

Abstract
Background
Small cell lung cancer (SCLC) is highly aggressive and is associated with a dismal prognosis. However, there are no clinically recognized biomarkers for early diagnosis. In this study, we used quantitative proteomics to build differential mitochondrial protein profiles that may be used for early diagnosis and investigated the pathogenesis of lung cancer.
Methods
We cultured SCLC cells (NCI‐H446) and normal human bronchial epithelial cells (16‐HBE); mitochondria were extracted and purified using differential and Percoll density gradient centrifugation. Subsequently, we used Western blot analysis to validate mitochondrial purity and labeled proteins/peptides from NCI‐H446 and 16‐HBE cells using relative and absolute quantification of ectopic tags. We then analyzed mixed samples and identified proteins using two‐dimensional liquid chromatography‐tandem mass spectrometry. Additionally, we performed subsequent bioinformatic proteome analyses using the programs ExPASy, GOA, and STRING. Finally, the relationship between ornithine aminotransferase expression and clinicopathological features in lung cancer patients was evaluated using immunohistochemistry.
Results
One hundred and fifty‐three mitochondrial proteins were differentially expressed between 16‐HBE and NCI‐H446 cells. The expression of 30 proteins between 16‐HBE and NCI‐H446 cells increased more than 1.3‐fold. The upregulation of ornithine aminotransferase was associated with pathological grade and clinical tumor node metastasis stage.
Conclusion
Our experiment represented a promising method for building differential mitochondrial protein profiles between NCI‐H446 and 16‐HBE cells. Such analysis may also help to identify novel biomarkers of lung cancer.
Keywords: Lung cancer, mitochondrial protein, ornithine aminotransferase, proteomics
Introduction
Small cell lung cancer (SCLC) accounts for 15% of all lung cancers.1 Because of its unclear pathogenesis and unpredictable progression, SCLC prognosis is poor. Early diagnosis and therapy have a close relationship to SCLC prognosis.2 Thus, the discovery of novel tumor markers, particularly for lung cancer, is an urgent research focus.
Mitochondria are ubiquitous cellular organelles that are responsible for energy metabolism in eukaryotic cells and have been associated with various conditions, including Alzheimer's disease, Parkinson's disease, diabetes mellitus, various cardiovascular diseases,3, 4 and lung cancer.5 Mitochondrial DNA mutations and associated disorders are also related to tumor development, growth, angiogenesis, and metastasis.6 However, although various mitochondrial protein functions have been defined,7 few of these proteins are considered tumor markers. The application of subcellular proteomics to mitochondrial proteomes may reveal novel tumor markers that play a role in the pathogenesis of lung carcinoma. Subcellular proteomics approaches using subcellular fractionation allow simultaneous identification of multiple proteins, rather than single specifically localized proteins. Moreover, these techniques are sensitive to mitochondrial proteins that have low copy numbers and may facilitate characterization of their functions. Thus, subcellular proteomics may help identify novel tumor markers in the mitochondrial proteome.
Recent developments in chromatography, mass spectrometry (MS), and bioinformatics have improved quantitative and comparative proteomic analysis and have provided powerful tools to globally evaluate protein expression. One of these tools, two‐dimensional fluorescence difference gel electrophoresis (2D‐DIGE), is widely used for quantitative and comparative protein expression studies but has several disadvantages, including: (i) poor reproducibility; (ii) poor representation of proteins with low abundance, high acidity, large size, or hydrophobicity; and (iii) automation difficulties for gel‐based techniques.8 Alternatives to this method include isotope‐coded affinity tag and isobaric tags for relative and absolute quantification (iTRAQ), which are quantitative MS‐based proteomics methods. These techniques employ differential labeling of up to eight protein samples using stable isotope tags for MS analyses.9 Although the major limitations of 2D‐DIGE can be overcome by both of these alternatives, iTRAQ reportedly provides more comprehensive proteomic quantification than the isotope‐coded affinity tag method.10
In the present study, we harnessed the power of iTRAQ coupled with liquid chromatography‐tandem mass spectrometry (LC–MS/MS) to investigate differential protein expression between NCI‐H446 and 16‐HBE cells. This method could facilitate the generation of differentially expressed protein profiles and the identification of novel biomarkers of lung cancer.
Methods
Cell culture
We obtained NCI‐H446 and 16‐HBE cells from the Chinese Academy of Medical Sciences (Beijing, China). Cells were cultured at 37°C with 5% CO2 in RPMI 1640 medium containing 10% fetal calf serum (Hyclone, Thermo Fisher Scientific Inc., Waltham, MA, USA).
Isolation and purification of mitochondria
Cells were cultured to 70–80% confluence in 150‐cm2 flasks and then resuspended in Well grout (pH 7.5) containing sucrose (0.25 M), 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) buffer (10 mM), and protease inhibitor cocktail (1 complete protease inhibitor in 50 mL medium; Roche, Basel, Switzerland). We then homogenized cells on ice using a motor‐driven, Teflon‐coated Dounce homogenizer (Wheaton, Millville, Il, USA) until more than 95% of the cells were disrupted. Following microscopic examination of the disrupted cells, we centrifuged the filtered homogenates at 1000 × g at 4°C for 10 minutes. We then collected and centrifuged the supernatants at 8000 × g at 4°C for 15 minutes. The resulting pellets were washed twice with 15 mL of mitochondria resuspension buffer containing 200 mM mannitol, 50 mM sucrose, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid (EGTA), 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.2 mM Na3VO4, 1 mM NaF, and 10 mM Tris‐HCl (pH 7.4). To remove Golgi and cytosolic proteins, we resuspended the pellets in 12 mL of 25% Nycodenz, which was carefully applied to the top of the solution gradient, and centrifuged the mixture at 52 000 × g for 90 minutes. Discontinuous Nycodenz gradient separation solutions were prepared in centrifuge tubes with a bottom‐to‐top gradient comprising 34% Nycodenz (5 mL), 30% Nycodenz (8 mL), 25% Nycodenz (12 mL), 23% Nycodenz (8 mL), and 20% Nycodenz (3 mL). Crude mitochondrial samples were collected from the 25%/30% Nycodenz layers, diluted with the same volume of mitochondria resuspension buffer, and centrifuged at 15 000 × g for 20 minutes. Purified mitochondria were then resuspended in feasible buffer and stored at −80°C. Protein concentrations were determined using the bicinchoninic acid method (Pierce; Thermo Fisher Scientific).
Western blot analysis
We analyzed purified mitochondria using Western blots with antibodies against mitochondrial cytochrome c oxidase subunit IV (COX IV). In these experiments, we separated 20 μg protein samples using 10% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred the samples to nitrocellulose membranes. Membranes were blocked for one hour using 5% non‐fat milk in Tris‐buffered saline with Tween (TTBS) and subsequently washed and incubated at 4°C overnight with rabbit polyclonal antibodies against COX IV. Membranes were then rinsed three to five times in TTBS and incubated for two hours with horseradish peroxidase‐labeled anti‐rabbit immunoglobulin G (1:2000). After washing in TTBS, blots were visualized using enhanced chemiluminescence, and images were quantified using Image J Software (NIH, Bethesda, MD, USA).
Isobaric tags for relative and absolute quantification labeling
Samples containing 100 μg of each protein were precipitated using four volumes of cold acetone, stored at −20°C for two hours, and dissolved in solution buffer. Proteins were denatured, and cysteine residues were blocked for iTRAQ LC–MS analyses, according to the manufacturer's instructions (Applied Biosystems, Foster City, CA, USA). Samples were then digested at 37°C overnight using 20 μL trypsin solution (0.25 μg/μL; Promega, Madison, WI, USA), and equal amounts of peptides were labeled with iTRAQ tags. We combined the labeled peptides for further analysis.
Strong cation exchange chromatography
Sample complexity was reduced for LC–MS/MS analysis using a 10‐fold dilution of pooled samples in strong cation exchange (SCX) buffer A containing 10 mM KH2PO4 (pH 3.0) in 25% acetonitrile. Samples were then separated using Phenomenex Luna 5u SCX columns (Los Angeles, CA, USA), which were eluted with a gradient of SCX buffer B containing 10 mM KH2PO4 (pH 3.0) and 2 M KCl in 25% acetonitrile. The following gradient was applied: 0–25% SCX buffer B for 20 minutes, followed by 25%–100% SCX buffer B for 40 minutes. Fractions were collected at one‐minute intervals, lyophilized in a vacuum concentrator, and subjected to C‐18 clean‐up using a C‐18 column (100 mm × 75 μm, 5 μm; Phenomenex, Torrance, CA, USA). Cleaned fractions were then lyophilized and stored at −20° for MS analysis.
Protein identification using electrospray ionization quadrupole time‐of‐flight tandem mass spectrometry
Liquid chromatography (LC) with tandem MS (LC–MS/MS) was performed using a nano‐LC coupled online to a QStar XL mass spectrometer (Proxeon, Odense, Denmark). Peptides were separated on a C‐18 capillary column (100 mm × 75 μm) with a mobile phase containing buffer A (0.1% formic acid in water) and buffer B (0.1% formic acid in acetonitrile) and then eluted with a gradient of buffer B from 5% to 80% over 120 minutes at a flow rate of 300 nL/minute. LC eluents were directed to an electrospray ionization (ESI) source for quadrupole time‐of‐flight (QTOF) tandem MS analysis using information‐dependent acquisition (IDA) MS in positive ion mode. MS spectra were collected in the m/z 200–3000 range, and peptides with +1 to +3 charge states were selected for tandem MS/MS.
Peptides were identified and quantified using ProteinPilot software (Applied Biosystems), and MS/MS spectra were searched in NCBInr. Relative protein quantification in iTRAQ was performed using MS/MS scans according to peak area ratios at m/z 114 and 116 Da. Error factors (EF) and P values were calculated using ProteinPilot software to indicate deviations and significant differences in protein levels.
Protein data analysis
Theoretical isoelectric point (pI) values and molecular weights (MWs) of identified proteins were analyzed according to primary amino acid sequences using the Expert Proteins Analysis System (ExPASy) proteomics web server (http://www.expasy.org/tools). Functional enrichment analyses were performed using Universal Gene Ontology Annotation (GOA) terms (http://www.ebi.ac.uk/GOA/), and protein–protein interaction networks were identified using the names of proteins and STRING software (http://string.embl.de/).
Immunohistochemistry and staining evaluation
For immunohistochemistry (IHC) analysis, we collected 92 lung cancer and 30 normal (> 5 cm adjacent to lung cancer tissue) tissue specimens from surgery cases at the Second Affiliated Hospital of Xi'an Jiao Tong University Medical College from 2014 to 2016. All of the procedures described herein were approved by the local ethics committee, and all patients who participated in the study provided informed consent. These patients included 65 men and 27 women, with a mean age of 59 ± 9.9 years. Tissue specimens were classified as follows: 53 cases of lung squamous cell carcinoma (LSCC), 34 lung adenocarcinoma (LAC), 5 SCLC, 49 without lymph node metastasis, and 43 cases with lymph node metastasis. Cases were classified according to the International Association for the Study of Lung Cancer (IASLC) eighth edition of the Tumor Node Metastasis (TNM) Staging System (2017): 53 cases were phase I/II and 39 were phase III/IV. None of the selected patients received chemotherapy or radiotherapy, and lung cancer was confirmed by pathology after surgery. We fixed tissue specimens (lung cancer and normal tissues) with formalin, embedded them in paraffin, and performed routine slicing of the embedded samples.
We performed immunohistochemical staining as follows: 4 μm thick paraffin‐embedded specimen sections were dewaxed and rehydrated in a series of ethanol solutions; antigens were retrieved using citric acid buffer in a microwave. Endogenous peroxidase activity was blocked using 3% H2O2 for 10 minutes. Non‐specific staining was blocked using normal goat serum for 15 minutes. The sections were incubated with anti‐ornithine aminotransferase (OAT) antibody (1:30) overnight at 4°C and then with the biotinylated secondary antibody for 30 minutes at 37°C. We added horseradish enzyme to label streptomycin and incubated the tissue sections for 30 minutes at 37°C. The immunoreaction was visualized using 3,3′‐diaminobenzidine (DAB) staining. The sections were counterstained with hematoxylin and eosin, dehydrated, and then mounted with coverslips. For the negative control, the primary antibody was replaced with phosphate‐buffered saline.
Two pathologists independently performed blind microscopic examination and evaluation of all sections. At least five high‐power fields were randomly selected, with > 200 cells counted per field. The immunohistochemical score (IHS) was used to evaluate OAT expression, based on the German immunoreactive score. The IHS was calculated by combining the staining intensity score with the quantity score (percentage of positively stained cells). Staining intensity was classified according to the following criteria: 0, no staining; 1, light yellow/weak staining; 2, yellow‐brown/moderate staining; and 3, brown/strong staining. The proportion of positive cells was scored as follows: 0, no positive tumor cells; 1, < 10% of cells stained; 2, 10–50% of cells stained; and 3, > 50% of cells stained. The final IHS (scores 0–9) was determined by combining these scores (staining intensity × quantity), which resulted in the following categories: 0, negative expression; 1–4, low expression; and > 4, high expression.
Results
Mitochondrial isolation and validation of purity
We isolated mitochondria from SCLC (NCI‐H446) and normal human bronchial epithelium (16‐HBE) cells, achieving high purity for proteomic analyses, as indicated by Western blot analysis. Rabbit polyclonal antibodies against COX IV were used to specifically identify the purified mitochondrial fraction in Western blotting experiments (Fig 1). These results demonstrated that we isolated high‐purity mitochondria using this subcellular isolation method.
Figure 1.
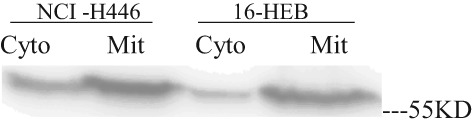
Western blot of purified mitochondrial fraction with rabbit polyclonal antibodies against mitochondrial cytochrome c oxidase subunit IV (COX IV).
Identification of differentially expressed proteins
Because the 2D‐LC–MS/MS method provides a powerful alternative to gel‐based analyses, particularly for the detection of hydrophobic proteins, we used the novel iTRAQ technique to compare mitochondrial protein expression between 16‐HBE and NCI‐H446 cells. Our 2D‐LC–MS/MS analyses identified 153 significantly differentially expressed proteins between these two cell types. Of the identified proteins, 41% (63/153) were identified by more than 10 peptides, 25% (38/153) were identified by five peptides, and 8% (13/153) were identified by two peptides. Moreover, of the 153 proteins, 80 exhibited higher expression and 73 lower expression in NCI‐H446 cells than in 16‐HBE cells. In particular, we found that expression of 14 proteins was > 1.5 times greater in NCI‐H446 cells than in 16‐HBE cells (Table 1).
Table 1.
Proteins with > 1.3‐fold differential expression between NCI‐H446 and 16‐HBE cells
| No. | Protein ID | Description | Unique peptides | Protein Mass | Score | Coverage Rate | Fold change |
|---|---|---|---|---|---|---|---|
| 1 | P31327 | Carbamoyl‐phosphate synthase (ammonia) (EC 6.3.4.16) precursor ‐ human | 32 | 165 923.11 | 580.17 | 20.13% | 2.55 |
| 2 | P04181 | ornithine aminotransferase precursor; Ornithine aminotransferase | 2 | 48 846.33 | 52.31 | 6.15% | 2.31 |
| 3 | P49753 | Mitochondrial Acyl‐CoA Thioesterase | 4 | 46 723.68 | 119.87 | 12.81% | 2.11 |
| 4 | P07954 | Fumarate hydratase, mitochondrial precursor (EC 4.2.1.2) (Fumarase) | 3 | 54 773.23 | 110.42 | 26.27% | 2.08 |
| 5 | P55809 | 3‐oxoacid CoA transferase precursor; Succinyl CoA:3‐oxoacid CoA transferase; succinyl‐CoA:3‐ketoacid‐CoA transferase precursor; 3‐oxoacid CoA transferase 1 | 12 | 56 578.1 | 234.34 | 25.00% | 1.99 |
| 6 | P15531 | Chain A, Nucleoside Triphosphate, Nucleoside Diphosphate Mol_id: 1; Molecule: Nucleoside Diphosphate Kinase; Chain: A, B, C, D, E, F; Ec: 2.7.4.6 | 2 | 17 269.94 | 76.54 | 23.84% | 1.93 |
| 7 | P50395 | GDP dissociation inhibitor 2; rab GDP‐dissociation inhibitor, beta | 3 | 51 087.05 | 106.85 | 18.65% | 1.84 |
| 8 | P50750 | tat‐associated protein | 13 | 31 364.49 | 276.95 | 33.33% | 1.83 |
| 9 | P08670 | (XM_042950) vimentin | 76 | 53 676.1 | 815.46 | 43.99% | 1.78 |
| 10 | Q13423 | NAD(P) transhydrogenase (B‐specific) (EC 1.6.1.1) precursor, mitochondrial | 8 | 114 591.75 | 210.75 | 6.91% | 1.67 |
| 11 | P60174 | triosephosphate isomerase 1 | 4 | 26 937.85 | 117.68 | 39.76% | 1.58 |
| 12 | P08133 | Annexin A6 (Annexin VI) (Lipocortin VI) (P68) (P70) (Protein III) (Chromobindin 20) (67 kDa calelectrin) (Calphobindin‐II) (CPB‐II) | 2 | 76 167.7 | 110.25 | 10.70% | 1.52 |
| 13 | Q02218 | OGDH protein | 4 | 117 058.85 | 160.97 | 9.97% | 1.51 |
| 14 | P51149 | small GTP binding protein Rab7 | 12 | 23 731.92 | 214.15 | 30.92% | 1.51 |
| 15 | O75431 | metaxin 2 | 3 | 30 086.2 | 77.78 | 17.49% | 1.48 |
| 16 | Q99714 | 3‐hydroxyacyl‐CoA dehydrogenase, isoform 2 | 5 | 42 438.94 | 108.26 | 13.59% | 1.47 |
| 17 | Q969X5 | transmembrane protein (63kD), endoplasmic reticulum/Golgi intermediate compartment; type‐II transmembrane protein p63 | 23 | 66 096.77 | 472.48 | 29.90% | 1.46 |
| 18 | Q9H9B4 | Sideroflexin 1 | 7 | 35 881.45 | 88.55 | 10.87% | 1.45 |
| 19 | O75390 | citrate synthase precursor isoform a; citrate synthase, mitochondrial | 4 | 51 907.6 | 76.40 | 5.58% | 1.42 |
| 20 | Q96TA2 | YME1‐like 1 isoform 3; ATP‐dependent metalloprotease FtsH1 homolog | 2 | 80 010.64 | 72.02 | 5.31% | 1.42 |
| 21 | P35613 | basigin isoform 1; OK blood group; emmprin; collagenase stimulatory factor; M6 antigen; extracellular matrix metalloproteinase inducer | 8 | 42 573.21 | 136.57 | 21.82% | 1.39 |
| 22 | Q9NS69 | mitochondrial import receptor Tom22 | 8 | 15 511.78 | 163.04 | 61.97% | 1.39 |
| 23 | P00558 | Phosphoglycerate kinase 1 (Primer recognition protein 2) (PRP 2) | 6 | 45 098.36 | 176.42 | 26.08% | 1.37 |
| 24 | P31937 | 3‐hydroxyisobutyrate dehydrogenase, mitochondrial precursor (EC 1.1.1.31) (HIBADH) | 3 | 35 704.91 | 130.92 | 11.01% | 1.37 |
| 25 | P62937 | peptidylprolyl isomerase A isoform 1; cyclophilin A; peptidyl‐prolyl cis‐trans isomerase A; T cell cyclophilin; rotamase; cyclosporin A‐binding protein | 29 | 18 228.97 | 304.08 | 51.52% | 1.36 |
| 26 | P04843 | Similar to ribophorin I | 9 | 64 655.75 | 187.58 | 22.54% | 1.35 |
| 27 | Q9HAV0 | guanine nucleotide‐binding protein, beta‐4 subunit; guanine nucleotide binding protein subunit beta 4; G protein beta‐4 subunit | 9 | 38 284.3 | 193.47 | 17.65% | 1.33 |
| 28 | O60313 | Dynamin‐like 120 kDa protein, mitochondrial, EC 3.6.5.5 | 3 | 114 017.03 | 128.97 | 11.25% | 1.32 |
| 29 | P22570 | ferredoxin reductase isoform 2 precursor; adrenodoxin reductase | 2 | 54 929.52 | 78.16 | 9.05% | 1.30 |
| 30 | P13995 | methylene tetrahydrofolate dehydrogenase 2 precursor; NAD‐dependent methylene tetrahydrofolate dehydrogenase cyclohydrolase | 3 | 37 467.98 | 80.97 | 11.92% | 1.30 |
Bioinformatics analysis of physicochemical characteristics of differentially expressed proteins
The MWs of the identified proteins in the present study ranged from 5 to 276 kDa; 13 (8.50%) had MWs < 20 kDa, and 19 (12.42%) had MWs > 100 kDa. Moreover, one protein (0.65%) had an MW > 200 kDa, and two proteins (1.31%) had MWs < 10 kDa. The pI of proteins ranged from 2.69 to 13.6. Furthermore, 6 (3.92%) were extremely basic with pI values > 10, and 3 (1.96%) were extremely acidic with pI values < 4.0. The differential expression of these 14 proteins increased > 1.5 fold in NCI‐H446 compared to 16‐HBE cells, and they were classified into eight functional categories using universal GOA terms (Fig 2). The top two molecular function categories were amino acid and lipid metabolism (28.57%) and signal transduction (21.43%).
Figure 2.
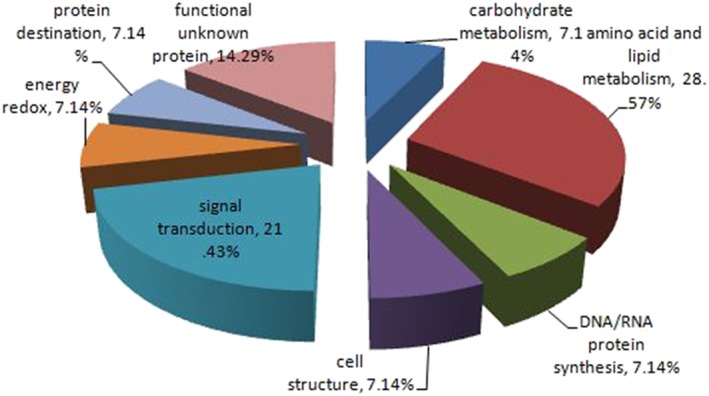
Functional categories of identified 14 proteins using universal Gene Ontology Annotation terms.
Protein–protein interaction analysis
Carcinogenesis is mediated by numerous protein interactions. Thus, to investigate protein–protein interactions, we performed searches using the STRING database. As shown in Figure 3, we found that 30 upregulated proteins were involved in protein–protein interactions. Among these, 25 were interlinked in a network. Five proteins, CDK9, RPN1, HSD17B10, MTX2, and ANXA6, were not involved in this network.
Figure 3.
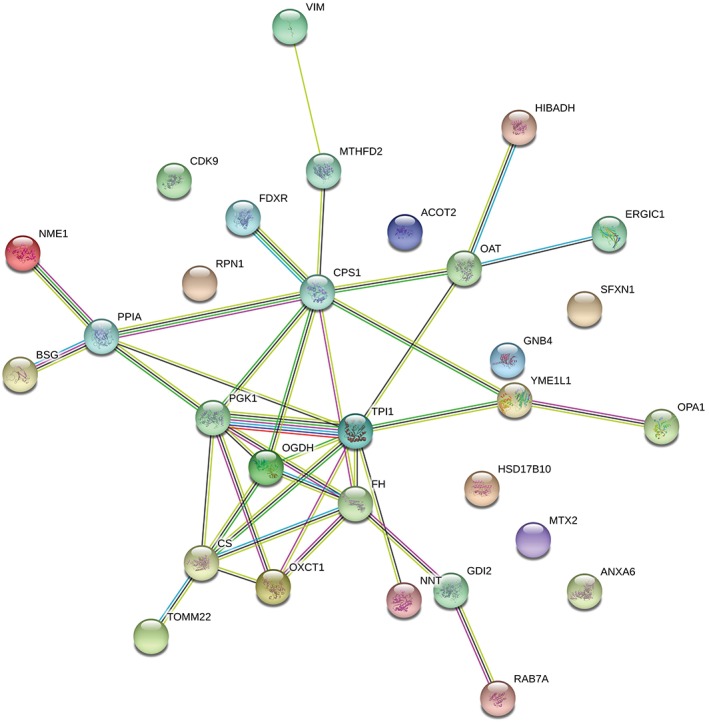
Protein–protein interaction network analyses using STRING software.
Ornithine aminotransferase expression in lung cancer and normal lung tissues
To verify the results obtained from quantitative proteomics analysis, we performed IHC to detect the expression of OAT in a series of 92 lung tissue specimens – including 53 LSCC, 34 LAC, and 5 SCLC – and 30 normal lung tissue specimens.
Increased OAT expression was observed in the cytoplasm of the tumor cells in most of the lung cancer specimens (Fig 4). However, negative or low OAT expression levels were observed in the majority of normal lung specimens (Fig 5). We found that OAT expression in LAC was significantly higher (P < 0.01) than in normal lung specimens (Table 2). Similarly, OAT expression was significantly higher in LSCC than in normal lung tissue specimens (P < 0.01). Although we only collected five SCLC specimens, the expression of OAT in these samples was significantly higher than in normal tissue specimens (P < 0.05).
Figure 4.
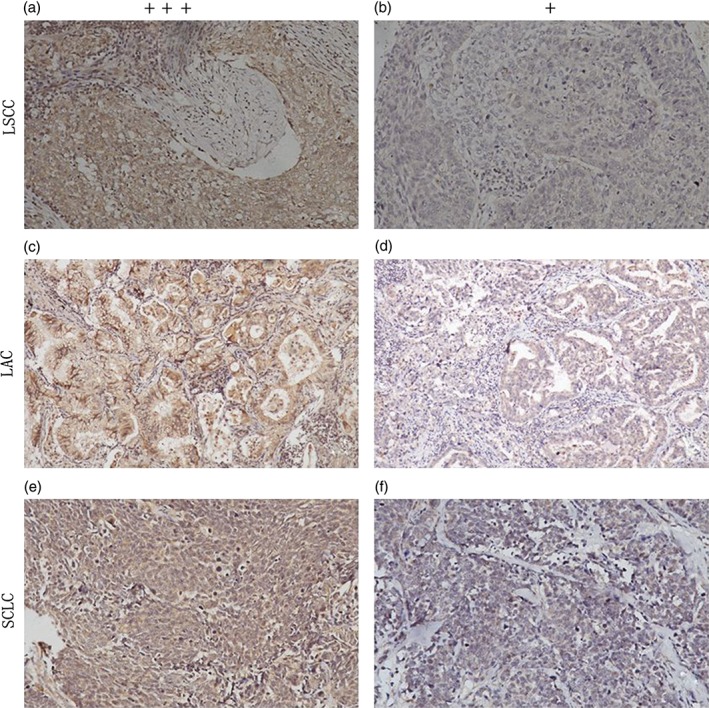
Ornithine aminotransferase expression in lung cancer: (a) Strong and (b) weak staining in lung squamous cell carcinoma (LSCC); (c) strong and (d) weak staining in lung adenocarcinoma (LAC); and (e) strong and (f) weak staining in small cell lung cancer (SCLC).
Figure 5.
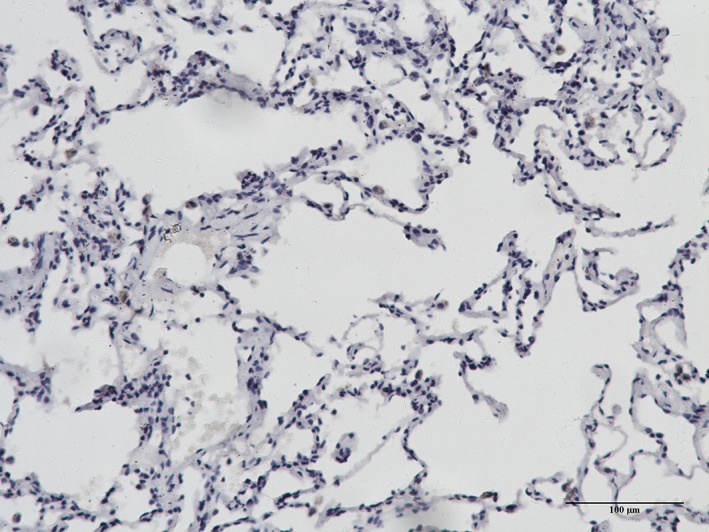
Ornithine aminotransferase expression in normal lung tissue.
Table 2.
OAT expression in lung cancer and normal lung tissues
| Group | No. | Expression level | P * | ||
|---|---|---|---|---|---|
| Neg | Low | High | |||
| LAC | 34 | 1 | 16 | 17 | < 0.01† |
| LSCC | 53 | 8 | 19 | 26 | < 0.01‡ |
| SCLC | 5 | 1 | 2 | 2 | < 0.05§ |
| LC | 92 | 10 | 37 | 45 | < 0.01¶ |
| Normal | 30 | 21 | 8 | 1 | |
Mann–Whitney U test.
Lung adenocarcinoma (LAC).
lung squamous cell carcinoma (LSCC).
small cell lung cancer (SCLC).
lung cancer (LC) versus normal lung tissue.
OAT, ornithine aminotransferase.
We also investigated the relationship between clinicopathological characteristics and OAT expression (Table 3). We found that OAT expression was higher in stage III/IV lung cancers than in stage I/II specimens (P < 0.05). However, we did not find any significant correlations between OAT expression and other clinicopathological characteristics, including pathological type, patient age, gender, or lymphatic invasion.
Table 3.
Association between OAT expression and clinicopathological characteristics of lung cancers
| Characteristics | No. | OAT expression | P | ||
|---|---|---|---|---|---|
| Negative | Low | High | |||
| Age | |||||
| ≤ 60 | 44 | 4 | 15 | 25 | 0.164 |
| > 60 | 48 | 6 | 22 | 20 | |
| Gender | |||||
| Male | 65 | 8 | 24 | 33 | |
| Female | 27 | 2 | 13 | 12 | 0.794 |
| Histology type | |||||
| LAC | 34 | 1 | 16 | 17 | 0.712 |
| LSCC | 53 | 8 | 19 | 26 | |
| SCLC | 5 | 1 | 2 | 2 | |
| TNM stage | |||||
| I/II | 53 | 6 | 29 | 18 | 0.004 |
| III/IV | 39 | 4 | 8 | 27 | |
| Lymphatic invasion | |||||
| N0 | 49 | 6 | 20 | 23 | 0.631 |
| N+ | 43 | 4 | 17 | 22 | |
P values by Mann–Whitney U or Kruskal‐Wallis test. LAC, lung adenocarcinoma; LSCC, lung squamous cell carcinoma; OAT, ornithine aminotransferase; SCLC, small cell lung cancer.
Discussion
Both relative and absolute high‐throughput protein quantitation are widely performed using iTRAQ, which enables simultaneous quantitation of up to eight different biological samples.11 In this study, we adopted iTRAQ combined with 2D‐LC–MS/MS,12 overcoming the shortcomings of traditional methods. Many proteins of extremely low and high MW and proteins with polar acids and bases were identified, with additional differentially expressed proteins identified for further experiments. With these data, we built more complete differential mitochondrial protein profiles. We identified 153 mitochondrial proteins differentially expressed between 16‐HBE and NCI‐H446 cells. Using the ExPASy proteomics web server, we identified proteins with MWs < 20 kDa and > 100 kDa and with pI < 4 and > 10, which is not easily achieved with other methods.
In further analyses, we identified 14 proteins with more than 1.5‐fold higher expression in SCLC cells. We classified these 14 proteins into eight functional categories according to GOA terms. Half of the differential proteins are associated with signal transduction pathways and amino acid and lipid metabolism. Signal transduction pathways13 and metabolism14 are associated with the pathogenesis of a variety of tumors. Through these links, we can determine possible tumor markers and trace the pathogenesis of cancer.
Using the STRING database, protein–protein interactions were identified between 30 upregulated proteins, providing evidence for the further study of protein function. In the literature, many proteins have been associated with tumors, such as CPS1,15 OAT,16 and VIM.17 Therefore, proteins associated with tumors provide a foundation for further study of the pathogenesis of lung cancer and screening of lung cancer markers.
Quantitative proteomics has previously been used to investigate the molecular mechanisms of cancer and identify novel tumor markers.18 For example, Zeng et al. performed quantitative proteomics and showed that glutathione S‐transferase pi 1 (GSTP1), heat shock protein beta‐1 (HSPB1), and creatine kinase B‐type (CKB) are potential novel biomarkers for the early detection of laryngeal squamous cell carcinoma and that GSTP1 downregulation is involved in human bronchial epithelial carcinogenesis.19 In addition, the novel cancer marker S100A14 has been identified and associated with well or moderately differentiated lung cancer.20
By quantitative proteomics, we established differential protein profiles between 16‐HBE and NCI‐H446 cells. In order to prove the reliability of the experimental results, we selected tumor‐related OAT with a > 1.5‐fold difference in expression and verified its reliability in clinical specimens. To build upon these findings, we further studied the expression of OAT in lung cancer tissues. The mitochondrial enzyme OAT catalyzes the formation of proline from ornithine and has been associated with cancers in multiple studies.16 In particular, Kobayashi et al. found 15‐fold higher expression of OAT in Morris hepatoma 44 cells than in normal liver cells.21 In addition, Tonack et al. found increased OAT expression after induction of CapG in tetracycline‐inducible pancreatic cancer cell lines.22 Similarly, elevated OAT expression has been observed in mammary carcinomas23 and prostate cancers.24 Moreover, OAT knockdown using RNA interference blocked cell division and led to cell death in human cervical carcinoma and osteosarcoma cells, suggesting the utility of OAT as a potential therapeutic target.25 As further evidence of this potential, Lee et al. identified multiple GABA‐AT‐binding molecules that could be used as selective inhibitors of OAT for cancer treatment.26
Our study showed that OAT expression was 2.31‐fold higher in NCI‐H446 cells than in 16‐HBE cells. Additionally, we found that OAT protein expression in SCLC, LAC, and LSCC was significantly higher than in adjacent non‐tumor lung tissues, and this difference was not correlated with gender, age, or lymphatic invasion. However, we observed significant differences in OAT protein expression that correlated with TNM stages, revealing higher expression in advanced‐stage than early‐stage patients. Collectively, these results strongly suggest that OAT is associated with the development of lung cancer. Nonetheless, further experiments are needed to confirm whether OAT can be used as a marker of lung cancer.
In summary, these data represent the first comprehensive evaluation of differential expression of mitochondrial proteins in lung cancers using iTRAQ–proteomics methods. We identified 153 unique proteins, 30 of which were differentially expressed by at least 1.3‐fold. Furthermore, subsequent bioinformatic proteome analyses demonstrated protein–protein interactions and identified GOA terms for 14 proteins that exhibited at least a 1.5‐fold difference in expression. Finally, OAT was differentially expressed between lung cancer and normal tissues. Future experiments will verify the mechanism of differential mitochondrial protein expression in the pathogenesis of lung cancer and whether these proteins can serve as biomarkers of lung cancer.
Disclosure
No authors report any conflict of interest.
Acknowledgments
This study was funded by the National Natural Science Foundation of China (grant no. 81672300) and the Public Relations Project of Social Development in Shaanxi Province (grant no. 2016SF‐249).
Contributor Information
Xuemei Zhang, Email: zxmzxs0417@163.com.
Shuanying Yang, Email: yangshuanying66@163.com.
References
- 1. Ahmed Z, Grover P, Kennedy KF, Masood A, Davis JR, Subramanian J. Predictors for chemotherapy in early stage small cell lung carcinoma (SCLC): A National Cancer Database (NCDB) analysis. Lung Cancer 2017; 113: 85–7. [DOI] [PubMed] [Google Scholar]
- 2. Glatzer M, Rittmeyer A, Müller J et al Treatment of limited disease small cell lung cancer: The multidisciplinary team. Eur Respir J 2017; 50: 1700422. [DOI] [PubMed] [Google Scholar]
- 3. Carri MT, Polster BM, Beart PM. Mitochondria in the nervous system: From Health to disease, part II. Neurochem Int 2018; 117: 1–4. [DOI] [PubMed] [Google Scholar]
- 4. Pan P, Wang X, Liu D. The potential mechanism of mitochondrial dysfunction in septic cardiomyopathy. J Int Med Res 2018; 46: 2157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang X, Chen C, Zhou G et al Sepia ink oligopeptide induces apoptosis of lung cancer cells via mitochondrial pathway. Cell Physiol Biochem 2018; 45: 2095–106. [DOI] [PubMed] [Google Scholar]
- 6. Mazzanti R, Giulivi C. Coordination of nuclear‐ and mitochondrial‐DNA encoded proteins in cancer and normal colon tissues. Biochim Biophys Acta 2006; 1757: 618–23. [DOI] [PubMed] [Google Scholar]
- 7. Jakupciak JP, Maragh S, Markowitz ME et al Performance of mitochondrial DNA mutations detecting early stage cancer. BMC Cancer 2008; 8: 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu WW, Wang G, Baek SJ, Shen RF. Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel‐ or LC‐MALDI TOF/TOF. J Proteome Res 2006; 5: 651–8. [DOI] [PubMed] [Google Scholar]
- 9. Butler GS, Dean RA, Morrison CJ, Overall CM. Identification of cellular MMP substrates using quantitative proteomics: Isotope‐coded affinity tags (ICAT) and isobaric tags for relative and absolute quantification (iTRAQ). Methods Mol Biol 2010; 622: 451–70. [DOI] [PubMed] [Google Scholar]
- 10. Tao WA, Aebersold R. Advances in quantitative proteomics via stable isotope tagging and mass spectrometry. Curr Opin Biotechnol 2003; 14: 110–8. [DOI] [PubMed] [Google Scholar]
- 11. Unwin RD. Quantification of proteins by iTRAQ. Methods Mol Biol 2010; 658: 205–15. [DOI] [PubMed] [Google Scholar]
- 12. Zhang W, Li Y, Yang S et al Differential mitochondrial proteome analysis of human lung adenocarcinoma and normal bronchial epithelium cell lines using quantitative mass spectrometry. Thorac Cancer 2013; 4: 373–9. [DOI] [PubMed] [Google Scholar]
- 13. Kalyanaraman B, Cheng G, Hardy M et al A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. (Published erratum appears in Redox Biol 2018;16:426–7). Redox Biol 2018; 14: 316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gu Y, Albuquerque CP, Braas D et al mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine‐glutamate antiporter xCT. Mol Cell 2017; 67: 128–38 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim J, Hu Z, Cai L et al CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1‐mutant lung cancer cells. Nature 2017; 546: 168–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zigmond E, Ben Ya'acov A, Lee H et al Suppression of hepatocellular carcinoma by inhibition of overexpressed ornithine aminotransferase. ACS Med Chem Lett 2015; 6: 840–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci 2011; 68: 3033–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li Y, Wang X, Ao M et al Aberrant Mucin5B expression in lung adenocarcinomas detected by iTRAQ labeling quantitative proteomics and immunohistochemistry. Clin Proteomics 2013; 10: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zeng GQ, Zhang PF, Deng X et al Identification of candidate biomarkers for early detection of human lung squamous cell cancer by quantitative proteomics. Mol Cell Proteomics 2012; 11: M111.013946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang X, Li W, Hou Y et al Comparative membrane proteomic analysis between lung adenocarcinoma and normal tissue by iTRAQ labeling mass spectrometry. Am J Transl Res 2014; 6: 267–80. [PMC free article] [PubMed] [Google Scholar]
- 21. Kobayashi K, Morris HP, Katunuma N. Studies on the turnover rates of ornithine aminotransferase in Morris hepatoma 44 and host liver. J Biochem 1976; 80: 1085–9. [DOI] [PubMed] [Google Scholar]
- 22. Tonack S, Patel S, Jalali M et al Tetracycline‐inducible protein expression in pancreatic cancer cells: Effects of CapG overexpression. World J Gastroenterol 2011; 17: 1947–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klopfleisch R, Klose P, Weise C et al Proteome of metastatic canine mammary carcinomas: Similarities to and differences from human breast cancer. J Proteome Res 2010; 9: 6380–91. [DOI] [PubMed] [Google Scholar]
- 24. Jariwala U, Prescott J, Jia L et al Identification of novel androgen receptor target genes in prostate cancer. Mol Cancer 2007; 6: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang G, Shang L, Burgett AW, Harran PG, Wang X. Diazonamide toxins reveal an unexpected function for ornithine delta‐amino transferase in mitotic cell division. Proc Natl Acad Sci U S A 2007; 104: 2068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee H, Juncosa JI, Silverman RB. Ornithine aminotransferase versus GABA aminotransferase: Implications for the design of new anticancer drugs. Med Res Rev 2015; 35: 286–305. [DOI] [PMC free article] [PubMed] [Google Scholar]