

Abstract
Key points
Ectopic action potentials (EAPs) arise at distal locations in axonal fibres and are often associated with neuronal pathologies such as epilepsy or nerve injury, but they also occur during physiological network conditions. This study investigates whether initiation of such EAPs is modulated by subthreshold synaptic activity.
Somatic subthreshold potentials invade the axonal compartment to considerable distances (>350 μm), whereas spread of axonal subthreshold potentials to the soma is inefficient.
Ectopic spike generation is entrained by conventional synaptic signalling mechanisms. Excitatory synaptic potentials promote EAPs, whereas inhibitory synaptic potentials block EAPs.
The modulation of ectopic excitability depends on propagation of somatic voltage deflections to the axonal EAP initiation site.
Synaptic modulation of EAP initiation challenges the view of the distal axon being independent of synaptic activity and may contribute to mechanisms underlying fast network oscillations and pathological network activity.
Abstract
While most action potentials are generated at the axon initial segment, they can also be triggered at more distal sites along the axon. Such ectopic action potentials (EAPs) occur during several neuronal pathologies such as epilepsy, nerve injuries and inflammation but have also been observed during physiological network activity. EAPs propagate antidromically towards the somato‐dendritic compartment where they modulate synaptic plasticity. Here we investigate the converse signal direction: do somato‐dendritic synaptic potentials affect the generation of ectopic spikes? We measured anti‐ and orthodromic spikes in the soma and axon of mouse hippocampal CA1 pyramidal cells. We found that synaptic potentials propagate reliably through the axon, causing significant voltage transients at distances >350 μm. At these sites, excitatory input efficiently facilitated EAP initiation in distal axons and, conversely, inhibitory input suppressed EAP initiation. Our data reveal a new mechanism by which ectopically generated spikes can be entrained by conventional synaptic signalling during normal and pathological network activity.
Keywords: ectopic action potential, axon, synaptic input, CA1 pyramidal neuron, input integration, hippocampus
Key points
Ectopic action potentials (EAPs) arise at distal locations in axonal fibres and are often associated with neuronal pathologies such as epilepsy or nerve injury, but they also occur during physiological network conditions. This study investigates whether initiation of such EAPs is modulated by subthreshold synaptic activity.
Somatic subthreshold potentials invade the axonal compartment to considerable distances (>350 μm), whereas spread of axonal subthreshold potentials to the soma is inefficient.
Ectopic spike generation is entrained by conventional synaptic signalling mechanisms. Excitatory synaptic potentials promote EAPs, whereas inhibitory synaptic potentials block EAPs.
The modulation of ectopic excitability depends on propagation of somatic voltage deflections to the axonal EAP initiation site.
Synaptic modulation of EAP initiation challenges the view of the distal axon being independent of synaptic activity and may contribute to mechanisms underlying fast network oscillations and pathological network activity.
Introduction
Cortical axons transmit neuronal information quickly and efficiently over long distances. The advent of axonal recording techniques identified the distal portion of the axon initial segment (AIS, ∼20–40 μm from the soma) as the main initiation site of most cortical action potentials (Kole et al. 2007; Shu et al. 2007; Hu et al. 2009 b; Popovic et al. 2011; Baranauskas et al. 2013). Starting from the AIS, action potentials propagate in a bidirectional manner towards the axon terminals as well as towards the somato‐dendritic compartment (Wong et al. 1979; Stuart et al. 1997;Palmer & Stuart, 2006). Besides the AIS, several compartments of cortical pyramidal cells contain voltage‐dependent ion channels at sufficient density to generate active voltage signals. Such spikes have been reported for apical and basal dendrites, soma, nodes of Ranvier and distal parts of the axonal arbor (Häusser et al. 1995; Colbert & Johnston, 1996; Hu et al. 2009 b; Remy et al. 2009).
Full ectopic action potentials (EAPs) are initiated in distal parts of axons and were shown to arise spontaneously during several pathological conditions such as epileptic seizures, nerve injury, pain, demyelination and inflammation (Pinault, 1995; Sasaki, 2013; Hamada & Kole, 2015). Yet, they have also been observed under physiological conditions. For example, intact peripheral fibres generate EAPs in response to heat stimuli, suggesting a role in physiological sensory signal transduction (Moalem et al. 2005; Hoffmann et al. 2008). In the rodent hippocampus, ectopic axonal activity was first observed in CA1 and CA3 pyramidal cells using in vivo seizure models (Rosen & Vastola, 1971; Gutnick & Prince, 1972). Under physiological conditions, both hippocampal interneurons and pyramidal cells were shown to receive ectopic activity during several different types of network activity in vitro and in vivo (Avoli et al. 1998; Bähner et al. 2011; Sheffield et al. 2011; Chorev & Brecht, 2012; Dugladze et al. 2012). In CA1, interneurons can enter a state of spontaneous “retro‐axonal barrage firing” following sustained generation of conventional action potentials (Sheffield et al. 2013). In CA1 pyramidal cells, EAPs occur during exploratory behaviour (Harvey et al. 2009; Epsztein et al. 2010) as well as during sharp wave ripple complexes in vitro (Bähner et al. 2011). Axons of CA3 pyramidal cells show ectopic activity at high frequencies during hippocampal gamma oscillations in vitro, during which the majority of EAPs do not propagate to the soma (Dugladze et al. 2012). Taken together, though less common than conventional action potentials, EAPs are observed in hippocampal neurons during different states of neuronal network activity, both in physiological and pathophysiological conditions (Pinault, 1995; Bucher & Goaillard, 2011).
The remote location of EAP initiation in the axon does not exclude modulation by synaptic activity. Indeed, previous work shows that synaptic potentials can propagate for considerable distances along the axon in several cortical cell types where they affect action potential waveform and transmitter release (Alle & Geiger, 2006; Shu et al. 2006; Apostolides et al. 2016). As a consequence, it has been suggested that analog synaptic signals interact with EAP initiation in remote axonal compartments (Vladimirov et al. 2013). This hypothesis has, however, not been addressed experimentally before.
In this study, we used patch clamp recordings from the soma and axon of CA1 pyramidal cells to investigate the propagation of passive and active potentials. We report that the generation of EAPs is strongly affected by subthreshold somatic signals over considerable distances of several hundred micrometres.
Methods
Ethical approval
The investigators understand the ethical principles under which The Journal of Physiology operates and their work complies with the animal ethics checklist as described by Grundy (2015). Experiments were approved by the animal welfare officer of the Institute of Physiology and Pathophysiology at Heidelberg University and by the responsible veterinarian at the University of Oslo. Animal maintenance and experimental procedures were performed in strict accordance with German and Norwegian law, the guidelines of the European Community Council and the state government of Baden‐Württemberg (Project T100/15). Male C57BL/6N mice were purchased from Charles River Laboratories (Sulzfeld, Germany, Strain Code: 027) aged 4 weeks (20–25 g) and either housed in the Interfaculty Biomedical Research Facility in Heidelberg (IBF) or they were imported by Scanbur (Nittedal, Norway) to Oslo and housed in the Department of Comparative Medicine at the Institute of Basic Medical Sciences. Animals were kept for up to 2 weeks in groups of up to four individuals, with nesting material made of cellulose, a 12 h:12 h light/dark cycle, with controlled temperature and humidity, and access to food and water ad libitum. In order to minimize the stress of euthanasia, mice were sedated by exposure to CO2 in a rising concentration (20–30 l/h sourced from a compressed gas cylinder, Heidelberg) or 12–15% desflurane (Oslo) until the animal fell unconscious. Mice were subsequently killed by decapitation.
Slice preparation
The brain was quickly removed and transferred to 4°C cold, carbogen buffered (95% O2, 5% CO2 at pH 7.4) artificial cerebrospinal fluid (ACSF) containing the following (in mM): 124 NaCl, 3 KCl, 1.8 MgSO4, 1.6 CaCl2, 10 glucose, 1.25 NaH2PO4, 26 NaH2CO3 with an osmolarity of 295 mOsm/l. The first third of frontal brain and the cerebellum were removed. Horizontal brain slices were cut at 300 μm using a VT1000s or VT1200s Vibratome (Leica, Nussloch, Germany). Cutting was performed using a specific cutting solution containing (in mM): 140 potassium gluconate, 10 HEPES, 15 sodium gluconate, 0.2 EGTA and 4 NaCl adjusted to pH 7.2 using KOH. Before recording, slices were incubated for 30 min in carbogen‐buffered ACSF at 34°C for recovery and then kept at room temperature (∼22°C) for at least 30 min before the start of experiments.
Electrophysiological recordings
Slices were transferred into a submerged type recording chamber with constant perfusion of carbogen‐buffered ACSF at 32°C. Dual soma/axon recordings were performed at the University of Oslo as described previously (Hu & Jonas, 2014). Briefly, CA1 pyramidal cells were filled with 50 μM Alexa Fluor 488 (Thermo Fisher Scientific, Oslo, Norway) through the somatic pipette to trace the axon using a Nipkow spinning disc confocal microscope in combination with IR‐DIC (BX50, Olympus, Asker, Norway; Ultraview live cell imager, PerkinElmer, Oslo, Norway). Axonal recordings were made from terminal spherical axon expansions (blebs), which form during the slicing procedure (Shu et al. 2006). Recording distances were determined using the Simple Neurite Tracer plugin in ImageJ (https://imagej.nih.gov/ij/). Pipettes from borosilicate glass had a resistance of 3–5 MΩ for somatic and 10–20 MΩ for axonal recordings. Current‐clamp recordings were performed using a Multiclamp 700B amplifier (Molecular Devices, Wokingham, Berkshire, UK). Signals were low‐pass filtered at 10 kHz and sampled at 100 kHz with a Digidata 1322 converter board using pCLAMP 9 (Molecular Devices).
Somatic recordings in combination with extracellular ectopic and synaptic stimulation were performed at Heidelberg University. CA1 pyramidal cells were visualized using an upright BX51 microscope (Olympus, Hamburg, Germany) with a 40× water‐immersion objective. Recording electrodes were pulled using borosilicate glass on a Flaming/Brown P‐97 Puller (Sutter Instrument, Novato, CA, USA) to yield a resistance of 3–6 MΩ. The electrode solution contained (in mM): 140 potassium gluconate, 3 KCl, 4 NaCl, 10 HEPES, 0.2 EGTA, 2 MgATP and 0.1 Na2GTP adjusted to pH 7.2 using KOH and to 288 mOsm/l by adding glucose. Recordings were obtained in current clamp mode with an ELC‐03XS amplifier (NPI electronic, Tamm, Germany). Signals were low‐pass filtered at 3 kHz and digitized with 20 kHz using a Micros 1401MKII AC‐converter (CED, Cambridge, UK). Data were collected using the Signal 4.10 software (CED). Voltages were not corrected for the calculated liquid‐junction potential of +14.5 mV. Test pulses of −100 pA and 200 ms were applied regularly to control for changes in series resistance. Bipolar platinum/iridium electrodes (100 kΩ at 1 kHz; 75 μm tip distance) were used to stimulate Schaffer collaterals (excitatory stimulus) and the alvear bundle (ectopic stimulus) using square pulses of 100 μs duration (Fig. 3 A arrows). Inhibitory potentials were evoked by stimulation in stratum pyramidale while excitatory transmission was blocked by bath application of dl‐2‐amino‐5‐phosphonovaleric acid (apv; 30 μM) and 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX; 10 μM). For orthodromic stimulation, the intensity was adjusted to evoke PSPs with average amplitudes of ∼5 mV at the somatic recording site. Ectopic stimuli were adjusted to be either just below the threshold for generating ectopic action potentials or to yield a firing probability between 30% (Figs 3 B–D, and 4 A–E and G) and 80% (Figs. 3 E and F, and 4 F) per train. Stimulation experiments were repeated at 20 s intervals to avoid activity‐dependent plasticity.
Figure 3. Synaptic input modulates EAP generation.
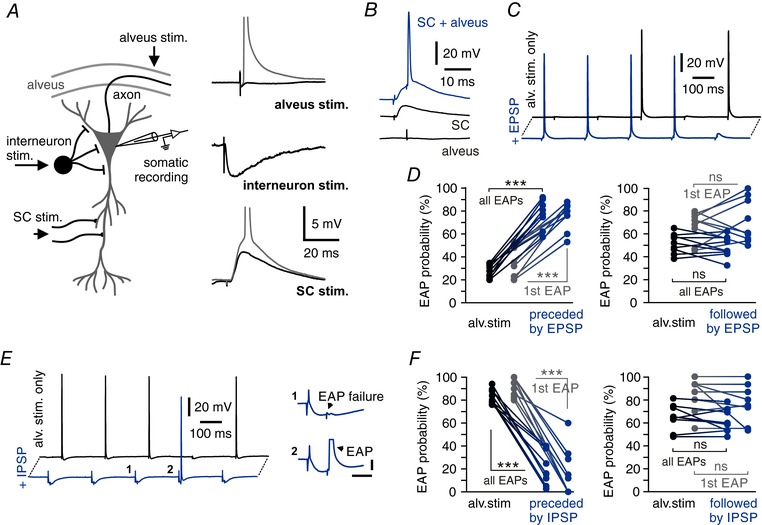
A, schematic illustration of recording and stimulating electrode positions. Stimulation electrodes (black arrows) activate either the alvear bundle (alveus), Schaffer collaterals (SC) or local interneurons in stratum pyramidale, respectively. Right: representative traces of subthreshold (black) and supra‐threshold responses (grey) to extracellular stimulations at the indicated locations. B, single EPSPs facilitate EAP generation. Paired stimulation of Schaffer collaterals and alvear fibres evoke EAPs (top, blue trace), but not the individual stimulations alone (bottom, black traces). C, EPSPs increase EAP firing probability. Black trace shows 5 ectopic stimulations of which two trigger an EAP. EPSPs preceding the ectopic stimulation by 10 ms increase EAP probability to 4 EAPs out of 5 stimulations (blue trace). D, left: EAP probability of ectopic stimulation alone and paired ectopic and preceding SC stimulation. Grey symbols indicate the EAP probability of the first ectopic stimulation of each 5 Hz train. Right: no facilitation of EAPs by EPSPs following the pulse by 10 ms. Grey symbols indicate EAP probability for the first ectopic stimulation of each 5 Hz train (n = 9 cells; *** P < 0.001; right panel: all EAPs P = 0.2570, first EAP P = 0.2498; ns = not significant; paired t test). E, IPSPs reduce EAP probability. Ectopic stimulation triggers 4 EAPs out of 5 stimulations (black trace). Single IPSPs preceding ectopic stimulation by 10 ms reduce EAP probability (blue trace). Insets show magnifications of an initiation failure and an EAP during a single IPSP, respectively. Horizontal bar: 10 ms, vertical bar: 5 mV. F, EAP probability is reduced for preceding IPSPs (left), but not for EAP stimulations followed by IPSPs (right, delay ±10 ms), compared to ectopic stimulation alone. Grey symbols indicate EAP probability only for the first ectopic stimulation of each 5 Hz train (n = 9 cells; *** P < 0.001; right panel: all EAPs P = 0.3257; first EAP P = 0.2189; ns = not significant; paired t test).
Figure 4. Repetitive synaptic input modulates axonal excitability via DC potential shifts.
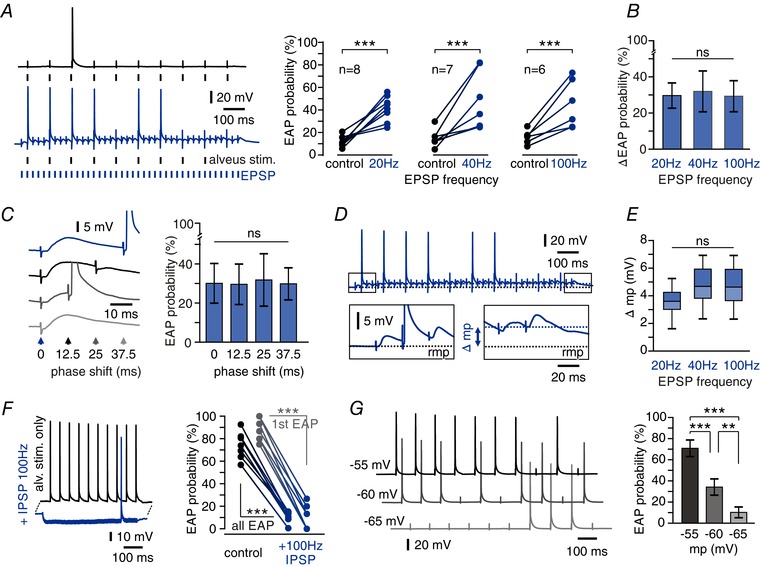
A, facilitation of EAP firing by repetitive synaptic stimulation. Left: ectopic stimulations (10 Hz) trigger more EAPs when paired with repetitive EPSP induction (40 Hz; blue trace) than alone (black trace). Right: EAP probability of ectopic stimulations alone and combined with repetitive EPSP induction at frequencies indicated. All frequencies exhibit robust facilitation of EAP firing (*** P < 0.001, paired t test). B, there is no significant difference in facilitation between synaptic stimulation at 20, 40 or 100 Hz (P = 0.9572, one‐way ANOVA test). C, a phase shift between synaptic and ectopic stimulation has no effect on EAP facilitation. Left: representative traces show synaptic stimulations (40 Hz) paired with ectopic stimulations (10 Hz) after varying delays as indicated by arrowheads. Right: EAP facilitation by EPSPs elicited with different phase relations to ectopic stimulation showed no significant differences (n = 6 cells, mean ± SD, P = 0.9980, one‐way ANOVA, ns = not significant). D, repetitive EPSPs caused long‐lasting membrane potential (mp) shifts. Boxes show expanded time scales of first and last stimulations. Repetitive stimulation produced a prolonged somatic depolarization reaching ∼5 mV between first and last stimulation (rmp, resting membrane potential). E, DC shift of membrane potential after orthodromic stimulation at different frequencies. No significant difference was found between stimulations at 20, 40 and 100 Hz (n = 7 cells, P = 0.0656, one‐way ANOVA). Box plots indicate median, 25th and 75th percentiles and 2.5th and 97.5th percentiles (whiskers). F, repetitive inhibitory input (100 Hz) hyperpolarizes CA1 pyramidal cells and strongly suppresses EAP generation. Left: example traces show 10 Hz ectopic stimulation yielding 10 EAPs under control conditions (black trace) and 1 EAP during repetitive induction of IPSPs (blue trace). Right: summary plot of EAP probability for control vs. 100 Hz IPSPs (black symbols: whole train, grey: first ectopic stimulation only; n = 6 cells, *** P < 0.001, paired t test). G, somatic membrane potentials directly modulate EAP probability. Left: traces show EAP induction at membrane potentials set to −55 mV (top), −60 mV (middle) and −65 mV (bottom) via somatic current injection. Right: mean EAP firing probability for the different membrane potentials (n = 9 cells, mean ± SD, *** P < 0.001, ** P < 0.01, one‐way ANOVA followed by Bonferroni's post hoc test).
Chemicals
Ionotropic glutamate‐receptor‐mediated neurotransmission was blocked by adding dl‐2‐amino‐5‐phosphonovaleric acid (APV; 30 μM) and 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX; 10 μM) to the bath solution where indicated. Bath application of muscimol (10 μM) was used to activate GABAA receptors in some experiments. All drugs were obtained from Tocris (Bristol, UK).
Statistical analysis
Data were analysed in Signal v. 4.10 (CED) or MATLAB (The MathWorks, Natick, MA, USA) using custom routines. Normally distributed data were tested by two‐tailed Student's t tests (two groups). Non‐normally distributed data were analysed using the non‐parametric Mann–Whitney test. For multiple comparisons, we used one‐way ANOVA followed by Bonferroni's post hoc test. Quantifications are given as mean ± standard deviation (SD). Box plots show median, 25% and 75% percentiles (boxes), as well as 2.5 and 97.5 percentiles (whiskers) of individual data sets. Significances are depicted as follows: not significant (ns); P < 0.05 (*); P < 0.01 (**); P < 0.001 (***). Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA) and Excel 2011 (Microsoft, Redmond, WA, USA).
Results
Signal propagation between soma and axon in CA1 pyramidal cells
We first investigated the axonal propagation of synaptic potentials using dual patch clamp recordings from somata and axons of CA1 pyramidal cells (Fig. 1 A). Somatic potential shifts did indeed invade distal axonal compartments, causing significant voltage deflections up to 358 μm away from the soma. This propagation was consistently observed for DC potentials following constant current injection to the soma, spontaneous postsynaptic potentials (PSPs) and artificial PSP‐like potentials created by brief current pulses applied to the soma (Fig. 1 B). The waveform and amplitude of somatic voltage changes had no influence on their ability to invade the axon, as all types of potentials showed the same amplitude ratio between soma and axon (Fig. 1 C). This included excitatory PSP‐like waveforms of different amplitudes with typical fast rise times (mean τrise(EPSP) = 1.7 ms; τrise(IPSP) = 5.7 ms). Surprisingly, signals were amplified by up to 50% at axonal recording sites close to the AIS (4 axons, distance 47–100 μm, median amplification +19.3%). At more remote sites, subthreshold depolarizing and hyperpolarizing potentials were attenuated but retained ∼80% of their somatic amplitude at distances between 106 and 205 μm (8 axons; median attenuation −18.9%; Fig. 1 D).
Figure 1. Synaptic potentials invade distal axonal compartments.
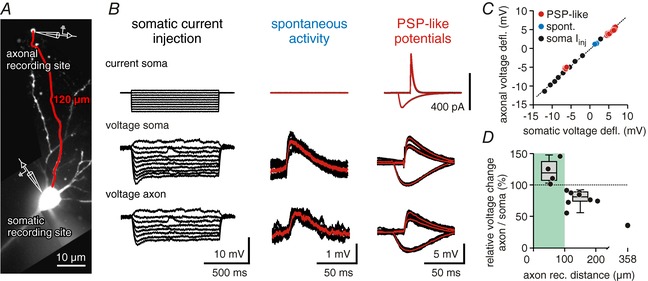
A, confocal image of a CA1 pyramidal cell used for dual soma/axon recordings (filled with Alexa 488). The axonal recording site was at a distance of 120 μm from the soma. B, somatic voltage deflections reach into distal axonal compartments. Traces show somatic current injection (top row), somatic voltage (middle row) and axonal voltage (bottom row). First column shows current/voltage relationships for somatic and axonal potentials, middle column shows spontaneous activity, and right column shows three postsynaptic‐potential like inputs induced by current injections to the soma (PSP‐like potentials). All traces were measured from the cell shown in A; average traces are shown in red. C, somatic vs. axonal voltage deflections for potentials shown in B. Hyperpolarizing and depolarizing potentials of different waveforms could be fitted with a linear function (dotted line). D, ratio of somatic to axonal voltage deflections vs. axonal recording distance. Within the first 100 μm, somatic potentials are amplified in the axon (median = +19.3%, left box plot; 4 cells). At soma–axon distances of 100–200 μm potentials in the axon retain ∼80% of the somatic amplitudes (median = −18.9%, right box plot; 8 cells).
We then studied signal propagation in the reverse direction, using the same recording configuration. We first elicited ectopic action potentials (EAPs) at distal axonal compartments. Somatic recordings showed different waveforms, characteristic for conventional orthodromic or ectopic action potentials, respectively. Orthodromic action potentials arose after a phase of somato‐dendritic depolarization until the firing threshold was reached at the AIS (Fig. 2 A, left panel). This pre‐depolarization was absent for EAPs which arose steeply from resting membrane potential in somatic recordings (Fig. 2 A, right panel) (Avoli et al. 1998; Hu et al. 2009 a; Bähner et al. 2011). The delay time of both orthodromic and ectopic action potentials showed strong linear correlations with the distance between soma and axonal recording site (simple linear regression, coefficient of determination r 2 ortho AP = 0.9471, r 2 EAP = 0.82241, respectively; Fig. 2 A and B). These data demonstrate that ectopically generated action potentials were triggered in close proximity to the recording pipette. All EAPs were detected first at the axonal recording site before appearing at the soma. Orthodromic action potentials were usually detected first at the soma and after a short delay at the axon. For very proximal axonal recordings (<100 μm away from the soma), action potentials could also occur near‐simultaneously, as the distance between AIS and each of the two recording sites was nearly identical. This is in line with the reported initiation site of orthodromic action potentials at the distal portion of the AIS (30–40 μm from the soma in cortical pyramidal cells) (Shu et al. 2006).
Figure 2. Ectopic action potentials reliably propagate back to the somatic compartment.
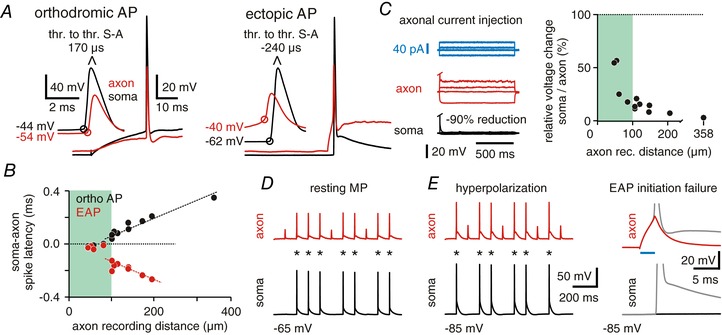
A, characteristics of orthodromic and ectopic action potentials (EAPs). Left panel: action potentials (APs) triggered by somatic current occur first in the soma and after a short delay at the axonal recording pipette; right panel: EAPs are first recorded in the axon and then at the somatic recording site where they arise directly from resting membrane potential. Both recordings are from the same cell, distance between somatic and axonal recording was 181 μm (voltage values indicate AP thresholds; resting membrane potential at soma and axon was −63 mV; thr. to thr.: interval between AP thresholds). B, scatter plot depicts latencies between somatic and axonal recordings of orthodromic and ectopic APs for all recordings. Note the near simultaneous occurrence of orthodromic APs at proximal recording distances, in line with spike initiation at the distal AIS. C, long current pulses (1 s) were injected into the axon through the patch pipette (blue trace). Resulting voltage deflections are very small in the soma (black trace, median = −85.6%, 13 cells) compared to the axon (red). Right panel: ratio of somatic to axonal voltage deflections vs. axonal recording distance. D, all EAPs evoked in the axon (red) reliably propagate back to the soma (black) at resting membrane potential (n = 2355 EAPs, 13 cells). Asterisks mark EAP induction. E, hyperpolarization of the soma induces no propagation failures (9 out of 10 cells). Asterisks mark EAP detection. Right: traces of EAPs (grey) and initiation failures taken from traces on the left (blue: duration of axonal current injection).
Thus, EAPs propagate towards the soma at similar velocities to orthodromic spikes. They may, however, be attenuated or blocked during antidromic propagation (Bucher & Goaillard, 2011; Debanne et al. 2011; Dugladze et al. 2012). We therefore tested how axonal subthreshold and supra‐threshold voltage transients are conducted towards the soma. Indeed, we found that the amplitude of axonal voltage deflections is strongly reduced in somatic recordings (13 cells, median attenuation −85.6%, Fig. 2 C). This amplitude loss is in line with the different capacitance of axonal and somatic compartments. Thin axonal fibres charge quickly, whereas the somatic membrane requires stronger current to yield similar voltage deflections. We then tested the propagation fidelity of EAPs, which were generated through the axonal pipette. In most cells (12/13), EAPs were recorded as full‐blown action potentials at the soma, indicating reliable propagation (Fig. 2 D, n = 2355 EAPs, 12 cells with axon recording distance <205 μm). In one very distal axonal recording (358 μm away from the soma) with three in‐between branch points, none of the EAPs propagated back to the soma, unless the cell was strongly depolarized (9 of 70 EAPs reached the soma following +10 mV somatic depolarization). In 12 of 13 cells, hyperpolarization of the soma to −80 mV had no effect on propagation fidelity (n = 480 EAPs, Fig. 2 E). In one cell, hyperpolarization caused propagation failures, and EAPs appeared as fast spikelets at the soma (134 EAPs, 57% failures, 111 μm recording distance). Finally, we tested the effect of GABA‐induced chloride conductance by bath application of the GABAA receptor agonist muscimol (10 μM). This led to a 9.2 ± 1.9 mV hyperpolarization of somatic membrane potential but had no effect on the propagation fidelity of EAPs, even when the soma was additionally hyperpolarized to −80 mV (n = 430 EAPs for muscimol at resting membrane potential, 6 cells; n = 268 EAPs for muscimol at −80 mV, 4 cells). Thus, EAP propagation failures appear to be rare under resting and hyperpolarized conditions in CA1 pyramidal cell axons up to 200 μm from the soma.
Synaptic input modulates EAP generation
We demonstrated that synaptic potentials invade the distal axon and that EAPs propagate reliably to the soma. To study the interaction between synaptic potentials and EAPs we then paired ectopic stimulation with activation of excitatory or inhibitory synaptic input (Fig. 3 A).
Extracellular stimulation of Schaffer collaterals in stratum radiatum efficiently evoked excitatory postsynaptic potentials (EPSPs). We paired low‐intensity, subthreshold stimulation of the axon with subthreshold EPSPs to test for possible interactions. Indeed, combined stimulation at both sites resulted in the generation of full‐blown EAPs (Fig. 3 B). EPSP‐facilitated EAPs exhibited the same time delay as EAPs evoked solely by stronger axonal stimulation (∼2 ms delay measured from stimulation onset). These action potentials arose from the peak of the somatic EPSP but their threshold was consistently more negative than that of orthodromic action potentials. EPSPs were elicited in trains of five stimuli at 5 Hz. When each EPSP preceded the axonal stimulation by 10 ms, generation of EAPs was significantly facilitated (27.6 ± 5.8% vs. 77.4 ± 11.7%, Fig. 3 C, blue trace, and Fig. 3 D). To control for adaptation, we analysed the first stimulation pulse of each train in separation and found a comparable EAP facilitation (36.7 ± 13.6% to 75.8 ± 11.7%; Fig. 3 D, grey symbols). Conversely, when ectopic stimulation preceded the EPSP by 10 ms, the EAP firing probability remained constant (Fig. 3 D, right panel). Thus, single EPSPs facilitate EAP generation in remote axonal locations when there is enough time for propagation towards the EAP initiation site, in line with predictions from computational studies (Vladimirov et al. 2013).
Ectopic firing has been observed during different hippocampal network oscillations (Traub et al. 2003; Bähner et al. 2011; Dugladze et al. 2012). Such patterns involve the precise timing of synaptic inputs, with a major synchronizing role of inhibition (Klausberger & Somogyi, 2008). We therefore tested whether inhibitory postsynaptic potentials (IPSPs) can modulate the firing pattern of EAPs. Inhibitory potentials were generated by local stimulation in stratum pyramidale of CA1 after blocking glutamatergic transmission via bath application of 10 μM CNQX and 30 μM APV (Fig. 3 A). EAPs were evoked by extracellular stimulation of the alvear fibre tract. IPSPs preceding ectopic stimulation by 10 ms significantly reduced the probability of EAP generation from 82.8 ± 6.4% to 21.3 ± 14.6% (Fig. 3 E, blue trace, and Fig. 3 F). The attenuation of EAP probability was already present for the first pulse in a 5 Hz train (92.5 ± 8.3% to 27.6 ± 33.6%; Fig. 3 F, grey symbols) and was absent when IPSPs were evoked 10 ms after ectopic stimulation (Fig. 3 F, right panel). In line with our findings using dual soma/axon recordings, these data suggest that IPSPs propagate into distal axonal compartments and locally modulate the initiation of EAPs.
Modulation of EAP initiation by somatic and axonal membrane potentials
Excitatory inputs often hit CA1 pyramidal cells repetitively, especially during network oscillations. To mimic these patterns, we evoked different series of EPSP trains at frequencies of 20, 40 and 100 Hz, while ectopic stimulations were always applied at 10 Hz (Fig. 4 A, bottom trace). As observed for single EPSPs, pairing of excitatory synaptic input and ectopic stimulation increased the number of EAPs (Fig. 4 A, bottom trace). This facilitation occurred already at the lowest frequency tested (20 Hz, Fig. 4 A right panel), and higher EPSP frequencies did not lead to further increase of EAP facilitation (Fig. 4 A and B). As synaptic integration in CA1 pyramidal cells is strongly dependent on the timing of converging inputs, we tested different delays between ectopic axonal and synaptic stimulation (Fig. 4 C, left panel). However, we found no correlation between EPSP timing and facilitation of ectopic spiking (Fig. 4 C, right panel).
Thus, repetitive synaptic stimulation altered EAP probability at all frequencies tested and independent of the stimulus phase. We therefore hypothesized that the effect was mainly due to a DC shift in somatic membrane potential which then propagates towards the axonal initiation site. Indeed, repetitive stimulation of excitatory afferents caused a steady‐state depolarization of the somatic membrane by ∼5 mV (Fig. 4 D), independent of stimulation frequency (20, 40 and 100 Hz; Fig. 4 E). Likewise, repetitive inhibitory input produced a sustained hyperpolarization that efficiently suppressed ectopic spiking in a voltage‐dependent manner (Fig. 4 F). We therefore tested the effect of changes in somatic membrane potential on EAP generation more directly by injecting constant depolarizing or hyperpolarizing currents through the recording pipette. Somatic depolarization to −55 mV caused a significant increase in firing probability by ∼70%, while hyperpolarization to −65 mV, on the other hand, reduced EAP firing efficiency to ∼10% (Fig. 4 G). In close agreement, dual soma/axon recordings showed a similar effect of hyperpolarization on EAP initiation (Fig. 5 A and B, compare Fig. 1 A–D). In these experiments, hyperpolarization of the soma by current injection led to hyperpolarization of the axon and reduced EAP induction in a voltage‐dependent manner (Fig. 5 A). Axonal current injection, adjusted to match the same negative potential difference evoked by somatic current injections, had the same effect on EAP initiation (Fig. 5 B).
Figure 5. Modulation of EAP initiation by somatic membrane potentials depends on local changes in axonal voltage.
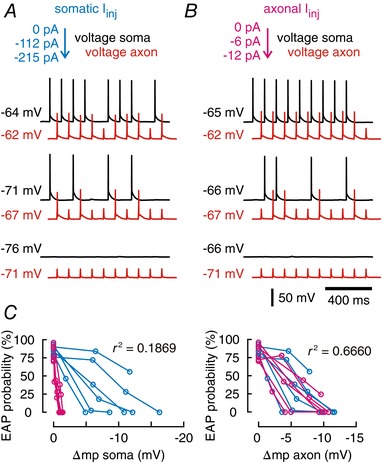
A, example traces for near‐threshold ectopic stimulation through axonal patch pipette (10 Hz, 3 ms) at resting membrane potential (top traces, black: somatic recording, red: axonal recording) vs. more hyperpolarized potentials (trace pairs below). Somatic hyperpolarization was controlled by current injections through the somatic pipette, while the axon was recorded current free. Soma–axon distance was 146 μm for this recording. B, the axon is hyperpolarized to the same axonal potentials as recorded in A by axonal current injection (black: somatic recording, red: axonal recording). The somatic potential was recorded current free (compare Fig. 2 C). C, mean EAP firing probability vs. change of somatic membrane potential (Δmp; left panel) and vs. axonal membrane potential (right panel). Data from experiments shown in A (magenta, somatic current injection) and B (cyan, axonal current injection; n = 6 cells).
When we correlated EAP initiation probability with somatic and axonal membrane potentials, we found that the axonal voltage constitutes a significantly better predictor of ectopic firing (r 2 axon = 0.6660) than somatic potentials (r 2 soma = 0.1869, n = 5 cells, axon distance = 106–205 μm; Fig. 5 C). We conclude that synaptic activity modulates ectopic activity by propagating far into the axon and locally changing the axonal membrane potential directly at the EAP initiation site.
Discussion
The main task of axons is the fast and reliable transmission of action potentials from the axon initial segment to all synaptic terminals. For this, axons carry a high density of voltage‐gated sodium and potassium channels which, together with their low capacitance, make them highly excitable. However, these properties enable the generation of action potentials at distal sites along the axonal fibre, i.e. ectopic action potentials. Such EAPs have been observed in various physiological and pathological conditions, but their origin and functional relevance is not entirely clear. One key question is whether EAPs are affected by somato‐dendritic signals and are thereby entrained by synaptic activity within the surrounding network. We report that somatic potential changes propagate deep into the axons of CA1 pyramidal cells where they strongly modulate axonal excitability. Excitatory synaptic input facilitates and inhibitory input suppresses the generation of ectopic action potentials. We conclude that ectopic activity can be modulated by chemical synaptic transmission and, thus, EAPs can be controlled by and integrated into the regular information flow during patterned activity in neuronal networks.
CA1 pyramidal cells constitute an ideal model to study orthodromic and antidromic signal interaction due to the clear anatomical separation of input and output domains. Defined excitatory pathways reach apical and basal dendrites, whereas the perisomatic region is targeted by strong inhibitory synapses. Axons run straight across stratum oriens into the alveus where they form an almost cell‐free fibre bundle (Cajal, 1911; Amaral & Lavenex, 2006). This separation minimizes co‐activation of afferent and efferent fibres by extracellular stimulation. As an even more unambiguous approach, we used the recently developed technique of dual somatic and axonal recordings, yielding direct access to both compartments within a single cell (Bischofberger et al. 2006; Shu et al. 2006). Direct recordings from axons have the limitation that they are mainly performed on unmyelinated sections that are accessible for the patch pipette. This was also the case in our recordings which were done in the unmyelinated axon section within stratum oriens. However, spontaneous EAPs were mostly described for un‐ or de‐myelinated fibres (Pinault, 1995; Hamada & Kole, 2015) and our results were in full accordance with data gained by extracellular stimulation in the alveus, indicating that the modulation of axonal excitability by somatic voltage changes does go even beyond the range tested.
Our data extend previous observations showing efficient propagation of somato‐dendritic signals into the axon or reliable backpropagation of EAPs. Such mechanisms have been reported for layer 5 neocortical pyramidal cells (Shu et al. 2006; Kole et al. 2007), hippocampal CA3 pyramidal cells (Sasaki et al. 2011; Kim et al. 2012) and dentate granule cells (Alle & Geiger, 2006; Schmidt‐Hieber et al. 2008). Our results show that somatic potential changes in CA1 pyramidal cells cause reliable potential changes in the axon, with little or no attenuation within the first 200 μm. This voltage transfer might be particularly efficient in axons emanating from a dendrite, rather than from the soma, as recently observed in ∼50% of CA1 pyramidal cells (Thome et al. 2014). Voltage transients were even amplified in the proximal axon (<100 μm), both for depolarizing and hyperpolarizing potentials. This is presumably caused by either positive or negative modulation of voltage‐gated sodium channels, which has been described for EPSPs and IPSPs in neocortical pyramidal neurons (Stuart & Sakmann, 1995; Stuart, 1999). It was suggested that the high sodium channel density at the AIS locally enhances the effect of channel modulation resulting in larger potentials in the proximal axon compared to the soma.
We found a linear relationship between axonal recording distance and temporal delay of EAPs between axonal and somatic recording sites. We therefore assume that EAPs were elicited very close to the axonal patch pipette, indicating that CA1 pyramidal cell axons are excitable at many sites (or continuously) along the first few hundred micrometres. When stimulating extracellularly, activation of axons could be more restricted to Na+ channel hot spots, e.g. nodes of Ranvier, which exhibit a high concentration of voltage‐gated Na+ and K+ channels (Poliak & Peles, 2003). In these experiments, we only used recordings with <2 ms delay time between stimulation onset and somatic EAPs, similar to the values found in dual soma/axon recordings. Again, this restriction makes us confident that stimulation site and the point of EAP initiation were not far apart.
Propagation vs. initiation failures
We found that EAPs propagate very reliably to the soma in CA1 pyramidal cells. This might seem unexpected considering previous observations in CA3 where axonal firing appeared largely independent from the somatic occurrence of action potentials during gamma oscillations in vitro (Dugladze et al. 2012). The latter observation has been explained by the strong activation of axo‐axonic inhibitory synapses during gamma network oscillations. In our recordings, however, propagation of EAPs was not impaired by bath application of the GABAA receptor agonist muscimol. It is thus feasible that ectopic spikes invade the soma of CA1 pyramidal cells more readily than in CA3. Nevertheless, we did see propagation failures for one very distal axonal recording (358 μm from the soma). It remains unclear whether strong activation of axo‐axonic inhibition in CA1 pyramidal cells can increase the failure rate of somatic EAP invasion. Under in vitro drug‐free conditions, however, spike transfer from the axon to the soma seems to be very stable.
When stimulating with extracellular stimulation electrodes at more remote sites, propagation failures may contribute to EAP loss between axon and soma, e.g. as a consequence of impedance mismatch between axon shafts and large en passant boutons or branch points (Debanne, 2004). However, axons of CA1 pyramidal cells have no boutons or large varicosities and generally show little arborization in the CA1 region (1–3 branch points, median = 1, n = 14 cells). Other possible causes for propagation failures may result from inactivation of Na+ channels. High‐frequency firing (30–40 Hz) causes frequency‐dependent propagation failures in CA3 pyramidal cell axons (Meeks & Mennerick, 2007), but there is considerable variability of action potential propagation fidelity amongst different cell types (Monsivais et al. 2005; Ritzau‐Jost et al. 2014). To prevent these effects, we used stimulation frequencies of 5–10 Hz in our experiments, outside the range of reported frequency‐dependent propagation failures. Together with the findings from dual soma/axon recordings, we conclude that somatic voltage modulates EAP generation, rather than propagation.
Functional implications
Is the observed crosstalk between synaptic input and ectopic signalling relevant for hippocampal network activity and information processing? Several studies revealed the occurrence of EAPs within the distal axon of hippocampal neurons (Avoli et al. 1998; Bähner et al. 2011; Sheffield et al. 2011). They may occur in different functional states of the network in vitro (Bähner et al. 2011; Dugladze et al. 2012) or in vivo (Harvey et al. 2009; Epsztein et al. 2010; Chorev & Brecht, 2012). Synaptic modulation of ectopic firing may therefore contribute to the high (sub‐millisecond) precision of action potential timing during fast network oscillations (Ellender et al. 2010; Bähner et al. 2011; Dugladze et al. 2012), which is in contrast to the notorious lack of temporal precision in EPSP‐to‐action‐potential coupling of pyramidal cells (Fricker & Miles, 2000). Axonal EAP generation and inter‐axonal signal propagation (Traub et al. 2012) may act together with the well‐established mechanism of network coordination by fast‐spiking inhibitory interneurons (Whittington & Traub, 2003; Mann & Paulsen, 2007; Buzsáki & Wang, 2012; English et al. 2014) to generate precisely timed fast network oscillations in the gamma and ripple frequency domain.
Notably, previous work has established another important impact of EAPs on hippocampal function: backpropagating ectopic spikes induce associative plasticity at dendritic excitatory synapses (Bukalo et al. 2013) and cause long‐term potentiation of EPSP‐to‐action‐potential coupling, i.e. increased spiking probability for a given EPSP amplitude (Jester et al. 1995). Thus, interactions between dendritic synaptic inputs and axonal firing probability are bi‐directional.
Under pathological conditions, EAP facilitation by excitatory synaptic activity might further aggravate neuronal hyperactivity and epileptic network discharges. However, synaptic control of EAP generation is likely to play a role in both excitatory and inhibitory neurons. Certain interneurons respond to strong excitatory input with prolonged antidromic spiking which further spreads ectopically to neighbouring inhibitory cells (Connors & Ahmed, 2011). Their activity might suppress faulty EAP generation and propagation out of the pathologically affected local network. This mechanism might then dampen epileptic activity and regulate EAP firing when seizures and hyperactivity threaten the network.
In summary, we report that synaptic activity of CA1 pyramidal cells travels far along the axon where it modulates EAP initiation. This mechanism links ectopic firing with regular synaptic input. Such interactions may be important for understanding the mechanisms underlying fast network oscillations (Dugladze et al. 2012; Traub et al. 2012; Vladimirov et al. 2013) and pathological network activity (Avoli et al. 1998) and challenge the view of the distal axon being independent of synaptic activity.
Additional information
Competing interests
None declared.
Authors contributions
C.T., A.D. and A.V.E. designed the research; C.T., A.Y. and J.O. performed experiments in Heidelberg and F.C.R. in Oslo; C.T. and J.O. analysed data. C.T., A.V.E. and A.D. wrote the article. F.C.R., J.O. and A.Y. contributed to drafting the article and revising it critically for important intellectual content. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DR 326/12‐1, to C.T. and A.D.; SFB1134 projects A01 and A03, to C.T., A.D. and A.V.E.), Deutscher Akademischer Austauschdienst (DAAD, PPP‐Norway, 57402121, to C.T., F.C.R., A.D. and A.V.E.), the Norwegian Research Council (NFR, IS‐DAAD, 281252; FRIMEDBIO, 250886, to F.C.R.) and the European Union's seventh framework programme (FP7‐PEOPLE‐2013‐COFUND, 60902 to F.C.R.) – Scientia Fellows.
Acknowledgements
We thank Dr Hua Hu (University of Oslo) for his support and helpful comments on the manuscript.
Biography
Christian Thome studied Biology and Philosophy at the Universities of Heidelberg and Glasgow. He specialized in the areas of Neuroscience and Philosophy of Mind and earned his PhD with pioneering work on the prevalence, anatomy and physiology of dendritic axon origins in hippocampal neurons. Currently, he is working on the development, plasticity and functional properties of non‐canonical axon morphologies at the Institute of Physiology and Pathophysiology at Heidelberg University.

Edited by: Ole Paulsen & Diego Contreras
Linked articles This article is highlighted in a Perspectives article by Fékété & Debanne. To read this article, visit https://doi.org/10.1113/JP277012.
References
- Alle H & Geiger JRP (2006). Combined analog and action potential coding in hippocampal mossy fibers. Science 311, 1290–1293. [DOI] [PubMed] [Google Scholar]
- Amaral D & Lavenex P (2006). Chapter 3. Hippocampal neuroanatomy In The Hippocampus Book, ed. Andersen P, Morris R, Amaral D, Bliss T & O'Keefe J. Oxford University Press, New York, Oxford. [Google Scholar]
- Apostolides PF, Milstein AD, Grienberger C, Bittner KC & Magee JC (2016). Axonal filtering allows reliable output during dendritic plateau‐driven complex spiking in CA1 neurons. Neuron 89, 770–783. [DOI] [PubMed] [Google Scholar]
- Avoli M, Methot M & Kawasaki H (1998). GABA‐dependent generation of ectopic action potentials in the rat hippocampus. Eur J Neurosci 10, 2714–2722. [DOI] [PubMed] [Google Scholar]
- Bähner F, Weiss EK, Birke G, Maier N, Schmitz D, Rudolph U, Frotscher M, Traub RD, Both M & Draguhn A (2011). Cellular correlate of assembly formation in oscillating hippocampal networks in vitro. Proc Natl Acad Sci U S A 108, E607–E616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranauskas G, David Y & Fleidervish IA (2013). Spatial mismatch between the Na+ flux and spike initiation in axon initial segment. Proc Natl Acad Sci U S A 110, 4051–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Li L, Geiger JR & Jonas P (2006). Patch‐clamp recording from mossy fiber terminals in hippocampal slices. Nat Protoc 1, 2075–2081. [DOI] [PubMed] [Google Scholar]
- Bucher D & Goaillard JM (2011). Beyond faithful conduction: Short‐term dynamics, neuromodulation, and long‐term regulation of spike propagation in the axon. Prog Neurobiol 94, 307–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukalo O, Campanac E, Hoffman DA & Fields RD (2013). Synaptic plasticity by antidromic firing during hippocampal network oscillations. Proc Natl Acad Sci U S A 110, 5175–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G & Wang X‐J (2012). Mechanisms of gamma oscillations. Annu Rev Neurosci 35, 203–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajal SRY (1911). Histologie du système nerveux de l'homme et des vertébrés. A. Maloine, Paris. [Google Scholar]
- Chorev E & Brecht M (2012). In vivo dual intra‐ and extracellular recordings suggest bidirectional coupling between CA1 pyramidal neurons. J Neurophysiol 108, 1584–1593. [DOI] [PubMed] [Google Scholar]
- Colbert CM & Johnston D (1996). Axonal action‐potential initiation and Na+ channel densities in the soma and axon initial segment of subicular pyramidal neurons. J Neurosci 16, 6676–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors BW & Ahmed OJ (2011). Integration and autonomy in axons. Nat Neurosci 14, 128–130. [DOI] [PubMed] [Google Scholar]
- Debanne D (2004). Information processing in the axon. Nat Rev Neurosci 5, 304–316. [DOI] [PubMed] [Google Scholar]
- Debanne D, Campanac E, Bialowas A & Carlier E (2011). Axon physiology. Physiol Rev 91, 555–602. [DOI] [PubMed] [Google Scholar]
- Dugladze T, Schmitz D, Whittington MA, Vida I & Gloveli T (2012). Segregation of axonal and somatic activity during fast network oscillations. Science 336, 1458–1461. [DOI] [PubMed] [Google Scholar]
- Ellender TJ, Nissen W, Colgin LL, Mann EO & Paulsen O (2010). Priming of hippocampal population bursts by individual perisomatic‐targeting interneurons. J Neurosci 30, 5979–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English DF, Peyrache A, Stark E, Roux L, Vallentin D, Long MA & Buzsaki G (2014). Excitation and inhibition compete to control spiking during hippocampal ripples: intracellular study in behaving mice. J Neurosci 34, 16509–16517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztein J, Lee AK, Chorev E & Brecht M (2010). Impact of spikelets on hippocampal CA1 pyramidal cell activity during spatial exploration. Science 327, 474–477. [DOI] [PubMed] [Google Scholar]
- Fricker D & Miles R (2000). EPSP amplification and the precision of spike timing in hippocampal neurons. Neuron 28, 559–569. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutnick MJ & Prince DA (1972). Thalamocortical relay neurons: antidromic invasion of spikes from a cortical epileptogenic focus. Science 176, 424–426. [DOI] [PubMed] [Google Scholar]
- Hamada MS & Kole MHP (2015). Myelin loss and axonal ion channel adaptations associated with gray matter neuronal hyperexcitability. J Neurosci 35, 7272–7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Collman F, Dombeck DA & Tank DW (2009). Intracellular dynamics of hippocampal place cells during virtual navigation. Nature 461, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusser M, Stuart G, Racca C & Sakmann B (1995). Axonal initiation and active dendritic propagation of action potentials in substantia nigra neurons. Neuron 15, 637–647. [DOI] [PubMed] [Google Scholar]
- Hoffmann T, Sauer SK, Horch RE & Reeh PW (2008). Sensory transduction in peripheral nerve axons elicits ectopic action potentials. J Neurosci 28, 6281–6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H & Jonas P (2014). A supercritical density of Na+ channels ensures fast signaling in GABAergic interneuron axons. Nat Neurosci 17, 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Graham LJ & Storm JF (2009. a). Complementary theta resonance filtering by two spatially segregated mechanisms in CA1 hippocampal pyramidal neurons. J Neurosci 29, 14472–14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H & Shu Y (2009. b). Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nat Neurosci 12, 996–1002. [DOI] [PubMed] [Google Scholar]
- Jester JM, Campbell LW & Sejnowski TJ (1995). Associative EPSP–spike potentiation induced by pairing orthodromic and antidromic stimulation in rat hippocampal slices. J Physiol 484, 689–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Guzman SJ, Hu H & Jonas P (2012). Active dendrites support efficient initiation of dendritic spikes in hippocampal CA3 pyramidal neurons. Nat Neurosci 15, 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T & Somogyi P (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MHP, Letzkus JJ & Stuart GJ (2007). Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron 55, 633–647. [DOI] [PubMed] [Google Scholar]
- Mann EO & Paulsen O (2007). Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci 30, 343–349. [DOI] [PubMed] [Google Scholar]
- Meeks JP & Mennerick S (2007). Action potential initiation and propagation in CA3 pyramidal axons. J Neurophysiol 97, 3460–3472. [DOI] [PubMed] [Google Scholar]
- Moalem G, Grafe P & Tracey DJ (2005). Chemical mediators enhance the excitability of unmyelinated sensory axons in normal and injured peripheral nerve of the rat. Neuroscience 134, 1399–1411. [DOI] [PubMed] [Google Scholar]
- Monsivais P, Clark BA, Roth A & Häusser M (2005). Determinants of action potential propagation in cerebellar Purkinje cell axons. J Neurosci 25, 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM & Stuart GJ (2006). Site of action potential initiation in layer 5 pyramidal neurons. J Neurosci 26, 1854–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D (1995). Backpropagation of action potentials generated at ectopic axonal loci: hypothesis that axon terminals integrate local environmental signals. Brain Res Brain Res Rev 21, 42–92. [DOI] [PubMed] [Google Scholar]
- Poliak S & Peles E (2003). The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci 4, 968–980. [DOI] [PubMed] [Google Scholar]
- Popovic MA, Foust AJ, McCormick DA & Zecevic D (2011). The spatio‐temporal characteristics of action potential initiation in layer 5 pyramidal neurons: a voltage imaging study. J Physiol 589, 4167–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy S, Csicsvari J & Beck H (2009). Activity‐dependent control of neuronal output by local and global dendritic spike attenuation. Neuron 61, 906–916. [DOI] [PubMed] [Google Scholar]
- Ritzau‐Jost A, Delvendahl I, Rings A, Byczkowicz N, Harada H, Shigemoto R, Hirrlinger J, Eilers J & Hallermann S (2014). Ultrafast action potentials mediate kilohertz signaling at a central synapse. Neuron 84, 152–163. [DOI] [PubMed] [Google Scholar]
- Rosen AD & Vastola EF (1971). Corticofugal antidromic activity in epileptogenic foci. Trans Am Neurol Assoc 96, 297–298. [PubMed] [Google Scholar]
- Sasaki T (2013). The axon as a unique computational unit in neurons. Neurosci Res 75, 83–88. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Matsuki N & Ikegaya Y (2011). Action‐potential modulation during axonal conduction. Science 331, 599–601. [DOI] [PubMed] [Google Scholar]
- Schmidt‐Hieber C, Jonas P & Bischofberger J (2008). Action potential initiation and propagation in hippocampal mossy fibre axons. J Physiol 586, 1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield MEJ, Best TK, Mensh BD, Kath WL & Spruston N (2011). Slow integration leads to persistent action potential firing in distal axons of coupled interneurons. Nat Neurosci 14, 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield MEJ, Edgerton GB, Heuermann RJ, Deemyad T, Mensh BD & Spruston N (2013). Mechanisms of retroaxonal barrage firing in hippocampal interneurons. J Physiol 591, 4793–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Duque A, Yu Y, Haider B & McCormick DA (2007). Properties of action‐potential initiation in neocortical pyramidal cells: evidence from whole cell axon recordings. J Neurophysiol 97, 746–760. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y & McCormick DA (2006). Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature 441, 761–765. [DOI] [PubMed] [Google Scholar]
- Stuart G (1999). Voltage‐activated sodium channels amplify inhibition in neocortical pyramidal neurons. Nat Neurosci 2, 144–150. [DOI] [PubMed] [Google Scholar]
- Stuart G & Sakmann B (1995). Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron 15, 1065–1076. [DOI] [PubMed] [Google Scholar]
- Stuart G, Schiller J & Sakmann B (1997). Action potential initiation and propagation in rat neocortical pyramidal neurons. J Physiol 505, 617–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome C, Kelly T, Yanez A, Schultz C, Engelhardt M, Cambridge SB, Both M, Draguhn A, Beck H & Egorov AV (2014). Axon‐carrying dendrites convey privileged synaptic input in hippocampal neurons. Neuron 83, 1418–1430. [DOI] [PubMed] [Google Scholar]
- Traub RD, Cunningham MO, Gloveli T, LeBeau FEN, Bibbig A, Buhl EH & Whittington MA (2003). GABA‐enhanced collective behavior in neuronal axons underlies persistent gamma‐frequency oscillations. Proc Natl Acad Sci U S A 100, 11047–11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Schmitz D, Maier N, Whittington MA & Draguhn A (2012). Axonal properties determine somatic firing in a model of in vitro CA1 hippocampal sharp wave/ripples and persistent gamma oscillations. Eur J Neurosci 36, 2650–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladimirov N, Tu Y & Traub RD (2013). Synaptic gating at axonal branches, and sharp‐wave ripples with replay: a simulation study. Eur J Neurosci 38, 3435–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington MA & Traub RD (2003). Interneuron diversity series: Inhibitory interneurons and network oscillations in vitro. Trends Neurosci 26, 676–682. [DOI] [PubMed] [Google Scholar]
- Wong RK, Prince DA & Basbaum AI (1979). Intradendritic recordings from hippocampal neurons. Proc Natl Acad Sci U S A 76, 986–990. [DOI] [PMC free article] [PubMed] [Google Scholar]