

Abstract
Key points
We developed a novel method to study sympatholysis in dogs.
We showed abolishment of sarcolemmal nNOS, and reduction of total nNOS and total eNOS in the canine Duchenne muscular dystrophy (DMD) model.
We showed sympatholysis in dogs involving both nNOS‐derived NO‐dependent and NO‐independent mechanisms.
We showed that the loss of sarcolemmal nNOS compromised sympatholysis in the canine DMD model.
We showed that NO‐independent sympatholysis was not affected in the canine DMD model.
Abstract
The absence of dystrophin in Duchenne muscular dystrophy (DMD) leads to the delocalization of neuronal nitric oxide synthase (nNOS) from the sarcolemma. Sarcolemmal nNOS plays an important role in sympatholysis, a process of attenuating reflex sympathetic vasoconstriction during exercise to ensure blood perfusion in working muscle. Delocalization of nNOS compromises sympatholysis resulting in functional ischaemia and muscle damage in DMD patients and mouse models. Little is known about the contribution of membrane‐associated nNOS to blood flow regulation in dystrophin‐deficient DMD dogs. We tested the hypothesis that the loss of sarcolemmal nNOS abolishes protective sympatholysis in contracting muscle of affected dogs. Haemodynamic responses to noradrenaline in the brachial artery were evaluated at rest and during contraction in the absence and presence of NOS inhibitors. We found sympatholysis was significantly compromised in DMD dogs, as well as in normal dogs treated with a selective nNOS inhibitor, suggesting that the absence of sarcolemmal nNOS underlies defective sympatholysis in the canine DMD model. Surprisingly, inhibition of all NOS isoforms did not completely abolish sympatholysis in normal dogs, suggesting sympatholysis in canine muscle also involves NO‐independent mechanism(s). Our study established a foundation for using the dog model to test therapies aimed at restoring nNOS homeostasis in DMD.
Keywords: Duchenne muscular dystrophy, Canine model, Functional sympatholysis, Nitric oxide synthase, noradrenaline, vasoconstriction, Acetylcholine, Vasodilatation
Key points
We developed a novel method to study sympatholysis in dogs.
We showed abolishment of sarcolemmal nNOS, and reduction of total nNOS and total eNOS in the canine Duchenne muscular dystrophy (DMD) model.
We showed sympatholysis in dogs involving both nNOS‐derived NO‐dependent and NO‐independent mechanisms.
We showed that the loss of sarcolemmal nNOS compromised sympatholysis in the canine DMD model.
We showed that NO‐independent sympatholysis was not affected in the canine DMD model.
Introduction
Duchenne muscular dystrophy (DMD) is the most common form of inherited muscle diseases in childhood, affecting around 1 in 5000 boys (Mendell et al. 2012). DMD is caused by null mutations in the dystrophin gene. The dystrophin gene encodes a critical structural protein that connects the actin cytoskeleton to the extracellular matrix (Hoffman et al. 1987). In normal muscle, this structural linkage protects muscle from mechanical stress‐induced damage by dissipating contractile force from the cytoskeleton to the extracellular matrix (Ervasti & Campbell, 1993). Absence of dystrophin abolishes this structural link. As a consequence, the muscle undergoes degeneration and necrosis (Goldstein & McNally, 2010; Allen et al. 2016).
Dystrophin assembles the membrane‐spanning dystrophin‐associated glycoprotein complex (DGC). The DGC contains a variety of intracellular, transmembrane and extracellular proteins. An important component of the skeletal muscle DGC is calcium/calmodulin‐dependent neuronal nitric oxide synthase μ (nNOSμ). During muscle contraction, increased cytosolic calcium activates nNOSμ to produce nitric oxide (NO). NO is a highly diffusible signalling molecule involved in many crucial biological processes in muscle (e.g. metabolism and mitochondrial bioenergetics) (Stamler & Meissner, 2001). NO‐mediated blood flow regulation is of particular importance to contracting muscle. Specifically, NO attenuates sympathetic vasoconstriction in exercising muscles via a cGMP‐mediated mechanism (Vo et al. 1992; Archer et al. 1994; Ishibashi et al. 1997). This process, termed functional sympatholysis, is required for cardiac output redistribution, ensuring adequate perfusion when a large muscle mass is active (Remensnyder et al. 1962). nNOSμ is anchored to the sarcolemma via interactions with the adaptor protein α‐syntrophin and spectrin‐like repeats 16 and 17 in the dystrophin rod domain (Adams et al. 2001; Lai et al. 2009, 2013). This sarcolemmal localization is critical for the normal function of nNOS as it ensures rapid diffusion of short‐lived NO to nearby blood vessels (Thomas et al. 2003; Lai et al. 2009). In the absence of dystrophin, nNOSμ delocalizes from the membrane and nNOS protein content is substantially reduced in dystrophic muscle, resulting in impaired nNOS activity and NO production (Thomas et al. 1998; Sander et al. 2000; Kasai et al. 2004). Disruption of nNOS homeostasis leads to unopposed sympathetic vasoconstriction during exercise, hence functional ischaemia and exercise‐induced fatigue in DMD patients (Kobayashi et al. 2008; Angelini & Tasca, 2012).
In addition to nNOS, NO can also be generated from constitutively expressed endothelial NOS (eNOS) and inducible NOS (iNOS). Similar to nNOS, eNOS also plays a role in the regulation of vascular tone (Lau et al. 2000; Duplain et al. 2001), muscle metabolism (Lee‐Young et al. 2010) and mitochondrial biogenesis and function (Le Gouill et al. 2007). It is currently unclear whether eNOS is involved in sympatholysis. While some studies suggest that eNOS‐derived NO may be involved in regulating blood flow to active muscles (Grange et al. 2001; Jendzjowsky & DeLorey, 2013), other studies suggest eNOS‐derived NO may not play an appreciable role in NO‐mediated attenuation of sympathetic vasoconstriction (Thomas & Victor, 1998; Thomas et al. 1998; Sato et al. 2008).
Dystrophin‐deficient dogs (referred to as DMD dogs in this article), similar to human DMD patients, display more severe clinical features than do dystrophin‐deficient mice (Valentine et al. 1988; Duan, 2015; McGreevy et al. 2015; Kornegay, 2017). Preclinical studies in the canine DMD model may thus provide a better understanding of DMD pathogenesis and a better indication of whether an experimental therapy can translate to human patients (Duan, 2011, 2015). The canine DMD model has been established since 1988 (Cooper et al. 1988) and DMD dogs have been used in numerous studies to evaluate pharmacological and genetic therapies for DMD. Yet, there has been no study on functional ischaemia in DMD dogs. To fill this knowledge gap, we developed a novel assay to quantify limb muscle blood flow in resting and contracting dog muscle. We applied our method to a large cohort of normal and DMD dogs. In conjunction with pharmacological inhibition of nNOS or all NOS isoforms, we profiled muscle perfusion, functional ischaemia and sympatholysis. We found that noradrenaline (norepinephrine, NE)‐induced vasoconstriction was significantly blunted in contracting muscles of both normal and DMD dogs. However, as a consequence of sarcolemmal nNOS‐derived NO, the sympatholysis response was significantly more pronounced in normal dogs.
Methods
Ethical approval
All animal experiments were approved by the Animal Care and Use Committee of the University of Missouri and were performed in accordance with National Institutes of Health guidelines. All steps were taken to minimize pain and suffering in experimental subjects. All experimental dogs were on a mixed genetic background of golden retriever, Labrador retriever, beagle and Welsh corgi and were generated in‐house by artificial insemination. All DMD dogs carry null mutations in the dystrophin gene. The genotype was determined by polymerase chain reaction according to published protocols (Fine et al. 2011; Smith et al. 2011).
All experimental dogs were housed in a specific‐pathogen‐free animal care facility and kept under a 12 h light/12 h dark cycle. DMD dogs were housed in a raised platform kennel while normal dogs were housed in a regular floor kennel. Depending on the age and size, two or more dogs were housed together to promote socialization. Normal dogs were fed dry Purina Lab Diet 5006 (LabDiet, St Louis, MO, USA) while DMD dogs were fed wet Purina Proplan Puppy food (Nestle Purina Petcare, St Louis, MO, USA). Dogs were given ad libitum access to clean drinking water. Toys were allowed in the kennel with dogs for enrichment. Dogs were monitored daily by the caregivers for overall health condition and activity. A full physical examination was performed by the veterinarian from the Office of Animal Research at the University of Missouri for any unusual changes in behaviour, activity, food and water consumption, or when clinical symptoms were noticed. The body weights of the dogs were measured periodically to monitor growth. Anaesthetized experimental subjects were killed at the end of the study according to the 2013 AVMA Guidelines for the Euthanasia of Animals. Specifically, the deeply anaesthetized dog was killed by exsanguination until ECG indicated the loss of a heartbeat. The exsanguinated dog further underwent bilateral pneumothorax and heart removal.
A total of 25 dogs were recruited for the study based on availability, out of which 11 were normal and 14 were DMD (Table 1). Both male and female dogs were used in the study. The age range of normal dogs was between 3.6 and 22.1 months. The age range of DMD dogs was between 5.3 and 21.8 months.
Table 1.
General characteristics of experimental subjects
| Normal | DMD | |
|---|---|---|
| Sample size | 11 | 14 |
| Age (months) | 14.2 ± 1.99* | 8.44 ± 1.11 |
| Age range (months) | 3.6–22.1 | 5.3–21.8 |
| Body weight (kg) | 17.08 ± 1.8* | 10.36 ± 0.84 |
| Heart rate (beats/min) | 102.44 ± 4.39* | 125.62 ± 5.29 |
| Forearm volume (mL) | 326.82 ± 42.07* | 193.57 ± 13.42 |
| ECU muscle P o (N) | 66.92 ± 10.96* | 31.80 ± 3.73 |
| ECU muscle sP o (N/cm2) | 6.01 ± 0.35* | 4.61 ± 0.50 |
Abbreviations: DMD, Duchenne muscular dystrophy; ECU, extensor carpi ulnaris. P o, maximum tetanic force; sP o, specific tetanic force. *Significantly different from the DMD group.
Chemicals
All drugs were purchased from Sigma‐Aldrich (St Louis, MO, USA). Noradrenaline (also called norepinephrine, NE, 10 μg/mL), acetylcholine (ACh, 10 μg/μL) and N ω‐nitro‐l‐arginine methyl ester hydrochloride (l‐NAME, 50 mg/mL) were dissolved in physiological saline. 7‐Nitroindazole (7‐NI) was dissolved in peanut oil (Sigma‐Aldrich) at 5 mg/mL.
Surgical preparation
Surgical preparation was carried out as described previously (Yang et al. 2012). Briefly, the experimental subject was sedated with ketamine (10 mg/kg body weight) and intubated with an endotracheal tube connected to a mechanical ventilator (Ohmeda 7000, Ohmeda, Madison, WI, USA). Anaesthesia was then induced and maintained with 1–5% isoflurane to ensure adequate depth of anaesthesia. The forearm was shaved and its volume measured by water displacement using the Archimedes principle. A catheter was placed in the saphenous vein for intravenous infusion of lactated saline. Core body temperature (rectal temperature) and electrocardiograph were monitored throughout the experiment. A schematic outline of the experimental procedures is provided in Fig. 1.
Figure 1. A schematic outline of the experimental protocol for studying sympatholysis in dogs.
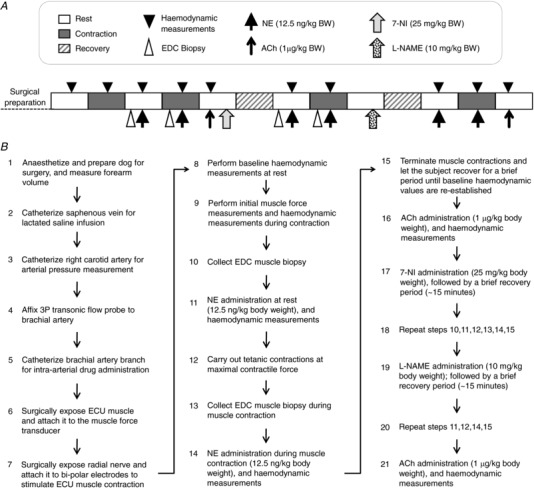
A, experimental steps are shown from left to right starting from surgical preparation of the subject. Boxes show the status of muscle (rest, contraction or recovery). B, a flow chart of the experimental protocol. BW, body weight; ECU, extensor carpi ulnaris muscle; EDC, extensor digitorum communis muscle.
Haemodynamic assay
The right carotid artery was surgically exposed and a catheter was inserted and advanced to the thoracic aorta for blood pressure monitoring. Next, a small incision was made on the medial side of the upper forelimb and the brachial artery was exposed and a 3P transonic flow probe (Module TS 420, Transonic systems, Ithaca, NY, USA) was affixed to the artery for blood flow rate measurement (mL/min). Finally, a brachial artery branch was carefully dissected proximal to the blood flow probe and a PE 50 catheter was inserted for delivering NE during the experiment.
The vascular conductance reflects blood pressure‐normalized blood flow rate. Hence, we used the vascular conductance as the readout to describe sympatholysis in this article. Since the muscle volume also influences the blood flow rate, we further normalized the vascular conductance by the forearm volume (per 100 mL). The final calculated brachial artery vascular conductance was determined using the formula: (measured blood flow rate (mL/min)/blood pressure (mmHg)]/[forearm volume/100]. It should be pointed out that normalization with the entire forelimb volume likely underestimated the actual conductance of the contracting muscles because we only elicited contraction of extensors (but not flexors) in the forelimb. However, this underestimation did not confound the comparisons between normal and DMD dogs because the relative contribution to the total forelimb volume by extensors and flexors was similar in these two groups of dogs. Specifically, the extensor muscles contributed to 35.7 ± 0.8% (n = 6) of the total forelimb volume in normal dogs and 33.9 ± 1.0% (n = 8) in DMD dogs (P > 0.05).
Muscle force measurement
Muscle force assay was performed as described before (Yang et al. 2012). Briefly, the extensor carpi ulnaris (ECU) muscle on the forearm was surgically exposed and connected to a muscle force transducer via the distal ECU tendon. The forearm incision was then extended until the radial nerve was clearly visible; the radial nerve was then carefully dissected and mounted on a bipolar electrode to stimulate muscle contraction as described previously (Yang et al. 2012). Stimulation of the radial nerve results in the contraction of all extensor muscles on the forearm. However, since only the ECU muscle was attached to the force transducer, only the force produced by the ECU muscle was recorded. The exposed ECU muscle and tendon were kept moist throughout the experiment with warm (37°C) saline and the temperature of the ECU muscle surface was also maintained at 37°C using an external heat source. Following preparation, the subject was allowed to stabilize for 10 min before muscle force measurements. For all experiments, electric stimulation was set at 8 V and 200 ms pulse duration (Grass S48 Stimulator, Grass Instruments, Quincy, MA, USA).
The optimal muscle length (L o) was first determined by applying single twitch stimulation while the muscle was held at different lengths. The muscle length that yielded the highest twitch force was selected as the L o. Next the force–frequency relationship was determined by applying 200 ms tetanic stimulations at different frequencies (20–120 Hz) while the muscle was held at the L o. The frequency that produced the largest force was selected as the optimal stimulation frequency. Next, three sets of tetanic contractions were performed at 15, 20 and 25 tetani per minute (TPM) by applying 200 ms tetanic stimulations for 5 min each to determine the TPM at which the highest blood flow rate was obtained without a change in blood pressure (Fig. 2). The optimal TPM, L o and stimulation frequency were used in all subsequent measurements.
Figure 2. Comparison of mean arterial pressure, mean arterial blood flow and absolute force production in response to tetanic contractions at 15, 20 and 25 tetani per minute (TPM).
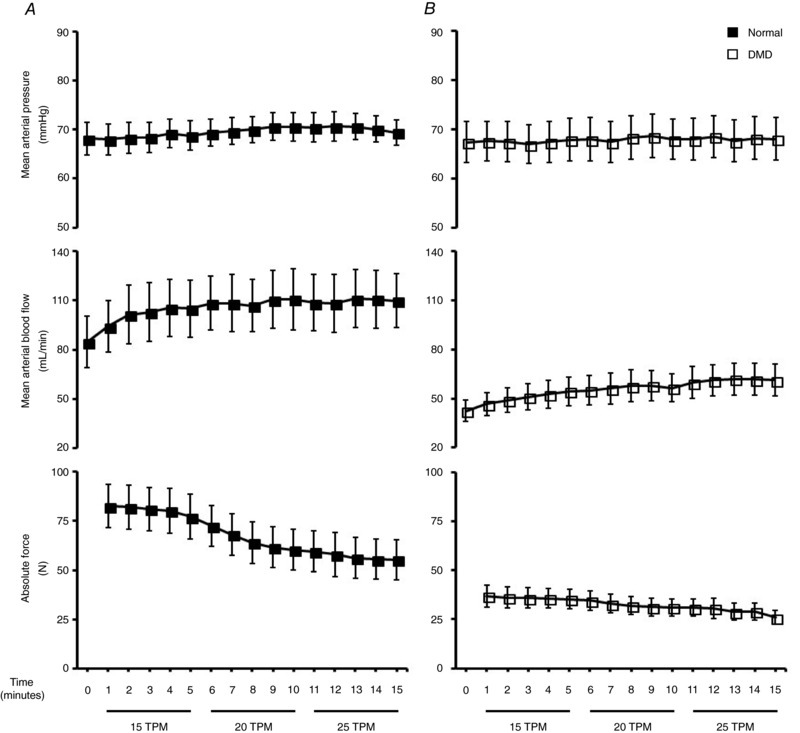
Mean arterial pressure and blood flow responses to three sets of tetanic contractions (15, 20, 25 TPM) performed at optimal muscle length and force frequency in normal (A) and DMD dogs (B) (n = 10 for normal and 13 for DMD dogs).
NE‐induced vasoconstriction
To mimic α‐adrenergic receptor‐mediated sympathetic vasoconstriction, we administrated NE intra‐arterially through a brachial artery branch (12.5 ng/kg body weight in 500 μL delivered over 30 s). Immediately after NE administration, the change in the conductance was determined. After ∼2–3 min, muscle contraction was initiated to achieve tetanic contraction at the maximal contractile force. About 2–3 min after stable tetanic force production was elicited, NE was injected again to determine the change in conductance during muscle contraction. Sympatholytic efficiency was calculated using the formula: [1 − (percentage change of conductance during contractions/percentage change of conductance at rest)] × 100.
Pharmacological NOS inhibition using selective and non‐selective NOS inhibitors
After collecting data from the resting and contracting muscle, the experimental subject was allowed to recover for ∼5 min and then the selective nNOS inhibitor 7‐NI (25 mg/kg body weight, i.p.) was injected. Following stabilization of haemodynamic values (∼15 min), sympathetic vasoconstriction was induced by administering NE as before, both at rest and during muscle contractions and changes in haemodynamic parameters were measured. After another ∼5 min of recovery, the non‐selective NOS antagonist l‐NAME (10 mg/kg body weight, i.v.) was injected. Following haemodynamic stabilization (∼15 min) sympathetic vasoconstriction was induced as before and the change in the vascular conductance was determined both at rest and during contraction (Fig. 1).
Evaluation of efficiency of NOS inhibition
The efficiency of 7‐NI‐mediated nNOS inhibition was determined by quantifying the level of phospho‐S1412‐nNOS. Briefly, the extensor digitorum communis (EDC) muscle in the forearm was biopsied at rest and during muscle contraction before 7‐NI administration. The EDC muscle was re‐biopsied at rest and during contraction following 7‐NI administration. Approximately 100–200 mg tissue was collected from each biopsy and snap‐frozen in liquid nitrogen. The biopsied site was immediately sewn with a surgical silk suture to prevent bleeding. The phospho‐S1412‐nNOS protein level in these muscle biopsies was determined by western blot (see below).
To determine the efficiency of the non‐selective NOS inhibitor l‐NAME, we quantified blood flow upon intra‐arterial injection of the eNOS‐dependent vasodilator ACh (1 μg/kg body weight in 1 mL) before administration of any NOS inhibitor and after l‐NAME administration. To avoid any systemic effects or flow‐mediated vasodilatation, ACh was injected gradually over a period of 60 s.
Brachial artery contractile response to NE
The log‐dose response to NE (10−9 to 10−4 M) was evaluated in the isolated brachial artery preparation according to a published protocol with modification (Delp et al. 1993). Briefly, the brachial artery rings (2–4 per dog) were mounted between two stainless steel wires connected to a force transducer and a micrometer device. The arterial rings were stretched to a passive tension of 1.5–2.0 g for 1 h at 37°C in oxygenated (95% O2–5% CO2) Krebs solution containing (in mM): 131.5 NaCl, 5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2, 11.2 glucose, 13.5 NaHCO3, 0.003 propranolol, 0.025 EDTA. Isometric tension was continuously recorded by a computer acquisition system.
To determine the optimal length for a given artery segment (L max), the artery ring was stimulated with 30 mM KCl and the length–tension curve was generated. The length that generated the highest tension was selected as L max. After L max determination, the arterial ring was washed until the tension returned to the resting level. The response to NE was then determined at L max by adding an increasing quantity of NE to the bath (from 10−9 to 10−4 M). The specific tension (g/mm2) was calculated using the formula: (contractile tension − resting tension)/ vessel area. The vessel area was calculated using the formula: (outer diameter of the artery − inner diameter of the artery) × (axial length of the vessel segment).
Morphology studies
Freshly collected muscle samples were embedded in Tissue‐Tek OCT (Sakura Finetek, Torrance, CA, USA) and snap‐frozen in 2‐methylbutane with liquid nitrogen. Immunofluorescence staining was carried out according to previously published protocols (Kodippili et al. 2014). Dystrophin was detected with a mouse monoclonal antibody against the C‐terminus of dystrophin (Novocastra, Newcastle upon Tyne, UK). nNOS was detected with a rabbit polyclonal antibody against the C‐terminus of nNOS (Sigma‐Aldrich). Histochemical evaluation of in situ nNOS activity was performed according to a published protocol (Lai et al. 2009). Positive nNOS activity staining appears blue under bright field microscopy.
Western blot
Western blot analysis was carried out for whole muscle lysate preparations as described before (Kodippili et al. 2014). The following primary antibodies were used including a rabbit polyclonal antibody against the C‐terminus of nNOS (Sigma‐Aldrich), a rabbit polyclonal antibody against the C‐terminus of eNOS (Abcam, Cambridge, MA, USA), a rabbit polyclonal antibody against Ser‐1412 phosphorylated nNOS (Abcam), a mouse monoclonal antibody against α‐tubulin (Sigma), and a rabbit polyclonal antibody against vinculin (Abcam).
Data collection and statistical analysis
Muscle force, brachial artery blood flow and arterial blood pressure were recorded using the Powerlab data acquisition software (ADInstruments, Castle Hill, Australia) interfaced with a Mac computer. Data are presented as mean ± standard error of the mean. Statistical analyses were performed using the SPSS statistical software (IBM, Chicago, IL, USA). Group differences in age and body weight were determined by Mann–Whitney U test due to asymmetric data distribution. Group differences in the heart rate, forearm volume and specific tetanic force of the ECU muscle were compared with the Student's t test. Western blot protein expression levels of nNOS and eNOS were compared with Student's t tests. Haemodynamics at rest and during muscle contraction, as well as haemodynamic responses to NE‐induced vasoconstriction were determined by three‐way repeated‐measures ANOVA (group × muscle contractile state × drug condition). Haemodynamic responses to ACh‐induced vasodilatation were analysed by two‐way ANOVA (group × drug condition). When significant interactions and main effects were established, a Bonferroni post hoc test or Tukey's multiple comparison test was carried out to determine group comparisons as appropriate. A P value of less than 0.05 was considered significant.
Results
Experimental animal characterization and basal haemodynamics
A total of 25 dogs (11 normal, 14 DMD) were used in the study (Table 1). Normal dogs had a significantly higher body weight, forearm volume and ECU muscle force (Table 1). DMD dogs had a significantly higher heart rate (Table 1) (Fine et al. 2011).
Normal and DMD dogs had a similar mean arterial pressure (MAP) at the baseline (Table 2). The baseline mean artery blood flow in the brachial artery (MABF) of DMD dogs was only ∼50% of that of normal dogs (Fig. 2, Table 2). Nevertheless, the forearm volume normalized mean vascular conductance (MVC) was similar between normal and DMD dogs at the baseline. Forelimb contraction at the maximal contractile force significantly increased MABF and MVC in both normal and DMD dogs (Table 2).
Table 2.
Blood flow haemodynamic profiles
| Drug condition | Muscle contractile status | Group | MAP (mmHg) | MABF (mL/min) | MVC (mL/min/mmHg/100 mL) |
|---|---|---|---|---|---|
| Baseline | |||||
| Rest | |||||
| Normal | 70.82 ± 3.27 | 102.00 ± 18.78 | 0.44 ± 0.06 | ||
| DMD | 70.29 ± 3.95 | 57.93 ± 7.56* | 0.42 ± 0.04 | ||
| Contraction | |||||
| Normal | 69.50 ± 4.06 | 123.00 ± 24.97† | 0.52 ± 0.07† | ||
| DMD | 69.93 ± 3.33 | 66.43 ± 7.70* , † | 0.50 ± 0.05† | ||
| After 7‐NI | |||||
| Rest | |||||
| Normal | 70.10 ± 3.33 | 100.60 ± 16.12 | 0.46 ± 0.06 | ||
| DMD | 67.14 ± 3.58 | 51.00 ± 5.43* | 0.41 ± 0.04 | ||
| Contraction | |||||
| Normal | 69.00 ± 3.19 | 126.56 ± 22.21† | 0.56 ± 0.06† | ||
| DMD | 66.86 ± 3.25 | 62.57 ± 6.07* , † | 0.51 ± 0.05† | ||
| After l‐NAME | |||||
| Rest | |||||
| Normal | 84.20 ± 3.83‡ | 132.10 ± 19.43‡ | 0.50 ± 0.06 | ||
| DMD | 80.21 ± 3.49‡ | 62.79 ± 6.40* , ‡ | 0.42 ± 0.04 | ||
| Contraction | |||||
| Normal | 84.90 ± 3.70‡ | 152.40 ± 21.90† , ‡ | 0.58 ± 0.07† | ||
| DMD | 81.93 ± 3.38‡ | 75.07 ± 6.81* , † , ‡ | 0.50 ± 0.05† | ||
Abbreviations: MAP, mean arterial pressure; MABF, mean arterial blood flow; MVC, mean vascular conductance; DMD, Duchenne muscular dystrophy; 7‐NI, nitroindazole. *Significantly different from normal. †Significantly different from rest. ‡Significantly different from baseline and after 7‐NI.
Neuronal and endothelial NOS expression is reduced in DMD dog skeletal muscle
Sarcolemmal expression of dystrophin was absent in DMD dogs on immunofluorescence staining (Fig. 3 A). A similar expression pattern was observed for nNOS by immunostaining and in situ nNOS activity staining (Fig. 3 A). On western blot, the total nNOS protein content in the ECU muscle was reduced by ∼90% in DMD dogs (Fig. 3 B). Total eNOS protein content in the ECU muscle was also significantly reduced in DMD dogs (Fig. 3 C).
Figure 3. Expression of nNOS and eNOS in the ECU muscle of normal and DMD dogs.
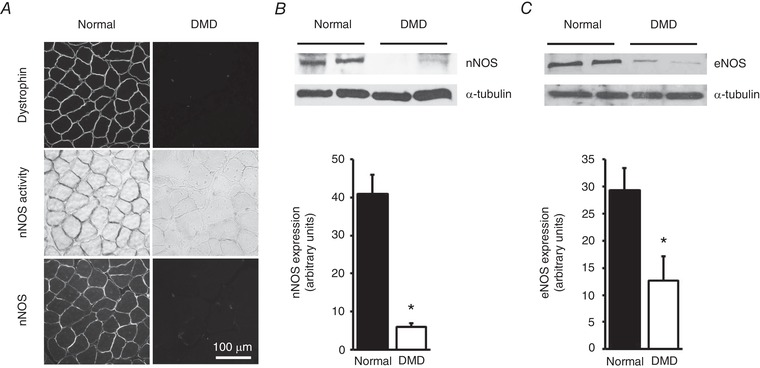
A, representative photomicrographs from dystrophin immunofluorescence staining (top panels), in situ nNOS activity staining (middle panels) and nNOS immunofluorescence staining (bottom panels). Dystrophin and nNOS are localized at the sarcolemma in normal dog muscle but are absent in DMD dog muscle. B, a representative western blot and densitometry results showing substantially downregulated expression of nNOS protein in DMD dog muscle (n = 7 for both normal and DMD). C, a representative western blot and densitometry quantification of eNOS expression in normal and DMD dog muscle (n = 4 for both normal and DMD). *Significantly different from normal dog muscle.
Contraction‐mediated attenuation of α‐adrenergic vasoconstriction is impaired in DMD dogs
We administered NE to induce vasoconstriction at rest and during muscle contraction (Figs 1 and 4, Tables 3, 4 and 5). In normal dogs, NE administration resulted in 63.76 ± 5.62% reduction of MVC at rest but only 29.46 ± 4.68% during contraction (P < 0.05, Fig. 4 C, Table 3). Sympatholysis efficiency reached 56.34 ± 5.07% (Fig. 4 E, Table 3). In DMD dogs, NE administration resulted in 76.89 ± 2.51% reduction of MVC at rest and 56.82 ± 3.15% reduction during contraction (P < 0.05, rest vs. contraction; Fig. 4 D, Table 3). Sympatholytic efficiency in DMD dogs (25.72 ± 3.77%) was significantly lower than that of normal dogs (P < 0.05, normal vs. DMD; Fig. 4 E, Table 3).
Figure 4. Attenuation of NE‐induced vasoconstriction in contracting skeletal muscle is impaired in DMD dogs.
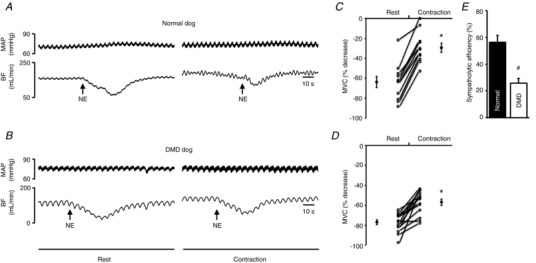
A and B, representative tracings of mean arterial pressure (MAP) and blood flow (BF) in response to noradrenaline (NE) administration at rest and during contraction in a normal (A) and a DMD (B) dog. Arrow marks the time of NE administration. C and D, percentage decrease in mean vascular conductance (MVC) in response to NE administration at rest and during contraction in normal (n = 11) and DMD dogs (n = 14). E, efficiency of functional sympatholysis in normal (n = 11) and DMD dogs (n = 14). *Significantly different from resting. #Significantly different from normal dogs.
Table 3.
NE‐mediated changes in MVC
| Drug condition | Group | MVC decrease at rest (%) | MVC decrease during contractions (%) | Sympatholytic efficiency (%) |
|---|---|---|---|---|
| Baseline | ||||
| Normal | 63.76 ± 5.62 | 29.46 ± 4.68* | 56.34 ± 5.07† | |
| DMD | 76.89 ± 2.51‡ | 56.82 ± 3.15* , ‡ | 25.72 ± 3.77 | |
| After 7‐NI | ||||
| Normal | 54.82 ± 5.51 | 40.01 ± 3.72* | 24.65 ± 5.17 | |
| DMD | 82.26 ± 2.50‡ | 61.18 ± 4.13* , ‡ | 26.17 ± 4.12 | |
| After l‐NAME | ||||
| Normal | 54.88 ± 5.17 | 43.18 ± 3.30* | 20.05 ± 2.94 | |
| DMD | 76.53 ± 3.04‡ | 57.59 ± 4.53* , ‡ | 25.68 ± 4.43 | |
Abbreviations: MVC, mean vascular conductance; DMD, Duchenne muscular dystrophy. *Significantly different from rest. †Baseline sympatholytic efficiency in normal dogs was significantly higher than that of all other conditions. ‡Significantly different from normal.
Table 4.
Haemodynamic responses to NE‐induced vasoconstriction
| Before NE | After NE | |||||||
|---|---|---|---|---|---|---|---|---|
| Drug condition | Muscle contractile status | Group | MAP (mmHg) | MABF (mL/min) | MVC (mL/min/mmHg/100 mL) | MAP (mmHg) | MABF (mL/min) | MVC (mL/min/mmHg/100 mL) |
| Baseline | ||||||||
| Rest | ||||||||
| Normal | 66.76 ± 3.86 | 99.87 ± 19.00 | 0.46 ± 0.05 | 69.19 ± 3.28 | 41.67 ± 13.00* | 0.19 ± 0.05* | ||
| DMD | 72.14 ± 4.21 | 52.60 ± 7.14† | 0.38 ± 0.04 | 71.40 ± 3.75 | 11.96 ± 2.12* , † | 0.08 ± 0.01* , † | ||
| Contraction | ||||||||
| Normal | 67.17 ± 4.86 | 123.18 ± 26.55 | 0.54 ± 0.07 | 67.84 ± 4.60 | 88.33 ± 21.72* | 0.37 ± 0.06* | ||
| DMD | 71.66 ± 4.04 | 63.99 ± 7.05† | 0.48 ± 0.05 | 71.89 ± 3.90 | 27.44 ± 3.51* , † | 0.21 ± 0.03* , † | ||
| After 7‐NI | ||||||||
| Rest | ||||||||
| Normal | 66.19 ± 4.66 | 95.76 ± 16.44 | 0.46 ± 0.05 | 66.58 ± 4.05 | 44.32 ± 10.79* | 0.22 ± 0.05* | ||
| DMD | 64.96 ± 3.33 | 48.66 ± 5.13† | 0.40 ± 0.04 | 64.99 ± 3.30 | 8.84 ± 1.41* , † | 0.08 ± 0.01* , † | ||
| Contraction | ||||||||
| Normal | 64.46 ± 3.28 | 119.83 ± 20.49 | 0.58 ± 0.06 | 65.52 ± 3.85 | 71.12 ± 13.65* | 0.34 ± 0.05* | ||
| DMD | 64.93 ± 3.48 | 59.29 ± 5.77† | 0.50 ± 0.05 | 64.42 ± 3.27 | 22.59 ± 2.76* , † | 0.20 ± 0.04* , † | ||
| After l‐NAME | ||||||||
| Rest | ||||||||
| Normal | 83.74 ± 3.31 | 126.98 ± 16.82 | 0.50 ± 0.06 | 84.26 ± 3.86 | 58.66 ± 12.10* | 0.23 ± 0.05* | ||
| DMD | 80.26 ± 4.17 | 59.99 ± 5.89† | 0.40 ± 0.04 | 81.41 ± 3.69 | 13.43 ± 1.75* , † | 0.10 ± 0.02* , † | ||
| Contraction | ||||||||
| Normal | 81.57 ± 3.72 | 158.43 ± 24.15 | 0.61 ± 0.08 | 82.43 ± 3.50 | 91.27 ± 15.04* | 0.35 ± 0.06* | ||
| DMD | 83.05 ± 3.75 | 73.13 ± 6.10† | 0.48 ± 0.05 | 83.40 ± 3.60 | 29.47 ± 2.90* , † | 0.21 ± 0.04* , † | ||
Abbreviations: NE, noradrenaline; MAP, mean arterial pressure; MABF, mean arterial blood flow; MVC, mean vascular conductance; DMD, Duchenne muscular dystrophy. *Significantly different from before NE. †Significantly different from normal.
Table 5.
Change in haemodynamic responses to NE‐induced vasoconstriction
| Drug condition | Muscle contractile status | Group | ∆MAP (mmHg) | ∆MABF (mL/min) | ∆MVC (mL/min/mmHg/100 mL) |
|---|---|---|---|---|---|
| Baseline | |||||
| Rest | |||||
| Normal | 2.43 ± 1.10 | −58.20 ± 10.76 | −0.27 ± 0.03 | ||
| DMD | −0.74 ± 0.87 | −40.64 ± 5.64 | −0.29 ± 0.03 | ||
| Contraction | |||||
| Normal | 0.67 ± 0.75 | −34.85 ± 8.97* | −0.16 ± 0.03* | ||
| DMD | 0.23 ± 0.77 | −36.56 ± 4.85 | −0.27 ± 0.03 | ||
| After 7‐NI | |||||
| Rest | |||||
| Normal | 0.39 ± 4.05 | −51.43 ± 9.76 | −0.24 ± 0.02 | ||
| DMD | 0.02 ± 1.22 | −39.83 ± 4.27 | −0.32 ± 0.03 | ||
| Contraction | |||||
| Normal | 1.07 ± 1.54 | −48.71 ± 8.48 | −0.24 ± 0.03 | ||
| DMD | −0.51 ± 1.47 | −36.70 ± 4.49 | −0.29 ± 0.03 | ||
| After l‐NAME | |||||
| Rest | |||||
| Normal | 0.52 ± 1.13 | −68.32 ± 9.73 | −0.27 ± 0.03 | ||
| DMD | 1.15 ± 1.20 | −46.56 ± 5.29 | −0.30 ± 0.03 | ||
| Contraction | |||||
| Normal | 0.87 ± 0.95 | −67.17 ± 12.47† | −0.26 ± 0.03 | ||
| DMD | 0.35 ± 1.16 | −43.66 ± 5.47 | −0.27 ± 0.03 | ||
Abbreviations: MAP, mean arterial pressure; MABF, mean arterial blood flow; MVC, mean vascular conductance; DMD, Duchenne muscular dystrophy. *Significantly different from normal at rest. †Significantly different from normal at baseline and after 7‐NI.
We next studied whether there was an inherent difference in the response of the brachial artery to NE in normal and DMD dogs. Despite the fact that NE induced significantly different levels of MVC changes in normal and DMD dogs (Fig. 4, Table 3), the isolated brachial artery rings showed a similar response to NE over a broad range of concentrations (1 × 10−9 to 1 × 10−4 M) irrespective of the diagnosis (normal or DMD) (Fig. 5).
Figure 5. Ex vivo evaluation of the contractile response of the brachial artery to NE stimulation.
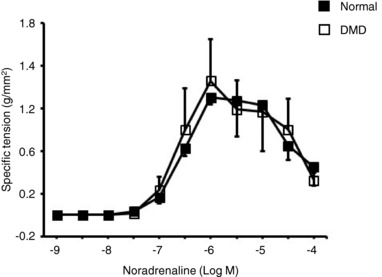
Specific tension at different NE concentrations from normal (n = 3) and DMD (n = 4) dogs. The assay was performed on 2–4 brachial artery rings for each dog.
Administration of selective nNOS inhibitor 7‐NI blocked nNOS activation and diminished functional sympatholysis in normal but not DMD dogs
To evaluate the contribution of nNOS to functional sympatholysis, we administered the nNOS‐specific inhibitor 7‐NI (Figs 1 and 6, Tables 2, 3, 4 and 5). To confirm 7‐NI indeed inhibited nNOS activity, we evaluated the level of phosphorylated nNOS, the active form of nNOS generated during muscle contraction (Fig. 6 A). Contraction resulted in a doubling of the level of phosphorylated nNOS in muscle (P < 0.05, rest vs. contraction; Fig. 6 A). 7‐NI administration abolished this effect, suggesting effective inhibition of nNOS by 7‐NI in our study (Fig. 6 A). In normal dogs, the efficiency of sympatholysis before 7‐NI administration was 56.34 ± 5.07% (Fig. 4 E, Table 3). The efficiency of sympatholysis dropped to 24.65 ± 5.17% after 7‐NI administration (P < 0.05, pre‐7‐NI vs. post‐7‐NI) (Fig. 6 F, Table 3). In DMD dogs, the efficiency of sympatholysis before 7‐NI administration was 25.72 ± 3.77% (Fig. 4 E, Table 3). The efficiency of sympatholysis remained at 26.17 ± 4.12% after 7‐NI administration (P > 0.05, pre‐7‐NI vs. post‐7‐NI) (Fig. 6 F, Table 3). These results suggest that treatment with 7‐NI had no impact on functional sympatholysis in DMD dogs. However, in normal dogs, treatment with 7‐NI led to a significant drop in the efficiency of functional sympatholysis to a level similar to that of DMD dogs.
Figure 6. Administration of nNOS inhibitor 7‐NI abolishes contraction‐induced nNOS activation and reduces sympatholytic efficiency in normal but not DMD dogs.
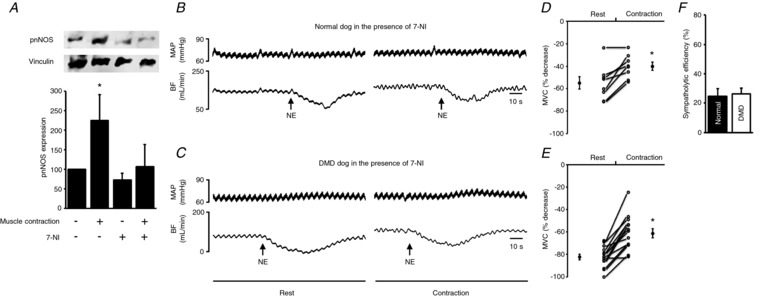
A, representative western blot and densitometry quantification results for phosphor‐S1412‐nNOS (pnNOS), an indicator of contraction‐induced nNOS activation (n = 4 normal dogs). *Significantly different from the remaining three experimental conditions. B and C, representative tracings of mean arterial pressure (MAP) and blood flow (BF) in response to noradrenaline (NE) administration at rest and during contraction in a normal (A) and a DMD (B) dog in the presence of 7‐NI. D and E, percentage decrease in mean vascular conductance (MVC) in response to NE administration at rest and during contraction in normal (n = 8) and DMD (n = 14) dogs in the presence of 7‐NI. F, efficiency of functional sympatholysis in normal (n = 8) and DMD dogs (n = 14) in the presence of 7‐NI. *Significantly different from resting.
Administration of non‐selective NOS inhibitor l‐NAME did not cause further change in functional sympatholysis in both normal and DMD dogs
To determine whether NO derived from other NOS isoforms (eNOS and inducible NOS) contributed to sympatholysis, we administered the non‐selective NOS inhibitor l‐NAME (Figs 1 and 7, Tables 2, 3, 4, 5 and 6). l‐NAME has been widely used to block all NOS isoforms in various studies (Vaupel et al. 1995; Pfeiffer et al. 1996). Here, we checked eNOS activity ablation as an indicator for l‐NAME inhibition (Fig. 7 A, Tables 6 and 7). Intra‐arterial administration of ACh induces vasodilatation through an eNOS‐mediated mechanism (Chen et al. 1996; Xu et al. 1996; Cohen et al. 1997). Following administration of ACh, the MVC in normal and DMD dogs increased by 67.89 ± 7.76% and 98.05 ± 10.66%, respectively (P < 0.05, pre‐ACh vs. post‐ACh) (Fig. 7 A, Table 6). l‐NAME treatment significantly impaired this ACh‐mediated vasodilatation (Fig. 7 A, Tables 6 and 7). Specifically, the percentage of MVC increase dropped to 29.39 ± 6.13% and 34.09 ± 3.72% for normal and DMD dogs, respectively (Fig. 7 A, Table 6). This suggests an effective inhibition of eNOS by l‐NAME in our study. l‐NAME has been shown to significantly increase the blood pressure (Sander et al. 1999). As an alternative method to validate l‐NAME inhibition in our study, we checked the blood pressure (Table 2). l‐NAME administration indeed resulted in a significant increase in the mean arterial pressure (MAP) in both normal (from 70.82 ± 3.27 mmHg at the baseline to 84.20 ± 3.83 mmHg post‐l‐NAME, P < 0.05) and DMD dogs (from 70.29 ± 3.95 mmHg at the baseline to 80.21 ± 3.49 mmHg post‐l‐NAME, P < 0.05) (Table 2). After validation of l‐NAME activity, we evaluated functional sympatholysis in the presence of l‐NAME (Fig. 7). The sympatholytic efficiency for normal and DMD dogs was 20.05 ± 2.94 and 25.68 ± 4.43, respectively (Fig. 7 F). This is essentially identical to what we saw following 7‐NI administration (P > 0.05, post‐7‐NI vs. post‐l‐NAME for both normal and DMD dogs) (Fig. 8, Table 3).
Figure 7. Modulation of NE‐induced vasoconstriction in normal and DMD dogs by l‐NAME.

A, percentage change of mean vascular conductance (MVC) in response to ACh administration in normal (n = 9) and DMD dogs (n = 14) in the absence and presence of l‐NAME. *Significantly different. B and C, representative tracings of mean arterial pressure (MAP) and blood flow (BF) in response to noradrenaline (NE) administration at rest and during contraction in a normal (B) and a DMD (C) dog in the presence of l‐NAME. D and E, percentage decrease in mean vascular conductance (MVC) in response to NE administration at rest and during contraction in normal (n = 9) and DMD (n = 14) dogs in the presence of l‐NAME. F, efficiency of functional sympatholysis in normal (n = 9) and DMD dogs (n = 14) in the presence of l‐NAME. *Significantly different from the resting condition.
Table 6.
ACh‐mediated changes in MVC before and after l‐NAME
| Drug condition | Group | MVC before ACh (mL/min/mmHg/100 mL) | MVC after ACh (mL/min/mmHg/100 mL) | MVC increase (%) |
|---|---|---|---|---|
| Before l‐NAME | ||||
| Normal | 0.51 ± 0.08 | 0.83 ± 0.11* | 67.89 ± 7.76 | |
| DMD | 0.42 ± 0.05 | 0.80 ± 0.08* | 98.05 ± 10.66 | |
| After l‐NAME | ||||
| Normal | 0.55 ± 0.09 | 0.71 ± 0.11* | 29.39 ± 6.13† | |
| DMD | 0.44 ± 0.04 | 0.58 ± 0.05* | 4.09 ± 3.72† | |
Abbreviations: MVC, mean vascular conductance; DMD, Duchenne muscular dystrophy; ACh, acetylcholine. *Significantly different from MVC before ACh. †Significantly different from before l‐NAME.
Table 7.
Change in haemodynamic responses to ACh‐induced vasodilatation
| Drug condition | Group | ∆MAP (mmHg) | ∆MABF (mL/min) | ∆MVC (mL/min/mmHg/100 mL) |
|---|---|---|---|---|
| Baseline | ||||
| Normal | 0.38 ± 0.91 | 78.46 ± 17.72 | 0.33 ± 0.04 | |
| DMD | 0.32 ± 0.72 | 49.70 ± 6.36* | 0.37 ± 0.04 | |
| After l‐NAME | ||||
| Normal | −0.91 ± 0.49 | 42.37 ± 9.56† | 0.16 ± 0.04† | |
| DMD | 1.46 ± 0.63 | 22.29 ± 3.13* , † | 0.15 ± 0.02† | |
Abbreviations: MAP, mean arterial pressure; MABF, mean arterial blood flow; MVC, mean vascular conductance; DMD, Duchenne muscular dystrophy. *Significantly different from normal. †Significantly different from baseline.
Figure 8. A summary of sympatholytic efficiency under various experimental conditions in normal and DMD dogs.
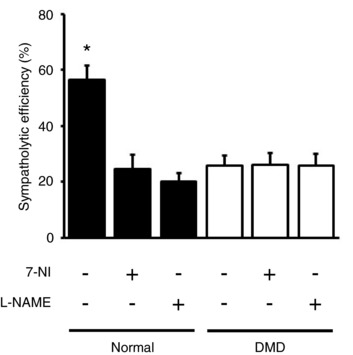
*Significantly different from the remaining experimental conditions.
Discussion
In this study, we examined nNOS and eNOS expression in normal and DMD dogs. We also developed a novel method for studying sympatholysis in dog skeletal muscle (Fig. 1). Importantly, we examined how the change in nNOS and eNOS expression/activity impacted blood perfusion in normal and DMD dog muscle at rest and during contraction. Consistent with what has been reported in mdx mice and human patients (Thomas et al. 1998; Sander et al. 2000), we found sarcolemmal nNOS was abolished and the total nNOS level was reduced in DMD dogs (Fig. 3). Previous studies have revealed different eNOS expression profiles in skeletal muscle from mdx mice and DMD patients (Thomas et al. 1998; Sander et al. 2000). In mdx muscle, eNOS expression was not altered (Thomas et al. 1998). However, in DMD patient muscle, the total eNOS level was reduced (Sander et al. 2000). In DMD dog muscle, we noticed a significant reduction of the total eNOS level, suggesting the dog model could better reproduce this human‐specific phenotype (Fig. 3).
Sympatholysis is an important protective mechanism to ensure sufficient blood perfusion during contraction. Defective sympatholysis not only contributes to exercise related muscle fatigue but also plays an important role in the pathogenesis of DMD. Methods for studying sympatholysis in humans and mice are well established. However, such a method is not available for dogs. Considering the existence of numerous dog models for neuromuscular diseases and the importance of blood perfusion for muscle function, there is an urgent need for an assay that can be used to study sympatholysis in dogs. To fill the gap, we developed a method (Fig. 1). Our protocol is developed based on our previously published in situ ECU muscle force assay (Yang et al. 2012). A similar set‐up was used except for the placement of a flow probe on the brachial artery for quantifying blood flow changes at rest and during contraction in the absence and presence of NE administration. Our protocol reliably portrayed a haemodynamic profile consistent with classic reflex sympathetic vasoconstriction during exercise. In normal dogs, administration of NE significantly reduced the artery conductance in resting muscle (Fig. 4 A and C, Table 4). This vasoconstriction effect was significantly blunted in contracting muscle (Fig. 4 A and C, Table 4). Establishment of this protocol opens the door to study the mechanisms of sympatholysis and to test therapeutic interventions aimed at improving sympatholysis in large animal models.
The molecular mechanisms of sympatholysis remain to be fully elucidated. The prevailing model suggests that NO generated by sarcolemmal nNOS plays a determining role in mice and humans (Thomas & Victor, 1998; Thomas et al. 1998, 2003; Sander et al. 2000; Martin et al. 2012; Nelson et al. 2014). To test if this is the case in dogs, we administered the nNOS‐specific inhibitor 7‐NI (Figs 1, 6 and 8, Tables 2, 3, 4 and 5) (Moore et al. 1993a,b; Li et al. 2011). A biochemical marker for nNOS activation is serine 1412 phosphorylation. The level of phosphorylated nNOS (pnNOS) positively correlates with NO synthesis (Rameau et al. 2007; Chiang et al. 2009; Hurt et al. 2012; Garbincius & Michele, 2015). We quantified the pnNOS level using a phospho‐S1412‐nNOS‐specific antibody by western blot. Muscle contraction resulted in the doubling of the pnNOS level (Fig. 6 A). 7‐NI administration abolished this effect, suggesting a complete inhibition of contraction‐induced nNOS activation by 7‐NI (Fig. 6 A). In normal dogs, the sympatholytic efficiency is ∼56% in the absence of 7‐NI (Figs 4 and 8, Table 3). Following 7‐NI administration, the sympatholytic efficiency dropped significantly (to ∼25%) (Figs 6 and 8, Table 3). This suggests that NO derived from nNOS accounts for ∼50% of the sympatholytic effect in contracting canine muscle.
Besides nNOS, evidence also exists for the involvement of other factors in sympatholysis such as adenosine, ATP, H+ , K+, oxygen, prostaglandins and NO derived from eNOS (Roach et al. 1999; Grange et al. 2001; Dinenno & Joyner, 2004; Rosenmeier et al. 2004; Mortensen et al. 2009; Crecelius et al. 2011; Jendzjowsky & DeLorey, 2013). Among these factors, eNOS is particularly intriguing because it is expressed in vascular endothelial cells and is well known for its role in the regulation of the vascular tone (Lau et al. 2000; Duplain et al. 2001). On western blot, we found abundant eNOS expression in normal dog muscle (Fig. 3 C). To study the role that eNOS plays in dog muscle sympatholysis, we applied l‐NAME, a compound that inhibits all NOS isoforms. Our rationales were (1) the baseline iNOS level is negligible in normal muscle, l‐NAME administration would mainly inhibit nNOS and eNOS, (2) we already knew the consequence of nNOS inhibition from 7‐NI administration, so any additional impact on sympatholysis from l‐NAME administration should come from eNOS inhibition. To confirm that we have effectively blocked eNOS activity, we tested ACh‐induced vasodilatation, a process mediated by eNOS (Chen et al. 1996; Xu et al. 1996; Cohen et al. 1997). ACh administration induced a ∼68% increase in artery conductance in normal dogs in the absence of l‐NAME (Fig. 7 A, Table 6). But this increase dropped to only ∼29% in the presence of l‐NAME, suggesting an effective inhibition of eNOS by l‐NAME (Fig. 7 A, Table 6). Compared to that of 7‐NI administration, treatment with l‐NAME resulted in slightly more decreased sympatholytic efficiency (∼25% following 7‐NI administration vs. ∼20% following l‐NAME administration) (Fig. 8, Table 3). However, this is not statistically significant. These results suggest that eNOS may play a nominal role in dog muscle sympatholysis. Future studies are needed to understand other cellular factors involved in sympatholysis in canine muscle and implications of NO‐independent sympatholysis with respect to dog muscle health.
A primary goal of our study is to better understand the pathogenic mechanisms of DMD in the canine model with the hope of more effectively using this model to test novel therapies in the future. While the full picture of DMD pathogenesis is still under investigation, several hypotheses have been proposed to explain how the loss of dystrophin leads to muscle pathology and force reduction. The vascular hypothesis is one of the first few hypotheses proposed in the early 1970s. This was prompted by the histological findings in muscle biopsy from early stage patients. At this stage, except for the presence of small groups of necrotic muscle fibres, the majority of myofibres appeared histologically normal. This was reminiscent of focal necrosis seen following ischaemic injury (Mendell et al. 1971; Engel & Hawley, 1977; Thomas, 2013). The discovery of nNOS‐derived NO as a key player in sympatholysis opened the door to test the vascular hypothesis experimentally (Thomas & Victor, 1998; Thomas et al. 1998, 2003; Sander et al. 2000; Martin et al. 2012; Nelson et al. 2014). Subsequent studies in mdx mice and DMD patients showed that dystrophin was required for sarcolemmal nNOS localization, membrane‐associated nNOS was lost and the total nNOS level was reduced in DMD patients and mdx mouse muscle (Brenman et al. 1995; Chang et al. 1996; Lai et al. 2009, 2013). Importantly, these changes resulted in functional ischaemia in dystrophic muscles in patients and mdx mice (Thomas & Victor, 1998; Thomas et al. 1998, 2003; Sander et al. 2000). Strategies aimed at improving blood perfusion in contracting muscle with PDE5 inhibitors significantly attenuated muscle disease in mdx mice (Asai et al. 2007; Percival et al. 2012). More recently, novel synthetic micro/mini‐dystrophin genes have been developed to anchor nNOS to the sarcolemma with the hope of providing more effective gene therapy for DMD (Lai et al. 2009; Zhang & Duan, 2012; Zhang et al. 2013; Hakim et al. 2017; Kodippili et al. 2018). Functional ischaemia is now not only an outcome measurement but also a therapeutic target for DMD.
The DMD dog is an excellent large animal model for scaling up experimental therapeutics due to the symptomatic similarity to human patients and the large body size. However, nNOS expression and its implication in functional ischaemia have never been systematically investigated in the canine DMD model. Considering the well‐known species differences in muscle physiology and disease pathology, it is essential to carefully document functional ischaemia as well as the involvement of nNOS in sympatholysis in the dog DMD model if we are to fully capitalize on this useful model in translational DMD research. With the establishment of the method for studying functional ischaemia in dogs and with the demonstration of reduced nNOS expression and nNOS sarcolemmal delocalization (Figs 1 and 3), we examined whether sympatholysis was compromised in DMD dogs.
We first quantified the baseline response to NE administration. Interestingly, NE resulted in a significantly higher decrease in MVC in DMD dogs even at rest (Table 3). This suggests that the brachial artery of the normal dog may be less sensitive to NE stimulation. To investigate this further, we quantified the NE response in the isolated brachial artery ex vivo. Surprisingly, no difference was detected (Fig. 5). Our results suggest that other yet undetermined factors may account for the baseline NE response difference between normal and DMD dogs.
Next, we studied sympatholysis. NE‐induced vasoconstriction was effectively blunted in contracting muscle in normal dogs with a sympatholytic efficiency that reached ∼56% (Figs 4 and 8, Table 3). However, the sympatholytic efficiency in contracting DMD dog muscle reached only ∼26%, a significant reduction compared to that of normal dogs (Figs 4 and 8, Table 3). Interestingly, selectively blocking nNOS activity with 7‐NI reduced the sympatholytic efficiency of contracting normal dog muscle to that of DMD dog muscle at the baseline (Figs 6 and 8, Table 3). Taken together, these data suggest that sympatholysis is significantly compromised in the canine DMD model. Further, the failure to effectively mount sympatholysis in DMD dogs can be attributed to the loss of sarcolemmal nNOS and reduction in total nNOS expression (Fig. 3). Future gene transfer studies with the nNOS‐recruiting synthetic dystrophin gene will be needed to further corroborate our conclusion. It should be noted that besides nNOS level reduction, we also observed a significant decrease of the total eNOS level in DMD dog muscle. Our results indicate that reduced eNOS expression did not contribute to the attenuated sympatholysis in DMD dog muscle because blocking all NOS isoforms with l‐NAME did not yield further change of the sympatholytic efficiency compared to that after 7‐NI treatment (Figs 7 and 8, Table 3).
An intriguing finding of our study is the preservation of ∼50% sympatholysis efficiency in DMD dog muscle (Figs 4 and 8, Table 3). This is in sharp contrast to what has been observed in DMD patients and mdx mice. In these studies, the protective sympatholytic effect was completely abolished due to the loss of sarcolemmal nNOS (Thomas et al. 1998; Sander et al. 2000). In DMD dog muscle, absence of sarcolemmal nNOS only eliminated sympatholytic contribution of nNOS. As discussed above, ∼50% of sympatholytic effect in normal dog muscle is due to yet undetermined cellular factors. Clearly, the physiological pathways mediating these cellular factors are not disturbed in DMD dog muscle. Considering the dramatic impact of DMD on muscle health, it will be interesting to further explore this nNOS‐independent sympatholytic pathway to figure out what these cellular factors are and why their functions are preserved in DMD. Similarly, future extension of our study to aged normal and DMD dogs may shed light on the kinetic changes of this unique biological process as a dog gets old and/or disease becomes more severe.
In summary, our results suggest that dog muscle utilizes both NO‐dependent and NO‐independent mechanisms to achieve protective sympatholysis during contraction. Dystrophin deficiency abolishes sarcolemmal nNOS localization and compromises sympatholysis and therefore muscle blood flow during exercise. The method we developed for studying canine muscle sympatholysis will become a handy tool to fully characterize the molecular mechanisms of sympatholysis in dog muscle and to evaluate novel therapies aimed at restoring nNOS homeostasis in DMD.
Additional information
Competing interests
D.D. is a member of the scientific advisory board for Solid Biosciences, LLC and an equity holder of Solid Biosciences, LLC. D.D. is an inventor on a patent (unrelated to the current study) licensed to Solid Biosciences, LLC. The Duan lab has received research support from Solid Biosciences, LLC.
Author contributions
All experiments were carried out at the Department of Molecular Microbiology and Immunology, School of Medicine, University of Missouri and the Department of Biomedical Sciences, College of Veterinary Medicine, University of Missouri. All authors contributed to the conception and/or design of the study. K.K., C.H.H., H.T.Y. and X.P. were involved in the acquisition of data. K.K., M.H.L., R.L.T. and D.D. were involved in the analysis and interpretation of data. K.K., N.N.Y. and D.D. were involved in drafting the manuscript. All authors were involved in revising the manuscript critically for important intellectual content, and have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the National Institutes of Health (NIH) NS‐90634 (D.D.), AR‐70517 (D.D.), AR‐67985 (D.D.), Department of Defense (MD130014), Jesse's Journey–The Foundation for Cell and Gene Therapy (D.D.) and the Jackson Freel DMD Research Fund.
Biography
Kasun Kodippili completed his BSc in Zoology at the University of Colombo, Sri Lanka in 2009, and his PhD in Professor Dongsheng Duan's laboratory at the University of Missouri, USA in 2017, where he currently works as a postdoctoral fellow. Dr Kodippili is interested in studying muscle biology/physiology and molecular pathogenesis/gene therapy of muscle diseases. His research has been recognized by multiple travel awards from the American Society of Gene and Cell Therapy as well as other organizations. This article stems from his thesis work investigating the impact of neuronal nitric oxide synthase deficiency in Duchenne muscular dystrophy dogs.

Edited by: Michael Hogan & Fernando Santana
References
- Adams ME, Mueller HA & Froehner SC (2001). In vivo requirement of the α‐syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin‐4. J Cell Biol 155, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DG, Whitehead NP & Froehner SC (2016). Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol Rev 96, 253–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini C & Tasca E (2012). Fatigue in muscular dystrophies. Neuromuscul Disord 22, Suppl. 3, S214–S220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ & Weir EK (1994). Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin‐sensitive K channel by cGMP‐dependent protein kinase. Proc Natl Acad Sci U S A 91, 7583–7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai A, Sahani N, Kaneki M, Ouchi Y, Martyn JA & Yasuhara SE (2007). Primary role of functional ischemia, quantitative evidence for the two‐hit mechanism, and phosphodiesterase‐5 inhibitor therapy in mouse muscular dystrophy. PLoS One 2, e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Xia H, Aldape K & Bredt DS (1995). Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82, 743–752. [DOI] [PubMed] [Google Scholar]
- Chang WJ, Iannaccone ST, Lau KS, Masters BS, McCabe TJ, McMillan K, Padre RC, Spencer MJ, Tidball JG & Stull JT (1996). Neuronal nitric oxide synthase and dystrophin‐deficient muscular dystrophy. Proc Natl Acad Sci U S A 93, 9142–9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H‐i, Chiang IP & Jen CJ (1996). Exercise training increases acetylcholine‐stimulated endothelium‐derived nitric oxide release in spontaneously hypertensive rats. J Biomed Sci 3, 454–460. [DOI] [PubMed] [Google Scholar]
- Chiang HT, Cheng WH, Lu PJ, Huang HN, Lo WC, Tseng YC, Wang JL, Hsiao M & Tseng CJ (2009). Neuronal nitric oxide synthase activation is involved in insulin‐mediated cardiovascular effects in the nucleus tractus solitarii of rats. Neuroscience 159, 727–734. [DOI] [PubMed] [Google Scholar]
- Cohen RA, Plane F, Najibi S, Huk I, Malinski T & Garland CJ (1997). Nitric oxide is the mediator of both endothelium‐dependent relaxation and hyperpolarization of the rabbit carotid artery. Proc Natl Acad Sci U S A 94, 4193–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper BJ, Winand NJ, Stedman H, Valentine BA, Hoffman EP, Kunkel LM, Scott MO, Fischbeck KH, Kornegay JN, Avery RJ, Williams JR, Schmickel RD & Sylvester JE (1988). The homologue of the Duchenne locus is defective in X‐linked muscular dystrophy of dogs. Nature 334, 154–156. [DOI] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Richards JC, Garcia LJ, Voyles WF, Larson DG, Luckasen GJ & Dinenno FA (2011). Mechanisms of ATP‐mediated vasodilation in humans: modest role for nitric oxide and vasodilating prostaglandins. Am J Physiol Heart Circ Physiol 301, H1302–H1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delp MD, Holder‐Binkley T, Laughlin MH & Hasser EM (1993). Vasoconstrictor properties of rat aorta are diminished by hindlimb unweighting. J Appl Physiol (1985) 75, 2620–2628. [DOI] [PubMed] [Google Scholar]
- Dinenno FA & Joyner MJ (2004). Combined NO and PG inhibition augments α‐adrenergic vasoconstriction in contracting human skeletal muscle. Am J Physiol Heart Circ Physiol 287, H2576–H2584. [DOI] [PubMed] [Google Scholar]
- Duan D (2011). Duchenne muscular dystrophy gene therapy: Lost in translation? Res Rep Biol 2011, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D (2015). Duchenne muscular dystrophy gene therapy in the canine model. Hum Gene Ther Clin Dev 26, 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B & Scherrer U (2001). Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 104, 342–345. [DOI] [PubMed] [Google Scholar]
- Engel WK & Hawley RJ (1977). Focal lesions of muscle in peripheral vascular disease. J Neurol 215, 161–168. [DOI] [PubMed] [Google Scholar]
- Ervasti JM & Campbell KP (1993). A role for the dystrophin‐glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol 122, 809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine DM, Shin JH, Yue Y, Volkmann D, Leach SB, Smith BF, McIntosh M & Duan D (2011). Age‐matched comparison reveals early electrocardiography and echocardiography changes in dystrophin‐deficient dogs. Neuromuscul Disord 21, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbincius JF & Michele DE (2015). Dystrophin‐glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of AMPK signaling. Proc Natl Acad Sci U S A 112, 13663–13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JA & McNally EM (2010). Mechanisms of muscle weakness in muscular dystrophy. J Gen Physiol 136, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange RW, Isotani E, Lau KS, Kamm KE, Huang PL & Stull JT (2001). Nitric oxide contributes to vascular smooth muscle relaxation in contracting fast‐twitch muscles. Physiol Genomics 5, 35–44. [DOI] [PubMed] [Google Scholar]
- Hakim CH, Wasala NB, Pan X, Kodippili K, Yue Y, Zhang K, Yao G, Haffner B, Duan SX, Ramos J, Schneider JS, Yang NN, Chamberlain JS & Duan D (2017). A five‐repeat micro‐dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J‐mdx model of Duchenne muscular dystrophy. Mol Ther Methods Clin Dev 6, 216–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH Jr & Kunkel LM (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928. [DOI] [PubMed] [Google Scholar]
- Hurt KJ, Sezen SF, Lagoda GF, Musicki B, Rameau GA, Snyder SH & Burnett AL (2012). Cyclic AMP‐dependent phosphorylation of neuronal nitric oxide synthase mediates penile erection. Proc Natl Acad Sci U S A 109, 16624–16629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi Y, Duncker DJ & Bache RJ (1997). Endogenous nitric oxide masks α2‐adrenergic coronary vasoconstriction during exercise in the ischemic heart. Circ Res 80, 196–207. [DOI] [PubMed] [Google Scholar]
- Jendzjowsky NG & DeLorey DS (2013). Role of neuronal nitric oxide in the inhibition of sympathetic vasoconstriction in resting and contracting skeletal muscle of healthy rats. J Appl Physiol (1985) 115, 97–106. [DOI] [PubMed] [Google Scholar]
- Kasai T, Abeyama K, Hashiguchi T, Fukunaga H, Osame M & Maruyama I (2004). Decreased total nitric oxide production in patients with Duchenne muscular dystrophy. J Biomed Sci 11, 534–537. [DOI] [PubMed] [Google Scholar]
- Kobayashi YM, Rader EP, Crawford RW, Iyengar NK, Thedens DR, Faulkner JA, Parikh SV, Weiss RM, Chamberlain JS, Moore SA & Campbell KP (2008). Sarcolemma‐localized nNOS is required to maintain activity after mild exercise. Nature 456, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodippili K, Hakim CH, Pan X, Yang HT, Yue Y, Zhang Y, Shin JH, Yang NN & Duan D (2018). Dual AAV gene therapy for Duchenne muscular dystrophy with a 7‐kb mini‐dystrophin gene in the canine model. Hum Gene Ther 29, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodippili K, Vince L, Shin JH, Yue Y, Morris GE, McIntosh MA & Duan D (2014). Characterization of 65 epitope‐specific dystrophin monoclonal antibodies in canine and murine models of Duchenne muscular dystrophy by immunostaining and western blot. PLoS One 9, e88280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornegay JN (2017). The golden retriever model of Duchenne muscular dystrophy. Skelet Muscle 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, Judge L, Bostick B, Chamberlain JS, Terjung RL & Duan D (2009). Dystrophins carrying spectrin‐like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest 119, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Zhao J, Yue Y & Duan D (2013). α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci U S A 110, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KS, Grange RW, Isotani E, Sarelius IH, Kamm KE, Huang PL & Stull JT (2000). nNOS and eNOS modulate cGMP formation and vascular response in contracting fast‐twitch skeletal muscle. Physiol Genomics 2, 21–27. [DOI] [PubMed] [Google Scholar]
- Le Gouill E, Jimenez M, Binnert C, Jayet PY, Thalmann S, Nicod P, Scherrer U & Vollenweider P (2007). Endothelial nitric oxide synthase (eNOS) knockout mice have defective mitochondrial β‐oxidation. Diabetes 56, 2690–2696. [DOI] [PubMed] [Google Scholar]
- Lee‐Young RS, Ayala JE, Hunley CF, James FD, Bracy DP, Kang L & Wasserman DH (2010). Endothelial nitric oxide synthase is central to skeletal muscle metabolic regulation and enzymatic signaling during exercise in vivo. Am J Physiol Regul Integr Comp Physiol 298, R1399–R1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Yue Y, Lai Y, Hakim CH & Duan D (2011). Nitrosative stress elicited by nNOSmicro delocalization inhibits muscle force in dystrophin‐null mice. J Pathol 223, 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGreevy JW, Hakim CH, McIntosh MA & Duan D (2015). Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech 8, 195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EA, Barresi R, Byrne BJ, Tsimerinov EI, Scott BL, Walker AE, Gurudevan SV, Anene F, Elashoff RM, Thomas GD & Victor RG (2012). Tadalafil alleviates muscle ischemia in patients with Becker muscular dystrophy. Sci Transl Med 4, 162ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Engel WK & Derrer EC (1971). Duchenne muscular dystrophy: functional ischemia reproduces its characteristic lesions. Science 172, 1143–1145. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Rodino‐Klapac L, Sahenk Z, Malik V, Kaspar BK, Walker CM & Clark KR (2012). Gene therapy for muscular dystrophy: lessons learned and path forward. Neurosci Lett 527, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PK, Babbedge RC, Wallace P, Gaffen ZA & Hart SL (1993a). 7‐Nitro indazole, an inhibitor of nitric oxide synthase, exhibits anti‐nociceptive activity in the mouse without increasing blood pressure. Br J Pharmacol 108, 296–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PK, Wallace P, Gaffen Z, Hart SL & Babbedge RC (1993b). Characterization of the novel nitric oxide synthase inhibitor 7‐nitro indazole and related indazoles: antinociceptive and cardiovascular effects. Br J Pharmacol 110, 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez‐Alonso J, Bune LT, Saltin B, Pilegaard H & Hellsten Y (2009). ATP‐induced vasodilation and purinergic receptors in the human leg: roles of nitric oxide, prostaglandins, and adenosine. Am J Physiol Regul Integr Comp Physiol 296, R1140–R1148. [DOI] [PubMed] [Google Scholar]
- Nelson MD, Rader F, Tang X, Tavyev J, Nelson SF, Miceli MC, Elashoff RM, Sweeney HL & Victor RG (2014). PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology 82, 2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival JM, Whitehead NP, Adams ME, Adamo CM, Beavo JA & Froehner SC (2012). Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J Pathol 228, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer S, Leopold E, Schmidt K, Brunner F & Mayer B (1996). Inhibition of nitric oxide synthesis by NG‐nitro‐L‐arginine methyl ester (L‐NAME): requirement for bioactivation to the free acid, NG‐nitro‐L‐arginine. Br J Pharmacol 118, 1433–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameau GA, Tukey DS, Garcin‐Hosfield ED, Titcombe RF, Misra C, Khatri L, Getzoff ED & Ziff EB (2007). Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J Neurosci 27, 3445–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remensnyder JP, Mitchell JH & Sarnoff SJ (1962). Functional sympatholysis during muscular activity. Observations on influence of carotid sinus on oxygen uptake. Circ Res 11, 370–380. [DOI] [PubMed] [Google Scholar]
- Roach RC, Koskolou MD, Calbet JA & Saltin B (1999). Arterial O2 content and tension in regulation of cardiac output and leg blood flow during exercise in humans. Am J Physiol 276, H438–H445. [DOI] [PubMed] [Google Scholar]
- Rosenmeier JB, Hansen J & Gonzalez‐Alonso J (2004). Circulating ATP‐induced vasodilatation overrides sympathetic vasoconstrictor activity in human skeletal muscle. J Physiol 558, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD & Victor RG (2000). Functional muscle ischemia in neuronal nitric oxide synthase‐deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 97, 13818–13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M, Chavoshan B & Victor RG (1999). A large blood pressure‐raising effect of nitric oxide synthase inhibition in humans. Hypertension 33, 937–942. [DOI] [PubMed] [Google Scholar]
- Sato K, Yokota T, Ichioka S, Shibata M & Takeda S (2008). Vasodilation of intramuscular arterioles under shear stress in dystrophin‐deficient skeletal muscle is impaired through decreased nNOS expression. Acta Myol 27, 30–36. [PMC free article] [PubMed] [Google Scholar]
- Smith BF, Yue Y, Woods PR, Kornegay JN, Shin JH, Williams RR & Duan D (2011). An intronic LINE‐1 element insertion in the dystrophin gene aborts dystrophin expression and results in Duchenne‐like muscular dystrophy in the corgi breed. Lab Invest 91, 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS & Meissner G (2001). Physiology of nitric oxide in skeletal muscle. Physiol Rev 81, 209–237. [DOI] [PubMed] [Google Scholar]
- Thomas GD (2013). Functional muscle ischemia in Duchenne and Becker muscular dystrophy. Front Physiol 4, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Sander M, Lau KS, Huang PL, Stull JT & Victor RG (1998). Impaired metabolic modulation of α‐adrenergic vasoconstriction in dystrophin‐deficient skeletal muscle. Proc Natl Acad Sci U S A 95, 15090–15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Shaul PW, Yuhanna IS, Froehner SC & Adams ME (2003). Vasomodulation by skeletal muscle‐derived nitric oxide requires α‐syntrophin‐mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ Res 92, 554–560. [DOI] [PubMed] [Google Scholar]
- Thomas GD & Victor RG (1998). Nitric oxide mediates contraction‐induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J Physiol 506, 817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine BA, Cooper BJ, de Lahunta A, O'Quinn R & Blue JT (1988). Canine X‐linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J Neurol Sci 88, 69–81. [DOI] [PubMed] [Google Scholar]
- Vaupel DB, Kimes AS & London ED (1995). Nitric oxide synthase inhibitors. Preclinical studies of potential use for treatment of opioid withdrawal. Neuropsychopharmacology 13, 315–322. [DOI] [PubMed] [Google Scholar]
- Vo PA, Reid JJ & Rand MJ (1992). Attenuation of vasoconstriction by endogenous nitric oxide in rat caudal artery. Br J Pharmacol 107, 1121–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Tong C & Eisenach JC (1996). Acetylcholine stimulates the release of nitric oxide from rat spinal cord. Anesthesiology 85, 107–111. [DOI] [PubMed] [Google Scholar]
- Yang HT, Shin JH, Hakim CH, Pan X, Terjung RL & Duan D (2012). Dystrophin deficiency compromises force production of the extensor carpi ulnaris muscle in the canine model of Duchenne muscular dystrophy. PLoS One 7, e44438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y & Duan D (2012). Novel mini‐dystrophin gene dual adeno‐associated virus vectors restore neuronal nitric oxide synthase expression at the sarcolemma. Hum Gene Ther 23, 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yue Y, Li L, Hakim CH, Zhang K, Thomas GD & Duan D (2013). Dual AAV therapy ameliorates exercise‐induced muscle injury and functional ischemia in murine models of Duchenne muscular dystrophy. Hum Mol Genet 22, 3720–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]