

Abstract
The 22q11.2 deletion syndrome is caused by non-allelic homologous recombination events during meiosis between low copy repeats (LCR22) termed A, B, C and D. Most patients have a typical LCR22A-D (AD) deletion of 3 million base pairs (Mb). In this report, we evaluated IQ scores in 1,478 subjects with 22q11.2DS. The mean of full scale IQ, verbal IQ and performance IQ scores in our cohort were 72.41 (standard deviation-SD of 13.72), 75.91(SD of 14.46) and 73.01(SD of 13.71), respectively. To investigate whether IQ scores are associated with deletion size, we examined individuals with the 3 Mb, AD (n = 1,353) and nested 1.5 Mb, AB (n = 74) deletions, since they comprised the largest subgroups. We found that full scale IQ was decreased by 6.25 points (P = 0.002), verbal IQ was decreased by 8.17 points (P = 0.0002) and performance IQ was decreased by 4.03 points (P = 0.028) in subjects with the AD versus AB deletion. Thus, individuals with the smaller, 1.5 Mb AB deletion have modestly higher IQ scores than those with the larger, 3 Mb AD deletion. Overall, the deletion of genes in the AB region largely explain the observed low IQ in the 22q11.2DS population. However, our results also indicate that haploinsufficiency of genes in the LCR22B-D region (BD) exert an additional negative impact on IQ. Furthermore, we did not find evidence of a confounding effect of severe congenital heart disease on IQ scores in our cohort.
Keywords: 22q11.2 deletion syndrome, intellectual disability, IQ, deletion size, low copy repeat, segmental duplication
INTRODUCTION
The 22q11.2 deletion syndrome (22q11.2DS; MIM188400; 192430) is the most common microdeletion syndrome in humans. Recent data has shown a frequency of 1:992 for deletions of 22q11.2 in low risk pregnancies (Grati et al., 2015). The syndrome has a frequency of 1:2,000 live births (Cancrini et al., 2014; McDonald-McGinn et al., 2015). Clinical manifestations of 22q11.2DS are highly heterogeneous and can range from mild to severe. Besides anomalies including congenital heart disease (CHD), palatal abnormalities, renal defects, immunodeficiency and hypocalcemia, almost all patients with 22q11.2DS have some degree of cognitive impairment. This is in part identified by the presence of reduced IQ scores (Oskarsdottir, Belfrage, Sandstedt, Viggedal, & Uvebrant, 2005; Swillen et al., 1997). A significant percentage of school age children have learning disabilities requiring placement in special education classes (Bassett et al., 2011; Bearden et al., 2001; De Smedt, Swillen, Ghesquiere, Devriendt, & Fryns, 2003; Duijff et al., 2012; Swillen et al., 1997; Swillen et al., 1999; Woodin et al., 2001). Approximately 50% of individuals with 22q112DS have intellectual disability (ID), as defined in part by an IQ score of less than 70 ± 5 (Swillen & McDonald-McGinn, 2015), while the rate of ID in the general population is 1-3%. Approximately one third of 22q11.2DS patients have relatively normal IQ levels within the range of 85 or higher. Overall genetic background can explain some of the ID variability in 22q11DS which was indicated by the cognitive functioning of first-degree relatives (Olszewski, Radoeva, Fremont, Kates, & Antshel, 2014). While the genetic influence on the variable expression of the cognitive phenotype in 22q11.2DS is still largely unknown, we hypothesized the size or position of the deletion on 22q11.2 may influence IQ scores.
The 22q11.2 deletion occurs due to meiotic non-allelic homologous recombination events between segmental duplications or low copy repeats (LCR22), termed LCR22A, B, C and D (Edelmann et al., 1999; Shaikh et al., 2000). The LCR22s do not contain functional genes and are comprised of blocks or modules of high sequence identity (Bailey et al., 2002). Over 90% of patients have a 3 million base pair (Mb) deletion between LCR22A-D (referred herein as AD). Approximately 5-8% have a nested, 1.5 Mb, LCR22A-B (AB) deletion, and the rest have deletions of other types (Shaikh et al., 2000). Consequently, haploinsufficiency of fewer genes in smaller or nested deletions may potentially result in a milder phenotype.
In a recent published study (Evers, Engelen, Houben, Curfs, & van Amelsvoort, 2016), they did not find a relationship between deletion size and IQ, however, the sample may have been underpowered (63 subjects). In the largest study to date, of 103 subjects with 22q11.2DS, there was also no evidence of a correlation between deletion size and IQ scores, however only four subjects in the study had an AB deletion (Michaelovsky et al., 2012). As part of the International 22q11.2 Brain and Behavior Consortium [IBBC; (Gur et al., 2017; Vorstman et al., 2015)], and prior to its establishment, we collected DNA samples from subjects with 22q11.2DS of age 6 years and older. We focused on the two most common types of 22q11.2 deletions, the AB (74 subjects) versus AD (1353 subjects) group. We test whether IQ levels in individuals with AB deletions (No. = 74) would differ from those with AD deletions (No. =1353), taking into account possible confounding effects of sex, age and severe CHD status.
MATERIALS AND METHODS
Study population
Recruitment of most of the subjects with 22q11.2DS in this study has been previously described (Gur et al., 2017). Briefly, a total of >2,000 de-identified DNA samples were collected with their informed consent (Internal Review Board, Committee of Clinical Investigation #1999-201). All individuals had a clinical diagnosis of 22q11.2DS. Fluorescence in situ hybridization (FISH) was done to make a molecular diagnosis in the majority of subjects, priori to inclusion. In addition, the presence of the deletion was confirmed by array comparative genome hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA). It is possible that the presence of severe CHD can influence neuro-cognition and could therefore act as a confounder in the analysis (see below). The majority of subjects had available cardiac phenotype information (Guo et al., 2018). After removing data with gender mismatch, duplicate or related samples, 1,807 remained for further analysis (Supplementary Fig. 1). The 22q11.2DS samples were recruited from 21 study sites, mostly in North America and Europe. We combined the 21 study sites into four groups including US and Canada, Northern Europe, Southern Europe and UK with Australia (Supplementary Table 2) according both to genetic proximity (not geographical proximity) (Nelis et al., 2009) and phenotypic proximity (IQ measure within a group should be more homogeneous than across groups).
Deletion type detection
Deletion types among 1,807 22q11.2DS patients were identified by copy number variation (CNV) analysis on raw intensity data obtained from Affymetrix 6.0 SNP array CEL files. Briefly, Univariate CNAM optimal segmenting algorithm implemented in Golden Helix SNP&VARIATION SUITE version 8.7.2 (http://www.goldenhelix.com/index.html) was employed to detect CNV segment boundaries as determined by log2 ratio [log2(observed intensity/reference intensity). In this study, controls included those processed at the same time period and at the same facility as those of the 22q11.2DS samples, and, in addition, we used pre-computed intensity data from 270 Hapmap samples. For 1,227 samples, the deletion type was also determined using MLPA (SALSA MLPA kit P250 DiGeorge; MRC Holland, The Netherlands (Vorstman et al., 2006)). Exclusion criteria were deletions that extended further centromeric (proximal) to LCR22A (unbalanced translocations) or deletions that extended further telomeric (distal) to LCR22D, such as LCR22E, F, G or H. This included the following subjects: LCR22A-G (1 subject, with a frequency of 0.05% among the population), DE (1, 0.01%), DF (1, 0.01%), EF (1, 0.01%) and unbalanced translocation (1, 0.05%). Atypical deletion types that occurred in ≤ 7 subjects were also excluded in our population, namely/specifically CD (2, 0.1%), nested AB (7, 0.39%), nested AD (4, 0.22%), proximal PRODH deletions (2, 0.1%). After removing these 20 samples, 1,787 subjects with a recurrent deletion within the AD region were included in this study. Detailed selection steps of patients with 22q11.2DS are shown in Supplementary Fig. 1. We identified 17 subjects with an atypical, nested 2.8 Mb deletion that occurred just telomeric to the PRODH gene, herein referred to as the A+-D deletion (Guo et al., 2018). This slightly smaller nested deletion type was validated by quantitative PCR analysis using primers in the 22q11.2 breakpoint region (Guo et al., 2018).
IQ assessment
There were 1,478 subjects with IQ scores available among the 1,787 subjects in this study. Wechsler Intelligence Scales (e.g., Wechsler preschool and Primary Scale of Intelligence, Wechsler Intelligence Scale for Children, Wechsler Adult Intelligence Scale and Wechsler Abbreviated Scale of Intelligence; WPPSI, WISC, WAIS, WASI) were employed to test IQ levels among 1,411 individuals with 22q11.2DS of age 6 years or older. The IQ test type for the remaining 67 subjects includes, CNB [Penn Computerized Neurocognitive Battery; (Yi et al., 2016); 6 subjects], GIT (General Intelligence Test-Canada; 1 subject), IQ National Adult Reading Test [(Willshire, Kinsella, & Prior, 1991); 25 subjects], McCarthy Scales [(Kaufman & Kaufman, 1974); 1 subject], RAVEN Matrices test [(Ballester-Plane et al., 2016); 2 subjects], RAIS [(Reynolds Intellectual Assessment Scales; 1 subject], SON [Nonverbal Intelligence Test; UK; (Geerlings, Laros, Tellegen, & Glas, 2014); 11 subjects] Stanford-Binet//Terman-Merrill [(Martinez-Cruz, Poblano, & Conde-Reyes, 2009); 6 subjects], UNIT (Universal Nonverbal Intelligence Test; 1 subject) and missing test type (13 subjects).
Statistical Analysis
All continuous variables including age, full scale IQ, verbal IQ and performance IQ were expressed as mean ± SD if not specified. Independent-samples t-test was employed to evaluate IQ differences between binary variables including gender and presence of severe CHD that could act as a confounder and must be considered. Severe CHD was defined as having a clinically recognized complex cardiac malformation, which usually requires surgery. In this study, it included, tetralogy of Fallot (TOF), persistent truncus arteriosus (PTA) or interrupted aortic arch type B (IAAB). Controls for severe CHD had 22q11.2DS but no detectable heart or aortic arch defect. Association between IQ and deletion size (AD versus AB) was assessed by multiple linear regression analyses with adjustment of sex and age of administration of the IQ test and research site group. Pearson chi-square test was used to assess the association between CHD status and deletion size. The one-sample Kolmogorov-Smirnov test was used to test whether age was normally distributed among patients with the AB and AD deletion, respectively. Independent-samples Mann-Whitney U test was employed to compare the age distribution between the AB and AD groups. Median Test, which is also a non-parametric statistical approach, was employed to compare age medians between the two groups. Significance was considered as P < 0.05.
RESULTS
Characterization of the 22q11.2DS study population
Basic demographic characteristics of the population is presented in Table I. Among the 1,478 subjects, gender is almost evenly distributed with 709 (48%) males and 769 (52%) females. The subjects were largely Caucasian, and retrospectively enrolled at 21 different research sites (Supplementary Table 2). The ethnicity of our cohort has been previously reported (Guo et al., 2017). We recently identified an atypical nested A+-D deletion in which the proximal deletion breakpoint was distal to the PRODH gene (Guo et al., 2018). Similar IQ score distributions were observed between A+-D and AD groups (Supplementary Fig. 2), nevertheless, we did not include the 17 A+-D samples in this study of IQ. A total of 307 (20.8%) had severe CHD, while 637 (43.1%) had a normal heart and/or aortic arch. We found that 1,353 (91.5%) had an AD deletion, 74 (5%) had an AB deletion, 27 (1.8%) had an AC deletion and 7 (0.5%) had a BD deletion.
Table I.
Demographic characteristics of 22q11.2DS population
| Total no. subjects = 1,478 | No. (%) for categorical variables |
|---|---|
| mean±SD for continuous variables | |
| Gender | |
| Male | 709(48.0) |
| Female | 769(52.0) |
| Study sites | |
| US & Canada | 618(41.8) |
| Northern_Europe | 430(29.1) |
| Southern_Europe | 265(17.9) |
| UK & Australia | 165(11.2) |
| CHDa | |
| No | 637(43.1) |
| Yes | 307(20.8) |
| Missing | 534(36.2) |
| Deletion type | |
| AD | 1353(91.5) |
| A+-D | 17(1.2) |
| AC | 27(1.8) |
| AB | 74(5) |
| BD | 7(0.5) |
| Age | 15.9±9.9 |
| FSIQb | 72.41±13.7 |
| VIQc | 75.91±14.5 |
| PIQd | 73.01±13.7 |
CHD, congenital heart disease, is defined as subjects with Tetralogy of Fallot (TOF), Persistent Truncus Arteriosus (PTA) or Interrupted Aortic Arch Type B (IAAB);
Full scale IQ;
Verbal IQ,
Performance IQ.
IQ score distribution among subjects with 22q11.2DS
Histogram plots depicting the frequency distribution of full scale IQ (72.41 ±13.72; Fig. 1A), verbal IQ (75.91±14.46; Fig. 1B) and performance IQ (73.01±13.71; Fig. 1C) of the entire cohort including all deletion sizes, are shown in Fig. 1. We found that verbal IQ was slightly higher (3 points) than performance IQ in this cohort. Histogram plots also included fitted normal curves, as well as the IQ distribution curve in the general population, respectively (Fig. 1A-C). Overall, in the general population, full scale IQ is normally distributed with a mean score of 100 and a SD of 15. Distribution of full scale IQ, verbal IQ and performance IQ was shifted to the left in subjects with 22q11.2DS as compared with those of the general population by almost two SDs (Fig. 1A-C) (all P values < 0.01 as determined by independent samples t-test; Fig. 1D), which is consistent with previous reports of smaller sized cohorts (Hooper et al., 2013; Niarchou et al., 2014).
Fig. 1. Distribution of full scale IQ, verbal IQ and performance IQ among subjects with 22q11.2DS.
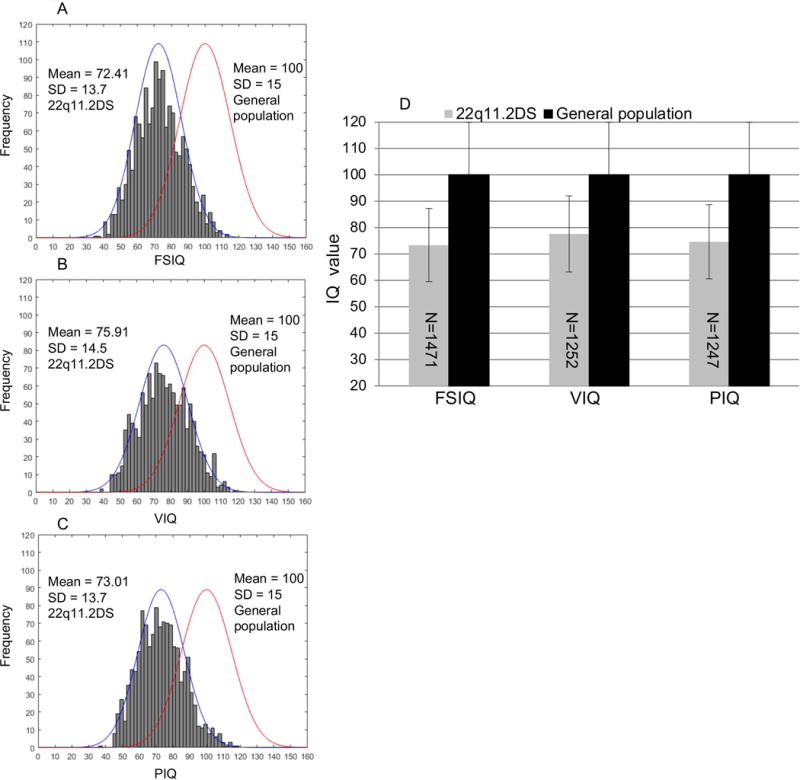
(A-C) Frequency distribution of Full scale IQ (FSIQ, 72.41 ±13.7), verbal IQ (VIQ, 75.91±14.5) and performance IQ (PIQ, 73.01±13.7) among the 22q11.2DS population and fitted normal curves to the right, respectively. The normal curves to the left denote the IQ distribution in general population (100 ±15). Distribution of IQ are shifted to the left as compared to those in general population by almost 2 SDs. (D) Differences of mean IQ scores were determined by independent samples t-test. ** P <0.01. Numbers in the bars denote the samples with FSIQ, VIQ and PIQ, available, respectively. FSIQ, VIQ and PIQ were all significantly lower than those of general population.
IQ score distribution among demographic variables in the cohort of subjects with 22q11.2DS
The AD and AB deletion type groups had the largest sample sizes. Statistical power calculations of the AD and AB groups is presented in Supplementary Table 4. We therefore limited the analysis of IQ and deletion type to the AD and AB groups. Compared with males, the full scale and performance IQ scores in females with 22q11.2DS with the AD and AB deletions were 1-2 points higher (Fig. 2, full scale IQ was 71.7±14.2 and performance IQ was 72.0±14.1; full scale IQ was 73.1±13.2 and performance IQ was 73.9±13.3 for males and females, respectively). These differences in both full scale and performance IQ scores, but not in verbal IQ, between males and females were significant P <0.05, despite their small effect sizes (Fig. 2).
Fig. 2. IQ distribution between males and females within the 22q11.2DS population.
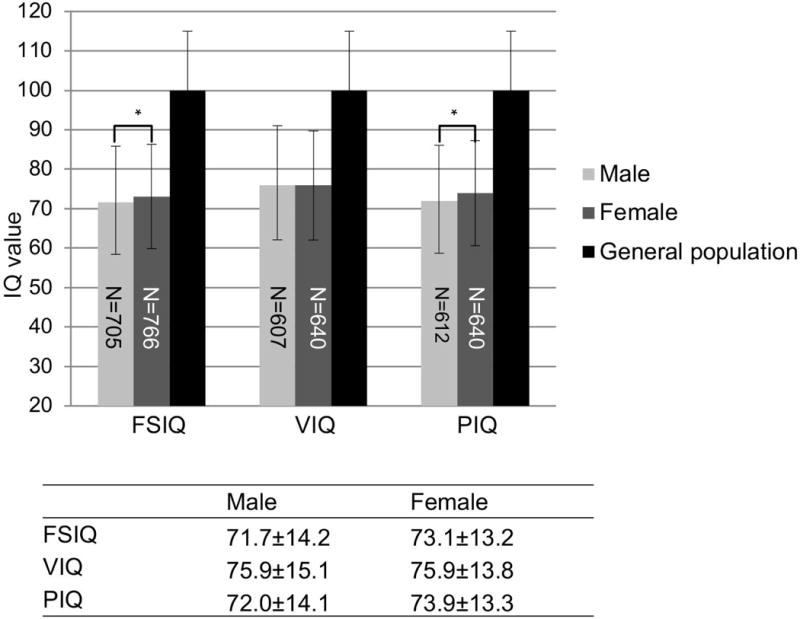
IQ values are expressed as mean±SD. An independent-samples t-test was conducted to compare the mean IQ scores in male versus female subjects. Significant differences are denoted by an asterisk in bar plots with P < 0.05. Numbers in the bars denote samples size of each category.
The age distribution in both the AD and AB groups were shifted towards having more than 50% of patients with an age between 6-20 years old and less than 50% with an age between 21-66 years (Supplementary Fig. 3, both P <0.05 for the one-sample Kolmogorov-Smirnov test for normality). However, the age distribution was not significantly different between the AD and AB groups (asymptotic P > 0.05 for independent-samples -Whitney U test). Moreover, no significant differences were observed for median ages between the two groups (age = 12.67±9.67 and 13.08±11.54 for AD and AB groups, respectively) according to the independent-samples non-parametric Median Test, as shown in Supplementary Fig. 3. We also evaluated the distributions of categorical variables between AD and AB groups (Supplementary Table 5). As is shown, the distribution of sex, CHD status, IQ test types and study sites are not significant (all P >0.05).
IQ score distribution in subjects with severe CHD as compared to those with a normal heart or aortic arch
Individuals in the general population with severe CHD that require surgery for survival, may have increased risk for neurodevelopmental deficits as measured, in part, by the presence of lower IQ scores (Latal, 2016; Marino et al., 2012). The prevalence of severe CHD in our cohort is shown in Table I. There was no significant difference between the prevalence of severe CHD in the AD (32.5%, 593 controls versus 286 severe CHD cases) or AB (36.2% 30 controls versus 17 severe CHD cases, asymptotic P>0.05) using the Pearson chi-square test. We found a decrease of <1 point in full scale IQ, verbal IQ and performance IQ in 22q11.2 subjects with severe CHD compared with 22q11.2 subjects with no detectable cardiac or aortic arch defects (Fig. 3). These differences were not significant (all three P >0.05).
Fig. 3. Full scale, verbal and performance IQ distribution between cases with severe CHD and control subjects with no CHD within the 22q11.2DS population.
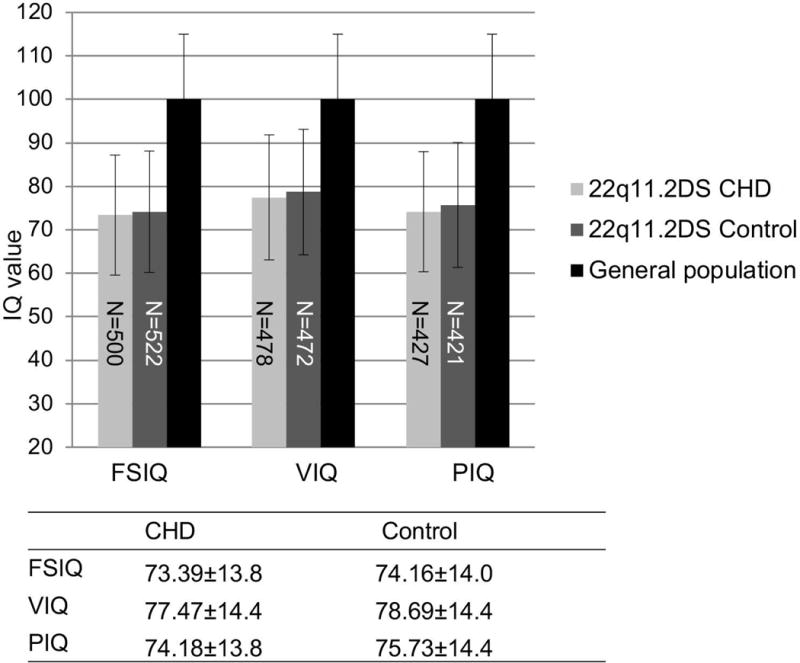
Severe CHD is defined as subjects with either tetralogy of Fallot, persistent truncus arteriosus or interrupted aortic arch type B. Control subjects for CHD cases had 22q11.2DS but no detectable heart or aortic arch defect. IQ values were expressed as mean±SD. An independent-samples t-test was conducted to compare IQ scores in subjects with the presence of severe CHD and controls within the 22q11.2DS cohort. All P values were > 0.05. Numbers in the bars denote the sample size of each category.
IQ distribution among 22q11.2DS patients with AB and AD deletion types
We found that full scale IQ, verbal IQ and performance IQ scores increased by 0.3-0.5 SD in the AB group compared to the AD group as shown in Fig. 4. We then conducted multiple linear regression analyses between IQ and deletion type with an adjustment of age, gender and collection site (Table II). We found an increase of 5.64, 7.06 and 4.42 points for full scale IQ, verbal IQ and performance IQ, respectively, all P <0.05. Adjusted R square/variance for full scale IQ, verbal IQ and performance IQ was 0.047, 0.037 and 0.05, respectively. Thus, while the presence of a deletion confers the highest influence on IQ, subjects with the smaller AB deletion were relatively less affected than those with the typical, larger AD deletion. Taking US and Canada as a reference, significantly lower IQ scores was observed for Northern Europe. Variances detected may reflect differences in health care structure/availability and/or education system. To check this further, we ran a stratified multivariable linear regression analysis of IQ and deletion size with adjustment of sex and age for each research site, respectively (Supplementary Table 3). Although the differences of IQ between AD and AB are not always significant for each of the 21 research sites, the direction of beta were consistent across the different groups.
Fig. 4. IQ score differences between AD and AB deletion types in subjects with 22q11.2DS.
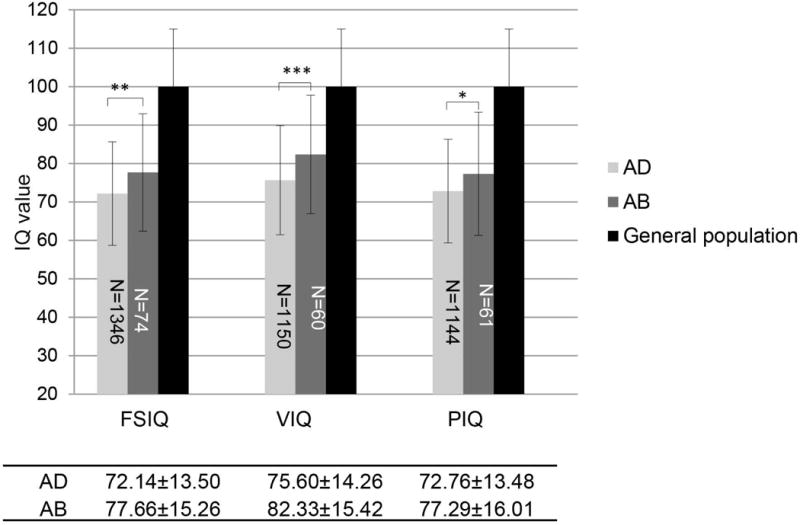
IQ values were expressed as mean±SD. An independent-samples t-test was conducted to compare mean IQ scores in subjects carrying the AB deletion with those carrying the AD deletion. ***P < 0.001, **P < 0.01, *P< 0.05. Numbers in the shaded bars denote the samples size in each category. Full scale IQ, verbal IQ and performance IQ decreased by 0.3-0.5 SD among those in the AD group as compared with those in the AB group.
Table II.
Multivariable linear regression analysis of IQ and deletion size with adjustment of covariates
| Full scale IQ | Verbal IQ | Performance IQ | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| β(SE)a | P | β(SE) | P | β(SE) | P | ||
| Deletion type | AD (reference) | ||||||
| AB | 6.25(2.02) | 0.002 | 8.17(2.22) | 2.31E-04 | 4.03(2.32) | 0.028 | |
| Age | −0.06(0.06) | 0.259 | −0.08(0.07) | 0.173 | −0.07(0.06) | 0.244 | |
| Gender | Male (reference) | ||||||
| Female | 1.84(0.94) | 0.05 | 1.20(0.99) | 0.23 | 3.40(1.03) | 0.001 | |
| Study site | Canada & US (reference) | ||||||
| Northern Europe | −5.85(1.15) | 1.00E-06 | −5.86(1.26) | 4.00E-06 | −4.92(1.24) | 7.60E-05 | |
| Southern Europe | −4.67(1.48) | 0.002 | −3.11(1.59) | 0.051 | −2.51(1.68) | 0.135 | |
| UK & Australia | −0.51(1.51) | 0.737 | −2.58(1.69) | 0.127 | 3.03(1.03) | 0.001 | |
β(SE) stands for regression coefficient (standard error) from multivariable linear regression analysis; CHD was not included in the model due to the finding that P >0.05 from the independent-samples t-test.
DISCUSSION
In this report, we examined IQ scores in a large cohort of 1,478 subjects with 22q11.2DS, obtained predominantly as part of the International 22q11.2 Brain and Behavior Consortium (Gur et al., 2017). Previously, it has been shown that verbal IQ scores are 6-8 points higher than performance IQ scores in individuals with 22q11.2DS (Antshel, Kates, Roizen, Fremont, & Shprintzen, 2005; De Smedt et al., 2007; Moss et al., 1999; Swillen et al., 1997). In this large cohort, we found that verbal IQ scores were only 3 points higher than performance IQ scores. Some previous reported data showed there was a small increase of IQ in females versus males, although in other studies, there was no increase found (Antshel et al., 2005; De Smedt et al., 2007; Klaassen et al., 2016; Moss et al., 1999; Niklasson & Gillberg, 2010; Van Aken et al., 2007). In this cohort, we found that full scale and performance IQ were only 1-2 points higher in females but this small difference was statistically significant. While a significant difference (P = 0.017) in age distribution between males (15.03±9.20) and females (16.78±10.43) was observed, the effect of gender on IQ may be slightly biased by this difference in age distribution. We found that among the different research sites for this study, our data was consistent with the finding that those with the AD deletion had a lower IQ than those with the AB deletion.
There is a consideration that individuals that inherit a 22q11.2 deletion tend to have a lower IQ than those with a de novo deletion (De Smedt et al., 2007; Swillen et al., 1997). When correcting for education, this difference disappeared (De Smedt et al., 2007). From the literature, >90% of individuals affected with 22q11.2DS have a de novo deletion, irrespective of deletion size (Driscoll, Budarf, & Emanuel, 1992; Lindsay et al., 1995; Morrow et al., 1995). One limitation of our study is that we have not yet obtained information on the parental deletion status or education level. We are currently in the process of getting such information for the cohort presented here, but as of yet it is quite incomplete.
It is well established using longitudinal studies (Duijff et al., 2012; Gothelf, Penniman, Gu, Eliez, & Reiss, 2007) and supported by cross-sectional studies (Antshel et al., 2010; Duijff et al., 2012; Golding-Kushner, Weller, & Shprintzen, 1985; Gothelf, Aviram-Goldring, et al., 2007; Gothelf et al., 2005; Niklasson & Gillberg, 2010; Shprintzen, 2000) that individuals with 22q11.2DS show decline in IQ scores, referred to as cognitive decline. In the largest study to date of 829 subjects with 22q11.2DS, ages 8-24 years, cognitive decline was quite notable. There was a reported decline of full scale IQ of 7.04 points, verbal IQ of 9.02 points and performance IQ of 5.09 points (Vorstman et al., 2015). We checked whether there was a bias of age between subjects in the AB group versus those with the AD group, and we didn’t find a significant difference. However, we are aware that our analysis has limitations in that the IQ scores represent summaries of cognitive abilities and may not reflect with the complete picture of possible cognitive differences between the deletion size cohorts.
CHD that requires surgery to repair were reported to be associated with neurodevelopmental deficits (Latal, 2016; Marino et al., 2012). In this report, we tested whether 22q11.2DS individuals with severe CHD that usually required surgical repair (tetralogy of Fallot, persistent truncus arteriosus and interrupted aortic arch type B; all complex cardiac outflow tract anomalies) had lower IQ scores. However, we did not find a correlation between severe CHD and altered IQ values for our cohort of 22q11.2DS subjects, suggesting that different mechanisms contribute to severe CHD and to cognition as measured by IQ tests in this 22q11.2DS group. Further, it suggests that surgical intervention to correct cardiac malformations do not further impair the IQ.
The phenotypic spectrum of 22q11.2DS is quite wide and variable. One possible mechanism contributing to the observed variable penetrance and expressivity could be the size of the deletion. It is only possible to evaluate deletion size with a large enough cohort because those with less common, nested deletions might be underpowered for statistical analyses. In this study, the only nested deletion type with sufficient statistical power to analyze in comparison to the typical 3 Mb, AD deletion was the nested 1.5 Mb, AB deletion. Our results indicate that the presence of the deletion, regardless of the AB or AD type, exerts most of the influence on IQ, resulting in ID in half the population. However, individuals with the AD deletion have a lower full scale IQ (6.25 points), lower verbal IQ (8.17 points) and lower performance IQ (4.03 points) than those with the AB deletion. This data suggests that genes in the AB deletion most likely account for the observed lowered IQ in 22q11DS relative to typical developing individuals, but that the BD deletion exerts an additional negative influence on IQ.
There are 30 known protein-coding genes that span the AB region on 22q11.2 (Supplementary Fig. 4). Many are implicated in brain development or function (Maynard et al., 2003; Meechan et al., 2015). Coding genes implicated in cognitive and/or neuropsychiatric findings include TBX1 (Cioffi et al., 2014; Hiroi, Hiramoto, Harper, Suzuki, & Boku, 2012), COMT, encoding catechol-O-methyltansferase (Dickinson & Elvevag, 2009; Guo et al., 2017; Radoeva et al., 2014; Vorstman et al., 2015), PRODH, encoding proline oxidase (Guo et al., 2017; Radoeva et al., 2014) and DGCR8, encoding an enzyme for miRNA processing (Forstner, Degenhardt, Schratt, & Nothen, 2013) among many others (Karayiorgou, Simon, & Gogos, 2010; Meechan et al., 2015). Thus, it is likely that haploinsufficiency of one or more of these genes is important for IQ and cognition in the AB region.
Upon examination of the literature, individuals with a BD or nested CD deletion have been identified with developmental delay and/or cognitive impairment (Rump et al., 2014). Amongst a set of 46 subjects combined with the literature, 9 of 17 individuals with a BD deletion (53%) and 16 of 29 with a CD deletion (55%) were affected with developmental delay and cognitive impairment (Rump et al., 2014). These data suggest that important genes may lie within the nested CD region. Among the nine coding genes in the CD region, SNAP29 (synaptosome associated protein 29, and CRKL (CRK like proto-oncogene, adaptor protein) are of particular interest. SNAP29 encodes a protein required for intracellular trafficking and homozygous inactivating mutations cause a rare disorder termed CEDNIK syndrome, associated with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma (Sprecher et al., 2005). Patients with 22q11.2DS and related brain malformations result from an inactivating point mutation in SNAP29 on the remaining allele (McDonald-McGinn et al., 2013). The CRKL gene encodes a cytoplasmic adaptor for RELN (Reelin) signaling important for neuronal migration and positioning (Ballif et al., 2004; Park & Curran, 2008). The function of the other genes in the brain, if any, are not well-established as of yet.
While this effect of deletion size is small, it is also significant. We must point out a few important limitations of the analysis and caveats that require additional investigation. The first and most obvious question relates to whether or not there is a qualitative difference in the type of cognitive impairment that results in the IQ difference. This is an important question that the study does not address. Another limitation is that IQ tests are constructed for the general population rather than a population with cognitive impairment like 22q11.2DS and therefore do not adequately capture their cognitive performance. This might have also confounded the analyses as individuals with IQ scores lower than 60, for example, might have missing values on this analysis. Other confounders like environmental interactions, socioeconomic status, education, genetic background and additional genomic changes, including CNV (Jensen et al., 2017) might have also played a role. Further, our study does not suggest that there could be a difference in the rehabilitation or treatment of patients with the AD versus AB deletion. We do not yet know if these findings can translate to the clinical realm. This current study’s size and scope cannot answer these questions based solely on IQ scores, but appropriate future translational research may yield important clues on how to focus specific treatment techniques to genomic data that will continue to emerge for this common genetic condition.
Conclusions
In this report, we compared the AD versus AB deletion type in a large cohort of subjects with 22q11.2DS to determine if there is a difference in IQ levels. Here we found that over and beyond the overall effect of having a 22q11.2 deletion, those with the larger, 3 Mb AD deletion have slightly lower IQ scores than those with the nested, 1.5 Mb AB deletion. Our results indicate that while genes in the AB region are critically important, genes within the distal BD region may exert an additional contribution to the overall cognitive phenotype in these patients. This is consistent with findings in the literature that patients with the non-overlapping BD or CD deletion also have cognitive impairment.
Supplementary Material
Acknowledgments
We would like to thank all patients with 22q11.2DS who enrolled in research and provided DNA as well as clinical records. We thank colleagues in the Molecular Cytogenetics core lab at Einstein for technical support. We thank Mark Zeffren, Nousin Haque, Antoneta Preldakaj, John Bruppacher, Daniel Arroyo, Michael Gleeson and Dominique Calandrillo for technical support at Einstein. We also greatly appreciate the laboratory effort of Dr. Frédérique Bena who works with SE and SEA (Institute of Genetics and Genomics of Geneva, Switzerland) and Oanh Tran and Andrea Jin who work with BSE (Division of Human Genetics, the Children’s Hospital of Philadelphia). This work was supported by grants National Institutes of Health R01 HL084410 (BSE, BEM, DMM, TG, AB), P01 HD070454 (EG, LEM, AJA, BSE, DMM, EEM, TG, TW, HN, CLC), U01 MH101720(BSE, BEM, DMM, GR, AB, EC, AS, DG, SE, FT, NP, CEB, TJS, EVD, TVA, WRK, TG, TW), R21HL118637 (TW, BEM, TG, EG), National Institutes of Health 5T32GM007491-41 (JHC), American Heart Association 14PRE199800006 (JHC). GR was supported by the FONDECYT-Chile (grants 1130392 and 1171014). ASB was supported by the Dalglish Chair in 22q11.2 Deletion Syndrome, the Canada Research Chair in Schizophrenia Genetics and Genomic Disorders, Canadian Institutes of Health Research funding (MOP-97800 and MOP-89066), and the University of Toronto McLaughlin Centre. CEB was supported by the National Institutes of Health (R01 MH085903). WRK was supported by the National Institutes of Health (R01 MH064824). SE was supported by the Swiss National Science Foundation (FNS 324730_121996; FNS 324730_144260) and the National Center of Competence in Research (NCCR) “Synapsy-The Synaptic bases of Mental Diseases” (51NF40-158776). JV was supported by the Flemish Science Foundation (FWO G.0E1117N). BSE was supported by funds from the Charles E.H. Upham chair in Pediatrics.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
Compliance with ethical standards: This work is in compliance with the ethical standards of the Committee of Clinical Investigation of Albert Einstein College of Medicine (Internal Review Board #1999-201).
References
- Antshel KM, Kates WR, Roizen N, Fremont W, Shprintzen RJ. 22q11.2 deletion syndrome: genetics, neuroanatomy and cognitive/behavioral features keywords. Child Neuropsychol. 2005;11(1):5–19. doi: 10.1080/09297040590911185. [DOI] [PubMed] [Google Scholar]
- Antshel KM, Shprintzen R, Fremont W, Higgins AM, Faraone SV, Kates WR. Cognitive and psychiatric predictors to psychosis in velocardiofacial syndrome: a 3-year follow-up study. J Am Acad Child Adolesc Psychiatry. 2010;49(4):333–344. [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Viggiano L, Misceo D, Horvath JE, Archidiacono N, Eichler EE. Human-specific duplication and mosaic transcripts: the recent paralogous structure of chromosome 22. Am J Hum Genet. 2002;70(1):83–100. doi: 10.1086/338458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester-Plane J, Laporta-Hoyos O, Macaya A, Poo P, Melendez-Plumed M, Vazquez E, Pueyo R. Measuring intellectual ability in cerebral palsy: The comparison of three tests and their neuroimaging correlates. Res Dev Disabil. 2016;56:83–98. doi: 10.1016/j.ridd.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Ballif BA, Arnaud L, Arthur WT, Guris D, Imamoto A, Cooper JA. Activation of a Dab1/CrkL/C3G/Rap1 pathway in Reelin-stimulated neurons. Curr Biol. 2004;14(7):606–610. doi: 10.1016/j.cub.2004.03.038. [DOI] [PubMed] [Google Scholar]
- Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, International 22q11.2 Deletion Syndrome, C Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159(2):332–339 e331. doi: 10.1016/j.jpeds.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden CE, Woodin MF, Wang PP, Moss E, McDonald-McGinn D, Zackai E, Cannon TD. The neurocognitive phenotype of the 22q11.2 deletion syndrome: selective deficit in visual-spatial memory. J Clin Exp Neuropsychol. 2001;23(4):447–464. doi: 10.1076/jcen.23.4.447.1228. [DOI] [PubMed] [Google Scholar]
- Cancrini C, Puliafito P, Digilio MC, Soresina A, Martino S, Rondelli R, Italian Network for Primary, I Clinical features and follow-up in patients with 22q11.2 deletion syndrome. J Pediatr. 2014;164(6):1475–1480 e1472. doi: 10.1016/j.jpeds.2014.01.056. [DOI] [PubMed] [Google Scholar]
- Cioffi S, Martucciello S, Fulcoli FG, Bilio M, Ferrentino R, Nusco E, Illingworth E. Tbx1 regulates brain vascularization. Hum Mol Genet. 2014;23(1):78–89. doi: 10.1093/hmg/ddt400. [DOI] [PubMed] [Google Scholar]
- De Smedt B, Devriendt K, Fryns JP, Vogels A, Gewillig M, Swillen A. Intellectual abilities in a large sample of children with Velo-Cardio-Facial Syndrome: an update. J Intellect Disabil Res. 2007;51(Pt 9):666–670. doi: 10.1111/j.1365-2788.2007.00955.x. [DOI] [PubMed] [Google Scholar]
- De Smedt B, Swillen A, Ghesquiere P, Devriendt K, Fryns JP. Pre-academic and early academic achievement in children with velocardiofacial syndrome (del22q11.2) of borderline or normal intelligence. Genet Couns. 2003;14(1):15–29. [PubMed] [Google Scholar]
- Dickinson D, Elvevag B. Genes, cognition and brain through a COMT lens. Neuroscience. 2009;164(1):72–87. doi: 10.1016/j.neuroscience.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Budarf ML, Emanuel BS. A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. Am J Hum Genet. 1992;50(5):924–933. [PMC free article] [PubMed] [Google Scholar]
- Duijff SN, Klaassen PW, de Veye HF, Beemer FA, Sinnema G, Vorstman JA. Cognitive development in children with 22q11.2 deletion syndrome. Br J Psychiatry. 2012;200(6):462–468. doi: 10.1192/bjp.bp.111.097139. [DOI] [PubMed] [Google Scholar]
- Edelmann L, Pandita RK, Spiteri E, Funke B, Goldberg R, Palanisamy N, Morrow BE. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum Mol Genet. 1999;8(7):1157–1167. doi: 10.1093/hmg/8.7.1157. [DOI] [PubMed] [Google Scholar]
- Evers LJ, Engelen JJ, Houben LM, Curfs LM, van Amelsvoort TA. The use of two different MLPA kits in 22q11.2 deletion syndrome. Eur J Med Genet. 2016;59(4):183–188. doi: 10.1016/j.ejmg.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstner AJ, Degenhardt F, Schratt G, Nothen MM. MicroRNAs as the cause of schizophrenia in 22q11.2 deletion carriers, and possible implications for idiopathic disease: a mini-review. Front Mol Neurosci. 2013;6:47. doi: 10.3389/fnmol.2013.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerlings H, Laros JA, Tellegen PJ, Glas CA. Testing the difficulty theory of the SON-R 5(1/2)-17, a non-verbal test of intelligence. Br J Math Stat Psychol. 2014;67(2):248–265. doi: 10.1111/bmsp.12017. [DOI] [PubMed] [Google Scholar]
- Golding-Kushner KJ, Weller G, Shprintzen RJ. Velo-cardio-facial syndrome: language and psychological profiles. J Craniofac Genet Dev Biol. 1985;5(3):259–266. [PubMed] [Google Scholar]
- Gothelf D, Aviram-Goldring A, Burg M, Steinberg T, Mahajnah M, Frisch A, Weizman A. Cognition, psychosocial adjustment and coping in familial cases of velocardiofacial syndrome. J Neural Transm (Vienna) 2007;114(11):1495–1501. doi: 10.1007/s00702-007-0766-9. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Eliez S, Thompson T, Hinard C, Penniman L, Feinstein C, Reiss AL. COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nat Neurosci. 2005;8(11):1500–1502. doi: 10.1038/nn1572. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Penniman L, Gu E, Eliez S, Reiss AL. Developmental trajectories of brain structure in adolescents with 22q11.2 deletion syndrome: a longitudinal study. Schizophr Res. 2007;96(1-3):72–81. doi: 10.1016/j.schres.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grati FR, Molina Gomes D, Ferreira JC, Dupont C, Alesi V, Gouas L, Vialard F. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat Diagn. 2015;35(8):801–809. doi: 10.1002/pd.4613. [DOI] [PubMed] [Google Scholar]
- Guo T, Diacou A, Hiroko N, McDonald-McGinn DM, Hestand M, Demaerel W, Behavior C. Deletion size analysis of 1,680 22q11.2DS subjects identifies a new recombination hotspot on chromosome 22q11.2. Hum Mol Genet. 2018 doi: 10.1093/hmg/ddy028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Repetto GM, McDonald-McGinn DM, Chung JH, Nomaru H, Campbell CL, Behavior C. Genome-Wide Association Study to Find Modifiers for Tetralogy of Fallot in the 22q11.2 Deletion Syndrome Identifies Variants in the GPR98 Locus on 5q14.3. Circ Cardiovasc Genet. 2017;10(5) doi: 10.1161/CIRCGENETICS.116.001690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur RE, Bassett AS, McDonald-McGinn DM, Bearden CE, Chow E, Emanuel BS, Morrow B. A neurogenetic model for the study of schizophrenia spectrum disorders: the International 22q11.2 Deletion Syndrome Brain Behavior Consortium. Mol Psychiatry. 2017;22(12):1664–1672. doi: 10.1038/mp.2017.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroi N, Hiramoto T, Harper KM, Suzuki G, Boku S. Mouse Models of 22q11.2-Associated Autism Spectrum Disorder. Autism Open Access. 2012;(Suppl 1):001. doi: 10.4172/2165-7890.S1-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper SR, Curtiss K, Schoch K, Keshavan MS, Allen A, Shashi V. A longitudinal examination of the psychoeducational, neurocognitive, and psychiatric functioning in children with 22q11.2 deletion syndrome. Res Dev Disabil. 2013;34(5):1758–1769. doi: 10.1016/j.ridd.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M, Kooy RF, Simon TJ, Reyniers E, Girirajan S, Tassone F. A higher rare CNV burden in the genetic background potentially contributes to intellectual disability phenotypes in 22q11.2 deletion syndrome. Eur J Med Genet. 2017 doi: 10.1016/j.ejmg.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayiorgou M, Simon TJ, Gogos JA. 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat Rev Neurosci. 2010;11(6):402–416. doi: 10.1038/nrn2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman NL, Kaufman AS. Comparison of normal and minimally brain dysfunctioned children on the McCarthy Scales of Children’s Abilities. J Clin Psychol. 1974;30(1):69–72. doi: 10.1002/1097-4679(197401)30:1<69::aid-jclp2270300120>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Klaassen P, Duijff S, Swanenburg de Veye H, Beemer F, Sinnema G, Breetvelt E, Vorstman J. Explaining the variable penetrance of CNVs: Parental intelligence modulates expression of intellectual impairment caused by the 22q11.2 deletion. Am J Med Genet B Neuropsychiatr Genet. 2016;171(6):790–796. doi: 10.1002/ajmg.b.32441. [DOI] [PubMed] [Google Scholar]
- Latal B. Neurodevelopmental Outcomes of the Child with Congenital Heart Disease. Clin Perinatol. 2016;43(1):173–185. doi: 10.1016/j.clp.2015.11.012. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Greenberg F, Shaffer LG, Shapira SK, Scambler PJ, Baldini A. Submicroscopic deletions at 22q11.2: variability of the clinical picture and delineation of a commonly deleted region. Am J Med Genet. 1995;56(2):191–197. doi: 10.1002/ajmg.1320560216. [DOI] [PubMed] [Google Scholar]
- Marino BS, Lipkin PH, Newburger JW, Peacock G, Gerdes M, Gaynor JW, Stroke C. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012;126(9):1143–1172. doi: 10.1161/CIR.0b013e318265ee8a. [DOI] [PubMed] [Google Scholar]
- Martinez-Cruz CF, Poblano A, Conde-Reyes MP. Cognitive performance of school children with unilateral sensorineural hearing loss. Arch Med Res. 2009;40(5):374–379. doi: 10.1016/j.arcmed.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Maynard TM, Haskell GT, Peters AZ, Sikich L, Lieberman JA, LaMantia AS. A comprehensive analysis of 22q11 gene expression in the developing and adult brain. Proc Natl Acad Sci U S A. 2003;100(24):14433–14438. doi: 10.1073/pnas.2235651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Fahiminiya S, Revil T, Nowakowska BA, Suhl J, Bailey A, Jerome-Majewska LA. Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to atypical findings in patients with 22q11.2DS. J Med Genet. 2013;50(2):80–90. doi: 10.1136/jmedgenet-2012-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Bassett AS. 22q11.2 deletion syndrome. Nat Rev Dis Primers. 2015;1:15071. doi: 10.1038/nrdp.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Maynard TM, Tucker ES, Fernandez A, Karpinski BA, Rothblat LA, LaMantia AS. Modeling a model: Mouse genetics, 22q11.2 Deletion Syndrome, and disorders of cortical circuit development. Prog Neurobiol. 2015;130:1–28. doi: 10.1016/j.pneurobio.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelovsky E, Frisch A, Carmel M, Patya M, Zarchi O, Green T, Gothelf D. Genotype-phenotype correlation in 22q11.2 deletion syndrome. BMC Med Genet. 2012;13:122. doi: 10.1186/1471-2350-13-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow B, Goldberg R, Carlson C, Das Gupta R, Sirotkin H, Collins J, et al. Molecular definition of the 22q11 deletions in velo-cardio-facial syndrome. Am J Hum Genet. 1995;56(6):1391–1403. [PMC free article] [PubMed] [Google Scholar]
- Moss EM, Batshaw ML, Solot CB, Gerdes M, McDonald-McGinn DM, Driscoll DA, Wang PP. Psychoeducational profile of the 22q11.2 microdeletion: A complex pattern. J Pediatr. 1999;134(2):193–198. doi: 10.1016/s0022-3476(99)70415-4. [DOI] [PubMed] [Google Scholar]
- Nelis M, Esko T, Magi R, Zimprich F, Zimprich A, Toncheva D, Metspalu A. Genetic structure of Europeans: a view from the North-East. PLoS One. 2009;4(5):e5472. doi: 10.1371/journal.pone.0005472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niarchou M, Zammit S, van Goozen SH, Thapar A, Tierling HM, Owen MJ, van den Bree MB. Psychopathology and cognition in children with 22q11.2 deletion syndrome. Br J Psychiatry. 2014;204(1):46–54. doi: 10.1192/bjp.bp.113.132324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niklasson L, Gillberg C. The neuropsychology of 22q11 deletion syndrome. A neuropsychiatric study of 100 individuals. Res Dev Disabil. 2010;31(1):185–194. doi: 10.1016/j.ridd.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Olszewski AK, Radoeva PD, Fremont W, Kates WR, Antshel KM. Is child intelligence associated with parent and sibling intelligence in individuals with developmental disorders? An investigation in youth with 22q11.2 deletion (velo-cardio-facial) syndrome. Res Dev Disabil. 2014;35(12):3582–3590. doi: 10.1016/j.ridd.2014.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsdottir S, Belfrage M, Sandstedt E, Viggedal G, Uvebrant P. Disabilities and cognition in children and adolescents with 22q11 deletion syndrome. Dev Med Child Neurol. 2005;47(3):177–184. doi: 10.1017/s0012162205000320. [DOI] [PubMed] [Google Scholar]
- Park TJ, Curran T. Crk and Crk-like play essential overlapping roles downstream of disabled-1 in the Reelin pathway. J Neurosci. 2008;28(50):13551–13562. doi: 10.1523/JNEUROSCI.4323-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radoeva PD, Coman IL, Salazar CA, Gentile KL, Higgins AM, Middleton FA, Kates WR. Association between autism spectrum disorder in individuals with velocardiofacial (22q11.2 deletion) syndrome and PRODH and COMT genotypes. Psychiatr Genet. 2014;24(6):269–272. doi: 10.1097/YPG.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rump P, de Leeuw N, van Essen AJ, Verschuuren-Bemelmans CC, Veenstra-Knol HE, Swinkels ME, van Ravenswaaij-Arts CM. Central 22q11.2 deletions. Am J Med Genet A. 2014;164A(11):2707–2723. doi: 10.1002/ajmg.a.36711. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC, O’Hare AM, Hu P, Roe BA, Emanuel BS. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9(4):489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- Shprintzen RJ. Velo-cardio-facial syndrome: a distinctive behavioral phenotype. Ment Retard Dev Disabil Res Rev. 2000;6(2):142–147. doi: 10.1002/1098-2779(2000)6:2<142∷AID-MRDD9>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Sprecher E, Ishida-Yamamoto A, Mizrahi-Koren M, Rapaport D, Goldsher D, Indelman M, Mandel H. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet. 2005;77(2):242–251. doi: 10.1086/432556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swillen A, Devriendt K, Legius E, Eyskens B, Dumoulin M, Gewillig M, Fryns JP. Intelligence and psychosocial adjustment in velocardiofacial syndrome: a study of 37 children and adolescents with VCFS. J Med Genet. 1997;34(6):453–458. doi: 10.1136/jmg.34.6.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swillen A, McDonald-McGinn D. Developmental trajectories in 22q11.2 deletion. Am J Med Genet C Semin Med Genet. 2015;169(2):172–181. doi: 10.1002/ajmg.c.31435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swillen A, Vandeputte L, Cracco J, Maes B, Ghesquiere P, Devriendt K, Fryns JP. Neuropsychological, learning and psychosocial profile of primary school aged children with the velo-cardio-facial syndrome (22q11 deletion): evidence for a nonverbal learning disability? Child Neuropsychol. 1999;5(4):230–241. doi: 10.1076/0929-7049(199912)05:04;1-R;FT230. [DOI] [PubMed] [Google Scholar]
- Van Aken K, De Smedt B, Van Roie A, Gewillig M, Devriendt K, Fryns JP, Swillen A. Motor development in school-aged children with 22q11 deletion (velocardiofacial/DiGeorge syndrome) Dev Med Child Neurol. 2007;49(3):210–213. doi: 10.1111/j.1469-8749.2007.00210.x. [DOI] [PubMed] [Google Scholar]
- Vorstman JA, Breetvelt EJ, Duijff SN, Eliez S, Schneider M, Jalbrzikowski M, Behavior in 22q11.2 Deletion, S Cognitive decline preceding the onset of psychosis in patients with 22q11.2 deletion syndrome. JAMA Psychiatry. 2015;72(4):377–385. doi: 10.1001/jamapsychiatry.2014.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JA, Jalali GR, Rappaport EF, Hacker AM, Scott C, Emanuel BS. MLPA: a rapid, reliable, and sensitive method for detection and analysis of abnormalities of 22q. Hum Mutat. 2006;27(8):814–821. doi: 10.1002/humu.20330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willshire D, Kinsella G, Prior M. Estimating WAIS-R IQ from the National Adult Reading Test: a cross-validation. J Clin Exp Neuropsychol. 1991;13(2):204–216. doi: 10.1080/01688639108401038. [DOI] [PubMed] [Google Scholar]
- Woodin M, Wang PP, Aleman D, McDonald-McGinn D, Zackai E, Moss E. Neuropsychological profile of children and adolescents with the 22q11.2 microdeletion. Genet Med. 2001;3(1):34–39. doi: 10.1097/00125817-200101000-00008. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Weinberger R, Moore TM, Calkins ME, Guri Y, McDonald-McGinn DM, Gur RC. Performance on a computerized neurocognitive battery in 22q11.2 deletion syndrome: A comparison between US and Israeli cohorts. Brain Cogn. 2016;106:33–41. doi: 10.1016/j.bandc.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.