

Abstract
A screen conducted with nearly 100,000 compounds and a surrogate functional assay for stimulation of an immune response that measured the release of TNF-α from treated human THP-1 myeloid cells differentiated along the macrophage line led to the discovery of the diprovocims. Unique to these efforts and of special interest, the screening leads for this new class of activators of an immune response came from a compound library designed to promote cell surface receptor dimerization. Subsequent comprehensive structure–activity relationship studies improved the potency 800-fold over that of the screening leads, providing diprovocim-1 and diprovocim-2. The diprovocims act by inducing cell surface toll-like receptor(TLR)-2 dimerization and activation with TLR1 (TLR1/TLR2 agonist), bear no structural similarity to any known natural or synthetic TLR agonist, are easy to prepare and synthetically modify, and selected members are active in both human and murine systems. The most potent diprovocim (3, diprovocim-1) elicits full agonist activity at extraordinarily low concentrations (EC50 = 110 pM) in human THP-1 cells, being more potent than the naturally-derived TLR1/TLR2 agonist Pam3CSK4 or any other known small molecule TLR agonist.
Graphical Abstract
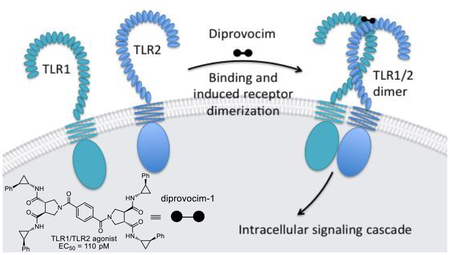
Introduction
The human immune system is composed of two components, the innate and adaptive immune systems. The innate immune system is encoded into the germline of an organism and constitutes the first line of defense in mammals. It responds to pathogens and abnormal cells through initiation of intracellular signaling cascades that lead to the activation of transcription factors which trigger the production of cytokines and chemokines with participation of multiple cell types, including dendritic cells, macrophages, neutrophils, and natural killer cells.1 The subsequent activation of the adaptive immune system involves antigen-specific T cell and B cell responses mediated by antigen-presenting dendritic cells. It serves to provide long term host protection through the action of T cells and antibodies to neutralize the pathogens and abnormal cells.1,2 The Toll-like receptors (TLRs)3–6 are the most widely recognized set of the pathogen-associated or damage-associated molecular pattern (PAMPs and DAMPs) receptors that recognize molecular components of pathogens or abnormal cells and marshal the initial innate immune response.3,5 They also induce the adaptive immune response,6 and the action of vaccines is often due in part to the activation of the TLR system.7 As such, TLR agonists are attractive as new vaccine adjuvants for both infectious diseases and oncology that act by well-defined mechanisms,7–10 as prophylactics against pathogen exposure (e.g.; biodefense),11 or as immunostimulators alone or in combination with other drugs,11 particularly in oncology.8–11 A recent exciting application lies in the field of cancer immunotherapy, where the adaptive immune system is exploited to kill cancer cells based on their expression of neo-antigens.12 The potential of such cancer immunotherapy results not only from the initial killing of tumor cells, but also from a long lasting systemic anti-tumor memory response (antigen-specific anti-tumor immunity). However, most known TLR agonists are mimics or modifications of microbial components that have unattractive structural, physical, or stability properties,13 and only a limited number of small molecule classes have been found to behave as TLR agonists.8–10 Notable examples of such small molecules include the TLR7 agonists imiquimod,14 isotoribine and 8-oxo-9-benzyladenine,14 as well as the TLR7/8 agonist resiquimod15 that today still serve as the inspiration for nearly all such work.16,17 Recent interest in the area has led to the discovery of a multi-Toll-like receptor agonist that acts on TLR 3, 8 and 9,18 to the intense study of the STING agonist cGAMP,19 and to our own disclosure of the neoseptins,20 a new class of mouse TLR4 agonists21–24 that bear no structural similarity to bacterial lipopolysaccharide (LPS) or its active core Lipid A (LPA).21
Herein, we report full details of the discovery of the diprovocims, a new and especially attractive synthetic small molecule class of human TLR agonists (Figure 1). Studies decades ago discovered the immune activating N-terminal segments of bacterial triacylated lipoproteins and lipopeptides (e.g.; Pam3CSK4,25,26 Figure 1), which were shown later to act by heterodimerization of TLR1/TLR2 at the cell surface.27,28 Such agonists based on the lipoproteins are effective vaccine adjuvants when admixed or covalently-linked with antigens25,26 and continue to be widely studied today.29 Among the TLRs, TLR2 requires heterodimerization with either TLR1 or TLR6 for activation. Bacterial triacylated lipoproteins or lipopeptides are the most widely recognized agonists that activate TLR1/TLR2 (e.g.; Pam3CSK4) and bacterial diacylated lipopolypeptides stimulate TLR2/TLR6 (e.g.; MALP-230, Figure 1).31 Complementary to recent studies that have disclosed the only other known small molecule TLR2/TLR1 agonists,32 the diprovocims are a new and exceptionally potent class of synthetic small molecule TLR2/TLR1 agonists and they emerged from screening a unique chemical library designed to promote cell surface receptor dimerization.33 The most potent of the diprovocims (compound 3) elicits full agonist activity at extraordinarily low concentrations (EC50 = 110 pM) in human cells, being more potent than Pam3CSK4 or any other known small molecule TLR agonist. The diprovocims act by a well-defined mechanism (TLR1/TLR2 agonist)34 and selected members are active in both human and murine systems. As shown herein, the class exhibits exquisite structure–activity (SAR) relationships. The efficacy of the class matches that of natural TLR agonists such as LPS or the lipopeptides and selected members exhibit a potency that matches or exceeds that of the natural agonists. Compound 3 (diprovocim-1) has been shown to be active as an adjuvant in mice when co-administered by conventional intramuscular (i.m.) injection (vaccination) with an antigen (OVA).34 It was also shown to act synergistically with a checkpoint inhibitor (anti-PD-L1), where the combination treatment cured or prolonged survival of mice implanted with an immunogen bearing murine melanoma (B16-OVA) and protected mice from tumor rechallenge.34 Notably, the latter impressive in vivo activity was observed with diprovocim/OVA co-administration i.m., rather than with intratumor adjuvant administration as has been a convention in recent studies. Finally, the class bears no structural similarity to the TLR1/TLR2 lipoprotein agonists or any other natural or synthetic TLR agonist, and members of the class are easy to prepare and synthetically modify.
Figure 1.

Structures of the known TLR1/TLR2 (Pam3CSK4) and TLR2/TLR6 (MALP-2) agonists and diprovocim-1 and diprovocim-2.
Results
Screen and Results.
The discovery of the role of the TLRs emerged in genetic studies in whole organisms with impaired innate immune responses caused by spontaneous or induced nucleic acid mutations in genes impacting the immune system (lack of sensitivity to LPS due to a mutated and disabled TLR4).5,35 Complementary to such efforts, we explored the use of an unbiased chemical genetics approach,35 screening libraries of compounds in surrogate cell-based functional assays for activation of an immune response. A screening campaign was conducted with nearly 100,000 compounds,36 using a functional assay that measured the stimulated release of TNF-α from treated human THP-1 myeloid cells partially differentiated along the macrophage line by PMA pretreatment (Supporting Information, Figure S1).34 The functional activity measured by this assay is both a rare activation event (activator/agonist vs inhibitor/antagonist) and is sensitive, such that even weak stimulation of TNF-α release proved easily detectable. By design, subsequent assay of active compounds against macrophages from available mice that bear germline genetic defects or knockouts of the genes encoding each of the TLRs, other molecular pattern recognition receptors, or downstream signaling molecules would be used to establish whether the activity was derived from known or presently unrecognized targets.20 The chemical libraries screened in the efforts represent a unique compound collection populated by nontraditional compounds36 designed to target protein–protein37,38 or protein–DNA interactions39 as well as the major enzyme classes.40 Within the full 100,000 compound library that was screened, the lead compounds detailed herein emerged from a previously unreported subset of the library designed to promote cell surface receptor dimerization (ca. 6,000 compounds), with each half monomer of the C2-symmetrical compounds designed to bind each protein receptor monomer.33 The sub-library from which the leads were identified was prepared as 100 mixtures of 10 compounds on the trans-pyrrolidine-3,4-dicarboxylate core template (Supporting Information Figure S2).38 It was composed of all individual combinations of ten defined R1 and another ten defined R2 substituents, and a mixture of ten linkers (denoted by X, Figure 2).33,41 The screening results were remarkable, providing pools of 10 compounds that displayed activity each and every time the R2 subunit was either 4-fluorophenethylamine (residue 4, well X4) or trans-2-phenylcyclopropylamine (residue 9, well X9) in the tested pools independent of the structure of R1 (Figure 2). Analogous libraries with different cores (e.g.; iminodiacetic acid and isoindoline-4,5-dicarboxylic acid vs pyrrolidine-3,4-dicrboxylic acid),33 other R1 and R2 substituents,33 alternative linker groupings,33 and even higher order displays (e.g.; trimer, tetramer)33 failed to provide activity in the screen. Even more remarkable was the fact that a large number of closely compounds with related R1 and R2 substituents within the sub-library were inactive, including phenethylamine, (4-methoxyphen)-ethylamine, (4-hydroxyphen)ethylamine, (3-methoxyphen)ethylamine, benzylamine (Supporting Information Figure S2). Thus, it was also determined that even small deviations from the structure resulted in no detection of activity, providing a first level structure–activity relationship (SAR) study. Not only was the activity robust and structurally specific, but the repetitive observation with each appearance of residue 4 or residue 9 in the compound mixtures regardless of the identity of R1 indicated that the activity was not the result of an unrecognized artefact (Figure 2).
Figure 2.

Top: Screening results for the receptor dimerization library (plate 39 G–H wells and plate 40 all wells) measuring stimulated TNF-α release from PMA-differentiated human THP-1 myeloid cells (10 compound mixtures tested at 50 μM). The compound mixtures in the starred wells were prepared as individual compounds. Bottom: Structures of active mixtures containing R2 residues 4 and 9 where R1 is single defined substituent and X is a mixture of ten linkers, see Supporting Information Figure S2.
Diprovocim-1.
The ten compounds in two of the active wells (plate 39G4 and plate 39G9), containing either the residue 4 or residue 9 substituent, were prepared and tested individually, identifying only two active linkers. Benzene-1,4-dicarboxylic acid was by far the most potent of the two active linkers and this is illustrated in Figure 3 with the individual compounds found in plate 39G9. Structurally specific activity was observed and even dicarboxylic acid linkers spatially or structurally related to benzene-1,4-dicarboxylic acid were inactive (e.g.; C5 vs C4 or C9 and C10).41 For simplicity, the same studies conducted concurrently with plate 39G4, containing the R2 residue 4 (4-fluorophen)ethylamine substituent are detailed separately following the discussion of chronological work conducted on plate 39G9.
Figure 3.

Top: Synthesis of the individual compounds in plate 39G9 used to identify the linker of active compounds. Bottom: Activity of individual compounds (labeled by the linker C1–C10) in the plate 39G9 mixture of the receptor dimerization library tested alongside controls (LPS and 0 = no compound) measuring stimulated TNF-α release from differentiated human THP-1 cells (tested at 50 μM unless indicated otherwise).
Because the R2 side chain was key to the activity, we prepared a set of five compounds, containing the identified linker (benzene-1,4-dicarboxylic acid), to systematically establish the importance of the presence and number of 2-phenylcyclopropylamine (R2) versus phenethylamine (R1) side chains present in the compounds in plate 39G9 (Figure 4).41 The five compounds were prepared from the three pyrrolidine-3,4-dicarboxamides 5–7 and simultaneous (8, 10 and 12) coupling with benzene-1,4-dicarboxylic acid (2 equiv PyBrOP, i-Pr2NEt, DMF, 23 °C) or sequential (9 and 11) couplings starting with the mono methyl ester of benzene-1,4-dicarboxylic acid with an intermediate hydrolysis of the methyl ester (4 equiv LiOH, THF:MeOH:H2O 4:1:1, 23 °C). The results of their assessment were stunning. Although the compounds that contained no or one 2-phenylcyclopropylamine substituents were inactive (8 and 9), each added 2-phenylcyclopropylamine substituent (two as found in library, three, and four) incrementally (3–5-fold each) increased the potency (potency with number of residue 9 side chains: 0 and 1 < 2 < 3 < 4). The compounds containing three and four 2-phenylcyclopropropylamine substituents (11 and 12) were more potent than the original lead 10 (EC50 = 80 nM) in the library and compound 12, containing four such substituents, was exceptionally potent (EC50 = 10 nM) and equally efficacious with LPS. Just as significantly and although the leads that emerged from the screening library were inactive in stimulating the release of TNF-α from mouse macrophages, compound 12 was active at doing so, albeit at a lower potency (EC50 = 100 nM). Although a lack of activity in the murine system would not be detrimental to their advancement for human studies, the observation of activity in mouse macrophages with 12 and subsequent related compounds is especially useful in conducting pharmacological in vivo studies in mouse models.
Figure 4.

Compounds that established the importance of the presence and number of 2-phenylcyclopropylamine residues derived from active lead (10) in plate 39G9. EC50’s derived from dose-response curves for the stimulated release of TNF-α from differentiated THP-1 cells or mouse macrophages by 8–12.
This activity of 12 is even more impressive since it was prepared with racemic trans-pyrrolidine-3,4-dicarboxylic acid and racemic trans-2-phenylcyclopropylamine as found in the original library. As a consequence and even though each trans stereochemical relationship in the pyrrolidine cores and the side chain substituents is fixed, 12 was still a mixture of all possible 21 diastereomers and enantiomers arising from the use of racemic materials. As daunting as this may seem, the unraveling of the activity within this complex diastereomeric mixture proved surprisingly straightforward. Each enantiomer of 2-phenylcyclopropylamine (1S,2R and 1R,2S) is commercially available42 and a reported resolution43 was used to obtain the two enantiomers (S,S and R,R) of N-Boc-pyrrolidine-3,4-dicaboxylic acid.43 These were used to prepare four pyrrolidine/side chain subunits 15–18, representing all combinations of each enantiomer of N-Boc-pyrrolidine-3,4-dicaboxylic acid independently substituted with each enantiomer of 2-phenylcyclopropylamine (same enantiomer at each of two sites) (Figure 5). Initially and for simplicity, we did not examine combinations where the two substitution sites on the pyrrolidine core contained the two different enantiomers (1S,2R and 1R,2S) of 2-phenylcyclopropylamine. The four substituted pyrrolidine core monomers 15–18 were then N-Boc deprotected (4 N HCl, dioxane, 23 °C, 2 h) and simultaneously or sequentially coupled to benzene-1,4-dicarboxylic acid to provide the linked dimers, containing each and all combinations of the individual enantiomerically defined monomers (10 compounds). Consistent with a specific receptor interaction, the results of their assessment were crystal clear. Activity was observed only in compounds that contained the (S,S)-pyrrolidine-3,4-dicarboxylic acid core and only when the (1S,2R)-2-phenylcyclopropylamine side chain was present. In fact, the activity increased as the number of (1S,2R)- versus (1R,2S)-2-phenylcyclopropylamine substituents increased in much the same manner as was observed in the side chain series 8–12. This further indicated that the incorporation of the inactive (1R,2S)-2-phenylcyclopropylamine enantiomer in pyrrolidine monomers also containing the active 1S,2R enantiomer would likely be unproductive (remaining 11 compounds). Moreover, this activity spanned a huge range with nearly all the compounds being less potent than the all racemate or inactive (Figure 5). Compound 3 (diprovocim-1), composed of a single enantiomer of the pyrrolidine core dicarboxylic acid (S,S) and a single enantiomer of the 2-phenylcyclopropylamine (1S,2R) proved to be by far the most active diastereomer, exhibiting a stunning potency (EC50 = 110 pM) for stimulating the release of TNF-α from human differentiated THP-1 cells. This activity proved to be 100-fold more potent than the all racemate 12 (EC50 = 10 nM), consistent with it being responsible for essentially all the activity observed in the original racemic mixture (21 diastereomers). Compound 3 was also found to be roughly >30-fold more potent than any other diastereomer. In addition, diprovocim-1 (3) was potent (EC50 = 1.3 nM) at stimulating the release of TNF-α from mouse macrophages. Here, it not only proved to be more potent than the diastereomeric mixture 12, but all other isomers were much less active and none approached or matched the activity of the all racemate (Figure 5). Finally, diprovocim-1 (3) proved to be equally efficacious but 5–10 times more potent than Pam3CSK4 (1, EC50 = 0.91 nM) in a direct side-by-side comparison in the assay for the stimulated release of TNF-α from differentiated human THP-1 cells (Supporting Information Figure S3).
Figure 5.

Ten Diastereomers prepared of the all racemate 12, identifying the active component. EC50’s derived from dose-response curves for the stimulated release of TNF-α from differentiated human THP-1 cells and mouse macrophages by 3 (diprovocim-1) and 19–27.
Compound 8 was also prepared as the single all (S)-enantiomer from 15 and was found to be inactive (EC50 >5 μM) like the all racemate mixture, failing to stimulate the release of TNF-α from either partially differentiated THP-1 cells or mouse macrophages. Just as significantly, it was found to be incapable of acting as an antagonist, inhibiting the activity of diprovocim-1 (3), when co-administered at 5 μM. This indicates that the lack of agonist activity with 8, and presumably related compounds, is due to ineffective binding to the target TLRs and is not due to effective binding but failure to induce the active dimer receptor conformation. To further establish the importance of each (1S,2R)-2-phenylcyclopropylamine amide substituent and to rule out an inhibitory effect of the presence an alternative substituent, we prepared a series of deletion analogues (29–34) of diprovocim-1 with sequential removal of each side chain or key structural feature (Supporting Information Figure S4).44 From these studies, it was established that the presence and stereochemistry of each (1S,2R)-2-phenylcyclopropylamine amide substituent, the presence and stereochemistry of the two intact (S,S)-pyrrolidine-3.4-dicarboxylate cores, and the benzene-1,4-dicarboxylate linker are all integral to the expressed activity of diprovocim-1 (3).
At this stage, we reexamined the compound linkers found in the original library (35–43), along with five additional closely related dicarboxylic acid linkers (44–48), but now with only the active enantiomer of the substituted pyrrolidine-3,4-dicarboxamide 15 and bearing four (1S,2R)-2-phenylcyclopropylamine amide substituents in place of the all racemate (Figure 6). The latter series 44–48 placed the two carboxylic acids on phenyl or naphthyl aromatic rings, but located spatially in positions slightly altered from that found in diprovocim-1 or at the same position but with an attached fused ring (45). Each was prepared in a single coupling step from 15 and the linker dicarboxylic acid (4 N HCl, dioxane–THF, 23 °C, 3 h; 2 equiv PyBrOP, i-Pr2NEt, DMF, 23 °C, 18 h). Not surprisingly, benzene-1,4-dicarboxylic acid remained the most potent linker in the original series (3 vs 35–43). Moreover, no compound in the series 44–48 displayed activity close to diprovocim-1. All except 44 were inactive, and even 44 was >10-fold less active than diprovocim-1. However, what is remarkable in these comparisons is how specific the activity remained for the benzene-1,4-dicarboxylic acid linker. All other linkers were ≥10-fold less active, with most displaying no activity even at concentrations >50,000-fold higher. Clearly, the linker contribution to the activity of 3 is just as specific and integral as that of the substituted pyrrolidine-3,4-dicarboxamide monomers.
Figure 6.

EC50’s derived from dose-response curves for the stimulated release of TNF-α from differentiated human THP-1 cells or mouse macrophages by 3 (diprovocim-1) and 35–48.
With this appreciation of the importance of the precise spacing of the dicarboxylic acid in the structure of the linker, a more targeted examination of alternative linkers was conducted (Figure 7). Isosteric replacements of the benzene ring (49), six-membered heterocyclic dicarboxylic acids (50, 51) that might alter the physical properties (e.g.; solubility) or define an orientation of the adjacent pyrrolidine-3,4-dicarboxamide, an acyclic unsaturated dicarboxylic acid linker that might flexibly substitute for benzene (52), a series of further substituted benzene-1,4-dicaboxylic acids introducing functionality for use in conjugation to candidate antigens (53–58), and those used to explore disubstitution (59–63) were examined. The latter series was especially interesting because of the substituents marked influence on the adoption of preferred conformational orientations of the adjacent pyrrolidine-3,4-dicarboxamides through destabilizing steric interactions or stabilizing hydrogen bonding. Each was prepared by acid-catalyzed Boc deprotection of 15 (4 N HCl, dioxane, 23 °C) and a single coupling step with the linker dicarboxylic acid (2 equiv PyBrOP, i-Pr2NEt, DMF, 23 °C, 18 h). Remarkably and as benign as many of the changes are, nearly all such compounds examined were less potent than diprovocim-1, highlighting the unique role the linker plays in the expression of the biological activity. The exception to this generalization was the phenol 53, which nearly matched the activity of diprovocom-1 itself. Although the number of compounds on which to base the conclusions is small, the potency appears to smoothly decrease as the size of the (adjacent) substituents increase (e.g.; potency: 3 > 53 > 55 > 58 > 54, 57 and 3 > 60 > 61 > 62 > 63). Although not required for their applications, compounds (53 and 55) within this series still exhibit excellent potency sufficient to confidently establish that functionalization sites on the linker core are available for conjugation with candidate antigens when warranted.
Figure 7.

Linker modifications. EC50’s were derived from dose-response curves for the stimulated release of TNF-α from differentiated human THP-1 cells and mouse macrophages by 49–63.
Diprovocim-2.
A more straightforward series of studies was conducted with the active plate 39G4 well. Examination of the 10 individual compounds in the well mixture identified the same active linker (benzene-1,4-dicarboxylic acid, not shown). Because the R2 side chain was key to the observation of the repetitive activity seen in the original screen independent of the structure of R1 (Figure 2), a set of five compounds was prepared with the active benzene-1,4-dicarboxylic acid linker to establish the importance of the presence and number of 4-fluorophenethylamine (R2) versus phenethylamine (R1) side chains present in the compounds in the plate 39G9 well (Figure 8).41 The five compounds were prepared from the three pyrrolidine-3,4-dicarboxamides 6, 64 and 65 and simultaneous (8, 67 and 69) or sequential (66 and 68) coupling with benzene-1,4-dicarboxylic acid or its mono methyl ester, respectively. The results of their assessment were clear. Activity was enhanced with the addition of each 4-fluorophenethylamine side chain (ca. 3–5-fold) and 69, containing four, was the most potent (EC50 = 100 nM, racemate) compound in the set.
Figure 8.

Synthesis of compounds that established the importance of the presence and number of 4-fluorophenethylamine side chains.
Since the 4-fluoro substituent on the 4-fluorophenethylamide uniquely conveyed activity to the compounds (phenethylamide inactive), a series of alternative substituents were examined in which the 4-fluoro group was moved to the 3- and 2-positions, conservatively replaced with other halogens (Cl and Br), replaced with a series of small electron-donating or electron-withdrawing groups with some capable of serving as hydrogen bond donors or acceptors, replaced with a large phenyl group, or with incorporation of a 2-naphthyl aromatic ring (Figure 9). Each was prepared as a racemate by coupling racemic N-Boc trans-pyrrolidine-3,4-dicarboxylic acid with the arylethylamine (2 equiv, EDCI, HOAt, 2,6-lutidine, DMF, 23 °C, 16 h) followed by N-Boc deprotection of 70–83 (4 N HCl, dioxane 23 °C, 3 h) and subsequent coupling with benzene-1,4-dicarboxylic acid (2 equiv PyBrOP, i-Pr2NEt, DMF, 23 °C, 18–24 h). Stunningly, all were inactive at stimulating the release of TNF-α from human differentiated THP-1 cells at concentrations up to 5 μM. The sole exception was 84 (EC50 = 1 μM, ≥10-fold less active), which incorporated the most conservative change of moving the fluorine substituent from the p- to m-position (3- vs 4-fluorophenethylamide). Clearly the fluorine substituent and the 4-fluorophenethylamide side chains are essential and remarkably specific to the expression of the biological activity of the compounds.
Figure 9.

Aryl substituent effects on activity of diprovocim-2 (4).
Because the library and 69 were prepared from racemic trans-pyrrolidine-3.4-dicarboxylic acid, they were a mixture of enantiomers and diastereomers. Compound 69 was a mixture of three compounds (two enantiomers and one meso compound), making the establishment of the active enantiomer in the mixture straightforward. Each of the two enantiomers (S,S and R,R) of N-Bocpyrrolidine-3,4-dicaboxylic acid43 was coupled with 4-fluorophenethylamine (EDCI, HOAt, 2,6-lutidine, 23 °C, 16 h) to prepare the two possible enantiomeric pyrrolidine-3,4-dicarboxamides (S,S)- and (R,R)-65 (Figure 10). These were independently but simultaneously coupled with benzene-1,4-dicarboxylic acid (2 equiv PyBrOP, i-Pr2NEt, DMF, 23 °C, 18–24 h) to provide the two enantiomers 4 and 98, bearing the all S or all R configurations, respectively. Similarly, sequential coupling (PyBrOP, i-Pr2NEt, DMF, 23 °C) of (S,S)- and (R,R)-65 with the mono methyl ester of benzene-1,4-dicarboxylic acid with an intermediate hydrolysis of the methyl ester (LiOH, THF/MeOH/H2O, 23 °C) provided the meso isomer 99. The all S enantiomer 4 (EC50 = 50 nM, diprovocim-2) proved to be 3-fold more potent than the all racemate (EC50 = 150 nM), whereas the all R enantiomer 98 was inactive (EC50 = >5 μM, >100-fold less potent). Interestingly, the meso isomer 99 (EC50 = 70 nM), containing both a S,S- and R,R-pyrrolidine-3,4-dicarboxamide, was also active and nearly matched the potency of the all S enantiomer. Despite this potent activity in human THP-1 cells, no compound in this series stimulated the release of TNF-α from mouse macrophages.
Figure 10.

Synthesis of the three enantiopure compounds found in the all racemate 69, identifying the active components. EC50’s were derived from dose-response curves for the stimulated release of TNF-α from differentiated human THP-1 cells by 4 (diprovocim-2) and 98–99.
With use of the enantiopure (S,S)-pyrrolidine-3,4-dicarboxamide 65, the dicarboxylic linkers in the original library (C1–C10) as well as a subset of those that proved most interesting with diprovocim-1 were re-examined now with compounds that bear all four 4-fluorophenethylamide side chains (Figure 11). Each was prepared in a single coupling step (2 equiv PyBrOP, i-Pr2NEt, DMF, 23 °C, 18–24 h) of the linker dicarboxylic acid following acid-catalyzed deprotection of enantiopure (S,S)-65. In this series, none of the alternative linkers provided compounds that were active in either human THP-1 cells or mouse macrophages and only the phenol 110, like the behavior of 53 versus diprovocim-1, nearly matched the activity diprovocim-2.
Figure 11.

Synthesis of compounds that contain the active pyrrolidine-3,4-dicarboxamide enantiomer (S,S)-65 and the original library 10 linkers or additional selected dicarboxylic acid linkers. Bottom: EC50’s derived from dose-response curves for the stimulated release of TNF-α from differentiated human THP-1 cells or mouse macrophages by 4 (diprovocim-2) and 100–110.
Hybrid structures of diprovocim-1 and diprovocim-2 and additional key compounds.
Given the unique behavior that only two of the examined side chains displayed and their importance to the functional activity of 3 and 4, compounds containing a combination of the two side chains were explored (111–113) and prepared as the all racemate diastereomeric mixtures (Supporting Information Figure S5). The compounds displayed a consistent trend where replacement of the phenethylamine side chains with 4-fluorophenethylamine sided chains improved activity and where a 2-phenylcyclopropylamine replacement of the 4-fluorophenethylamine side chains further improved activity. Thus, the two side chains can be used interchangeably with predictable influences on the biological potency (trans-2-phenylcyclopropylamine > 4-fluorophenethylamine) indicating they are binding at the same site influencing activity in a similar manner at its target. The most potent of these were prepared as single enantiomers, bearing the identified active enantiomer derived from 15 and either (S,S)- and (R,R)-65 (Figure 12). These studies established that the hybrid structure 114 (diprovocim-4), possessing the two (S,S)-pyrrolidine cores, is an effective TLR1/TLR2 agonist active in both human THP-1 cells and mouse macrophages. Thus, 114 displays properties like those of diprovocim-1. In addition, it is less potent than diprovocim-1 but more active than diprovocim-2, displaying a potency between that of the compounds from it is derived. The analogous hybrid structure 115 (diprovocim-5), containing one subunit derived from (R,R)-65, was 5-fold less active in human THP-1 cells and inactive in mouse macrophages, reflecting both a lower potency and greater species difference.
Figure 12.

Diprovocim-4 and diprovocim-5. EC50’s derived from dose-response curves for the stimulated release of TNF-α from differentiated human THP-1 cells or mouse macrophages.
Among the most interesting of the modifications made to the diprovocims was the incorporation of the fluorine group found in diprovocim-2 (4) into the side chain substituent of diprovocim-1 (3) (Figure 13). Thus, coupling of (1S,2R)-2-(4-fluorophenyl)cyclopropylamine45 (116, 2 equiv) with (S,S)-14 (EDCI, HOAt, 2,6-lutidine, DMF, 23 °C, 16 h) followed by Boc deprotection of 117 (4 N HCl, dioxane, 23 °C, 3 h) and coupling of the resulting amine with benzene-1,4-dicarboxylic acid (PyBrOP, i-Pr2NEt, DMF, 23 °C, 18–24 h) provided 118 (diprovocim-3). Its assessment for the stimulated release of TNF-α from differentiated human THP-1 cells (EC50 = 130 pM) and mouse macrophages (EC50 = 1.2 nM) revealed that it was indistinguishable from diprovocim-1 (3), displaying exceptionally potent activity. Thus, the addition of the p-fluoro group did not further enhance the potency of 3, but its presence also did not disrupt the activity of 3. Another way of viewing this result is that introduction of the four rigidifying and stereochemically-defined cyclopropanes into diprovocim-2 (4) improved the activity 400-fold. The unique behavior of the 4-fluoro substituent in 118 was established with the examination of a small but key series of additional compounds that contain alternative C4 phenyl substituents (X = Cl, Br, Me, OMe and CN vs F, Figure 13). The set contains conservative but larger halogen replacements (X = Cl, Br), a conservative but larger hydrophobic substituent replacement (X = Me), and both strong yet small electron-donating (X = OMe) and electron-withdrawing (X = CN) substituent replacements. Compounds 119–123 were prepared by the route outlined for 118, using the corresponding 4-substituted (1S,2R)-2-(phenyl)cyclopropylamines.45 Remarkably, all compounds in this expanded series exhibited substantially reduced activity that correlates with the progressively increased size or extended length of the phenyl C4 substituent (activity: H = F > Cl > Me > Br > OMe, CN). The most active of these compounds (119, X = Cl) proved to be 300-fold less potent in THP-1 cells and >1000-fold less potent in mouse macrophages.
Figure 13.

Diprovocim-3 (118) and related aryl substitution analogues 119–123 of diprovocim-1 (3).
Hybrid structures with incorporation of the lipid side chains found in natural TLR2/TLR1 and TLR2/TLR6 agonists.
Among the TLRs, TLR2 requires heterodimerization with either TLR1 or TLR6 for activation. Bacterial triacylated lipoproteins are the most widely-recognized agonists activating TLR1/TLR2 (e.g.; Pam3CSK425–29), whereas bacterial diacylated lipopolypeptides bind and activate TLR2/TLR6 (e.g.; MALP-230, see Figure 1).31 TLR1/TLR2 preferentially binds triacyl lipopeptides and binds diacyl lipopeptides only weakly, whereas TLR2/TLR6 only binds diacyl lipopeptides. X-ray crystallographic structures of Pam3CSK4 (three lipid chains) bound to hTLR1/TLR2 and Pam2CSK4 (two lipid chains) bound to mTRL2/TLR6 have been disclosed.28 These studies revealed that the amide lipid chain of Pam3CSK4 inserts into a hydrophobic pocket in TLR1, while the remaining two ester lipid chains bind in a TLR2 hydrophobic channel, filling a long continuous hydrophobic pocket spanning both proteins at the TLR1/TLR2 heterodimer interface. In comparison, Pam2CSK4 binds with both ester lipid chains also bound to TLR2, but does not contain or require the amide lipid binding to TLR6 to promote active TLR2/TLR6 dimer formation. In fact, TLR6 lacks the lipid binding pocket needed to accommodate the third amide lipid chain of the triacyl lipopeptides and proteins. Within the diprovocims, the amide side chains are serving the role of the lipid chains, extending into the hydrophobic pockets spanning TLR1/TLR2. The distinction being that the symmetrical diprovocims contain four, not three, such groups. As a result, we examined a range of lipid amide replacements for the trans-2-phenylcyclopropylamine and 4-fluorophenethylamine derived amides and explored hybrid structures containing three as well as four side chains. The first series examined was composed of compounds that contain three side chains, linking one half of diprovocim-1 with a single lipid side chain amide attached either directly to the benzene-1,4-dicarboxylic acid linker (126–129) or as the amide substituent on (R)- or (S)-pyrrolidine-3-carboxylic acid (130–137) (Figure 14). These were prepared by coupling 125 with the corresponding alkyl amine or the corresponding (R)- or (S)-pyrrolidine-3-carboxamide (EDCI, HOAt, 2,6-lutidine, DMF–CH2Cl2, 25 °C, 3–4 h). Their assessment for the stimulated release of TNF-α from differentiated human THP-1 cells and mouse macrophages revealed that no compound in the series 126–129 exhibited agonist activity. In contrast, the series in which the lipid amide substituent was attached via a pyrrolidine-3-carboxamide stimulated the release of TNF-α from both human THP-1 cells and mouse macrophages (Figure 14). Like diprovocim-1, each was more effective in the human THP-1 cells than mouse macrophages (>10-fold, typically ca. 100-fold). Little distinction was observed whether the lipid chain was attached via the (R)- or (S)-pyrrolidine-3-carboxamide. A preference for the lipid chain length was observed with the intermediate lengths displaying the greatest potency (131/135 > 132/136) and the longest chain examined (130/134) seemed to display a uniquely detrimental effect in mouse versus human cells, likely representing species distinctions in the signaling receptors. The best in the series were ≥20-fold less potent than diprovocim-1 in human THP-1 cells and >200-fold less effective in mouse macrophages, comparable in their activity with diprovocim-4 or Pam2CSK4 and more effective than diprovocim-2.
Figure 14.

Hybrid structures containing one lipid side chain and a total of three hydrophobic amide side chains. EC50’s derived from dose-response curves for the stimulated release of TNF-α from differentiated human THP-1 cells or mouse macrophages.
An even more significant set of observations were made with hybrid structures that contained two such lipid side chains. The first such series examined is composed of compounds that contain four side chains, linking one half of diprovocim-1 with symmetrical diamides derived from (S,S)-pyrrolidine-3,4-dicarboxylic acid that bear two lipid side chain amides (Figure 15). Single step preparation of the (S,S)-pyrrolidine-3,4-dicarboxamides, obtained by coupling (S,S)-14 with the corresponding alkyl amines (EDCI, HOAt, 2,6-lutidine, DMF–CH2Cl2, 25 °C), followed by Boc removal (4 N HCl, dioxane, 25 °C) and coupling of the resultant amine with 125 (EDCI, HOAt, 2,6-lutidine, DMF–CH2Cl2, 25 °C, 3–4 h) provided the series of hybrid structures 138–144 containing two lipid side chains. Because of the activity observed, a comprehensive set of lipid side chain lengths was prepared and examined. Three compounds in this series (141–143), containing the intermediate lipid chain lengths (6–10 carbons), exhibited remarkably potent and robust activity (EC50 = 280–170 pM, THP-1 cells), being more potent than Pam3CSK4 and approaching the activity of diprovocim-1. The compounds with the longer lipid chain lengths, although still very potent, displayed progressively less potent activity (140 > 139 > 138) and the compound with the shortest chain length (144) was notably even less potent. The potency difference between stimulation of human versus mouse cell release of TNF-α smoothly and progressively diminished as the chain length was shortened with 143 exhibiting the most potent activity in both cell lines and a 20-fold differential. The behavior of 143, containing two C6 lipid side chains that closely approximates the length and number of carbon atoms found the diprovocim-1 and diprovocim-2 side chains, nearly matches that of diprovocim-1 in both the human THP-1 cells and mouse macrophages (EC50 = 180 pM and 4 nM, respectively), being only 2-fold and 4-fold less potent. Because of this lipid chain length of 6 carbons, and its incidental chronological discovery, it is a compound we now refer to as diprovocim-6 (143).
Figure 15.

Hybrid structures containing two lipid side chains and a total of four hydrophobic amide side chains.
A second series of such hybrid structures was prepared and examined in which two lipid side chain amides were incorporated at opposite ends of the diprovocim-1 structure, each replacing one of the (1S,2R)-2-phenylcyclopropylamine amide substituents. The compound in this series (147) that contained the intermediate lipid chain length (six carbons), most closely approximating the length of the 2-phenylcyclopropylamine amide substituents, exhibited excellent activity (EC50 = 800 pM, THP-1 cells), being slightly more potent than Pam3CSK4 and only 8-fold less active than diprovocim-1 (Figure 15). This compound series also exhibited a smooth and pronounced trend of diminished activity as the chain length of the lipid amide substituent increased (potency: 147 > 148 > 149). In addition, the activity of 147–149 in mouse macrophages was more significantly reduced relative to diprovocim-1 (ca. 1000-fold) and the activity differential in human THP-1 cells versus mouse macrophages increased (30–1000 vs 10-fold). Notably, the synthetic precursors 145 and 146 from which 147–149 were prepared, which lack two of the four hydrophobic side chain amides, were found to be inactive.
Additionally, a series of compounds in which all four side chains of diprovocim-1 were replaced with a comprehensive set of lipid side chain amides was prepared and examined (Figure 16). The (S,S)-pyrrolidine-3,4-dicarboxamides that contained the two amide lipid side chains (16–4 carbons) were coupled in a single step with benzene-1,4-dicarboxylic acid (EDCI, HOAt, 2,6-lutidine, DMF–CH2Cl2, 25 °C, 4–5 h) to provide 150–156. No compound in this series proved capable of stimulating the release of TNF-α from either in the human THP-1 cells or mouse macrophages (EC50 >5 μM). However, this series of compounds also displayed especially poor solubility properties and we cannot rule out that this contributed to the lack of activity observed in the cell-based functional assays.
Figure 16.

Compounds that contain a full set of four lipid chain amides.
A final series of diprovocim-1 analogues was examined that was composed of compounds in which a single amide substituent was replaced with a lipid amide (Figure 17). These were prepared by coupling of 125 (EDCI, HOAt, 2,6-lutidine, DMF–CH2Cl2, 25 °C, 3–4 h, 51%) to provide the mono methyl ester 157. Hydrolysis of the methyl ester to provide the carboxylic acid 158 (LiOH, THF/MeOH/H2O, 25 °C, 2.5 h, 99%) followed by its coupling with the corresponding alkyl amines provided 159–166. These compounds also displayed well-defined activity trends, but one where the stimulated release of TNF-α from human THP-1 cells increased as the lipid chain length increased through and up to C14 (potency: 166 (C16) < 165 (C14) > 164 (C12), 163 (C10) > 162 (C8), 161 (C6) > 160 (C4) > 159 (C2)). Compound 164 matches and 165 slightly exceeds the stunning potency of diprovocim-1.
Figure 17.

Hybrid diprovocims incorporating a single lipid chain amide.
Discussion and Conclusions
Diprovocim-1 has been examined in vitro and in vivo and the results of these studies have been reported elsewhere.34 Although the target identification for leads that emerge from an undirected functional assay such as the one used in this work is usually challenging, especially if the lead compounds bear no structural resemblance to known effector molecules, it proved straightforward for 3 and 4. By design, it was recognized that subsequent assay of active compounds against macrophages from available mice that bear germline genetic defects or knockouts of the genes encoding each of the TLRs, other pattern recognition receptors, or downstream signaling molecules could be used to establish whether the activity was derived from known or unrecognized targets.46 By using this approach, the TNF-α production induced by 3 was established to be solely dependent on both TLR1 and TLR2, independent of all other TLRs including TLR6 (Figure 18), and independent of other molecular pattern recognition receptors (not shown).34 The activity of diprovocim-1 was further shown to be dependent on TLR1/TLR2 signaling through MyD88, TIRAP, and IRAK4 (Figure 18) and 3 has been shown to induce the phosphorylation and activation of IKKα, IKKβ, p38, JNK, and ERK, activating conventional TLR1/TLR2 downstream signaling through both the MAPK and NF-κB pathways.34 Diprovocim-1 (3) acts as an adjuvant in mice when administered by conventional means (intramuscular, i.m.) with an antigen (OVA) and it was established that this in vivo activity (i.e.; vaccination) is dependent on both TLR1 and TLR2 (inactive in both TLR1 and TLR2 deficient mice).34 Most significantly, diprovocim-1 was shown to act synergistically with checkpoint blockade (anti-PDL1) where the combined treatment, but neither alone, cured or prolonged the survival of mice implanted with antigen bearing murine melanoma (B16-OVA) and systemically protected mice from tumor re-challenge in an antigen specific manner (antigen-specific anti-tumor immunity).34 The in vivo activity was observed with the anti-cancer vaccine (diprovocim-adjuvanted OVA) administration intramuscularly (i.m.) and distal from the tumor, not intratumoral as has been a convention in recent related studies.19d,48 Diprovocim-1 displayed no intrinsic cytotoxic activity in cell culture (IC50 >10 μM), not only exhibiting no cell growth inhibition against mouse macrophages or human THP-1 cells used in the assays, but also when tested against L1210 mouse leukemia, HCT116 human colon cancer, B16 mouse melanoma, and human foreskin (HFF) or mouse (NIH/3T3) fibroblast cells in culture (not shown).
Figure 18.

Top: Assay for stimulated TNF-α release upon treatment with 3 (500 nM) using macrophages from mice containing disabling germline mutations or knockouts of genes encoding key mTLRs and downstream signaling proteins. Bottom: Assay for stimulated TNF-α release upon treatment with 3 (250 pM) versus controls without compound using differentiated THP-1 cells that is blocked by antibodies that bind either TLR1 or TLR2. All results are representative of two independent experiments and error bars represent SEM.
As detailed herein, it was a screen conducted with nearly 100,000 compounds and a surrogate functional assay for stimulation of an immune response that led to the discovery of the diprovocims. Unique to these efforts, the screening leads for this new class of activators of an immune response came from a compound library specifically designed to promote cell surface receptor dimerization.33 Such receptor dimerizers,33 a subclass of chemical inducers of dimerization (CID),49 are composed of two linked monomers, where each monomer is incorporated and functions to bind one of the two receptor proteins, promoting or stabilizing receptor dimerization (Figure 19). With the diprovocims (dimer provocation of an immune response) and TLR1/2, this is also accomplished with receptor dimer adoption of an activated, signaling receptor complex (agonist vs antagonist). X-ray crystallographic studies of a (diprovocim-1)2/(TLR2)2 complex, computational modeling of diprovocim-1 binding to TLR1/TLR2 heterodimer, and subsequent TLR1 and TLR2 mutagenesis studies indicate binding of diprovocim-1 at the site of and in a manner analogous to Pam3CSK4.47 One amide side chain substituent extends into the hydrophobic pocket in TLR1 and two adjacent amide substituents bind in the TLR2 hydrophobic channel, filling a long continuous hydrophobic pocket spanning both proteins at the TLR1/TLR2 heterodimer interface analogous to the three lipid chains of Pam3CSK4. The remaining fourth amide substituent of diprovocim-1 binds along and further stabilizes the TLR1/TLR2 dimer interface. This is further supported by the results of the studies detailed herein where certain lipid amides can effectively replace the diprovocim side chain amides in selected instances (e.g.; 165).
Figure 19.

One half of diprovocim each binds a receptor protein, promoting receptor dimerization and activation.
The systematic SAR studies detailed herein improved the potency 800-fold over that of the lead compounds, providing diprovocim-1 and diprovocim-2. The diprovocims act by a well-defined mechanism (TLR1/TLR2 agonist), bear no structural similarity to any known natural or synthetic TLR agonist, are easy to prepare and synthetically modify, and selected members are active in both human and murine systems. The most potent diprovocim (3, diprovocim-1) elicits full agonist activity at extraordinarily low concentrations (EC50 = 110 pM) in human THP-1 cells, being more potent than any other known small molecule TLR agonist, including the naturally-derived TLR1/TLR2 agonist Pam3CSK4. Complementary to the studies conducted to date with diprovocim-1,34 further studies of this class of synthetic TLR1/TLR2 agonists as new organic vaccine adjuvants in infectious diseases and oncology, as adjuvants in vaccine prophylactics against pathogen exposure (e.g.; biodefense), and as immunostimulators alone or in combination with other treatments, particularly in oncology, are underway. It is worth highlighting that the characteristics of such an immune stimulator are expectedly different than those of a conventional drug.17 It is administered infrequently as part of prime and boost vaccinations, it is typically administered i.m., the vaccination not only provides long term systemic protection but does so with a localized low dose administration, and the acceptable chemical (e.g.; Mwt), physical (e.g.; low solubility and high cLogP), PK (e.g.; short vs long circulating half-life in blood), permeability (low vs high), and metabolic stability properties are distinct from those of a traditional drug.17 Most notably and because of the importance of the field, the diprovocims represent a welcomed new addition to the limited number of known TLR agonists. Most are mimics or modifications of microbial components (e.g.; LPS, lipopeptides, nucleic acids) and represent unattractive starting points for drug discovery, many are difficult to structurally modify (e.g.; covalent antigen-adjuvant construct) and chemically prepare, and only a limited number of other synthetic small molecule classes have been found to behave as TLR agonists. As a result, we expect many others will find the diprovocims especially attractive to work with.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge the financial support of the National Institutes of Health (AI082657 and CA042056, D.L.B.; AI082657, AI100627, and AI125581, B.B.; GM104496, H.Z.). We thank Aleksandar Radakovic and Jelena Momirov for conducting the screening for cytotoxic activity and Kevin Litwin for the Figure 19 and TOC graphics.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Full experimental details. This material is available free of charge via the internet at http://pubs.acs.org.
Competing interests: B.B., D.L.B. and H.Z. have financial interests in Tollbridge Therapeutics, which has licensed the patent on the diprovocims.
References
- 1.Beutler B Mol. Immunol 2004, 40, 845–859. [DOI] [PubMed] [Google Scholar]
- 2.Hoebe K; Janssen E; Beutler B Nat. Immunol 2004, 5, 971–974. [DOI] [PubMed] [Google Scholar]
- 3.(a) Moresco EMY; LaVine D; Beutler B Curr. Biol 2011, 21, R488–R493. [DOI] [PubMed] [Google Scholar]; (b) Blasius AL; Beutler B Immunity 2010, 32, 305–315. [DOI] [PubMed] [Google Scholar]; (c) Kawai T; Akira S Nat. Immunol 2010, 13, 373–384. [DOI] [PubMed] [Google Scholar]; (d) Beutler B Blood 2009, 113, 1399–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Hashimoto C; Hudson KL; Anderson KV Cell 1988, 52, 269–279. [DOI] [PubMed] [Google Scholar]; (b) Lemaitre B; Nicolas E; Michaut L; Reichhart JM; Hoffmann JA Cell 1996, 86, 973–983. [DOI] [PubMed] [Google Scholar]
- 5.(a) Poltorak A; He X; Smirnova I; Liu M-Y; Van Huffel C; Du X; Birdwell D; Alejos E; Silva M; Galanos C; Freudenberg MA; Ricciardi-Castagnoli P; Layton B; Beutler B Science 1998, 282, 2085–2088. [DOI] [PubMed] [Google Scholar]; (b) Beutler B; Du X; Poltorak AJ Endotoxin Res. 2001, 7, 277–280. [PubMed] [Google Scholar]; (c) Beutler B; Rietschel ET Nat. Rev. Immunol 2003, 3, 169–176. [DOI] [PubMed] [Google Scholar]
- 6.Medzhitov R; Preston-Hurlburt P; Janeway CA Jr. Nature 1997, 388, 394–397. [DOI] [PubMed] [Google Scholar]
- 7.(a) Vogel FR Clin. Infect. Dis 2000, 30 (Suppl 3), S266–S270. [DOI] [PubMed] [Google Scholar]; (b) Guy B Nat. Rev. Microbiol 2007, 5, 505–517. [DOI] [PubMed] [Google Scholar]; (c) Coffman RL; Sher A; Seder RA Immunity 2010, 33, 492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Czarniecki M J. Med. Chem 2008, 51, 6621–6626. [DOI] [PubMed] [Google Scholar]; (b) Peri F; Calabrese VJ Med. Chem 2014, 57, 3612–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X; Smith C; Yin H Chem. Soc. Rev 2013, 42, 4859–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Hennessy EJ; Parker AE; O’Neill LAJ Nat. Rev. Drug Discovery 2010, 9, 293–301. [DOI] [PubMed] [Google Scholar]; (b) Meyer T; Stockfleth E Expert Opin. Invest. Drugs 2008, 17, 1051–1065. [DOI] [PubMed] [Google Scholar]; (c) Hoebe K; Jiang Z; Georgel P; Tabeta K; Janssen E; Du X; Beutler B Curr. Pharmaceut. Des 2006, 12, 4123–4134. [DOI] [PubMed] [Google Scholar]; (d) Kanzler H; Barrat FJ; Hessel EM; Coffman RL Nat. Med 2007, 13, 552–559. [DOI] [PubMed] [Google Scholar]
- 11.(a) Amlie-Lefond C; Paz DA; Connelly MP; Huffnagle GB; Whelan NT; Whelan HT J. Allergy Clin. Immunol 2005, 116, 1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) O’Neill LA; Bryant CE; Doyle SL Pharmacol. Rev 2009, 61, 177–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Aldous AR; Dong JZ Bioorg. Med. Chem 2018, 26, 2842–2849. [DOI] [PubMed] [Google Scholar]; (b) Sahin U; Tureci O Science 2018, 359, 1355–1360. [DOI] [PubMed] [Google Scholar]; (c) Ribas A; Wolchok JD Science 2018, 359, 1350–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engel AL; Holt GE; Lu H Expert Rev. Clin. Pharmacol 2011, 4, 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Prins RM; Craft N; Bruhn KW; Khan-Farooqi H; Koya RC; Stripecke R; Miller JF; Liau LM J. Immunol 2005, 176, 157–164. [DOI] [PubMed] [Google Scholar]; (b) Lee J; Chuang T-H; Redecke V; She L; Pitha PM; Carson DA; Raz E; Cottam HB Proc. Natl. Acad. Sci. USA 2003, 100, 6646–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee J; Wu CCN; Lee KJ; Chuang T-H; Katakura K; Liu YT; Chan M; Tawatao R; Chung M; Shen C; Cottam HB; Lai MMC; Raz E; Carson DA Proc. Natl. Acad. Sci. USA 2006, 103, 1828–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smits ELJM; Cools N; Lion E; Camp K; Ponsaerts P; Berneman ZN; Van Tendeloo VF I. Cancer Immunol. Immunother 2010, 59, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beesu M; Malladi SS; Fox LM; Jones CD; Dixit A; David SA J. Med. Chem 2014, 57, 7325–7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu TYH; Singh M; Miller AT; De Gregorio E; Doro F; D’Oro U; Skibinski DAG; Mbow ML; Bufali S; Herman AE; Cortez A; Li Y; Nayak BP; Tritto E; Filippi CM; Otten GR; Brito LA; Monaci E; Li C; Aprea S; Valentini S; Calabro S; Laera D; Brunelli B; Caproni E; Malyala P; Panchal RG; Warren TK; Bavari S; O’Hagan DT; Cooke MP; Valiante NM Sci. Transl. Med 2014, 6(263), 263ra160. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L; Dewan V; Yin HJ Med. Chem 2017, 60, 5029–5044. [DOI] [PubMed] [Google Scholar]
- 19.(a) Ishikawa H; Barber GN Nature 2008, 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Burdette DL; Monroe KM; Sotelo-Troha K; Iwig JS; Eckert B; Hyodo M; Hayakawa Y; Vance RE Nature 2011, 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sun L; Wu J; Du F; Chen X; Chen ZJ Science 2013, 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fu J; Kanne DB; Leong M; Glickman LH; McWhirter SM; Lemmens E; Mechette K; Leong JJ; Lauer P; Liu W; Sivick KE; Zhen Q; Soares KC; Zheng L; Portnoy DA; Woodward JJ; Pardoll DM; Dubensky TW; Kim Y Sci. Transl. Med 2015, 7(283), 283ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Wang Y; Su L; Morin MD; Jones BT; Whitby LR; Surakattula MMRP; Huang H; Shi H; Choi JH; Wang K; Moresco EMY; Berger M; Zhan X; Zhang H; Boger DL; Beutler B Proc. Natl. Acad. Sci. USA 2016, 113, E884–E893. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Morin MD; Wang Y; Jones BT; Su L; Surakattula MMRP; Berger M; Huang H; Beutler EK; Zhang H; Beutler B; Boger DL J. Med. Chem 2016, 59, 4812–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Johnson DA Curr. Top. Med. Chem 2008, 8, 64–79. [DOI] [PubMed] [Google Scholar]; (b) Casella CR; Mitchell TC Cell Mol. Life Sci 2008, 65, 3231–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Persing DH; Coler RN; Lacy MJ; Johnson DA; Baldridge JR; Hershberg RM; Reed SG Trends Microbiol. 2002, 10, S32–S37. [DOI] [PubMed] [Google Scholar]
- 22.(a) Neve JE; Wijesekera HP; Duffy S; Jenkins ID; Ripper JA; Teague SJ; Campitelli M; Garavelas A; Nikolakopoulos G; Le PV; de A. Leone P; Pham NB; Shelton P; Fraser N; Carroll AR; Avery VM; McCrae C; Williams N; Quinn RJ J. Med. Chem 2014, 57, 1252––1275. [DOI] [PubMed] [Google Scholar]; (b) Chan M; Hayashi T; Mathewson RD; Nour A; Hayashi Y; Yao S; Tawatao RI; Crain B; Tsigelny IF; Kouznetsova VL; Messer K; Pu M; Corr M; Carson DA; Cottam HB J. Med. Chem 2013, 56, 4206–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan M; Hayashi T; Mathewson RD; Nour A; Hayashi Y; Yao S; Tawatao RI; Crain B; Tsigelny IF; Kouznetsova VL; Messer K; Pu M; Corr M; Carson DA; Cottam HB J. Med. Chem 2013, 56, 4206–4223, DOI: 10.1021/jm301694x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmer SM; Liu J; Clayton JL; Stephens DS; Snyder JP J. Biol. Chem 2008, 283, 27916–27926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Metzger J; Wiesmuller K-H; Schaude R; Bessler WG; Jung G Int. J. Peptide Protein Res 1991, 37, 46–57. [DOI] [PubMed] [Google Scholar]; (b) Bessler WG; Cox M; Lex A; Suhr B; Wiesmuller K-H; Jung GJ Immunol 1985, 135, 1900–1905. [PubMed] [Google Scholar]
- 26.Deres K; Schild H; Wiesmuller K-H; Jung G; Rammensee H-G Nature 1989, 342, 561–564. [DOI] [PubMed] [Google Scholar]
- 27.(a) Alexopoulou L; Thomas V; Schnare M; Lobet Y; Anguita J; Schoen RT; Medzhitov R; Fikrig E; Flavell RA Nat. Med 2002, 8, 878–884. [DOI] [PubMed] [Google Scholar]; (b) Takeuchi O; Sato S; Horiuchi T; Hoshino K; Takeda K; Dong Z; Modlin RL; Akira SJ Immunol 2002, 169, 10–14. [DOI] [PubMed] [Google Scholar]
- 28.(a) Jin MS; Kim SE; Heo JY; Lee ME; Kim HM; Paik SG; Lee H; Lee JO Cell 2007, 130, 1071–1081. [DOI] [PubMed] [Google Scholar]; (b) Kang JY; Nan XH; Jin MS; Youn SJ; Ryu YH; Mah S; Han SH; Lee H; Paik SG; Lee JO Immunity 2009, 31, 873–884. [DOI] [PubMed] [Google Scholar]; (c) Jin MS; Lee JO Curr. Opin. Immunol 2008, 20, 414–419. [DOI] [PubMed] [Google Scholar]
- 29.(a) Guo X; Wu N; Shang Y; Liu X; Wu T; Zhou Y; Liu X; Huang J; Liao X; Wu L Front. Immunol 2017, 8, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Santone M; Aprea S; Wu TYH; Cooke MP; Mbow ML; Valiante NM; Rush JS; Dougan S; Avalos A; Ploegh H; De Gregorio E; Buonsanti C; D’Oro U Hum. Vaccin. Immunother 2015, 11, 2038–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Salunke DB; Connelly SW; Shukla NM; Hermanson AR; Fox LM; David SA J. Med. Chem 2013, 56, 5885–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Salunke DB; Shukla NM; Yoo E; Crall BM; Balakrishna R; Malladi SS; David SAJ Med. Chem 2012, 55, 3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Agnihotri G; Crall BM; Lewis TC; Day TP; Balakrishna R; Warshakoon HJ; Malladi SS; David SA J. Med. Chem 2011, 54, 8148–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muhlradt PF; Kiess M; Meyer H; Sussmuth R; Jung GJ Exp. Med 1997, 185, 1951–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buwitt-Beckmann U; Heine H; Wiesmuller K-H; Jung G; Brock R; Akira S; Ulmer AJ J. Biol. Chem 2006, 281, 9049–9057. [DOI] [PubMed] [Google Scholar]
- 32.(a) Guan Y; Omueti-Ayoade K; Mutha SK; Hergenrother PJ; Tapping RI J. Biol. Chem 2010, 285, 23755–23762. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cheng K; Gao M; Godfroy JI; Brown PN; Kastelowitz N; Yin H Sci. Adv 2015, 1, e1400139. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Murgueitio MS; Ebner S; Hortnagl P; Rakers C; Bruckner R; Henneke P; Wolber G; Santos-Sierra S Biochim. Biophys. Acta – Gen. Subj 2017, 1861, 2680–2681. [DOI] [PubMed] [Google Scholar]
- 33.Goldberg J; Jin Q; Ambroise Y; Satoh S; Desharnais J; Capps K; Boger DL J. Am. Chem. Soc 2002, 124, 544–555. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y; Su L; Morin MD; Jones BT; Mifune Y; Shi H; Wang KW; Zhan X; Liu A; Wang J; Li X; Tang M; Ludwig S; Hilderbrand S; Zhou K; Siegwart D; Moresco EMY; Zhang H; Boger DL; Beutler B Proc. Natl. Acad. Sci. USA 2018, 115, E8698–8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Beutler B Immunogen. 2005, 57, 385–392. [DOI] [PubMed] [Google Scholar]; (b) O’Connor CJ; Laraiaa L; Spring DR Chem. Soc. Rev 2011, 40, 4332–4345. [DOI] [PubMed] [Google Scholar]
- 36.(a) Whitby LR; Boger DL Acc. Chem. Res 2012, 45, 1698–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boger DL; Desharnais J; Capps K Angew. Chem. Int. Ed 2003, 42, 4138–4176. [DOI] [PubMed] [Google Scholar]
- 37.Shaginian A; Whitby LR; Hong S; Hwang I; Faroogi B; Chen J; Searcey M; Vogt PK; Boger DL J. Am. Chem. Soc 2009, 131, 5564–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whitby LR; Ando Y; Setola V; Vogt PK; Roth BL; Boger DL J. Am. Chem. Soc 2011, 133, 10184–10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stover JS; Shi J; Jin W; Vogt PK; Boger DL J. Am. Chem. Soc 2009, 131, 3342–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otrubova K; Srinivasan V; Boger DL Bioorg. Med. Chem. Lett 2014, 24, 3807–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. At this stage, the compounds were assessed as a mixture of diastereomers, having been prepared using racemic trans-pyrrolidine-3,4-dicarboxylic acid and racemic trans-2-phenylcyclopropylamine as found in the original library.
- 42. Purchased from D-L Chiral Chemicals.
- 43.Bao J; Baker RK; Parsons WH; Rupprecht K U.S. Patent 6,489,354 B1. After chromatographic resolution of the diastereomers of ethyl 1-benzyl-4-((S)-4-benzyl-2-oxooxazolidine-3-carbonyl)pyrrolidine-3-carboxylate as reported, hydrogenolysis (Pd(OH)2/C, EtOH, 23 °C, 18 h, 84%) of the N-benzyl group in the presence of Boc2O (1.05 equiv) and subsequent hydrolysis of the resulting N-Boc derivative (2.5–5 equiv LiOH, 4 equiv H2O2, THF–H2O, 23 °C, 5 h, 94%) provided each enantiomer (S,S and R,R) of N-Boc-pyrrolidine-3,4-dicaboxylic acid (see Supporting Information).
- 44. Prepared from commercially available (3S)-pyrrolidine-3-carboxylic acid ((S)-β-proline, see Supporting Information).
- 45.Benelkebir H; Hodgkinson C; Duriez PJ; Hayden AL; Bulleid RA; Crabb SJ; Packham G; Ganesan A Bioorg. Med. Chem 2011, 19, 3709–3716. The (1R,2S)-116 enantiomer was prepared in reference 45 and we employed the opposite enantiomer of the reported ligand for preparation of the desired (1S,2R)-116. [DOI] [PubMed] [Google Scholar]
- 46.Beutler B; Du X; Xia Y Nat. Immunol 2007, 8, 659–664. [DOI] [PubMed] [Google Scholar]
- 47.Su L; Wang Y; Wang J; Mifune Y; Morin MD; Jones BJ; Moresco EMY; Boger DL; Beutler. B; Zhang H Manuscript in preparation.
- 48.(a) Marabelle A; Kohrt H; Caux C; Levy R Clin. Cancer Res. 2014, 20, 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang S; Campos J; Gallotta M; Gong M; Crain C; Naik E; Coffman RL; Guiducci C Proc. Natl. Acad. Sci. USA 2016, 113, E7240–E7249. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Takeda Y; Kataoka K; Yamagishi J; Ogawa S; Seya T; Matsumoto M Cell Rep 2017, 19, 1874–1887. [DOI] [PubMed] [Google Scholar]; (d) Sagiv-Barfi I; Czerwinski DK; Levy S; Alam IS; Mayer AT; Gambhir SS; Levy. R. Sci. Transl. Med 2018, 10, eaan4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diver ST; Schreiber SL J. Am. Chem. Soc 1997, 119, 5106–5109. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.