes can differ considerably between individuals, and differences exist across age groups. This variability is consistent with the phenomenon of phenotypic pleiotropy observed in many rare CNV’s [Bassett and others 2010; Vorstman and Ophoff 2013], whereby one specific genetic variant can result in independent phenotypic expressions. For example, in the context of 22q11DS, the presence or absence of congenital cardiac problems does not seem to be associated with an altered risk for psychotic disorders [Bassett and Chow 2008]. Therefore, congenital cardiac defects and psychotic disorders can be considered as true pleiotropic manifestations of the 22q11.2 deletion.
Observations from numerous studies suggest that in 22q11DS different psychiatric phenotypes can also emerge independently, as pleiotropic conditions. The distinction between true pleiotropy and “pseudopleiotropy” is important in this regard. The latter refers to phenotypes that are observed as separate manifestations, whereas in reality they represent the same pathological process, for instance at different developmental stages [Vorstman and others 2013]. Prospective longitudinal studies can provide insight in this regard. For example, a recent study found evidence for true pleiotropy regarding ASD and psychotic disorders, given that individuals with 22q11DS with ASD at a young age were not at an increased risk for developing psychotic disorders later in life compared to those without ASD [Fiksinski and others 2017] [Vorstman and others 2013]. On the other hand, cognitive decline [Duijff and others 2012] and psychotic disorders in 22q11DS were initially reported as two different phenotypes, while subsequent prospective studies indicated that these two phenotypes represent, at least in a subset of individuals, the same pathological process but at different developmental stages [Vorstman and others 2015], thereby providing an example of pseudopleiotropy. Such findings underline the importance of considering additional factors that may influence the observed phenotypic variability. Here, we will briefly discuss such factors, including the phenomena of gene-environment interactions, cross-site ascertainment and assessment differences, age-dependent phenotypes, and diagnostic classification artifacts that impact the observed comorbidity.
2.2.2. Environmental factors as a source of inter-individual variability
Several studies point towards the impact of environmental factors on the observed behavioral phenotype in individuals with 22q11DS. For example, studies have shown that lower parental socio-economic-status (SES) and intrusive parenting style [Barber 2002] were associated with worse social functioning and other clinically significant problems in children with 22q11DS [Allen and others 2014; Shashi and others 2010]. Additional evidence is provided by a study examining the effect of Sept5 deficiency (one of the genes in the 22q11.2 region) on social functioning in a mice model of 22q11DS[Harper and others 2012]. This study showed that Sept5 deficient mice had decreased affiliative social interactions compared to wild type mice. Interestingly, Sept5 deficient male mice exposed to individual housing were characterized by reduced anxiety and increased affiliative social interactions compared to mice exposed to group-housing, thereby showing a significant gene-environment interaction. Although more research is needed in this regard, such emerging evidence points to complex interactions between genetic and environmental factors as a source of phenotypic variability among individuals with 22q11DS.
2.2.3. Ascertainment and assessment bias
The IBBC studies[Schneider and others 2014a] that contributed to the overview in Figure 1 varied considerably in terms of ascertainment. Some sites are child psychiatry clinics, others are non-clinical research settings and still others function primarily as adult specialty clinics. Studies conducted in clinical sites may bias against individuals with 22q11DS who function well, while clinical treatment may also reduce the “true natural” occurrence of psychiatric phenotypes in an untreated population. A recent epidemiological Danish nation-wide study of 22q11DS and 22q11.2 duplication syndrome offers a different perspective, as a population-based study. This study included 244 adult individuals with 22q11DS identified through case registration in a population of 3,768,943. The reported patterns of developmental and psychiatric disorders in 22q11DS, compared to 24,400 age- and gender-matched population controls, were similar to what is described in clinically ascertained 22q11DS cohorts, but, as expected, with somewhat lower prevalence rates [Hoeffding and others 2017].
The IBBC studies also varied with regard to the diagnostic assessment tools they employed and comorbid mental health problems screened [Schneider and others 2014a]. For example, not all sites used instruments to assess at risk status for psychotic disorders or standardized assessment for autism. Consequently, some of the differences in prevalence rates reported from different sites may be, at least partly, explained by differences in ascertainment and assessment (biases) across sites.
2.2.4. Age-dependent phenotypes
In individuals with 22q11DS, the variability observed across age groups can be inherent to phenotypic characteristics such as typical age of onset, consistent with observations in the general population. For example, in patient cohorts under the age of 18, one would not expect to find a high prevalence of schizophrenia spectrum disorders, as the first psychotic episode typically emerges in late adolescence/early adulthood [Owen and others 2016]. Differences in typically conducted psychiatric assessments across age groups may also contribute to the phenotypic variability reported across different ages. Standard adult psychiatric assessment often does not include screening for developmental disorders such as ASD or ADHD. In cohorts assessed as adults, one would therefore not expect to find a high prevalence of such developmental disorders, while in reality a portion of these individuals might have been diagnosed as such, had their psychiatric assessment – either as children or adults – included screening for these developmental disorders. With increasing awareness of ASD and ADHD in adults and associated clinical service developments however [Murphy and others 2016], the reported prevalence of ASD and ADHD in adults with 22q11DS may change in future reports.
Not only can psychiatric conditions emerge as an individual matures, certain psychiatric disorders or neurodevelopmental presentations may also improve over time. Indeed, our clinical impression suggests that in some individuals with 22q11DS with a previous childhood neurodevelopmental diagnosis, improvement occurs to such extent that a diagnostic classification may no longer be justified in adulthood (e.g. ADHD or ASD), which is consistent with what is observed in idiopathic populations with such neurodevelopmental disorders (e.g.[Fein and others 2013; Woolfenden and others 2012] ). The finding that in a sample of 70 individuals with 22q11DS 30% of those previously diagnosed with attention deficit disorder (ADD) did not manifest sufficient symptoms justifying an ADD diagnosis at follow-up assessment [Antshel and others 2010] provides preliminary evidence in this regard. Although such late-maturation trajectories are observed only in a minority of individuals with 22q11DS, they are relevant and warrant further study.
2.2.5. The pitfalls of a categorical approach to the psychiatric phenotype in 22q11DS
From both a clinical and a research perspective, employing a categorical diagnostic classification system has merits, as it provides clinicians and researchers with a shared vocabulary. However, a too stringent adherence to such a categorical, dichotomous approach to psychopathology also has substantial limitations. While this is relevant to all psychiatric populations, several observations in the 22q11DS population render the consideration of such potential limitations particularly salient for these individuals. On the one hand, it may result in the application of several diagnostic labels to account for a relatively small set of symptoms, while on the other hand clinically relevant but isolated symptoms may not readily fit in any diagnostic category and thus be overlooked [Baker and Vorstman 2012].
From a categorical perspective, a substantial portion of 22q11DS individuals is diagnosed with more than one psychiatric disorder [Jolin and others 2009; Schneider and others 2014a; Tang and others 2014]. In addition, categorical classifications may be influenced by different interpretations of the same symptom domains. For instance, repetitive behaviors may be classified as an obsessive-compulsive disorder by one clinician, while the same symptoms in the same patient may be considered as part of an autistic spectrum disorder by another. The same diagnostic ambiguity may exist with regard to anxiety symptoms, which can justify an anxiety disorder but may not always be considered as such when occurring in the context of a psychotic disorder. Such ambiguities are inevitable as symptoms belonging to different diagnostic categories are frequently observed within the same individual with 22q11DS [Baker and Vorstman 2012]. Moreover, even in those children not meeting formal criteria for a psychiatric disorder, clinically relevant psychiatric symptoms are often present. Even within one symptom domain the use of categorical diagnoses may fall short in describing the reality of psychiatric symptoms with clinical relevance. For example, much higher prevalence rates of psychotic symptoms (25%) than of psychotic disorders (10%) are observed in adolescents with 22q11DS [Schneider and others 2016; Vorstman and others 2006]. Importantly, such symptoms, though not reflected in an individual’s psychiatric diagnosis, may still be relevant in understanding an individual’s profile of difficulties and competencies, as well as in implementing adequate treatment and preventive strategies.
3. IMPLICATIONS FOR RESEARCH
3.1. 22q11DS as a genetic model for schizophrenia
Several aspects of 22q11DS make this genetic disorder a highly appealing model to investigate neuroscientific questions, particularly the etiology of schizophrenia[Van and others 2017]. The highly increased risk for this illness in 22q11DS patients, and the opportunity to identify individuals early in life based on their genetic diagnosis has allowed investigators to study clinical and biological correlates of the developmental trajectory of schizophrenia. Moreover, a unique and easily overlooked aspect of these studies is that they are conducted in a genetically relatively homogeneous context, i.e. all individuals share the same 22q11.2 deletion, which can be assumed to be the cause of their high vulnerability for schizophrenia. In contrast, the broad and largely unknown genetic heterogeneity of schizophrenia hampers studies in unselected general population cohorts. Recent studies confirm the usefulness of 22q11DS as a human genetic model to unravel the gene x environment interactions leading to schizophrenia[Insel 2010]. Importantly, the psychopathological path leading to transition to psychosis in 22q11DS [Schneider and others 2016; Weisman and others 2017] is broadly comparable to that observed in other clinical high risk samples[Fusar-Poli and others 2013]. More specifically, the sub-threshold psychotic symptoms and the Clinical High Risk (CHR) for psychosis criteria, previously developed in the general population[Fusar-Poli and others 2013], are reliable and also applicable in the 22q11DS population.
3.2. 22q11DS exemplifies the need for dimensional and repeated assessment approaches
Up until now, subclassifying individuals with 22q11DS by psychiatric diagnoses has not proven particularly useful in delineating underlying neurogenetic mechanisms. Indeed, it has been proposed that despite the divergence in diagnostic classifications, observations from different studies in 22q11DS converge into a limited number of symptom domains[Baker and Vorstman 2012]. Furthermore, preliminary associations have been described between genetic factors and symptoms dimensions that cut across existing diagnostic categories[Bassett and others 2007; Raux and others 2007; Shashi and others 2006]. These and other studies underscore the potential added value of broad dimensional, quantitative and repeated assessments as a means towards a more dimensional perspective on mental health equilibrium and the risk of psychopathology[Nelson and others 2017].
3.3. 22q11DS as a model for neuropsychiatric pleiotropy in rare copy number variants
The high variability of neuropsychiatric phenotypes observed in 22q11DS represents another unique research opportunity. The past decade has witnessed the discovery of a growing list of pathogenic genetic variants, including Copy Number Variants (CNVs)[Malhotra and Sebat 2012] and rare Single Nucleotide Variants (SNVs)[RK and others 2017]. Typically, the prevalence of each of these pathogenic variants is rare, but when present in an individual they can confer a substantial risk. The picture emerging from neuropsychiatric studies in individuals carrying these rare high impact variants is remarkably similar to what is observed in 22q11DS: a high degree of neuropsychiatric variability with increased rates of different disorders including schizophrenia, anxiety, ADHD, ASD and affective disorders and varying levels of cognitive impairment[Vorstman and Ophoff 2013]. Examples include, but are not limited to, the 3q29 deletion, which is associated with various neuropsychiatric disorders including anxiety and mood disorders and schizophrenia [Glassford and others 2016], and the 16p11.2 deletion, associated with intellectual disability, ASD, and schizophrenia [Moreno-De-Luca and others 2015]. Quantifying the risk of the different associated disorders and understanding how they emerge across the lifespan is essential to improve counseling and clinical care for individuals carrying these rare but high impact genetic variants[Vorstman and others 2017]. To this end, studies are needed to investigate for each of these rare pathogenic genetic variants to what extent there is true neuropsychiatric pleiotropy, the age-dependent emergence of certain phenotypes, and the possible influence of classification artifacts and ascertainment bias. In addition, it will be key to identify factors, both genetic and environmental, that modulate the different neuropsychiatric outcomes. Similarly, early phenotypic characteristics should be studied as potential markers for poor functional outcome. However, such studies are severely hampered by the fact that each of these variants occurs at extremely low rates in the population, impeding the collection of sufficiently powered samples.
It is in this context that the 22q11.2 deletion syndrome provides a unique research opportunity. Its prevalence is high (estimated 1/2,000–4,000)[McDonald-McGinn and others 2015] and its genetic description in the early 1980s[McDonald-McGinn and others 2015] has preceded by approximately two decades the much more recent discovery of the majority of other rare pathogenic genetic variants associated with psychiatric phenotypes[Malhotra and Sebat 2012]. This has afforded 22q11DS studies a significant advance over studies on other pathogenic variants. Indeed, at present, multiple cohorts of several hundreds of individuals with 22q11DS are examined in research institutes across the world. Findings of 22q11DS studies show the potential of using early phenotypic characteristics to identify subgroups with poor functional outcome[Schneider and others 2014b], and indicate several early potential biomarkers of psychotic disorder[Bakker and others 2016; Padula and others 2017; Ramanathan and others 2016; Raux and others 2007; Scariati and others 2016; Tomescu and others 2014]. The IBBC [Gur and others 2017] has pooled phenotypic data from over 22 sites which together amount to well over 1,800 individuals with 22q11DS. This sample size, unprecedented in any other study on rare genetic variants with neuropsychiatric impact, allows the investigation of aforementioned questions with sufficient statistical power.
4. IMPLICATIONS FOR CLINICAL CARE
4.1. Need for regular psychiatric assessments
International guidelines for clinical care for individuals with 22q11DS mandate regular psychiatric and cognitive assessment[Bassett and others 2011], understandably so, considering the overall high rates of psychopathology in this patient population. The observations reviewed in this article provide several directions in this regard. Psychiatric symptoms and disorders in 22q11DS may either remain constant over time, they may emerge or intensify (e.g. psychotic disorders), or they may be outgrown and no longer be valid as individuals mature. Cognitive abilities may not be stable in a subgroup of patients and a decline in verbal IQ may indicate increased risk of developing a psychotic disorder[Vorstman and others 2015]. In order to maintain accuracy in describing an individual’s neuropsychiatric profile (and thereby allowing individualized mental healthcare) this time-related phenotypic variability needs to be considered and, consequently, repeated psychiatric and neuropsychological assessments are required.
Emerging mental, rather than physical, health concerns are more likely to bring adolescents and young adults with 22q11DS to medical attention[Fung and others 2015; McDonald-McGinn and others 2015; Schneider and others 2016; Swillen 2016]. Despite increasing awareness of the importance of planned transitions for young people with neurodevelopmental disorders to adult health and social care[Medicine. 2002; Stewart 2009; Young and others 2011], there is limited investigation of how best to do this[Belling and others 2014; Paul and others 2015] and future research is warranted. However, best practice guidelines for young people with 22q11DS should include a planned transition of mental health care (including psychiatric and cognitive assessments if resources allow) across different life stages and stressors. Examples include when moving from primary to secondary school and from adolescent to adult health care.
With respect to all clinical recommendations for individuals with 22q11DS, including the need for regular psychiatric assessments and the importance of a guided transition to adulthood, there is the necessity to acknowledge, investigate and work towards overcoming the obstacles to implementing such recommendations. While there is consensus regarding the importance of discussing risk for psychiatric disorders in these individuals repeatedly throughout different stages of life [Bassett and others 2011; Hercher and Bruenner 2008], studies have reported that, for various possible reasons including stigma, other medical issues requiring attention and a young age at time of counseling, at present such discussions are insufficiently implemented in most clinical settings [Baughman and others 2015; Morris and others 2013].
4.2. Need to look beyond diagnostic categories
Full-blown developmental and psychiatric disorders occur and may necessitate pharmacological or cognitive-behavioral therapeutic interventions in individuals with 22q11DS. However, the myriad of clinically relevant symptoms in these individuals is not always sufficiently captured in diagnostic classification categories. When symptoms are under-recognized or occur in isolation (i.e. in the absence of usually co-occurring symptoms critical for a diagnostic classification), they will not be reflected in an individual’s psychiatric diagnosis. Such symptoms may have significant impact on an individual’s daily functioning, but will remain unrecognized if only diagnostic categories are taken into account. In psychiatric assessments of individuals with 22q11DS, careful attention should therefore be paid not only to psychiatric diagnoses, but also to symptomatology as this may be important for lifestyle adaptations and professional care and (symptomatic) treatment. Taking symptom domains into consideration is pivotal in understanding an individual’s profile of competencies and difficulties[Baker and Vorstman 2012; Beaton and Simon 2011]. Delineating such a profile is highly informative in finding and/or creating an environment that is optimally adapted to the individual.
4.3. Emerging symptoms may represent imbalance abilities and demands; the importance of stress
In all instances where severe psychopathology becomes manifest, adequate psychiatric treatment, often including pharmacological treatment, is required. In some instances where new symptoms emerge in a developing child, such as anxiety, depression, or psychosis, they appear to be related to an emerging discrepancy between the individual’s competencies and difficulties, and environmental demands. Clinically relevant psychiatric symptoms of mood, anxiety or psychosis may be indicative of a mismatch between an individual’s abilities and environmental demands and symptoms may improve when this balance is recovered. In some instances an accurate understanding of an individual’s neuropsychiatric phenotype, taking into consideration subclinical symptoms and fluctuations over time, allows for adequate and timely environmental adaptations, without exposing individuals to psychopharmacological compounds and their related side-effects.
If a mismatch between an individual’s capacities and difficulties and their environmental demands exists for a prolonged period of time, the individual is likely to experience chronic stress. Stress has been identified as a risk factor for psychopathology in the general population[Sommer and others 2016] as well as a trigger to manifestation of a psychotic episode in idiopathic schizophrenia populations[Yui and others 1999]. Indeed, high levels of anxiety in youth with 22q11DS have been proposed as a predictor of transition to psychosis[Gothelf and others 2013], which supports the hypothesized importance of stress. In light of the 20–25% risk for developing schizophrenia and related psychotic disorders that individuals with 22q11DS have, optimal care should be taken to avoid stress. Creating and maintaining a balance between their neurocognitive, social and behavioral profile and environmental demands is essential in this regard.
5. CONCLUSION
The 22q11.2 deletion exemplifies the fast emerging novel class of rare pathogenic genetic variants as identifiable etiologies in the field of psychiatry, which raises important new challenges with immediate relevance for both researchers and clinicians. The early discovery of this rare CNV in the 1980’s however, has allowed a time advantage over the majority of the other, more recently identified pathogenic rare variants. Consequently, implications from studies on 22q11DS are not limited to this genetic disorder, but can also contribute to the understanding of phenomena observed in other rare pathogenic variants, including the high variability and variable expression of associated phenotypes.
In addition to the well-established risk for schizophrenia, individuals with 22q11DS are at increased risk for a wide range of psychopathology from early childhood onwards. To some extent, this phenotypic variability may be an artifact of forcing the observed symptoms into our current diagnostic classifications. In addition, variable assessment and ascertainment methods are bound to further contribute to differences between studies. However, notwithstanding these caveats, several observations from 22q11DS studies begin to stand out.
First, the prevalence of some pediatric psychiatric phenotypes, in particular anxiety, ASD, ADHD, and mood disorders, clearly exceeds what is observed in idiopathic ID. Together with the increased risk of schizophrenia in adolescents and (young) adults with 22q11DS, these observations strongly suggest a phenotypic effect that is specific to this genetic variant. Second, some neuropsychiatric phenotypes in 22q11DS are independent of others, indicating true pleiotropy, while others may represent time-dependent expressions of the same disease trajectory. Third, the disparity of reported phenotypes is mostly manifested in the limited framework of categorical diagnostic classifications. When focusing on the observed symptom domains, regardless of the classification used, a stronger coherence between different studies becomes readily apparent.
The clinical implications follow from these observations. In any child with 22q11DS a thorough psychiatric evaluation is mandated, regardless of intellectual level. Furthermore, expression of psychiatric phenotypes may vary over time even within the same individual, such as the decline in cognitive functioning observed in a subgroup of 22q11DS individuals. Taken together, these observations underline the importance of repeated clinical evaluations in this population. In the context of a genetic predisposition for developing schizophrenia, it is important to maintain an optimal balance between individual abilities and environmental expectations. A global low cognitive level (IQ), but also specific (and sometimes covert) relative weaknesses in cognitive domains and neurodevelopmental functions (e.g. attention, information processing, social and communicative abilities and sensitivity to sensory input) can contribute to chronic stress due to demands that exceed abilities. The clinical importance is twofold. First, such a situation is undesirable in itself as it causes discomfort and stress to any person. Second, numerous studies indicate that stress may play a role in the course of schizophrenia. While such evidence is not yet robustly available in the 22q11DS population, it is possible that in the context of a high genetic risk, high levels of stress may contribute to the expression of schizophrenia.
The genetic predisposition for psychotic disorders conferred by 22q11DS provides strong impetus to obtain detailed insight into an individual’s cognitive and neurodevelopmental profile to avoid or reduce (chronic) situations of stress. Interventions to correct such situations may have direct clinical impact but can also serve as a model to study the feasibility of primary and secondary intervention strategies in a population at risk for schizophrenia. However, the full scope of clinically relevant symptoms may often not be accurately represented in diagnostic classifications. Findings from studies in the 22q11DS population indicate that a dimensional, quantitative symptom assessment of psychopathology may be required in order to obtain the most accurate picture, both for clinical care and for scientific research.
ACKNOWLEDGEMENTS
This study was supported by the NIMH International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome (grant number 5U01MH101722–02). The Ter Meulen Grant of the Royal Netherlands Academy of Arts and Sciences (A.M. Fiksinski)
Footnotes
All authors declare that there are no conflicts of interest in relation to the subject of this study.
REFERENCES
- Allen TM, Hersh J, Schoch K, Curtiss K, Hooper SR, Shashi V. 2014. Association of the family environment with behavioural and cognitive outcomes in children with chromosome 22q11.2 deletion syndrome. J Intellect Disabil Res 58(1):31–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antshel KM, Fremont W, Roizen NJ, Shprintzen R, Higgins AM, Dhamoon A, Kates WR. 2006. ADHD, major depressive disorder, and simple phobias are prevalent psychiatric conditions in youth with velocardiofacial syndrome. J Am Acad Child Adolesc Psychiatry 45(5):596–603. [DOI] [PubMed] [Google Scholar]
- Antshel KM, Shprintzen R, Fremont W, Higgins AM, Faraone SV, Kates WR. 2010. Cognitive and psychiatric predictors to psychosis in velocardiofacial syndrome: a 3-year follow-up study. J Am Acad Child Adolesc Psychiatry 49(4):333–344. [PMC free article] [PubMed] [Google Scholar]
- Autism, Developmental Disabilities Monitoring Network Surveillance Year Principal I, Centers for Disease C, Prevention. 2009. Prevalence of autism spectrum disorders - Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill Summ 58(10):1–20. [PubMed] [Google Scholar]
- Baker K, Vorstman JA. 2012. Is there a core neuropsychiatric phenotype in 22q11.2 deletion syndrome? Current opinion in neurology 25(2):131–137. [DOI] [PubMed] [Google Scholar]
- Bakker G, Caan MW, Schluter RS, Bloemen OJ, da Silva-Alves F, de Koning MB, Boot E, Vingerhoets WA, Nieman DH, de Haan L, Booij J, van Amelsvoort TA. 2016. Distinct white-matter aberrations in 22q11.2 deletion syndrome and patients at ultra-high risk for psychosis. Psychol Med 46(11):2299–2311. [DOI] [PubMed] [Google Scholar]
- Barber BK. 2002. Intrusive Parenting: How Psychological Control Affects Children and Adolescents. Washington, DC, USA: American Psychological Association. [Google Scholar]
- Bassett AS, Caluseriu O, Weksberg R, Young DA, Chow EW. 2007. Catechol-O-methyl transferase and expression of schizophrenia in 73 adults with 22q11 deletion syndrome. BiolPsychiatry 61(10):1135–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW. 2008. Schizophrenia and 22q11.2 deletion syndrome. Curr Psychiatry Rep 10(2):148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, AbdelMalik P, Gheorghiu M, Husted J, Weksberg R. 2003. The schizophrenia phenotype in 22q11 deletion syndrome. The American journal of psychiatry 160(9):1580–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA. 2005. Clinical features of 78 adults with 22q11 Deletion Syndrome. Am J Med Genet A 138(4):307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Hodgkinson K, Chow EW, Correia S, Scutt LE, Weksberg R. 1998. 22q11 deletion syndrome in adults with schizophrenia. AmJMedGenet 81(4):328–337. [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J, International 22q11.2 Deletion Syndrome C. 2011. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr 159(2):332–339 e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Scherer SW, Brzustowicz LM. 2010. Copy number variations in schizophrenia: critical review and new perspectives on concepts of genetics and disease. Am J Psychiatry 167(8):899–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman ST, Morris E, Jensen K, Austin J. 2015. Disclosure of psychiatric manifestations of 22q11.2 deletion syndrome in medical genetics: A 12-year retrospective chart review. Am J Med Genet A 167A(10):2350–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaton EA, Simon TJ. 2011. How might stress contribute to increased risk for schizophrenia in children with chromosome 22q11.2 deletion syndrome? J Neurodev Disord 3(1):68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belling R, McLaren S, Paul M, Ford T, Kramer T, Weaver T, Hovish K, Islam Z, White S, Singh SP. 2014. The effect of organisational resources and eligibility issues on transition from child and adolescent to adult mental health services. J Health Serv Res Policy 19(3):169–176. [DOI] [PubMed] [Google Scholar]
- Bruining H, Eijkemans MJ, Kas MJ, Curran SR, Vorstman JA, Bolton PF. 2014. Behavioral signatures related to genetic disorders in autism. Mol Autism 5(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd L, Kerbeshian J. 1987. A North Dakota prevalence study of schizophrenia presenting in childhood. J Am Acad Child Adolesc Psychiatry 26(3):347–350. [DOI] [PubMed] [Google Scholar]
- Carroll Chapman SL, Wu LT. 2012. Substance abuse among individuals with intellectual disabilities. Res Dev Disabil 33(4):1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow EW, Watson M, Young DA, Bassett AS. 2006. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophr Res 87(1–3):270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland WE, Angold A, Shanahan L, Costello EJ. 2014. Longitudinal patterns of anxiety from childhood to adulthood: the Great Smoky Mountains Study. Journal of the American Academy of Child and Adolescent Psychiatry 53(1):21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EJ, Mustillo S, Erkanli A, Keeler G, Angold A. 2003. Prevalence and development of psychiatric disorders in childhood and adolescence. Archives of general psychiatry 60(8):837–844. [DOI] [PubMed] [Google Scholar]
- De Smedt B, Devriendt K, Fryns JP, Vogels A, Gewillig M, Swillen A. 2007. Intellectual abilities in a large sample of children with Velo-Cardio-Facial Syndrome: an update. J Intellect Disabil Res 51(Pt 9):666–670. [DOI] [PubMed] [Google Scholar]
- Developmental Disabilities Monitoring Network Surveillance Year Principal I, Centers for Disease C, Prevention . 2014. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ 63(2):1–21. [PubMed] [Google Scholar]
- Duijff SN, Klaassen PW, de Veye HF, Beemer FA, Sinnema G, Vorstman JA. 2012. Cognitive development in children with 22q11.2 deletion syndrome. Br J Psychiatry 200(6):462–468. [DOI] [PubMed] [Google Scholar]
- Einfeld SL, Ellis LA, Emerson E. 2011. Comorbidity of intellectual disability and mental disorder in children and adolescents: a systematic review. J Intellect Dev Disabil 36(2):137–143. [DOI] [PubMed] [Google Scholar]
- Emerson E, Hatton C. 2007. Mental health of children and adolescents with intellectual disabilities in Britain. Br J Psychiatry 191:493–499. [DOI] [PubMed] [Google Scholar]
- Evers LJ, De Die-Smulders CE, Smeets EE, Clerkx MG, Curfs LM. 2009. The velo-cardio-facial syndrome: the spectrum of psychiatric problems and cognitive deterioration at adult age. Genet Couns 20(4):307–315. [PubMed] [Google Scholar]
- Evers LJ, van Amelsvoort TA, Candel MJ, Boer H, Engelen JJ, Curfs LM. 2014. Psychopathology in adults with 22q11 deletion syndrome and moderate and severe intellectual disability. J Intellect Disabil Res 58(10):915–925. [DOI] [PubMed] [Google Scholar]
- Fein D, Barton M, Eigsti IM, Kelley E, Naigles L, Schultz RT, Stevens M, Helt M, Orinstein A, Rosenthal M, Troyb E, Tyson K. 2013. Optimal outcome in individuals with a history of autism. J Child Psychol Psychiatry 54(2):195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiksinski AM, Breetvelt EJ, Duijff SN, Bassett AS, Kahn RS, Vorstman JA. 2017. Autism Spectrum and psychosis risk in the 22q11.2 deletion syndrome. Findings from a prospective longitudinal study. Schizophr Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung WL, Butcher NJ, Costain G, Andrade DM, Boot E, Chow EW, Chung B, Cytrynbaum C, Faghfoury H, Fishman L, Garcia-Minaur S, George S, Lang AE, Repetto G, Shugar A, Silversides C, Swillen A, van Amelsvoort T, McDonald-McGinn DM, Bassett AS. 2015. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet Med 17(8):599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusar-Poli P, Borgwardt S, Bechdolf A, Addington J, Riecher-Rossler A, Schultze-Lutter F, Keshavan M, Wood S, Ruhrmann S, Seidman LJ, Valmaggia L, Cannon T, Velthorst E, De Haan L, Cornblatt B, Bonoldi I, Birchwood M, McGlashan T, Carpenter W, McGorry P, Klosterkotter J, McGuire P, Yung A. 2013. The psychosis high-risk state: a comprehensive state-of-the-art review. JAMA Psychiatry 70(1):107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillberg C 2001. Epidemiology of early onset schizophrenia. Cambridge: Cambridge University Press. [Google Scholar]
- Glassford MR, Rosenfeld JA, Freedman AA, Zwick ME, Mulle JG, Unique Rare Chromosome Disorder Support G. 2016. Novel features of 3q29 deletion syndrome: Results from the 3q29 registry. Am J Med Genet A 170A(4):999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothelf D, Schneider M, Green T, Debbane M, Frisch A, Glaser B, Zilkha H, Schaer M, Weizman A, Eliez S. 2013. Risk factors and the evolution of psychosis in 22q11.2 deletion syndrome: a longitudinal 2-site study. Journal of the American Academy of Child and Adolescent Psychiatry 52(11):1192–1203 e1193. [DOI] [PubMed] [Google Scholar]
- Gur RE, Bassett AS, McDonald-McGinn DM, Bearden CE, Chow E, Emanuel BS, Owen M, Swillen A, Van den Bree M, Vermeesch J, Vorstman JAS, Warren S, Lehner T, Morrow B. 2017. A neurogenetic model for the study of schizophrenia spectrum disorders: the International 22q11.2 Deletion Syndrome Brain Behavior Consortium. Mol Psychiatry 22(12):1664–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper KM, Hiramoto T, Tanigaki K, Kang G, Suzuki G, Trimble W, Hiroi N. 2012. Alterations of social interaction through genetic and environmental manipulation of the 22q11.2 gene Sept5 in the mouse brain. Hum Mol Genet 21(15):3489–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellgren L, Gillberg C, Enerskog I. 1987. Antecedents of adolescent psychoses: a population-based study of school health problems in children who develop psychosis in adolescence. J Am Acad Child Adolesc Psychiatry 26(3):351–355. [DOI] [PubMed] [Google Scholar]
- Hercher L, Bruenner G. 2008. Living with a child at risk for psychotic illness: the experience of parents coping with 22q11 deletion syndrome: an exploratory study. Am J Med Genet A 146A(18):2355–2360. [DOI] [PubMed] [Google Scholar]
- Hoeffding LK, Trabjerg BB, Olsen L, Mazin W, Sparso T, Vangkilde A, Mortensen PB, Pedersen CB, Werge T. 2017. Risk of Psychiatric Disorders Among Individuals With the 22q11.2 Deletion or Duplication: A Danish Nationwide, Register-Based Study. JAMA psychiatry 74(3):282–290. [DOI] [PubMed] [Google Scholar]
- Insel TR. 2010. Rethinking schizophrenia. Nature 468(7321):187–193. [DOI] [PubMed] [Google Scholar]
- Jansen PW, Duijff SN, Beemer FA, Vorstman JA, Klaassen PW, Morcus ME, Heineman-de Boer JA. 2007. Behavioral problems in relation to intelligence in children with 22q11.2 deletion syndrome: a matched control study. AmJMedGenetA 143(6):574–580. [DOI] [PubMed] [Google Scholar]
- Jolin EM, Weller RA, Jessani NR, Zackai EH, McDonald-McGinn DM, Weller EB. 2009. Affective disorders and other psychiatric diagnoses in children and adolescents with 22q11.2 Deletion Syndrome. J Affect Disord 119(1–3):177–180. [DOI] [PubMed] [Google Scholar]
- Lipari RN, Hughes A, Williams M. 2013. State Estimates of Major Depressive Episode among Adolescents: 2013 and 2014 The CBHSQ Report; Rockville (MD). [PubMed] [Google Scholar]
- Malhotra D, Sebat J. 2012. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148(6):1223–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS. 2015. 22q11.2 deletion syndrome. Nat Rev Dis Primers 1:15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Saha S, Chant D, Welham J. 2008. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev 30:67–76. [DOI] [PubMed] [Google Scholar]
- Medicine. AAoPAAoFPACoP-ASoI. 2002. A consensus statement on health care transitions for young adults with special health care needs. Pediatrics 110:1304–1306. [PubMed] [Google Scholar]
- Moreno-De-Luca A, Evans DW, Boomer KB, Hanson E, Bernier R, Goin-Kochel RP, Myers SM, Challman TD, Moreno-De-Luca D, Slane MM, Hare AE, Chung WK, Spiro JE, Faucett WA, Martin CL, Ledbetter DH. 2015. The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry 72(2):119–126. [DOI] [PubMed] [Google Scholar]
- Morris E, Inglis A, Friedman J, Austin J. 2013. Discussing the psychiatric manifestations of 22q11.2 deletion syndrome: an exploration of clinical practice among medical geneticists. Genet Med 15(9):713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CM, Wilson CE, Robertson DM, Ecker C, Daly EM, Hammond N, Galanopoulos A, Dud I, Murphy DG, McAlonan GM. 2016. Autism spectrum disorder in adults: diagnosis, management, and health services development. Neuropsychiatr Dis Treat 12:1669–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KC, Jones LA, Owen MJ. 1999. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry 56(10):940–945. [DOI] [PubMed] [Google Scholar]
- Nelson B, McGorry PD, Wichers M, Wigman JT, Hartmann JA. 2017. Moving From Static to Dynamic Models of the Onset of Mental Disorder: A Review. JAMA Psychiatry. [DOI] [PubMed] [Google Scholar]
- Niarchou M, Zammit S, van Goozen SH, Thapar A, Tierling HM, Owen MJ, van den Bree MB. 2014. Psychopathology and cognition in children with 22q11.2 deletion syndrome. Br J Psychiatry 204(1):46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niklasson L, Rasmussen P, Oskarsdottir S, Gillberg C. 2009. Autism, ADHD, mental retardation and behavior problems in 100 individuals with 22q11 deletion syndrome. Res Dev Disabil 30(4):763–773. [DOI] [PubMed] [Google Scholar]
- Owen MJ, Sawa A, Mortensen PB. 2016. Schizophrenia. Lancet 388(10039):86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padula MC, Schaer M, Scariati E, Maeder J, Schneider M, Eliez S. 2017. Multimodal investigation of triple network connectivity in patients with 22q11DS and association with executive functions. Hum Brain Mapp 38(4):2177–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul M, Street C, Wheeler N, Singh SP. 2015. Transition to adult services for young people with mental health needs: A systematic review. Clin Child Psychol Psychiatry 20(3):436–457. [DOI] [PubMed] [Google Scholar]
- Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. 2007. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. The American journal of psychiatry 164(6):942–948. [DOI] [PubMed] [Google Scholar]
- Pulver AE, Nestadt G, Goldberg R, Shprintzen RJ, Lamacz M, Wolyniec PS, Morrow B, Karayiorgou M, Antonarakis SE, Housman D. 1994. Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. JNervMentDis 182(8):476–478. [DOI] [PubMed] [Google Scholar]
- Ramanathan S, Mattiaccio LM, Coman IL, Botti JC, Fremont W, Faraone SV, Antshel KM, Kates WR. 2016. Longitudinal trajectories of cortical thickness as a biomarker for psychosis in individuals with 22q11.2 deletion syndrome. Schizophr Res. [DOI] [PubMed] [Google Scholar]
- Raux G, Bumsel E, Hecketsweiler B, van Amelsvoort T, Zinkstok J, Manouvrier-Hanu S, Fantini C, Breviere GM, Di Rosa G, Pustorino G, Vogels A, Swillen A, Legallic S, Bou J, Opolczynski G, Drouin-Garraud V, Lemarchand M, Philip N, Gerard-Desplanches A, Carlier M, Philippe A, Nolen MC, Heron D, Sarda P, Lacombe D, Coizet C, Alembik Y, Layet V, Afenjar A, Hannequin D, Demily C, Petit M, Thibaut F, Frebourg T, Campion D. 2007. Involvement of hyperprolinemia in cognitive and psychiatric features of the 22q11 deletion syndrome. Hum Mol Genet 16(1):83–91. [DOI] [PubMed] [Google Scholar]
- RK CY, Merico D, Bookman M, J LH, Thiruvahindrapuram B, Patel RV, Whitney J, Deflaux N, Bingham J, Wang Z, Pellecchia G, Buchanan JA, Walker S, Marshall CR, Uddin M, Zarrei M, Deneault E, D'Abate L, Chan AJ, Koyanagi S, Paton T, Pereira SL, Hoang N, Engchuan W, Higginbotham EJ, Ho K, Lamoureux S, Li W, MacDonald JR, Nalpathamkalam T, Sung WW, Tsoi FJ, Wei J, Xu L, Tasse AM, Kirby E, Van Etten W, Twigger S, Roberts W, Drmic I, Jilderda S, Modi BM, Kellam B, Szego M, Cytrynbaum C, Weksberg R, Zwaigenbaum L, Woodbury-Smith M, Brian J, Senman L, Iaboni A, Doyle-Thomas K, Thompson A, Chrysler C, Leef J, Savion-Lemieux T, Smith IM, Liu X, Nicolson R, Seifer V, Fedele A, Cook EH, Dager S, Estes A, Gallagher L, Malow BA, Parr JR, Spence SJ, Vorstman J, Frey BJ, Robinson JT, Strug LJ, Fernandez BA, Elsabbagh M, Carter MT, Hallmayer J, Knoppers BM, Anagnostou E, Szatmari P, Ring RH, Glazer D, Pletcher MT, Scherer SW. 2017. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scariati E, Padula MC, Schaer M, Eliez S. 2016. Long-range dysconnectivity in frontal and midline structures is associated to psychosis in 22q11.2 deletion syndrome. J Neural Transm (Vienna) 123(8):823–839. [DOI] [PubMed] [Google Scholar]
- Schneider M, Armando M, Pontillo M, Vicari S, Debbane M, Schultze-Lutter F, Eliez S. 2016. Ultra high risk status and transition to psychosis in 22q11.2 deletion syndrome. World Psychiatry 15(3):259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Debbane M, Bassett AS, Chow EW, Fung WL, van den Bree M, Owen M, Murphy KC, Niarchou M, Kates WR, Antshel KM, Fremont W, McDonald-McGinn DM, Gur RE, Zackai EH, Vorstman J, Duijff SN, Klaassen PW, Swillen A, Gothelf D, Green T, Weizman A, Van Amelsvoort T, Evers L, Boot E, Shashi V, Hooper SR, Bearden CE, Jalbrzikowski M, Armando M, Vicari S, Murphy DG, Ousley O, Campbell LE, Simon TJ, Eliez S, International Consortium on B, Behavior in 22q11.2 Deletion S. 2014a. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am J Psychiatry 171(6):627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Van der Linden M, Menghetti S, Glaser B, Debbane M, Eliez S. 2014b. Predominant negative symptoms in 22q11.2 deletion syndrome and their associations with cognitive functioning and functional outcome. J Psychiatr Res 48(1):86–93. [DOI] [PubMed] [Google Scholar]
- Shashi V, Keshavan M, Kaczorowski J, Schoch K, Lewandowski KE, McConkie-Rosell A, Hooper SR, Kwapil TR. 2010. Socioeconomic status and psychological function in children with chromosome 22q11.2 deletion syndrome: implications for genetic counseling. J Genet Couns 19(5):535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashi V, Keshavan MS, Howard TD, Berry MN, Basehore MJ, Lewandowski E, Kwapil TR. 2006. Cognitive correlates of a functional COMT polymorphism in children with 22q11.2 deletion syndrome. ClinGenet 69(3):234–238. [DOI] [PubMed] [Google Scholar]
- Shprintzen RJ, Goldberg R, Golding-Kushner KJ, Marion RW. 1992. Late-onset psychosis in the velo-cardio-facial syndrome. AmJMedGenet 42(1):141–142. [DOI] [PubMed] [Google Scholar]
- Smith LE, Barker ET, Seltzer MM, Abbeduto L, Greenberg JS. 2012. Behavioral phenotype of fragile X syndrome in adolescence and adulthood. American journal on intellectual and developmental disabilities 117(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer IE, Bearden CE, van Dellen E, Breetvelt EJ, Duijff SN, Maijer K, van Amelsvoort T, de Haan L, Gur RE, Arango C, Diaz-Caneja CM, Vinkers CH, Vorstman JA. 2016. Early interventions in risk groups for schizophrenia: what are we waiting for? NPJ Schizophr 2:16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart D 2009. Transition to adult services for young people with disabilities: current evidence to guide future research. Dev Med Child Neurol 51 Suppl 4:169–173. [DOI] [PubMed] [Google Scholar]
- Swerts C, Van de Velde S, van der Nagel J, van der Plasschen W, Claes C, De Maeyer J. 2016. Substance use among individuals with intellectual disabilities living independently in Flanders . Research in Developmental Disabilities 63:107. [DOI] [PubMed] [Google Scholar]
- Swillen A 2016. The importance of understanding cognitive trajectories: the case of 22q11.2 deletion syndrome. Curr Opin Psychiatry 29(2):133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swillen A, Vandeputte L, Cracco J, Maes B, Ghesquiere P, Devriendt K, Fryns JP. 1999. Neuropsychological, learning and psychosocial profile of primary school aged children with the velo-cardio-facial syndrome (22q11 deletion): evidence for a nonverbal learning disability? Child Neuropsychol 5(4):230–241. [DOI] [PubMed] [Google Scholar]
- Tang SX, Yi JJ, Calkins ME, Whinna DA, Kohler CG, Souders MC, McDonald-McGinn DM, Zackai EH, Emanuel BS, Gur RC, Gur RE. 2014. Psychiatric disorders in 22q11.2 deletion syndrome are prevalent but undertreated. Psychol Med 44(6):1267–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomescu MI, Rihs TA, Becker R, Britz J, Custo A, Grouiller F, Schneider M, Debbane M, Eliez S, Michel CM. 2014. Deviant dynamics of EEG resting state pattern in 22q11.2 deletion syndrome adolescents: A vulnerability marker of schizophrenia? Schizophr Res 157(1–3):175–181. [DOI] [PubMed] [Google Scholar]
- Van L, Boot E, Bassett AS. 2017. Update on the 22q11.2 deletion syndrome and its relevance to schizophrenia. Curr Opin Psychiatry. [DOI] [PubMed] [Google Scholar]
- Vorstman JA, Breetvelt EJ, Duijff SN, Eliez S, Schneider M, Jalbrzikowski M, Armando M, Vicari S, Shashi V, Hooper SR, Chow EW, Fung WL, Butcher NJ, Young DA, McDonald-McGinn DM, Vogels A, van Amelsvoort T, Gothelf D, Weinberger R, Weizman A, Klaassen PW, Koops S, Kates WR, Antshel KM, Simon TJ, Ousley OY, Swillen A, Gur RE, Bearden CE, Kahn RS, Bassett AS, International Consortium on B, Behavior in 22q11.2 Deletion S. 2015. Cognitive decline preceding the onset of psychosis in patients with 22q11.2 deletion syndrome. JAMA psychiatry 72(4):377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JA, Breetvelt EJ, Thode KI, Chow EW, Bassett AS. 2013. Expression of autism spectrum and schizophrenia in patients with a 22q11.2 deletion. Schizophr Res 143(1):55–59. [DOI] [PubMed] [Google Scholar]
- Vorstman JA, Morcus ME, Duijff SN, Klaassen PW, Heineman-de Boer JA, Beemer FA, Swaab H, Kahn RS, van Engeland H. 2006. The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. Journal of the American Academy of Child and Adolescent Psychiatry 45(9):1104–1113. [DOI] [PubMed] [Google Scholar]
- Vorstman JA, Ophoff RA. 2013. Genetic causes of developmental disorders. Current opinion in neurology 26(2):128–136. [DOI] [PubMed] [Google Scholar]
- Vorstman JA, Parr JR, Moreno-De-Luca D, Anney RJ, Nurnberger JI Jr., Hallmayer JF. 2017. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet. [DOI] [PubMed] [Google Scholar]
- Weisman O, Guri Y, Gur RE, McDonald-McGinn DM, Calkins ME, Tang SX, Emanuel B, Zackai EH, Eliez S, Schneider M, Schaer M, Kates WR, Antshel KM, Fremont W, Shashi V, Hooper SR, Armando M, Vicari S, Pontillo M, Kushan L, Jalbrzikowski M, Bearden CE, Cubells JF, Ousley OY, Walker EF, Simon TJ, Stoddard J, Niendam TA, van den Bree MB, Gothelf D, International Consortium on B, Behavior in 22q11.2 Deletion S. 2017. Subthreshold Psychosis in 22q11.2 Deletion Syndrome: Multisite Naturalistic Study. Schizophr Bull [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfenden S, Sarkozy V, Ridley G, Williams K. 2012. A systematic review of the diagnostic stability of autism spectrum disorder. . Research in Autism Spectrum Disorders 6:345–354. [Google Scholar]
- Young S, Murphy CM, Coghill D. 2011. Avoiding the 'twilight zone': recommendations for the transition of services from adolescence to adulthood for young people with ADHD. BMC Psychiatry 11:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui K, Goto K, Ikemoto S, Ishiguro T, Angrist B, Duncan GE, Sheitman BB, Lieberman JA, Bracha SH, Ali SF. 1999. Neurobiological basis of relapse prediction in stimulant-induced psychosis and schizophrenia: the role of sensitization. Mol Psychiatry 4(6):512–523. [DOI] [PubMed] [Google Scholar]