

Abstract
Phages have attracted a renewed interest as alternative to chemical antibiotics. Although the number of phages is 10-fold higher than that of bacteria, the number of genomically characterized phages is far less than that of bacteria. In this study, phage TC6, a novel lytic virus of Pseudomonas aeruginosa, was isolated and characterized. TC6 consists of an icosahedral head with a diameter of approximately 54 nm and a short tail with a length of about 17 nm, which are characteristics of the family Podoviridae. TC6 can lyse 86 out of 233 clinically isolated P. aeruginosa strains, thus showing application potentials for phage therapy. The linear double-stranded genomic DNA of TC6 consisted of 49796 base pairs and was predicted to contain 71 protein-coding genes. A total of 11 TC6 structural proteins were identified by mass spectrometry. Comparative analysis revealed that the P. aeruginosa phages TC6, O4, PA11, and IME180 shared high similarity at DNA sequence and proteome levels, among which PA11 was the first phage discovered and published. Meanwhile, these phages contain 54 core genes and have very close phylogenetic relationships, which distinguish them from other known phage genera. We therefore proposed that these four phages can be classified as Pa11virus, comprising a new phage genus of Podoviridae that infects Pseudomonas spp. The results of this work promoted our understanding of phage biology, classification, and diversity.
Keywords: Pseudomonas aeruginosa, phage TC6, Pa11virus, structural protein, comparative genomic analysis, podovirus
Introduction
Bacteriophages (phages) are natural components of all ecosystems, and the estimated numbers of phages are approximately 10-fold higher than that of bacteria (Breitbart and Rohwer, 2005). Phages have been exploited by scientists to understand basic biological mechanisms, such as genetic recombination, horizontal gene transfer (HGT), molecular biology, and evolution (Henry and Debarbieux, 2012). As natural killers of bacteria, phages have been used to treat human bacterial infectious diseases at the early stage of discovery (Chanishvili, 2012). Considering that the problem of drug resistance has become increasingly serious, both the fundamental research and therapeutic application of phages are re-evaluated (Cisek et al., 2017).
As a non-fermentative Gram-negative bacteria, Pseudomonas aeruginosa are widely distributed in soil, sewage, medical institutions, and other environments (Stover et al., 2000). It is also an important opportunistic pathogen, which is the main Gram-negative bacteria causing nosocomial infections; infection caused by this pathogen can be fatal (Hilker et al., 2015; Crull et al., 2018). P. aeruginosa has a strong ability to form biofilm and possesses intrinsic drug resistance (Hauser, 2009). The emerging of multidrug-resistant (MDR) and pan-drug-resistant P. aeruginosa strains makes the treatment of P. aeruginosa infection very difficult (Ang et al., 2016).
The study of P. aeruginosa phages plays an increasingly promising role in combating this notorious pathogen. The use of phages to challenge clinically isolated MDR P. aeruginosa strains and biofilms in vivo and in vitro has continued for many years, and some of the results are very promising (Alemayehu et al., 2012; Krylov, 2014; Pires et al., 2015; Verbeken et al., 2016). However, phage therapy still faces multiple challenges. The main obstacles are that phage’s host spectrum is excessively narrow, bacteria have several pathways to resist phage predation (Labrie et al., 2010), and our understanding of phages is limited and shallow.
As of July 20, 2018, 2294 phages (2203 infecting bacteria and 91 infecting archaea) have been completely sequenced based on the data from NCBI Viral genome browser1. A total of 155 are sequenced phages infecting Pseudomonas spp. (including 132 P. aeruginosa strains), among which 93 are lytic phages, 34 are temperate phages, and 28 are not yet determined. Out of the lytic phages of P. aeruginosa, 41% belong to the family Myoviridae, 38% belong to the family Podoviridae, 20% belong to the family Siphoviridae, and approximately 1% cannot be classified at present (Ceyssens and Lavigne, 2010). The variation of the genome size of the phages in each family is large. The genome size distribution of the phages in the family Myoviridae is in the range of 64.1–309.2 kb, while the phage genomes of the family Podoviridae and Siphoviridae are distributed between 41.6–74.9 and 34.5–61.1 kb, respectively. The tailed phages of Pseudomonas spp. can be divided into 11 genera (Shen et al., 2016). However, many Pseudomonas phages are currently unassigned or unclassified. Given the difficulties in the implementation of phage therapy in medical practice related to the insufficient number and diversity of lytic phages that are active against P. aeruginosa (Krylov, 2014), the characterization and classification of novel P. aeruginosa phages will not only be important in understanding the interactions between P. aeruginosa and its phages but will also aid in the development of novel approaches against this notorious pathogen.
Materials and Methods
Bacterial Strains and Culture Conditions
Pseudomonas aeruginosa PA1 was originally isolated from a patient with respiratory tract infection (Lu et al., 2013, 2015; Li et al., 2016). A total of 233 clinically isolated P. aeruginosa strains were previously collected from 7 different Chinese Hospitals (Yang et al., 2016). All the above-mentioned P. aeruginosa strains are stored in our laboratory. Bacteria were grown in Luria–Bertani (LB) liquid medium with shaking or plated onto LB medium containing 1.5% (w/v) agar. The incubation temperature for all bacterial strains was 37°C.
Phage Isolation, Propagation, and Purification
Phage TC6 was isolated from the sewage of Southwest Hospital (Chongqing, China) on the basis of a standard lambda phage isolation protocol (Sambrook and Russell, 2006). Briefly, 1000 mL sewage samples were centrifuged at 10000 g for 10 min. Subsequently, the supernatant was filtered through a 0.22 μm filter. Then, 3 mL PA1 culture at the log phase was added to the supernatant and cultured at 37°C for 12 h. The culture was filtered again through a 0.22 μm filter to remove the bacteria. The supernatant was serially diluted and spotted on the bacterial lawn according to the double-agar layer method. After incubation at 37°C overnight, single plaque was separated, and its phages were obtained and added to a culture of PA1 at the log phase. After incubation at 37°C for 6 h, a few drops of chloroform were added to the culture. Then, the culture was centrifuged for 5 min at 10000 g and filtered through a 0.22 μm filter. The resulting phage lysate was stored at 4°C for further analysis. TC6 crude phage suspensions were concentrated and purified by PEG8000 precipitation according to the method described previously (Govind et al., 2006). The purified TC6 particles were further purified using CsCl gradient ultracentrifugation (Casas and Rohwer, 2007). The optimum multiplicity of infectivity (MOI) of TC6 was observed using the method reported before (Huang et al., 2013).
One-Step Growth Curve
The one-step growth curve of TC6 was performed as described previously (Lu et al., 2013; Latino et al., 2014). Briefly, the early logarithmic growth phase cultures of P. aeruginosa PA1 (OD600 = 0.2) were continuously diluted 10 times. Bacteria cells were calculated by CFU plate counts. Phage samples were added to 3 mL PA1 cultures (OD600 = 0.1) at a MOI of 10:1. After incubation at 37°C for 15 min to allow the adsorption of phages, the mixture was then centrifuged for 30 s at 13000 g. The supernatant containing any un-adsorbed phages was removed by washing twice with LB medium. Pellets were re-suspended in 3 mL LB and the cultures were then grown at 37°C, 180 rpm shaking. Ten microliters of samples were taken every 10 min, and the titer of TC6 particles was determined by the double-layer agar plate method. The burst time and burst size were calculated based on the one-step growth curve.
Host Range Analysis
Host range analysis was performed as described previously (Yang et al., 2016). Phage sensitivity of 233 clinically isolated P. aeruginosa strains was determined by dot plaque assay. Briefly, 5 mL of molten 0.7% LB agar containing 100 μL of each test bacterial culture was overlaid on 1.5% LB agar plates. Subsequently, 1 μL of TC6 (∼1010 PFU/mL) was spotted on the soft agar. Phage resistance/susceptibility was determined by the formation of clear plaques after overnight culture at 37°C.
Transmission Electron Microscopy (TEM)
Transmission Electron Microscopy analysis was performed as described previously (Shen et al., 2016). Briefly, filtered phage lysates (∼1010 PFU/mL) were placed on copper grids to allow adsorption for 10 min, then negatively stained with 2% phosphotungstic acid (pH of 4.5) for 1 min and subsequently air dried. Phage particles were observed using TECNAI 10 electron microscope (Philips, Netherlands) at a voltage of 80 kV and with a magnification of 200000. Images were acquired digitally with a camera (Gatan, Model 785) inside the microscope.
Phage Genome Sequencing
DNA extraction and purification were performed as described previously (Lu et al., 2013). Briefly, TC6 particles purified by CsCl gradient ultracentrifugation were digested by proteinase K (50 μg mL-1) at 56°C for 1 h to release phage DNA. Then, the mixture was purified with phenol/chloroform/isoamyl alcohol (25:24:1) at 5000 g for 10 min. The aqueous layer was extracted again with chloroform at 5000 g for 10 min. Isopropanol was used to precipitate DNA, and the precipitated DNA was collected and washed with ethanol. The obtained TC6 DNA was suspended in TE buffer (pH of 8.0) for further use. Genomic DNA sequencing was carried out at Shanghai Biotechnology Corporation (Shanghai, China) by using PacBio sequencing technology (Rothberg et al., 2011; Gochez et al., 2018). Briefly, about 10 μg purified TC6 genomic DNA was subjected to sequencing by using PacBio RS (Pacific Biosciences, Menlo Park, CA, United States). SMRTbell template libraries with DNA fragments of 5 kb were prepared. TC6 genomic DNA was fragmented using Covaris microTUBE (ThermoFisher Scientific, MA, United States) and then purified by AMPure PB Beads2. Sequencing was then performed using one SMRT cells and zero-mode waveguide (ZMW) signals were obtained. After raw reads were filtered (filter criteria: minimum polymerase read quality: 0.75; minimum polymerase read length: 3,500), subreads were filtered (filter criteria: minimum subread length: 3,500); then read quality was calculated as 0.86. Approximately 78% of the filtered subreads were TC6 related. Filtered subreads were de novo assembled using the SPAdes v. 3.5.0 software package (Bankevich et al., 2012) with default parameters, and single contig with an average sequence coverage of 5700-fold revealed.
Sequence Analysis and Genome Annotation
The software package DNAStar (Rosseel et al., 2012) was used to analyze the general features of the TC6 genome sequence. TC6 genes were predicted using RAST3 (Aziz et al., 2008; McNair et al., 2018). DNA sequences and protein sequences were scanned for homologs by using BLAST (Boratyn et al., 2013; Singh and Raghava, 2016). The software tRNAscan-SE4 was used to predict tRNA genes (Lowe and Chan, 2016). The circular presentation of the TC6 genome was performed using BLAST ring image generator (BRIG)5 (Alikhan et al., 2011) and CGView6 (Grant and Stothard, 2008), and the results of BRIG and CGView were combined to present the genome map of TC6.
Identification of the TC6 Structural Proteins
Identification of the TC6 structural proteins was performed as described previously (Lu et al., 2013). Briefly, purified TC6 particles were heat denaturized and loaded onto a 10% (w/v) polyacrylamide gel to visualize TC6 structural proteins. Proteins were stained with Coomassie Brilliant Blue R250 dye and washed with methanol/acetic acid/water. Then, distinct protein bands were excised from the gel for high-performance liquid chromatography-mass spectrometry (HPLC-MS) analysis by using an HPLC-CHIP-MS/MS ION TRAP 6330 system (Agilent, Santa Clara, CA, United States). Data generated by HPLC-MS analysis were processed using Agilent Spectrum Mill proteomics software (Rev A.03.02.060; Agilent, Santa Clara, CA, United States) to allocate each protein band to its corresponding gene.
Comparative Genomic Analysis
The Circos diagram of nucleotide sequence alignment of Pseudomonas phages TC6, O4 (Li et al., 2010; Zhang et al., 2018), PA11 (Kwan et al., 2006), IME180 (GenBank accession no. MF788075) and PaP2 (GenBank accession no. NC_005884) were constructed using the software Circoletto7 (Darzentas, 2010). TC6 genome sequence was used as reference. The tBlastX analysis of phage genomes was performed using EasyFig8 (Sullivan et al., 2011). The major capsid protein sequences of podoviruses infecting Pseudomonas spp. were downloaded from GenBank (Benson et al., 2018). Multiple sequence alignments of major capsid protein sequences were conducted using ClustalW (Hung et al., 2015) with default parameters, and phylogenetic tree was constructed by MEGA 7.0.149 (Kumar et al., 2016) with the neighbor-joining method (Telles et al., 2018). Core genes were analyzed using CoreGenes3.5 (Zafar et al., 2002; Mahadevan et al., 2009). Venn diagrams were made by using an online tool10.
Results and Discussion
Morphology of TC6
Among the sequenced phages, approximately 90% belonged to the order Caudovirales (Ceyssens and Lavigne, 2010). Tailed phages have double-stranded DNA and can be divided into three different families according to different tail structures, as follows: Podoviridae, short and inflexible tail; Myoviridae, long and contractile tail; and Siphoviridae, long, flexible, but noncontractile tail (Grose and Casjens, 2014; Buttner et al., 2016). TEM analysis indicated that TC6 head structure was an icosahedron with a diameter of approximately 54 nm (Figure 1). Short and inflexible tail had a length of approximately 17 nm. The morphological characteristics of TC6 suggested that it should be assigned to Podoviridae family. TC6 had a structure and dimensions identical to that of Pseudomonas phage O4, which was isolated from the sea water of the coastal seashore of China Bohai inner sea, and it was also classified as a member of the Podoviridae family (Li et al., 2010).
FIGURE 1.

Electron micrograph of TC6 phage particles. Sample was stained with phosphotungstate. Scale bar represents 100 nm. White arrows indicate short tails.
Biological Characteristics of TC6
Lytic phages of P. aeruginosa can be isolated from a variety of environments, and approximately 56% of phages are derived from hospital sewage and wastewater of wastewater treatment plants (Ceyssens and Lavigne, 2010). Separating valuable phages was more feasible for hospital sewage than for the wastewater of wastewater treatment plants because it contains many pathogenic and drug-resistant bacteria. TC6 was isolated from hospital sewage by using P. aeruginosa PA1 as the host bacterium (Li et al., 2016). After being cultured overnight (∼12 h) on double agar plate with 0.7% agar for upper LB broth, TC6 formed pin-sized plaques (∼0.5 mm in diameter) with haloes (Figure 2A). The plaque morphology of TC6 is identical to that of O4 (Li et al., 2010). In rich liquid medium, TC6 can reach high titers (∼1010 PFU/mL). TC6 particles are resistant to chloroform and stable for storage at 4°C. The optimum MOI of TC6 was 10:1 (i.e., phages:hosts). The one-step growth curve of TC6 showed that its latent period is approximately 40 min, and its burst period is approximately 60 min (Figure 2B). The average burst size of TC6 was estimated to be 95, that is, one host bacterium can produce approximately 95 TC6 progenies. The efficient replication of TC6 progenies suggested that TC6 was a lytic phage. A total of 233 clinically isolated P. aeruginosa strains (Yang et al., 2016) were challenged by TC6, and 86 strains can be lysed, thereby indicating potential application value of TC6 in phage therapy (Nobrega et al., 2015; Rossitto et al., 2018).
FIGURE 2.

Plaques and one-step growth curve of TC6. (A) TC6 plaques on double agar plate. Phages were incubated at 37°C for 12 h. (B) One-step growth curve of phage TC6. Experiment was repeated thrice with duplicate samples at a MOI of 10:1.
Genomic Identification of TC6
TC6 genome is a linear dsDNA molecule containing 49796 base pairs (bp) with a G + C content of 45.06%, which is significantly less than that of its host PA1 (66.35%) (Supplementary Table S1). No intact tRNA genes were predicted in the TC6 genome. A total of 71 protein coding genes were predicted, and only 5.69% of the genome were noncoding regions (Supplementary Table S1). BlastP analysis revealed that 64 TC6 proteins had homologs in other viruses and/or bacteria, while only 38 proteins were functionally identified. A large proportion of the TC6 proteins were assigned as hypothetical proteins, which is common among phage populations (Kwan et al., 2006; Li et al., 2010; Shen et al., 2016). Numerous functionally unknown phage proteins formed the so-called viral dark matter, which are largely involved in the adaptation to host-specific conditions and the formation of tail fibers and may represent valuable information of molecular tools for biotechnological or antimicrobial applications (Filee et al., 2005; Cazares et al., 2014; Hatfull, 2015; De Smet et al., 2017).
The detailed annotation and organization of the TC6 genome is illustrated in Figure 3. TC6 genome can be divided into four functional modules, of which one functionally unknown module situated near the 3′ ends of the TC6 genome, and 12 small genes with unknown functions clustered in this module (Figure 3). The gene TC6_018 encoded a putative hemagglutinin (HA) protein of 592 aa, which is homologous to streptococcal hemagglutinin protein of Pseudomonas phage GP100 genome assembly, plasmid: I (GenBank accession no. LT968167) with an amino acid sequence coverage of 100% and an identity of 39%. As the major surface glycoprotein of influenza viruses, HA is a toxic protein that can agglutinate red blood cells (Byrd-Leotis et al., 2015). The putative HA protein of TC6 should be further investigated because phage-generated toxic proteins must be extensively considered during the practice of phage therapy (Watts, 2017). No homologs of phage integrases, excisionases, repressors, or transposases were predicted in the TC6 genome, thereby supporting our conclusion that TC6 is a lytic phage.
FIGURE 3.

Circular presentation of the TC6 genome. The products of predicted genes were indicated in the outermost region. The outermost ring denotes genes on the plus strand, followed by rings that depict genes on the minus strand, GC content (black), GC skew (purple/green), functional modules, and position scales.
Analysis of the Structural Proteins of TC6
A tailed phage particle was mainly made up of a series of structural proteins and a genomic DNA molecule inside its head. Genes encoding TC6 structural proteins clustered in a functional module that is situated near the 5′ ends of the TC6 genome (Figure 3). To identify the structural proteins of TC6 experimentally, we used sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to visualize the main structural proteins in the denatured gel (Figure 4). Approximately 19 protein bands with molecular weights ranging from 18 to 190 kDa were resolved. Protein bands were excised for HPLC-MS analysis. A total of 11 protein bands were identified as the products of different TC6 genes, leaving several protein bands unassigned (Figure 4). This result may be caused by the experimental errors during HPLC-MS. The predominant band was the major capsid protein (TC6_008, ∼35 kDa), which was the same as the predicted result. The major capsid protein of TC6 shared similar amino acid sequences and molecular weights to those of the P. aeruginosa phages O4 (Li et al., 2010; Zhang et al., 2018), PA11 (Kwan et al., 2006), and IME180 (GenBank accession no. MF788075), thereby suggesting a close relationship among these phages. Four hypothetical proteins, including TC6_001, TC6_011, TC6_016, and TC6_017, were separated by SDS-PAGE (Figure 4), thereby indicating that they are actually structural proteins. The gene TC6_002 encoded a putative pectate lyase of 464 aa, which was identified as a structural component of the TC6 virion (Figure 4). TC6_002 possessed a conserved domain homologous to pectate_lyase_3, which belongs to the pectate lyase superfamily that possesses a beta helical structure. This family is most closely related to glycosyl hydrolase family 28. As a structural protein, TC6_002 may participate in host cell wall lysis to promote DNA injection.
FIGURE 4.

Identification of TC6 structural proteins. Proteins were visualized by SDS-PAGE analysis in a 10% (w/v) gel. The detailed results of HPLC-MS analysis were indicated on the right side of the figure. aMW value was theoretically calculated. bMW value was experimentally estimated. cDistinct summed MS/MS search score.
Whole Genome Comparison Analyses of TC6
TC6 genome sequence was searched as a query in the nucleotide database of National Center for Biotechnology Information (NCBI11; Bethesda, MD, United States) (Sharma et al., 2018). The result showed that the genome sequences of O4 (Li et al., 2010; Zhang et al., 2018), PA11 (Kwan et al., 2006), and IME180 (GenBank accession no. MF788075) shared similar query coverage above 85% and identity above 96% with TC6 genome (Supplementary Table S2). Similar phages were isolated in different areas around the world, including Asia and North America, thereby suggesting complex evolutionary relationships among these phages. The genomic similarities of TC6, O4, PA11, IME180, and the control podovirus PaP2 (GenBank accession no. NC_005884) are illustrated in Figure 5. The whole genome sequences of TC6, O4, PA11, and IME180 showed remarkable similarities, thereby indicating that these phages may descend from a common ancestral phage. PaP2 was also isolated and sequenced in our laboratory, but it did not share genome sequence similarity with TC6. The genomic regions situated near the 3′ ends of these similar genomes showed lower similarities than the regions situated near the 5′ ends (Figure 5). This result indicated that the 3′ ends regions of these similar genomes may be highly variable during phage evolution, and some of them can be exchanged by HGT (Hafstrom et al., 2011).
FIGURE 5.

Circos diagram of nucleotide sequence alignment. Ribbons represent the similarity levels among the linked phage genome sequences. Different ranges of BLAST alignment bitscores are shown with distinct colors.
tBlastX analysis provided the translated comparisons of genome sequences; it revealed that TC6, O4, PA11, and IME180 showed remarkable similarities at the protein level (Figure 6). The identity of regions near the 3′ ends of these genomes were relatively lower than the average level (Figure 6), and the 3′ end regions were functionally unknown modules with small proteins of unknown functions. This phenomenon may have resulted from phage mutation accumulation to adapt to the changes in their living environment.
FIGURE 6.

Visualized tBlastX comparison of phage genomes. Arrowheads denote genes, and the identity cut-off is set at 43%. Other function indicates putative pectate lyase, putative DNA cytosine methyltransferase, thioredoxin-like protein, PhoH-like protein, DUF3310 domain-containing protein, putative 3′–5′ exonuclease, putative 5′–3′ exonuclease, putative endonuclease, putative ATP-grasp protein, putative amidoligase, putative COOH-NH2 ligase, putative amidotransferase, putative glutamine amidotransferase, homing endonuclease, or putative serine/threonine protein phosphatase.
Establishment of Genus Pa11virus
Taxonomically similar phages can be clustered by their major capsid proteins because they are considered to have similar head structural components (Bamford et al., 2005). Therefore, to determine the taxonomical location of TC6, we performed phylogenetic analysis on the basis on the major capsid proteins. As of July 20, 2018, 349 phages of the family Podoviridae have been completely sequenced based on the data from the GenBank database, including 53 phages infected Pseudomonas spp. Among 53 Pseudomonas phages, 38 of which infected P. aeruginosa, 5 infected P. fluorescens, 5 infected P. putida, 3 infected P. syringae, 1 infected P. tolaasii, and 1 infected P. plecoglossicida. We selected 53 Pseudomonas phages to analyze the phylogenetic relationships on the basis of their major capsid protein sequences, and a phylogenetic tree was constructed (Figure 7). The result showed that phages of the same genus were clustered into one clade, thereby dividing the phylogenetic tree into several distinct clades (Figure 7). TC6, O4, PA11, and IME180 were closely clustered, distinguishing them from other Pseudomonas podoviruses. This finding further indicated that these four phages were descendants of the same ancestor.
FIGURE 7.

Phylogenetic relationships of Podoviridae phages infecting Pseudomonas spp. Major capsid proteins and neighbor-joining method were used to construct the phylogenetic tree. Different clades are marked with different colors. The name of each clade is identical with the GenBank database, except that the Pa11virus (marked in red) was proposed in this work. The scale length of relative evolution distance is 0.2.
Phages TC6, O4, PA11, and IME180 shared remarkable similarity (Figures 5, 6), of which average nucleotide identity (ANI) reached to 97.25% (Supplementary Table S2). These phages also had very close phylogenetic relationships, which differentiated them from other known phage genera (Figure 7). Therefore, these phages should be grouped as a new phage genus. We named this novel phage genus according to the publication date and genomic sequence release date of the first phage discovered, that is, “PA11-like phages” (the publication date and genomic sequence release date of PA11 were 2006 and 2009, respectively). We then renamed “PA11-like phages” as “Pa11virus” according to the suggestions of Krupovic et al. (2016), as shown in Figure 7.
The genus Pa11virus had a mean genome size of 49860 bp (SD = 390 bp), a mean G + C% of 44.76% (SD = 0.19%), and share 54 core genes (Figure 8). A total of 45 accessory genes were predicted, and 18 of which are unique (only belonging to one of the four phages). In the future, increasing number of newly characterized phages will be assigned to the genus Pa11virus, thereby improving our understanding of phage biology and diversity.
FIGURE 8.
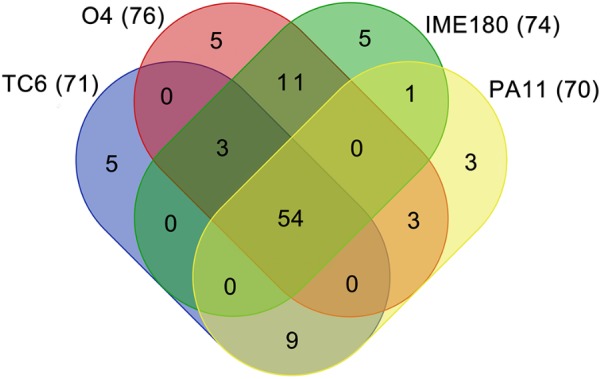
Venn diagram of genomes of Pa11virus. The results of core genes analysis are shown. The number of proteins were indicated in parenthesis.
Conclusion
This work focused on the morphological, genomic, proteomic, and comparative analyses of P. aeruginosa phage TC6, which is a lytic phage that belongs to the family Podoviridae. TC6 can lyse 86 out of 233 clinically isolated P. aeruginosa strains, thereby showing potential application for phage therapy. A total of 4 hypothetical proteins were determined as structural proteins, which improved the understanding on phage proteome. P. aeruginosa phages TC6, O4, PA11, and IME180 share remarkable similarity and have very close phylogenetic relationships, which differentiate them from other known phage genera. Accordingly, we proposed that they should be classified as a new phage genus of Podoviridae that infects Pseudomonas spp., which was named as Pa11virus according to the publication date of the first phage that was discovered (PA11). The results of this work will contribute to the diversity and classification of tailed phages.
Data Availability Statement
The complete genome sequence and annotations of TC6 have been deposited in GenBank under the accession number MG676466.
Author Contributions
SL and CT conceived and designed the experiments. CT, CD, YZ, and CX performed the experiments. SL and JW analyzed the data. JW, XR, and FH contributed reagents, materials, and analysis tools. SL wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding. This work was supported by the National Natural Science Foundation of China (31871251 and 31400163 to SL, 31570173 to FH).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02561/full#supplementary-material
References
- Alemayehu D., Casey P. G., Mcauliffe O., Guinane C. M., Martin J. G., Shanahan F., et al. (2012). Bacteriophages phiMR299-2 and phiNH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. mBio 3:e0029-12. 10.1128/mBio.00029-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alikhan N. F., Petty N. K., Ben Zakour N. L., Beatson S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. 10.1186/1471-2164-12-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang J. Y., Abdel-Haq N., Zhu F., Thabit A. K., Nicolau D. P., Satlin M. J., et al. (2016). Multidrug-Resistant Pseudomonas aeruginosa infection in a child with cystic fibrosis. Antimicrob. Agents Chemother. 60 5627–5630. 10.1128/AAC.00705-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz R. K., Bartels D., Best A. A., Dejongh M., Disz T., Edwards R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford D. H., Grimes J. M., Stuart D. I. (2005). What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 15 655–663. 10.1016/j.sbi.2005.10.012 [DOI] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D. A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Ostell J., Pruitt K. D., et al. (2018). GenBank. Nucleic Acids Res. 46 D41–D47. 10.1093/nar/gkx1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boratyn G. M., Camacho C., Cooper P. S., Coulouris G., Fong A., Ma N., et al. (2013). BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 41 W29–W33. 10.1093/nar/gkt282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M., Rohwer F. (2005). Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 13 278–284. [DOI] [PubMed] [Google Scholar]
- Buttner C. R., Wu Y., Maxwell K. L., Davidson A. R. (2016). Baseplate assembly of phage Mu: defining the conserved core components of contractile-tailed phages and related bacterial systems. Proc. Natl. Acad. Sci. U.S.A. 113 10174–10179. 10.1073/pnas.1607966113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd-Leotis L., Galloway S. E., Agbogu E., Steinhauer D. A. (2015). Influenza hemagglutinin (HA) stem region mutations that stabilize or destabilize the structure of multiple HA subtypes. J. Virol. 89 4504–4516. 10.1128/JVI.00057-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas V., Rohwer F. (2007). Phage metagenomics. Methods Enzymol. 421 259–268. 10.1016/S0076-6879(06)21020-6 [DOI] [PubMed] [Google Scholar]
- Cazares A., Mendoza-Hernandez G., Guarneros G. (2014). Core and accessory genome architecture in a group of Pseudomonas aeruginosa Mu-like phages. BMC Genomics 15:1146. 10.1186/1471-2164-15-1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceyssens P. J., Lavigne R. (2010). Bacteriophages of Pseudomonas. Future Microbiol. 5 1041–1055. 10.2217/fmb.10.66 [DOI] [PubMed] [Google Scholar]
- Chanishvili N. (2012). Phage therapy–history from Twort and d’Herelle through Soviet experience to current approaches. Adv. Virus Res. 83 3–40. 10.1016/B978-0-12-394438-2.00001-3 [DOI] [PubMed] [Google Scholar]
- Cisek A. A., Dabrowska I., Gregorczyk K. P., Wyzewski Z. (2017). Phage therapy in bacterial infections treatment: one hundred years after the discovery of Bacteriophages. Curr. Microbiol. 74 277–283. 10.1007/s00284-016-1166-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crull M. R., Somayaji R., Ramos K. J., Caldwell E., Mayer-Hamblett N., Aitken M. L., et al. (2018). Changing rates of chronic Pseudomonas aeruginosa infections in cystic fibrosis: a population-based cohort study. Clin. Infect Dis. 67 1089–1095. 10.1093/cid/ciy215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzentas N. (2010). Circoletto: visualizing sequence similarity with Circos. Bioinformatics 26 2620–2621. 10.1093/bioinformatics/btq484 [DOI] [PubMed] [Google Scholar]
- De Smet J., Hendrix H., Blasdel B. G., Danis-Wlodarczyk K., Lavigne R. (2017). Pseudomonas predators: understanding and exploiting phage-host interactions. Nat. Rev. Microbiol. 15 517–530. 10.1038/nrmicro.2017.61 [DOI] [PubMed] [Google Scholar]
- Filee J., Tetart F., Suttle C. A., Krisch H. M. (2005). Marine T4-type bacteriophages, a ubiquitous component of the dark matter of the biosphere. Proc. Natl. Acad. Sci. U.S.A. 102 12471–12476. 10.1073/pnas.0503404102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gochez A. M., Huguet-Tapia J. C., Minsavage G. V., Shantaraj D., Jalan N., Strauss A., et al. (2018). Pacbio sequencing of copper-tolerant Xanthomonas citri reveals presence of a chimeric plasmid structure and provides insights into reassortment and shuffling of transcription activator-like effectors among X. citri strains. BMC Genomics 19:16. 10.1186/s12864-017-4408-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind R., Fralick J. A., Rolfe R. D. (2006). Genomic organization and molecular characterization of Clostridium difficile bacteriophage PhiCD119. J. Bacteriol. 188 2568–2577. 10.1128/JB.188.7.2568-2577.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant J. R., Stothard P. (2008). The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36 W181–W184. 10.1093/nar/gkn179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grose J. H., Casjens S. R. (2014). Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae. Virology 46 421–443. 10.1016/j.virol.2014.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafstrom T., Jansson D. S., Segerman B. (2011). Complete genome sequence of Brachyspira intermedia reveals unique genomic features in Brachyspira species and phage-mediated horizontal gene transfer. BMC Genomics 12:395. 10.1186/1471-2164-12-395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull G. F. (2015). Dark matter of the biosphere: the amazing world of Bacteriophage diversity. J. Virol. 89 8107–8110. 10.1128/JVI.01340-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A. R. (2009). The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7 654–665. 10.1038/nrmicro2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry M., Debarbieux L. (2012). Tools from viruses: bacteriophage successes and beyond. Virology 434 151–161. 10.1016/j.virol.2012.09.017 [DOI] [PubMed] [Google Scholar]
- Hilker R., Munder A., Klockgether J., Losada P. M., Chouvarine P., Cramer N., et al. (2015). Interclonal gradient of virulence in the Pseudomonas aeruginosa pangenome from disease and environment. Environ. Microbiol. 17 29–46. 10.1111/1462-2920.12606 [DOI] [PubMed] [Google Scholar]
- Huang G., Le S., Peng Y., Zhao Y., Yin S., Zhang L., et al. (2013). Characterization and genome sequencing of phage Abp1, a new phiKMV-like virus infecting multidrug-resistant Acinetobacter baumannii. Curr. Microbiol. 66 535–543. 10.1007/s00284-013-0308-7 [DOI] [PubMed] [Google Scholar]
- Hung C. L., Lin Y. S., Lin C. Y., Chung Y. C., Chung Y. F. (2015). CUDA ClustalW: an efficient parallel algorithm for progressive multiple sequence alignment on Multi-GPUs. Comput. Biol. Chem. 58 62–68. 10.1016/j.compbiolchem.2015.05.004 [DOI] [PubMed] [Google Scholar]
- Krupovic M., Dutilh B. E., Adriaenssens E. M., Wittmann J., Vogensen F. K., Sullivan M. B., et al. (2016). Taxonomy of prokaryotic viruses: update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 161 1095–1099. 10.1007/s00705-015-2728-0 [DOI] [PubMed] [Google Scholar]
- Krylov V. N. (2014). Bacteriophages of Pseudomonas aeruginosa: long-term prospects for use in phage therapy. Adv. Virus Res. 88 227–278. 10.1016/B978-0-12-800098-4.00005-2 [DOI] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Tamura K. (2016). MEGA7: molecular evolutionary genetics analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 33 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan T., Liu J., Dubow M., Gros P., Pelletier J. (2006). Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J. Bacteriol. 188 1184–1187. 10.1128/JB.188.3.1184-1187.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie S. J., Samson J. E., Moineau S. (2010). Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8 317–327. 10.1038/nrmicro2315 [DOI] [PubMed] [Google Scholar]
- Latino L., Essoh C., Blouin Y., Vu Thien H., Pourcel C. (2014). A novel Pseudomonas aeruginosa bacteriophage, Ab31, a chimera formed from temperate phage PAJU2 and P. putida lytic phage AF: characteristics and mechanism of bacterial resistance. PLoS One 9:e93777. 10.1371/journal.pone.0093777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Shen M., Le S., Tan Y., Li M., Zhao X., et al. (2016). Genomic analyses of multidrug resistant Pseudomonas aeruginosa PA1 resequenced by single-molecule real-time sequencing. Biosci. Rep. 36:e00418. 10.1042/BSR20160282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Yang H., Lin S., Jia S. (2010). Classification of 17 newly isolated virulent bacteriophages of Pseudomonas aeruginosa. Can. J. Microbiol. 56 925–933. 10.1139/w10-075 [DOI] [PubMed] [Google Scholar]
- Lowe T. M., Chan P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44 W54–W57. 10.1093/nar/gkw413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S., Le S., Li G., Shen M., Tan Y., Zhao X., et al. (2015). Complete genome sequence of Pseudomonas aeruginosa PA1, isolated from a patient with a respiratory tract infection. Genome Announc. 3:e01453-15. 10.1128/genomeA.01453-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S., Le S., Tan Y., Zhu J., Li M., Rao X., et al. (2013). Genomic and proteomic analyses of the terminally redundant genome of the Pseudomonas aeruginosa phage PaP1: establishment of genus PaP1-like phages. PLoS One 8:e62933. 10.1371/journal.pone.0062933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan P., King J. F., Seto D. (2009). Data mining pathogen genomes using GeneOrder and CoreGenes and CGUG: gene order, synteny and in silico proteomes. Int. J. Comput. Biol. Drug Des. 2 100–114. 10.1504/IJCBDD.2009.027586 [DOI] [PubMed] [Google Scholar]
- McNair K., Aziz R. K., Pusch G. D., Overbeek R., Dutilh B. E., Edwards R. (2018). Phage genome annotation using the RAST pipeline. Methods Mol. Biol. 1681 231–238. 10.1007/978-1-4939-7343-9_17 [DOI] [PubMed] [Google Scholar]
- Nobrega F. L., Costa A. R., Kluskens L. D., Azeredo J. (2015). Revisiting phage therapy: new applications for old resources. Trends Microbiol. 23 185–191. 10.1016/j.tim.2015.01.006 [DOI] [PubMed] [Google Scholar]
- Pires D. P., Vilas Boas D., Sillankorva S., Azeredo J. (2015). Phage therapy: a step forward in the treatment of Pseudomonas aeruginosa infections. J. Virol. 89 7449–7456. 10.1128/JVI.00385-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosseel T., Scheuch M., Hoper D., De Regge N., Caij A. B., Vandenbussche F., et al. (2012). DNase SISPA-next generation sequencing confirms Schmallenberg virus in Belgian field samples and identifies genetic variation in Europe. PLoS One 7:e41967. 10.1371/journal.pone.0041967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossitto M., Fiscarelli E. V., Rosati P. (2018). Challenges and promises for planning future clinical research into bacteriophage therapy against Pseudomonas aeruginosa in cystic fibrosis. An argumentative review. Front. Microbiol. 9:775. 10.3389/fmicb.2018.00775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg J. M., Hinz W., Rearick T. M., Schultz J., Mileski W., Davey M., et al. (2011). An integrated semiconductor device enabling non-optical genome sequencing. Nature 475 348–352. 10.1038/nature10242 [DOI] [PubMed] [Google Scholar]
- Sambrook J., Russell D. W. (2006). The Condensed Protocols from Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Sharma S., Ciufo S., Starchenko E., Darji D., Chlumsky L., Karsch-Mizrachi I., et al. (2018). The NCBI BioCollections Database. Database 2018:bay006. 10.1093/database/bay006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M., Le S., Jin X., Li G., Tan Y., Li M., et al. (2016). Characterization and comparative genomic analyses of Pseudomonas aeruginosa phage PaoP5: new members assigned to PAK_P1-like viruses. Sci. Rep. 6:34067. 10.1038/srep34067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H., Raghava G. P. (2016). BLAST-based structural annotation of protein residues using Protein Data Bank. Biol. Direct. 11:4. 10.1186/s13062-016-0106-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover C. K., Pham X. Q., Erwin A. L., Mizoguchi S. D., Warrener P., Hickey M. J., et al. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406 959–964. 10.1038/35023079 [DOI] [PubMed] [Google Scholar]
- Sullivan M. J., Petty N. K., Beatson S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27 1009–1010. 10.1093/bioinformatics/btr039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telles G. P., Araujo G. S., Walter M., Brigido M. M., Almeida N. F. (2018). Live neighbor-joining. BMC Bioinformatics 19:172. 10.1186/s12859-018-2162-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeken G., Huys I., Ceulemans C., Jennes S., De Vos D., Pirnay J. P. (2016). Bacteriophage therapy: fast-forward to the past lessons identified from the advanced therapy regulation. Burns 42 11–12. 10.1016/j.burns.2015.10.022 [DOI] [PubMed] [Google Scholar]
- Watts G. (2017). Phage therapy: revival of the bygone antimicrobial. Lancet 390 2539–2540. 10.1016/S0140-6736(17)33249-X [DOI] [PubMed] [Google Scholar]
- Yang Y., Lu S., Shen W., Zhao X., Shen M., Tan Y., et al. (2016). Characterization of the first double-stranded RNA bacteriophage infecting Pseudomonas aeruginosa. Sci. Rep. 6:38795. 10.1038/srep38795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafar N., Mazumder R., Seto D. (2002). CoreGenes: a computational tool for identifying and cataloging “core” genes in a set of small genomes. BMC Bioinformatics 3:12. 10.1186/1471-2105-3-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Huang K., Yang X., Sun L., You J., Pan X., et al. (2018). Characterization of a novel lytic podovirus O4 of Pseudomonas aeruginosa. Arch. Virol. 163 2377–2383. 10.1007/s00705-018-3866-y [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The complete genome sequence and annotations of TC6 have been deposited in GenBank under the accession number MG676466.