

Abstract
HIV persists despite effective antiretroviral therapy in long lived cells, posing a major barrier towards a cure. A key step in the HIV replication cycle and a hallmark of the Retroviridae family is the integration of the viral DNA into the host genome. Once integrated, HIV expression is regulated by host machinery and the provirus persists until the cell dies. A reservoir of cells harboring replication competent proviruses can survive for years, and mechanisms that maintain that reservoir are under investigation. The majority of integrated proviruses, however, are defective or have large deletions, and the composition of the proviral landscape during therapy remains unknown. Methods to quantify HIV proviruses are useful in investigating HIV persistence, and presented in this unit is a method for total HIV DNA quantification of various HIV genome targets that utilizes the next generation PCR platform, digital PCR. The abundance of various HIV gene targets reflects the overall proviral composition. In this protocol, total genomic DNA is isolated from patient derived cells, which is used as a template for droplet digital PCR in which the PCR reaction is partitioned into approximately 20,000 individual droplets, PCR amplified to an end point, and subjected to absolute quantification by counting the number of positive and negative droplets. Copy number is directly calculated using straightforward Poisson correction. Additionally, this methodological approach can be used to obtain absolute quantification of other DNA targets.
Keywords: Human immunodeficiency virus (HIV), viral DNA integration, HIV DNA quantification
INTRODUCTION
Human immunodeficiency virus (HIV) persists despite effective combination antiretroviral treatment (cART) in long-lived reservoirs of cells and can rebound if treatment is interrupted, thereby necessitating lifelong therapy [1–3]. Although the majority of all integrated proviruses are defective or deleted, a small fraction of inducible replication competent proviruses persist for years in most individuals on cART [4, 5]. Furthermore, accurately measuring the size of the latent HIV reservoir is critical to assess the effectiveness of curative strategies aimed at HIV remission, but the proviral landscape during the course of cART remains poorly characterized. A universal, precise, efficient, scalable, and high throughput assay to confirm the complete elimination of the latent reservoir in vivo would be ideal, as it would be a key assay to identify HIV eradication and determine timing of discontinuation of cART without experiencing a rebound in viremia [6]. In practice, no assay accurately quantifies the reservoir of infectious HIV, and it is likely that input from a variety of assays will be necessary in developing strategies to eradicate or control HIV without cART.
A number of assays have been developed to measure the size of the reservoir in infected individuals (Reviewed in Han et al. [7]). Investigating the size of the reservoir and the composition of the proviral landscape is limited by the ability to find rare HIV infected cells in a background of uninfected cells, much like finding a needle in a haystack. The quantitative viral outgrowth assay (qVOA) measures the frequency of resting CD4+ T-cells that produce infectious virus after a single round of maximum in vitro global T-cell activation but underestimates the true size of the reservoir [2–4]. PCR based assays to measure HIV DNA overestimate the true size of the latent reservoir of replication competent proviruses, as they will also amplify defective or deleted proviruses which are not capable of producing virus [8]. Other methods, such as identifying cell surface markers specific for HIV infected cells have not been sufficiently sensitive or specific, but this area of research is under active investigation and some T-cell subsets have been reported to be enriched in HIV DNA [9–15]. Identifying the population of HIV infected cells remains a major challenge towards HIV eradication.
Droplet Digital PCR (ddPCR) is a next generation PCR platform that enables absolute target quantification of a DNA target molecule, and it offers several advantages over traditional quantitative PCR approaches. First, ddPCR provides an absolute quantification of target molecules, and does not require use of a standard curve; in contrast traditional qPCR methods employ a relative quantification, using a standard curve to determine DNA copies. Second, the sensitivity of ddPCR makes it an ideal platform for studying rare events molecules in the presence of large background of unaffected molecules. This level of sensitivity has made ddPCR an attractive modality in detecting rare mutations associated with cancer phenotypes in liquid biopsies [16] as well as low level pathogen detection [17–22]. Indeed, ddPCR has been used to detect HIV DNA, with single targets of 60-80 nucleotides (reviewed in Rutsaert et al and Trypsteen et al [23, 24]). In particular, ddPCR has been implemented to quantify total HIV DNA in peripheral blood cells including PBMCs, CD4+ T cells [8, 22, 25–27] and regulatory T cells (Treg) [28] as well as in cells from the central nervous system [27]. Moreover, ddPCR has been utilized to assess impacts of intervention strategies on the HIV reservoir size in peripheral cells [8, 22, 29–32] and in cells from the gut-associated lymphoid tissue [33]. Third, ddPCR has a broad linear range of quantification, with sensitive two-fold detection of differences, permitting resolution of small changes in abundance [34]; standard qPCR has been useful in detecting 5-10 fold changes in abundance, but small changes in are not well quantified. Finally, ddPCR detection can be multiplexed for simultaneous detection of two targets. This advantage is especially useful for estimates of the proportion of HIV proviruses that contain deletions; for instance, the finding of HIV LTR without detection of an internal portion of the genome (e.g., gag, tat, and rev) immediately determines that the HIV provirus is deleted.
HIV exhibits substantial intra- and inter patient genetic variability, and optimal primer-probe combinations to detect different HIV DNA targets among patients have not been defined. Here, we have developed novel multiplexed quantitative ddPCR assays to explore mechanisms of HIV persistence by investigating the reservoir structure during the course of cART, that have been useful for a spectrum of patients. The protocol presented here describes isolation of total genomic DNA from patient derived cells (see Basic Protocol 1) and quantification of the levels of HIV DNA in those samples using ddPCR in order to assess the total viral DNA burden as well as the overall structure of the proviral landscape (see Basic Protocol 2). Principles of detection and quantification as well as strategies for single and multiplexed assays are described.
CAUTION: Proper biosafety practices must be followed when working with human blood, cells, or infectious agents. HIV has a BSL2* biosafety level and requires appropriate containment for the following assay.
Basic Protocol 1: TOTAL GENOMIC DNA EXTRACTION FROM PATIENT DERIVED CELLS
This protocol describes how to extract total genomic and HIV DNA from patient derived cells. Presumably DNA isolated from HIV infected cells will be in a background of genomic DNA from uninfected cells in the same sample. This protocol will also describe how to properly resuspend and fragment the extracted genomic DNA to prepare the sample for accurate quantification by droplet digital PCR (ddPCR).
Materials
1M Calcium Chloride (SIGMA #21115)
0.5M EDTA (AMBION #AM9260G)
100% Ethanol (SIGMA)
Glycogen (20mg/mL, ROCHE #10901393001)
8M Guanidinium Hydrochloride (SIGMA #G9284)
6M Guanidinium Isothiocyanate (SIGMA #50983)
GuHCL+/Prot K Solution (see Reagents and Solutions)
GuSCN+/Gly Solution (see Reagents and Solutions)
100% Isopropanol (SIGMA #I9516-4X25ML)
Proteinase K (20mg/mL, APPLIED BIOSYSTEMS #AM2548)
DNase/RNase Free Molecular Grade Water (GIBCO #10977)
RPMI 1640 with L-glutamine and phenol red indicator (LONZA #12-702F)
1M Trizma-HCl, ph 7.6 (SIGMA #T2444-100ML)
1M Tris-HCl, pH 8.0 (INVITROGEN #15568025)
37° C water bath in a biosafety cabinet
42°C Water bath
100°C Thermomixer (EPPENDORF)
Thin Tipped Transfer Pipettes (THERMOFISHER #23120S)
1.5 mL screw cap polypropylene micro centrifuge tube (SARSTEDT #2.692.005)
Cup horn sonifier (BRANSON)
Cryopreserved cells
Genomic DNA Extraction
Prepare reagent stocks and the GuHCL+/Prot K and GuSCN+/Gly mixes outlined in the reagents and solutions section of this article.
If extracting nucleic acids from cryopreserved cells stored in Liquid Nitrogen (LN2), remove cells from the LN2 tank and equilibrate in a −80°C freezer overnight before following the steps below.
Warm up RPMI media in a 37°C water bath (approximately 10-20 minutes for full 500 mL bottle.
Thaw cryopreserved cells individually; retrieve one vial at a time from the −80°C freezer and place on dry ice. The following was developed for 1 mL cryo tubes; steps 1-6, and 8-11 must be completed in a biocontainment cabinet. After the addition of guanidinium samples may be safely manipulated on a benchtop, although the biocontainment hood is frequently more convenient.
When ready, place the vial of cells from dry ice in the 37°C water bath in a biosafety cabinet for 2 minutes and gently rock back and forth until nearly thawed. Do not leave cells unattended, as they will thaw quickly. Only thaw one vial at a time to reduce amount of time for thawing.
Add 1mL of RPMI drop-wise using a 1000 μL pipette to the vial of cells in cryopreservation solution. Slowly pipette up cells and transfer a maximum of 2x106 cells to individual 1.5mL tubes. Bring the final volume up to 1mL with RPMI. For instance, if you started with 10 million cells/mL then transfer 400μL into 5 tubes each and add 600μL of RPMI.
Pellet cells by centrifuging the tubes at 500xg for 5 minutes at room temperature.
Carefully, completely remove the supernatant by aspiration using a small transfer pipette, and recap tubes.
Place cell pellets on dry ice until you are ready to proceed to the next step. Extra frozen cell pellets can be stored at −80°C or in LN2.
Allow cell pellets to begin to thaw (20-30 seconds at room temperature) and gently flick to resuspend the cell pellet in residual RPMI media.
Add 100μL of the GuHCl+/ProK solution to each sample and vortex immediately for 10 seconds. Inspect sample and if necessary, vortex for an additional 5 seconds to completely disperse.
Incubate the samples on a heat block or in a water bath at 42°C for 1 hour.
Add 400μL of the GuSCN+/Gly solution to each sample. Pulse vortex three times to mix and incubate at 42°C for an additional 10 mins.
Add 500μL of 100% isopropanol at room temperature to each tube and vortex for 10 seconds at high intensity to precipitate the nucleic acids.
Pellet nucleic acids by centrifugation at 21,000xg for 10 minutes at room temperature.
Carefully, completely remove the supernatant without disturbing the pellet.
Add 750μL of 70% Ethanol at room temperature to each tube, invert the sample multiple times to wash the pellet.
Spin at 21,000 × g for 10 minutes at room temperature.
Carefully remove the supernatant using thin tipped transfer pipettes without disturbing the pellet.
Spin tubes in bench-top centrifuge at full speed for 15 seconds. Carefully remove residual ethanol using 10μl pipette tip.
Allow the pellets to air dry until just translucent it is important not to have residual EtOH as it can inhibit PCR.
Add 150μl of 5mM Tris-HCL (pH 8.0) to the nucleic acid pellet.
Resuspend genomic DNA using a cup horn sonicator (Branson) set to 60% amplitude. Pulse sonicate tube for 5 seconds, then vortex and spin down, repeat for a total of three times per sample.
Transfer sonicated sample to screw cap tube (Sarstedt) and heat in the ThermoMixer (Eppendorf) on 100°C for 15 minutes, snap cool on ice.
Spec DNA (Denovix) and record concentration on tube (Optional).
Temporarily store the gDNA at +4°C or −20°C until ready to proceed to DNA quantification.
Basic Protocol 2: ABSOLUTE QUANTIFICATION OF DNA USING DROPLET DIGITAL PCR
This protocol describes how to quantify total genomic DNA and HIV DNA copy numbers from genomic DNA extract from patient derived cells using the droplet digital PCR (ddPCR) platform using the Bio-Rad QX200 ddPCR instruments (droplet generator and reader) and accompanying Quantasoft software. The general ddPCR workflow is shown in Figure 1, in which a PCR reaction is first partitioned into around 20,000 nanoliter-sized water in oil droplets, those droplets are then thermal cycled to an endpoint and finally the droplets are read by a FACS-like droplet reader. Data generated from the droplet reader can then be analyzed in the Quantasoft software. Total HIV DNA copies can be normalized to 1e6 CD4+ T-cells based on input measured using CCR5 copies per well and normalizing to CD4% measured by flow cytometry.
Figure 1.
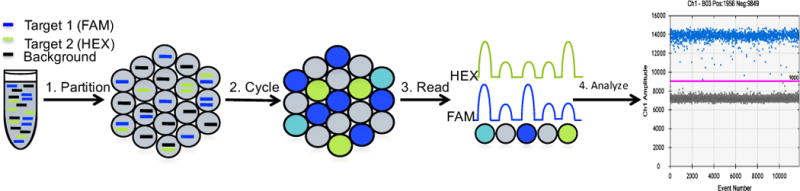
ddPCR workflow: First a PCR mix containing target DNA along with primers and fluorescently labeled probes is partitioned into around 20,000 nanoliter sized droplets with the QX200 droplet generator. Then these droplets are cycled to an endpoint on a thermal cycler. Next the droplets are read with the QX200 Droplet Reader which sips each well, singulates the droplets and passes them by a two-color detection system. The data generated can finally be analyzed in the quantasoft software where thresholds should be set between the negative and positive droplets.
Materials
Total genomic DNA resuspended in 150μl Tris-HCL pH 8.0 (Basic Protocol 1)
100 μM primers and probes (TABLE 1)
ddPCR Supermix for Probes (Bio-Rad #186-3010)
DG8 Cartridges and Gaskets (Bio-Rad #186-3051)
Droplet generator oil for probes (Bio-Rad #186-3005)
Pierceble Foil Heat Seal (Bio-Rad #181-4040)
Semi-skirted 96-well PCR plate (EPPENDORF #951020346)
5-50μl RAININ 8 channel pipette (RAININ # 17013804)
Heat Sealer at 180°C (Bio-Rad)
QX200 Droplet Generator (Bio-Rad)
Thermal Cycler
QX200 Droplet Reader (Bio-Rad)
Table 1.
ddPCR oligo sequences for DNA quantification
| Target | Primer Name | Sequence | Reference |
|---|---|---|---|
|
| |||
| CCR5 | hCCR5-F1 | 5′-CCA GAA GAG CTG AGA CAT CCG-3′ | Thomas, J.A. et al., 2006, Virology |
| hCCR5-R1 | 5′-GCC AAG CAG CTG AGA GGT TAC T-3′ | ||
| FAM-hCCR5-P01 | 5′-/56-FAM/-TCC CCT ACA AGA AAC TCT CCC CGG-/3BHQ_1/-3′ | ||
|
| |||
| LTR | RU5-F | 5′-CTT AAG CCT CAA TAA AGC TTG CC-3′ | |
| RU5-R | 5′-GGA TCT CTA GTT ACC AGA GTC-3′ | ||
| RU5-Probe | 5′-/5HEX/-AGT AGT GTG TGC CCG TCT G-/ZEN//3IaBkFQ/-3′ | ||
|
| |||
| gag | HIV SCA 6F | 5′-CAT GTT TTC AGC ATT ATC AGA AGG A-3′ | Palmer, S. et al., 2003, J. Clin Microbiol |
| HIV SCA 84R | 5′-TGC TTG ATG TCC CCC CAC T-3′ | ||
| HIV SCA probe 32t | 5′-/56-FAM/-CCA CCC CAC AAG ATT TAA ACA CCA TGC TAA-/ZEN//3IaBkFQ/-3′ | ||
|
| |||
| tat Exon 1 | TatRev1_F | 5′-GCA TCC AGG AAG TCA GCC TA-3′ | |
| TatRev1_R | 5′-CTT CGT CGC TGT CTC CGC-3′ | ||
| TatRev1_probe | 5′-/5HEX/-TC TTC CTG CCA TAG GAG ATG C-/3BHQ_1/-3′ | ||
|
| |||
| tat/rev Exon 2 | TatRev2_F | 5′- GTG AAT AGA GTT AGG CAG GGA TA-3′ | |
| msRNA-R | 5′-GTC TCT GTC TCT CTC TCC ACC-3′ | ||
| msRNA Probe | 5′-/56-FAM/-TCG GGC CTG TCG GGT CC-/3BHQ_1/-3′ | ||
Droplet generation
Plan the 96-well plate layout. Samples should be run in triplicate by columns of 8 since the droplet generator gaskets accommodate 8 samples per run. See Figure 2 for a typical plate layout.
Prepare ddPCR master mixes. All primer sequences can be found in Table 1. Table 2: Singleplexed mix for total DNA input (CCR5 copies) and Table 3: duplexed mix to measure HIV DNA (gag/RU5). A schematic of the primer probe target regions within the HIV genome are shown in Figure 3.
Distribute 16.5μl of master mix per well into a 96 well plate.
Add 5.5μl of resuspended sample per reaction.
Cover the plate and spin it down briefly in a table top centrifuge to get rid of any bubbles.
-
Transfer 20ul of the PCR reaction to the DG8 cartridge using 20μl multichannel pipette. See Figure 4 for a layout of the DG8 cartridges.
Middle Row: 20μl of the PCR reaction avoiding bubbles.
Bottom Row: 70μl of droplet generator oil for probes.
Seal DG8 cartridge with DG8 gasket.
Load the sealed DG8 cartridge into the QX200 droplet generator.
Close lid to start droplet generator, light will flash while running and stay solid when done.
Transfer cartridge generator back to workstation and remove gasket carefully as to not cross contaminate samples.
-
Slowly transfer droplets from the cartridge which have now collected in the last row of wells to a new Eppendorf 96-well PCR plate using a 50μl RAININ multichannel set to 43μl being careful not to shear the fragile droplets by following these guidelines:
Do not smash pipette tips down into the wells
Hold pipette at an angle as to not shear the droplets
Slowly count to 7 while drawing up and aspirating droplets to prevent shearing
Repeat steps 6 to 11 with the next row of samples.
After all droplets have been made and transferred to the 96-well PCR plate, heat seal with pierceable foil (Bio-Rad) at 180°C for 5 seconds.
-
Transfer plate to a thermal cycler and cycle droplets to endpoint using the following conditions:
Step 1: 95°C for 10 minutes
Step 2: 94°C for 30 seconds
Step 3: 55°C for 1 minute with ramp rate of 2
Step 4: Repeat steps 2 – 3 for 39 cycles
Step 5: 98°C for 10 minutes
Hold: 12 °C forever
Transfer plate of cycled droplets to QX200 droplet reader, load and run samples.
Analyze the data generated using the Quantasoft software.
Figure 2.
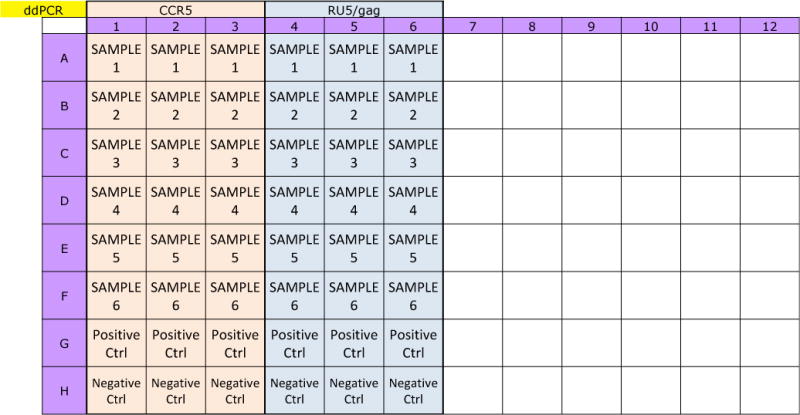
ddPCR Plate Layout: A typical layout for droplet generation and amplification. Samples should be run in triplicate. DG8 cartridge can accommodate 8 samples at a time so it is convenient to organize samples by columns. All runs should include positive and negative controls, which could be DNA extracted from HIV negative cells such as the cell line CEM and DNA extracted from HIV positive cells such as the ACH2 cell line.
Table 2.
Singleplexed ddPCR master mix for total DNA input (CCR5 copies)
| ddPCR Singleplexed Master Mix
| ||
|---|---|---|
| Reagent | Final Concentration | Per Rxn (ul) |
|
| ||
| dH20 | 4.65 | |
| 2× BIO-RAD Supermix | 1× | 10 |
| 100∝M CCR5 Forward | 750nM | 0.15 |
| 100∝M CCR5 Reverse | 750nM | 0.15 |
| 100∝M CCR5 Probe | 250nM | 0.05 |
|
| ||
| Total | 15 | |
| Template | 5 | |
|
| ||
| Rxn Volume | 20 | |
Table 3.
Duplexed ddPCR master mix to measure HIV DNA (HIV gag/RU5 copies)
| ddPCR Duplexed Master Mix
| ||
|---|---|---|
| Reagent | Final Concentration | Per Rxn (ul) |
|
| ||
| dH20 | 4.65 | |
| 2× BIO-RAD Supermix | 1× | 10 |
| 100um gag Forward | 750nm | 0.15 |
| 100um gag Reverse | 750nm | 0.15 |
| 100 ∝M gag FAM-probe | 200nM | 0.05 |
| 100∝M RU5 Forward | 750nm | 0.15 |
| 100∝M RU5 Reverse | 750nm | 0.15 |
| 100∝M RU5 HEX-probe | 200nM | 0.05 |
|
| ||
| Total | 15 | |
| Template | 5 | |
|
| ||
| Rxn Volume | 20 | |
Figure 3.
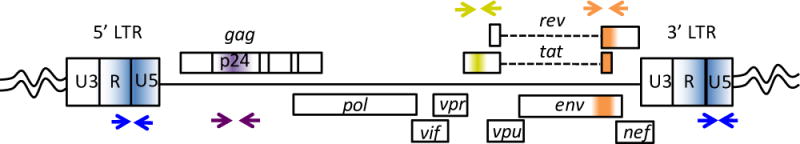
Schematic of ddPCR primer/probe target regions in the HIV genome for HIV DNA quantification.
Figure 4.
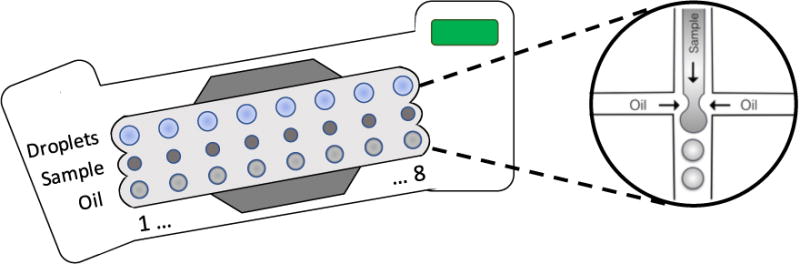
Schematic of DG8 cartridge for droplet generation: The droplet cartridges are composed of microfluidic channels. Sample is loaded into the center chamber of the cartridge, and oil is loaded into the bottom chamber then a gasket is applied and the cartridge plus holder are transferred to the droplet generator. The droplet generator applies vacuum pressure to pull samples and oil though these microfluidic channels. The samples and oil meet at a T-junction where they form water-in-oil droplets which then collect in the last chamber.
REAGENTS AND SOLUTIONS
-
70% Ethanol
35ml of 100% Ethanol
15ml of dH20
-
100mM CaCl2
1ml of 1M CaCl2
9ml of dH20
-
GuHCl+ stock (Store at room temperature and away from light)
18.75ml of 8M GuHCl
2.5ml of 1M Tris-HCL pH 7.6
0.5ml of 100mM CaCl2
28.25ml of dH20
-
GuHCl+/Proteinase K Solution (Make fresh before each use)
Add 50μL of Proteinase K per 1ml of GuHCl/ProK as required
(100μL of GuHCl/ProtK are needed per sample)
-
GUSCN+ stock (Store at room temperature and away from light)
50ml of 6M GuSCN
2.5ml of 1M Tris-HCL pH 7.6
106μl of 0.5M EDTA
-
GuSCN+/Glycogen Solution (Make fresh before each use)
Add 30μl of glycogen per 1ml of GuSCN+/Gly as required
(400μl of GuSCN/Gly is required per sample).
COMMENTARY
Background Information
Digital PCR utilizes limiting dilution of target molecules to achieve absolute quantification of specific nucleic acids by counting amplified molecules [35]. Since this method does not rely on the extrapolation from a standard curve generated by serial dilutions of known standards, it is less susceptible to user error and therefore quantification with digital PCR is more precise and reproducible than standard quantitative PCR (qPCR) [22]. To date, digital PCR is used for quantification in many fields, and numerous platforms have been developed that differ in the number of partitions generated from a single bulk PCR reaction and the partitioning method. For instance, the droplet digital PCR (ddPCR) platform produced by Bio-Rad (QX200) uses emulsion chemistry to partition a single PCR reaction into approximately 20,000 nanoliter water in oil droplets.
As in qPCR, a fluorescently labeled probe is used in ddPCR. However, the key principle of ddPCR is the partitioning of a single bulk PCR reaction into c. 20,000 nanoliter droplets each could contain, in theory, only one molecule per droplet. Since the sample is randomly distributed into these droplets ddPCR essential allows 20,000 parallel PCR reactions from one initial sample. The individual droplets are then pooled and thermal cycled using a standard thermal cycler until the sample reaches saturation, corresponding to the plateau phase of a sigmoidal amplification curve in qPCR. The resulting positive fluorescence is then read with a FACs-like droplet reader, which sips each well and passes the droplets individually by a two-color detection system. Since each droplet contains approximately one, or less, copies of the target DNA template the droplet reader can simply count the number of positive and negative droplets. Then a threshold is set between the negative droplets that did not contain target DNA, and the positive droplets that contained target DNA. The goal is to ensure that each droplet contains no more than a single DNA molecule, and the proportion of positive partitions and the copy number per well is calculated according to a Poisson distribution, with higher concentrations having increased chance of multiple molecules per droplet. Since accurate quantification depends on a clean separation between positive and negative droplets, the dynamic range of the QX200 system is from less than 100,000 copies per well, near complete saturation of positive droplets, down to theoretically 1 copy per well. However, erroneous false positive droplets and a phenomenon known as ‘rain’ complicate low level detection abilities and are discussed in the critical parameters section below. As a general rule, we routinely require an average of 5 positive droplets per well run in triplicate for reliable and reproducible quantification of DNA.
Critical Parameters and Troubleshooting
Genomic DNA extraction
This protocol has been optimized for the extraction and quantification of genomic DNA from a starting cell count of up to 2e6 cells. Overloading the extraction with greater number of cells has been shown to greatly reduce the recovery efficiency of genomic DNA. Further, saturation of CCR5 DNA is seen when assaying greater numbers of cells as the dynamic range of this platform reaches 100,000 copies per well. This protocol can be performed on total peripheral blood mononuclear cells (PBMCs) as well as cell subsets enriched by negative or positive selection using magnetic bead kits (STEM CELL technologies) or by fluorescence-activated cell sorting (FACS).
It is of critical importance to always flash spin to collect liquids before opening tubes to avoid aerosol and cross-contamination of specimens. HIV negative cell lines (such as CEM) should also be extracted and run for each assay to ensure no HIV DNA contamination has been introduced which could result in false positive results.
Droplet generation
Proper droplet generation is crucial for successful and accurate DNA quantification. Full resuspension of genomic DNA samples is required to ensure even partitioning of sample into droplets. Stubborn DNA samples can be fully resuspended with the help of sonication or restriction digestion. Overloading the cartridges with too much genomic DNA input or DNA that is not in resuspension will clog the microfluidic channels and result in decreased droplet formation and complete run failure. Furthermore, overloading DNA could lead to the saturation of target and the inability to accurately quantify target DNA, this can particularly be a problem when quantifying cellular genes such as CCR5 (every cell has 2 copies of the gene). We tested the dynamic range of the Bio-Rad QX200 platform using cDNA stocks of known quantity obtained from serially diluted and reverse transcribed RNA transcripts, and found that this system is accurate up to 100,000 copies over which saturation occurs and quantification is inaccurate (Figure 5). Low end quantification is accurate down to a single copy with Poisson distribution error (Figure 5). however, erroneous detection of false positive wells remain an issue and is discussed below.
Figure 5.
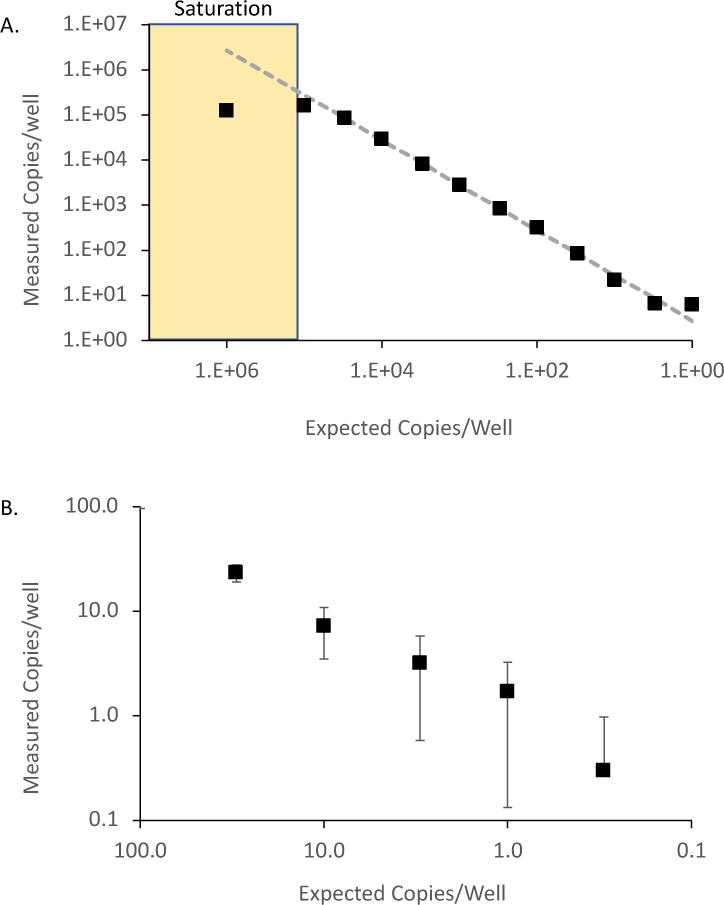
Dynamic Range of ddPCR platform: A.) ddPCR accurately quantifies targets from 0.3 to 1x105 copies (mean 2.2-fold variation from expected) but was inaccurate above 1x105 copies due to saturation of positive droplets. RNA transcripts were serially diluted, reversed transcribed, and quantified using ddPCR. Measured copies/well are compared to the expected line. B.) RNA transcripts were serially diluted from 100 to 0.3cps/well, reversed transcribed, and quantified using ddPCR. The number next to each point shows the average of the 10 replicates; the error bar is ±1 standard deviation (SD). Note that the error bar is very close to that predicted for a Poisson distribution, an irreducible minimum.
Avoiding bubbles that may form prior to droplet generation is essential for proper droplet formation. When loading sample and master mix into the DG8 cartridges, be sure to avoid any bubbles. Increasing the master mix and sample volume (e.g. 25μl instead of 20μl) can help ensure ample template and mix is available to prevent bubble formation. If bubbles are loaded into the cartridges they should be carefully removed prior to the generation of droplets. Pockets of air in the microfluidic channels can prevent proper formation and partitioning of sample into droplets.
Prior to PCR amplification droplets are extremely fragile and must be handled with care. Shearing of droplets can lead to increased intermediate fluorescence droplets, known as ‘rain’ (Figure 6), and may lead to inaccurate quantification of target DNA. When transferring droplets from the generation cartridge to the PCR plate it is best to angle the pipettor to prevent smashing the pipette tip on the bottom of the well and to give ample room for the droplets to enter the pipette tip. It is also important to draw the droplets up into the pipette tips and to aspirate the droplets slowly counting to 7 for both processes. Using a 5-50μl 8-channel pipette (RAININ) which has a long plunger length allows slow and smooth pipetting and will prevent shearing of droplets. Another option is to purchase the Bio-Rad Auto DG robotic droplet generator which bypasses the need for manual handling of the droplets as they are generated and transferred within the machine. See Figure 6 for an example of sheared droplets as a result of fast pipetting compared to intact droplets from proper pipetting. Other considerations for maintaining droplet integrity extend beyond droplet formation but prior to PCR amplification, such as gentle transferring of the plate containing droplets and time considerations as droplets are fragile prior to amplification. The final step in PCR amplification heats the droplets to 98°C for 10 minutes which inactivates the enzyme and stabilizes the droplets.
Figure 6.
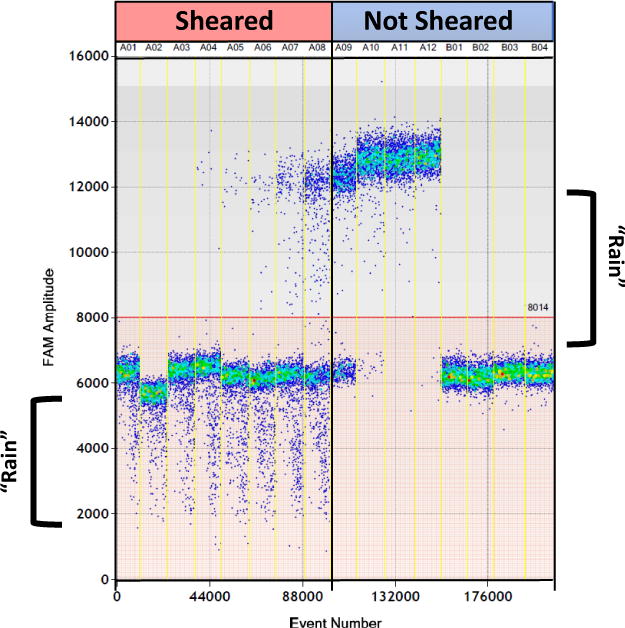
Trouble shooting example of sheared droplets: HIV RNA transcript standards were serially diluted and droplets were generate for each dilution. Droplets in the 8 wells to the left were sheared due abrupt and fast pipetting. Sheared droplets are easily distinguished with ‘rain’ under the negative droplet populations (bracket). Rain under the positive droplets is typically seen and may be attributed to inefficient PCR amplification. The 8 wells on the right show properly handled droplets having minimal rain under both the positive and negative droplets.
Setting the Threshold
Setting the threshold cut-off is a critical step in the analysis process as it determines the fraction of positive and negative droplets which is ultimately used to calculate the target abundance after applying a Poisson correction. In most cases, minor changes in the threshold set point will not influence the overall quantification greatly as there are relatively few droplets confined to the regions between negative and positive droplets. Droplets that are registered in the area between negative and positive droplets are known as ‘rain’(Figure 6). The ‘rain’ phenomenon is not clearly understood [23, 24, 26] but could be attributed to multiple droplets coalescing leading to a higher background fluorescence, inefficient PCR amplification that could be due to sequence variation, or poor signal-to-noise separation which could be improved by optimizing the assay or using dual-quenched probes (such as ZEN quenched probes from IDT). Still, issues with ‘rain’ as well as false positive droplets [22, 26, 27, 34], a common problem across multiple Digital PCR platforms, continues to complicate low level detection abilities. False positive droplets can be either the result of contamination of reagents or the environment, or erroneous ‘rain’. Estimations of the false-positive rate is recommended when attempting low copy number determinations. To estimate the false positive rate, a minimum of three negative template controls (NTC) should be tested and algorithm approaches can be applied as discussed below.
High copy number samples are less affected by minor adjustments in threshold levels. Setting the threshold level at 10 evenly spaced increments between the mean fluorescence of the negative and positive droplets from our HIV gag ddPCR assay performed on a dilution series of known standards did not significantly influence the concentrations obtained (Figure 7). However, it is possible that patient-to-patient mismatches in the target sequence can lead to lower overall positive fluorescence and therefore the threshold limit must be taken into careful consideration. Figure 8 shows three individual participants with different numbers of mismatches in the probe region of the HIV gag primer set. The individual with the most mismatches has the lowest peak positive fluorescence compared to the other two individuals and if the threshold was set too high, these positive droplets would be missed completely.
Figure 7.
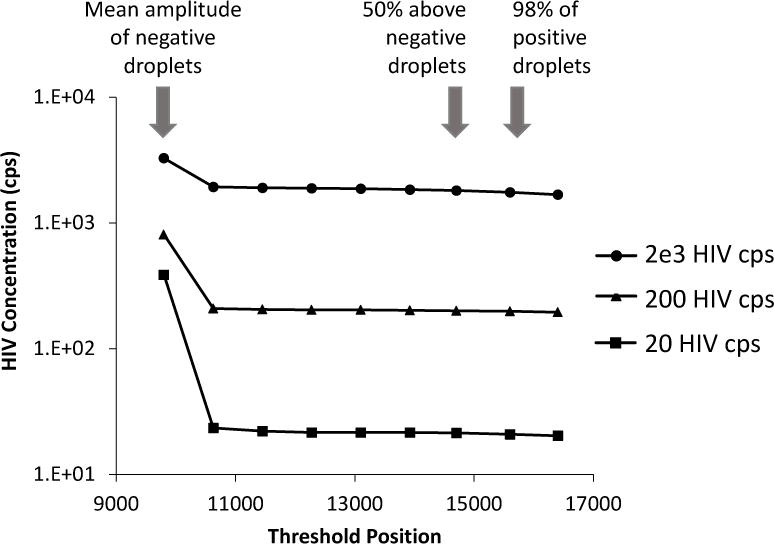
Setting the threshold: HIV RNA transcript standards were serially diluted and ddPCR was performed for each dilution. The data generated was then analyzed by setting varying thresholds. 10 increments were determined to set threshold bars ranging from the mean of fluorescence intensity of the negative droplets (mean: 9800) to under the mean fluorescence intensity of the positive droplets (mean: 17832). Increments also included a value that was 50% above the negative fluoresce (14700) and a value that would capture 98% of all positives (15600). Once above the negative droplets the overall HIV concentration measured with ddPCR does not significantly change even when adjusting the threshold level.
Figure 8.
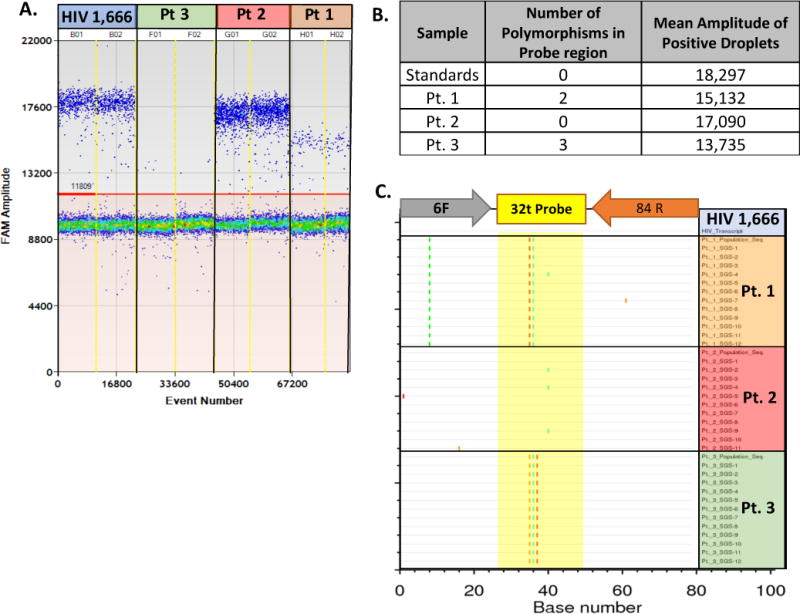
Trouble shooting example variance in mean fluorescence due to primer/probe mismatches: Primer/probe mismatches lead to a decrease in the mean fluorescence amplitude of positive droplets. A. Quantification schematic from a ddPCR run on patient plasma from three individuals compared to an RNA transcript control. B. The number of polymorphisms in the probe target region obtained by population sequencing and the corresponding mean amplitude of fluorescence in positive droplets obtained by ddPCR. C. Highlighter plot showing the population based and single genome sequences obtained from patient derived plasma from three individuals with the PCR region of interest compared to the primer sequences.
There are numerous ways to set the threshold cut-off. The Quantasoft software automatically assigns a threshold cut-off using an undisclosed method or allows a user-defined manual threshold to be set. The automatic threshold generated by the software is typically too low and falsely calls droplets positive even if they have a low fluorescence and appear negative to the user, this necessitates a manual change of the threshold. Since setting thresholds can be challenging, multiple algorithms have been developed to offer unbiased methods to generate threshold cut-offs. K-nearest clustering approaches were developed to determine the median and variance of negative and positive droplet fluorescence, then the statistical likelihood that an outlier should be grouped in either the negative or positive cluster is determined [22, 36]. Many user friendly bioinformatics approaches have been developed to aid researchers in the analysis of both one and two-channel ddPCR experiments, especially in defining cutoffs for positive and negative signals. An open access bioinformatics pipeline called ‘definetherain’ uses a positive control to identify the two clusters with a k-means algorithm, and then excludes droplets with intermediate fluorescence (http://definetherain.org.uk/) [36]. Another approach, ‘ddpcRquant’, uses negative controls to model the extreme values using extreme value theory (http://statapps.ugent.be/dPCR/ddpcrquant/) [38]. This approach does not make assumptions about the distribution of droplet fluorescence and provides an automated threshold determination without discarding any droplets from the analysis. For two channel duplexed experiments, ‘twoddpcr’, implements k-clustering and a Mahalanobis distance-based approach to identify double positive droplets from single target positive and double negative droplets (http://shiny.cruk.manchester.ac.uk/twoddpcr/) [37]. To date, setting the threshold cut-off remains a challenge and should be carefully considered particularly for low copy number determinations.
Anticipated Results
The outcome of this protocol is to obtain HIV DNA copy numbers normalized to cellular input. Running these duplexed assays will provide information on the abundance of various HIV genes and will enable accurate determination of the prevalence of different genes to provide information on the composition of the proviral landscape at the time of the sample by comparing the ratios of LTR to internal HIV DNA sequences such as gag, tat exon 1 and tat/rev exon 2. A high ratio of HIV LTR DNA to internal HIV DNA (>2) indicates that the proviral landscape is dominated by deleted proviruses. The data obtained from this protocol can be used to determine viral DNA burden as well as to look at the change in the various proviruses overtime. As the ddPCR platform enables absolute quantification small changes can be observed, an achievement that is not feasible using standard real time qPCR assays which have inherent day-to-day and operator variation. Therefore, these assays could be used to quantify decreases in total HIV DNA after various eradication interventions.
Time Considerations
This protocol from extraction to DNA quantification can be performed in 1 to 2 days. The DNA extraction requires a 1-hour proteinase k digestion. Samples can be extracted in advance and multiple samples can be quantified concurrently. Since droplets are fragile prior to PCR amplification, generation of droplets and PCR amplification should be carried out on the same day. Droplet generation cartridges accommodate 8 samples at a time, corresponding to 1 row on a 96-well plate format. It takes about 30 minutes to generate and transfer droplets for an entire 96-well plate (12 cartridges run sequentially). The PCR amplification protocol takes approximately 2 hours, after amplification droplets are stable and can be stored overnight until they are read. The droplet reader draws up the samples well-by-well, singulates the droplets, and passes them by a 2-color detection system to count positive and negative droplets in a total of two channels (HEX and FAM). The droplet reader processes droplets from an entire 96-well plate and generates data in under 1 hour. Data analysis is then performed in the Quantasoft software and data can be exported into comma separated files and pdfs.
Acknowledgments
This work was supported by the IATAP program from the office of the Director, NIH.
RESEARCH FUNDING: Yes,
FUNDING DETAILS: NCI Intramural Funding
Footnotes
SERIES EDITOR: Dr. Michelle Kloc
References
- 1.Finzi D, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5(5):512–7. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 2.Finzi D, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278(5341):1295–300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 3.Siliciano JD, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. 2003;9(6):727–8. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 4.Ho YC, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155(3):540–51. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruner KM, et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med. 2016;22(9):1043–9. doi: 10.1038/nm.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perelson AS, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387(6629):188–91. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 7.Han Y, et al. Experimental approaches to the study of HIV-1 latency. Nat Rev Microbiol. 2007;5(2):95–106. doi: 10.1038/nrmicro1580. [DOI] [PubMed] [Google Scholar]
- 8.Eriksson S, et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 2013;9(2):e1003174. doi: 10.1371/journal.ppat.1003174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Descours B, et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature. 2017;543(7646):564–567. doi: 10.1038/nature21710. [DOI] [PubMed] [Google Scholar]
- 10.Buzon MJ, et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med. 2014;20(2):139–42. doi: 10.1038/nm.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chomont N, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15(8):893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fromentin R, et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog. 2016;12(7):e1005761. doi: 10.1371/journal.ppat.1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaafoura S, et al. Progressive contraction of the latent HIV reservoir around a core of less-differentiated CD4(+) memory T Cells. Nat Commun. 2014;5:5407. doi: 10.1038/ncomms6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perreau M, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med. 2013;210(1):143–56. doi: 10.1084/jem.20121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tran TA, et al. Resting regulatory CD4 T cells: a site of HIV persistence in patients on long-term effective antiretroviral therapy. PLoS One. 2008;3(10):e3305. doi: 10.1371/journal.pone.0003305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oellerich M, et al. Using circulating cell-free DNA to monitor personalized cancer therapy. Crit Rev Clin Lab Sci. 2017;54(3):205–218. doi: 10.1080/10408363.2017.1299683. [DOI] [PubMed] [Google Scholar]
- 17.Srisutham S, et al. Four human Plasmodium species quantification using droplet digital PCR. PLoS One. 2017;12(4):e0175771. doi: 10.1371/journal.pone.0175771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mu D, et al. A sensitive and accurate quantification method for the detection of hepatitis B virus covalently closed circular DNA by the application of a droplet digital polymerase chain reaction amplification system. Biotechnol Lett. 2015;37(10):2063–73. doi: 10.1007/s10529-015-1890-5. [DOI] [PubMed] [Google Scholar]
- 19.Sedlak RH, et al. Clinical utility of droplet digital PCR for human cytomegalovirus. J Clin Microbiol. 2014;52(8):2844–8. doi: 10.1128/JCM.00803-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts CH, et al. Development and evaluation of a next-generation digital PCR diagnostic assay for ocular Chlamydia trachomatis infections. J Clin Microbiol. 2013;51(7):2195–203. doi: 10.1128/JCM.00622-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson M, et al. Development of droplet digital PCR for the detection of Babesia microti and Babesia duncani. Exp Parasitol. 2015;149:24–31. doi: 10.1016/j.exppara.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strain MC, et al. Highly precise measurement of HIV DNA by droplet digital PCR. PLoS One. 2013;8(4):e55943. doi: 10.1371/journal.pone.0055943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rutsaert S, et al. Digital PCR as a tool to measure HIV persistence. Retrovirology. 2018;15(1):16. doi: 10.1186/s12977-018-0399-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trypsteen W, et al. Diagnostic utility of droplet digital PCR for HIV reservoir quantification. J Virus Erad. 2016;2(3):162–9. doi: 10.1016/S2055-6640(20)30460-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henrich TJ, et al. Low-level detection and quantitation of cellular HIV-1 DNA and 2-LTR circles using droplet digital PCR. J Virol Methods. 2012;186(1–2):68–72. doi: 10.1016/j.jviromet.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiselinova M, et al. Comparison of droplet digital PCR and seminested real-time PCR for quantification of cell-associated HIV-1 RNA. PLoS One. 2014;9(1):e85999. doi: 10.1371/journal.pone.0085999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Oliveira MF, et al. Comparative Analysis of Cell-Associated HIV DNA Levels in Cerebrospinal Fluid and Peripheral Blood by Droplet Digital PCR. PLoS One. 2015;10(10):e0139510. doi: 10.1371/journal.pone.0139510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunay GA, et al. Assessment of the HIV-1 reservoir in CD4+ regulatory T cells by a Droplet Digital PCR based approach. Virus Res. 2017;240:107–111. doi: 10.1016/j.virusres.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 29.Henrich TJ, et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PLoS Med. 2017;14(11):e1002417. doi: 10.1371/journal.pmed.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosas-Umbert M, et al. Virological and immunological outcome of treatment interruption in HIV-1-infected subjects vaccinated with MVA-B. PLoS One. 2017;12(9):e0184929. doi: 10.1371/journal.pone.0184929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mothe B, et al. Safety and immunogenicity of a modified vaccinia Ankara-based HIV-1 vaccine (MVA-B) in HIV-1-infected patients alone or in combination with a drug to reactivate latent HIV-1. J Antimicrob Chemother. 2015;70(6):1833–42. doi: 10.1093/jac/dkv046. [DOI] [PubMed] [Google Scholar]
- 32.Tebas P, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370(10):901–10. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moron-Lopez S, et al. Sensitive quantification of the HIV-1 reservoir in gut-associated lymphoid tissue. PLoS One. 2017;12(4):e0175899. doi: 10.1371/journal.pone.0175899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hindson BJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604–10. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96(16):9236–41. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones M, et al. Low copy target detection by Droplet Digital PCR through application of a novel open access bioinformatic pipeline, ‘definetherain’. J Virol Methods. 2014;202:46–53. doi: 10.1016/j.jviromet.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiu A, et al. twoddpcr: an R/Bioconductor package and Shiny app for Droplet Digital PCR analysis. Bioinformatics. 2017;33(17):2743–2745. doi: 10.1093/bioinformatics/btx308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trypsteen W, et al. ddpcRquant: threshold determination for single channel droplet digital PCR experiments. Anal Bioanal Chem. 2015;407(19):5827–34. doi: 10.1007/s00216-015-8773-4. [DOI] [PubMed] [Google Scholar]