

Summary
Preclinical pharmacology studies in animal models of seizures and epilepsy have provided a platform to identify more than 20 antiseizure drugs in recent decades. To minimize variability in lab‐to‐lab studies and to harmonize approaches to data collection and reporting methodology in pharmacologic evaluations of the next generation of therapies, we present common data elements (CDEs), case report forms (CRFs), and this companion manuscript to help with the implementation of methods for studies in established preclinical seizure and epilepsy models in adult rodents. The development of and advocacy for CDEs in preclinical research has been encouraged previously by both clinical and preclinical groups. It is anticipated that adoption and implementation of these CDEs in preclinical studies may help standardize approaches to minimize variability and increase the reproducibility of preclinical studies. Moreover, they may provide a methodologic framework for pharmacology studies in atypical animal models or models in development, which may ultimately promote novel therapy development. In the present document, we refer selectively to animal models that have a long history of preclinical use, and in some cases, are clinically validated.
Keywords: Chemoconvulsive models, Electroconvulsive models, Kindled rodent models, Animal models of epilepsy, Antiseizure drug discovery, Status epilepticus models, Phenytoin
Key Points.
There is increasing demand for greater reproducibility in preclinical research; harmonized approaches may reduce lab‐to‐lab variability
Common data elements and case report forms were developed for preclinical pharmacologic studies for epilepsy to address this need
Common data elements are core components that are essential for pharmacologic research in preclinical models of seizure and epilepsy
Common data elements were developed for the models that have a long preclinical history, and in some cases, are clinically validated
To promote the harmonization of preclinical approaches to pharmacologic studies in animal models of seizures and epilepsy, and to promote data comparability, sharing, and reproducibility, the Pharmacology Working Group of the TASK3 group of the Joint Translational Task Force of the International League Against Epilepsy (ILAE)/American Epilepsy Society (AES) sought to identify common data elements (CDEs; Appendix S1), develop case report forms (CRFs; Appendix S1), and draft companion papers to assist with the CRF/CDE implementation. These endeavors were motivated by the concerns that have been raised lately about the reproducibility and the translational value of preclinical drug discovery.1 Although some have advocated that the clinical failures of investigational therapies also rely, in part, on preclinical animal models that do not simulate the human disease well,2 many investigators would say that preclinical studies are valuable and that the problem is instead related to differences in procedures or other aspects of the conduct of preclinical studies. CDEs/CRFs would therefore standardize some aspects of data collection and reporting methodology to better support well‐controlled, rigorous pharmacologic evaluations of investigational therapies or treatment approaches for the management of seizures and epilepsy. Thus, CDEs, whether in the preclinical or clinical realm,3, 4 promote standardized approaches across groups and laboratories, and thus facilitate systematic data collection and analyses. The Task Force efforts are consistent with efforts by other groups to develop and encourage the use of CDEs in preclinical research for epilepsy.4
Rationale for model selection
A Core CRF and CDE document has already been released by the AES/ILAE, which comprises the most critical information to be applied to all preclinical studies for epilepsy3, 4 (e.g., physiology, behavior, electroencephalography [EEG] analysis, and pharmacology). This Core document includes valuable information pertaining to animal diet, strain, age, gender, and so on.5 The present manuscript now details accompanying CDEs and CRFs that extend that prior report to include information determined necessary for sound pharmacologic evaluations in preclinical models of seizure and epilepsy. It is critical to note that the animal models and approaches that are described in this present document do not represent the entirety of available animal models of seizures and epilepsy,6 whether for pediatric, adult, or geriatric epilepsies. We present selected models as representative of those that have been used predominately for antiseizure drug (ASD) development ever since Merritt and Putnam first identified phenytoin in the maximal electroshock test (MES) in 1937.4 Many of the models presented herein are also those in use at the National Institute of Neurological Disorders and Stroke (NINDS)–sponsored Epilepsy Therapy Screening Program (ETSP). These models have been historically used by the NINDS ETSP and other investigators to identify numerous ASDs for clinical use6; there is thus well‐established pharmacology with approved ASDs and strong consensus in the field about anticipated strain/species differences and applicability of the specific model to ASD pharmacology studies.7 Furthermore, because many of the models described in the present manuscript have such significant historical data to support pharmacologic evaluations of investigational agents, we provide herein detailed information to guide CDE/CRF implementation. Our intention is that these CDE and CRF documents can serve as a primer for individuals who are new to pharmacology or studies in animal models of seizures or epilepsy; these CDEs will thus inform on best practices in pharmacologic studies, whether for evaluations of acute anticonvulsant (or antiseizure) efficacy, or for chronic evaluations of disease modification or antiepileptogenesis. We attempted to broadly classify the models described herein into those that present with the following: (1) acute, electrically induced seizures; (2) acute, chemically induced seizures; (3) chronic, electrically induced seizures; (4) chronic, chemically induced seizures; and (5) spontaneous seizures after insult. We omitted detailed description of studies in genetic epilepsy models because the core principles of pharmacology evaluations in these models rely on the principles that are encompassed in one of these 5 categories. For example, thermally induced seizure threshold is a common outcome measure for Scn1a −/− mice evaluations,8, 9 but the principles of group size and threshold to seizure event are consistent with any chemically or electrically induced seizure threshold test described in this document. In this regard, the core principles of this document and accompanying CDEs/CRFs should be broadly applicable to most pharmacologic evaluations for animal seizure or epilepsy models.
Limitations of selected models
We have primarily selected models for evaluation in adult rodent populations that attempt to simulate acute seizures and/or the epilepsies in the general population (Table 1). However, these models do not represent special patient populations, such as pediatric epilepsies, genetic epilepsies, or epilepsy in the elderly. Furthermore, we have provided limited guidance on designing anti‐epileptogenesis and disease‐modification studies, primarily due to the complexity that surrounds such models, as well as the limited consensus in the field with regard to how best conduct these studies. The core principles for pharmacologic evaluations in any model or therapeutic indication, however, are provided within these accompanying CRFs/CDEs. In designing the CDEs and CRFs for pharmacologic studies for epilepsy, this Task Force focused on minimizing the overall burden on researchers who want to use CDEs while still promoting rigor and reproducibility. Herein, we outline the characteristics and components of sound pharmacologic research that are based, in a large part, on the success of the NINDS‐sponsored ETSP.10, 11 For investigators wishing to evaluate investigational agents or pursue pharmacologic studies for specific patient populations, we encourage the implementation of these CRFs and CDEs with adjustments for the specific model at hand. Moreover, although every effort is made to be thorough in these model summaries and CDEs/CRFs, we understand that variability can arise because of region, diet, humidity, animal vendor, and so on, and can contribute to lab‐to‐lab variability.12, 13, 14 Thus, in all aspects of model implementation and development, we strongly encourage that the criteria for any model be independently reviewed and refined within each laboratory, using these CDE/CRFs to guide implementation. This is to say, for example, if a median convulsant dose of pentylenetetrazol (PTZ) is needed by an individual lab, we suggest first evaluating the time and dose response to the desired endpoint in the animal strain of interest prior to selecting the PTZ dose and timing of administration. For basic principles on establishing time‐ or dose‐response relationships, the reader is referred to other manuscripts15 or a practical application.16 Finally, some of the models presented herein are appropriate for both acute antiseizure studies, as well as antiepileptogenesis studies; thus, we have outlined recommendations for CDEs for either study wherever appropriate. The following text provides guidance to implementation and use of the accompanying CRFs/CDES. Wherever possible, examples and background information is included to assist a user with application of these documents to her research.
Table 1.
Human correlate and pharmacology of acute animal model CRFs
| Animal model | Seizure phenotype | Human correlate | Predictive validity | Acutely effective antiseizure drugs |
|---|---|---|---|---|
| Maximal electroshock | Tonic‐extension seizure | Generalized tonic–clonic seizures | Yes | PHT, CBZ, OxCBZ, VPA, PB, FBM, GBP, LTG, LCM, TPM, ZNS, EZG |
| 6 Hz test | Limbic seizures secondarily generalized | Pharmacoresistant focal seizures secondarily generalized | Undefined; suggested model of pharmacoresistant seizures | CBZ, FBM, LCM, LEV, EZG, VPA |
| Subcutaneous pentylenetetrazol | Minimal clonic seizure | Generalized myoclonic seizure | Yes | ESM, VPA, BZD, EZG, FBM, GBP, PBa, TGBa, VGBa |
| Kindled rodent | Limbic seizures secondarily generalized | Focal seizures secondarily generalized | Yes | CBZ, OxCBZ, PHT, VPA, PB, BZD, FBM, GBP, PGB, LCM, LTG, TPM, TGB, ZNS, LVT, VGB, EZG |
| Chemonconvulsant and electrically induced status epilepticus models (acute) | Acute status epilepticus | Acute status epilepticus | Yes | BDZ, CBZ, VPA, PB |
| Post‐status epilepticus models | Spontaneous recurrent seizures | Temporal lobe epilepsy | Unknown | CBZ,97, 98 TPM,99 VPA,97 PB100 |
BDZ, benzodiazepine; CBZ, carbamazepine; ESM, ethosuximide; EZG, ezogabine; FBM, felbamate; GBP, gabapentin; LCM, lacosamide; LTG, lamotrigine; LVT, levetiracetam; OxCBZ, oxcarbazepine; PB, phenobarbital; PGB, pregabalin; PHT, phenytoin; TGB, tiagabine; TPM, topiramate; VPA, valproic acid; VGB, vigabatrin; ZNS, zonisamide.
PB, TGB, and VGB block clonic seizures induced by s.c. PTZ but are inactive against generalized absence seizures and may exacerbate spike wave seizures.
CRFs and CDE Guidance
CRF: pharmacology general core
CRF Filename: 1 General core pharmacology CRF.docx
CDE chart Filename: 1 General core pharmacology CDE Chart.xlsx
The General Core CRF (Appendix S1) covers the recommended core and supplemental elements to support a pharmacologic evaluation in preclinical models of epilepsy. However, this does not mean that every option meets the standard that is required to conduct a valid pharmacologic study. In essence, it is possible that a study is performed “not blinded to treatment” or that the animals are “not randomized to treatment groups”; however, to design a study in this way could confound the interpretation of the data. The form is to be filled in for one individual animal. This CRF only collects data specific to drug characteristics and administration, study design, and so on. For information about each individual animal used in the study, the specific physiological or behavioral outcome measures, and other aspects of testing (e.g., formulation of the investigational drug), please fill in the appropriate additional CRFs listed in Table 2.
Table 2.
List of CRFs/CDEs discussed in this companion manuscript. Files can be located online as Supplemental Material
| CRF/CDE module name | Primary study objective | Route of seizure induction | Outcome measures |
|---|---|---|---|
| 1 General core pharmacology | Pharmacologic testing information | N/A | Drug information (route of administration, frequency, vehicle, solubility, etc.) |
| 2 MES and MEST | Antiseizure test | Electroconvulsant | Maximal electroshock seizure and maximal electroshock seizure threshold ‐ individual seizure response; population seizure threshold |
| 3 Acute 6 Hz | Antiseizure test | Electroconvulsant | 6 Hz seizure and 6 Hz seizure threshold – individual seizure response; population seizure threshold |
| 4 Subcutaneous chemoconvulsant | Antiseizure test | Chemoconvulsant | Chemonconvulsant seizure and chemoconvulsant seizure threshold – individual seizure response; population seizure threshold |
| 5 Timed intravenous chemoconvulsant infusion test | Antiseizure test | Chemoconvulsant | Chemoconvulsant seizure threshold – individual seizure threshold response |
| 6 Intracerebral electrical kindling | Antiseizure test; antiepileptogenesis test | Electroconvulsant | Antiseizure testing: acute protection from seizures; antiepileptogenesis testing: modification of kindling acquisition or behavioral comorbidities |
| 7 Corneal kindling | Antiseizure test; antiepileptogenesis test | Electroconvulsant | Antiseizure testing: acute protection from seizures; antiepileptogenesis testing: modification of kindling acquisition or behavioral comorbidities |
| 8 Chemoconvulsant kindling | Antiseizure test; antiepileptogenesis test | Chemoconvulsant | Antiseizure testing: acute protection from seizures; antiepileptogenesis testing: modification of kindling acquisition or behavioral comorbidities |
| 9 CS_SE | Antiseizure test; antiepileptogenesis test | Chemoconvulsant | Antiseizure testing: modification of SE, or protection from acute symptomatic seizures; antiepileptogenesis testing: modification of SRS frequency, or behavioral comorbidities, or neuropathology |
| 10 CS_ESE | Antiseizure test; antiepileptogenesis test | Electroconvulsant (self‐sustained) | Antiseizure testing: modification of SE, or protection acute symptomatic seizures; antiepileptogenesis testing: modification of SRS frequency, or behavioral comorbidities, or neuropathology |
| 11 CS_fKASE | Antiseizure test; antiepileptogenesis test | Chemoconvulsant | Antiseizure testing: modification of SE, or protection from acute symptomatic seizures; antiepileptogenesis testing: modification of SRS frequency, or behavioral comorbidities, or neuropathology |
The General Core CRF contains specific information for data collection on Drug Characteristics and Administration and Study Design. The specific information for each subsection of this Core CRF is detailed below:
Model: There are specific modules that can be used to fill out CRFs for each acute and chronic seizure and epilepsy model. The following information should be used when conducting an in vivo pharmacology study with any of the CRF/CDEs provided:
Drug characteristics and administration: For every pharmacologic study, the characteristics of a drug (e.g., active pharmaceutical ingredient – API), whether a prototypical ASD or investigational agent, are essential parameters that must be considered for any preclinical study. The drug characteristics, for example, chemical formula (salt factor), manufacturer information, lot number, solubility, half‐life, and brain penetration, determine the route of administration and several aspects of the study design, so this should be considered before the study begins. It is also essential to include drug formulation information (e.g., solution vs. suspension and steps for such formulation) and ensure that vehicle treatment alone does not confer anticonvulsant effect.17
Study design: This section is intended to allow for either pharmacologic interventions against acute seizures (whether through single dose or sub‐chronic/chronic drug administration) or chronic seizures (e.g., antiepileptogenesis or disease modification). In this regard, pretreatment vs. post‐treatment of the drug in relation to the seizure test should be recorded. In most cases, acute seizure suppression studies would rely on a pre‐treatment paradigm, whereas disease‐modification and antiepileptogenesis studies may rely on pre‐treatment or a treatment paradigm after the seizure event (e.g., post‐status epilepticus, see following discussion). This section of the CRF also includes information regarding animal disposition, for example, treatment groups, investigator blinding, and pharmacokinetic samplings. For all acute drug administration studies, we recommend that rodents acclimate to the housing facility for at least 3 (better 7) days and to the testing room for at least 1 h before testing. Some evidence of room acclimatization may also be notable, which should thus be included in the CRF. In some cases it may be valuable to acclimate animals longer, and also acclimate them to the person who will handle them. When possible, investigators may even need to pilot their studies to determine how much acclimation is needed.
- ED50 calculations: For all models, median effective dose (ED50) of an investigational compound is the dose needed to suppress a seizure in 50% of the animals in the treated group (Fig. 1). Dose selection is typically best achieved by first demonstrating activity at a range of doses (e.g., logarithmic steps) and range of time points to define the time of peak activity of a selected agent. Binary outcome measures, e.g., “protected/not protected,” can facilitate determination of an ED50 by the Probit method.18 The same principles can be applied to calculate behaviorally impairing doses (e.g., TD50) or lethal doses (e.g., LD50) for general determination of safety margin of a compound (i.e., protective index19). If the Probit method is included, it is essential that the 95% confidence intervals (CIs) are within the dose range tested. Group sizes should be appropriately powered, that is, 8 animals per group gives 80% power at 95% significance to detect a difference between the groups that is 1.5‐fold greater than the determined standard deviation. In our experience, 5–6 groups (n = 8/group) is sufficient to accurately calculate an ED50 10 (or TD50/LD50) with 95% confidence intervals within the dose range tested. Wherever possible, pharmacokinetic information (e.g., plasma concentrations of API) should be correlated to pharmacodynamic endpoints (e.g., protection from seizures) as well.
Figure 1.
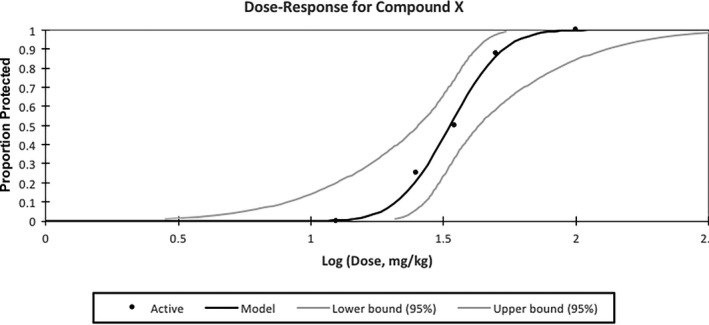 Hypothetical dose‐response curve calculation for Compound X by the Probit method. The ED50 is calculated to be 33.46 mg/kg [95% confidence intervals 25.42–43.03]. The black line indicates the calculated regression line and black dots represent actual data points for each treatment group. Line slope of 6.32 and standard error of 2.08. For an accurate ED50 calculation, the 95% confidence intervals should fall within the dose range tested.
Hypothetical dose‐response curve calculation for Compound X by the Probit method. The ED50 is calculated to be 33.46 mg/kg [95% confidence intervals 25.42–43.03]. The black line indicates the calculated regression line and black dots represent actual data points for each treatment group. Line slope of 6.32 and standard error of 2.08. For an accurate ED50 calculation, the 95% confidence intervals should fall within the dose range tested.
Additional CRFs and CDEs
With the use of the Pharmacology General Core CRF, the majority of the essential information to support a basic pharmacologic evaluation in any preclinical model, whether provided herein or otherwise, will be collected. The following guidelines for the selected models are sufficient to support a basic ASD screening and evaluation platform, similar in scope to the long‐established NINDS ETSP,10, 11 and other approaches that have been able to identify numerous symptomatic treatments for epilepsy.20 Furthermore, the principles for each model should be sufficient to expand to other models of similar etiology (e.g., acute electroconvulsive seizures, acute chemoconvulsant seizures, chronic electroconvulsive seizures, chronic chemoconvulsant seizures, and spontaneous seizures).
Example CRFs
Electroconvulsant models
CRF Filenames: 2 MES and MEST CRF.docx; 3 Acute 6 Hz CRF.docx
CDE chart Filename: 2 MES and MEST CDE Chart.docx; 3 Acute 6 Hz CDE Chart.xlsx
The maximal electroshock test (or MES), maximal electroshock threshold test (MEST), minimal clonic threshold test (MCT), and 6 Hz test all represent frequently used models of electrically evoked acute seizures (Table 1) that are used widely in ASD discovery and can be performed in rats and mice,6, 16, 21, 22, 23 but other animal species are also susceptible to electrically evoked seizures.7 The MES test was first used by Merritt and Putnam in 1937 to characterize the response of cats treated with phenytoin7; 1 year later, phenytoin was clinically available, thus validating the MES test. The MES test represents a model of primary generalized tonic–clonic seizures (Table 1), and probably identifies agents that can reduce seizure spread, as opposed to agents that affect the seizure focus directly.24 Administration of investigational compounds prior to testing in this model provides an indication of an ASD's ability to prevent seizure spread when all neuronal circuits in the brain are maximally active. MES seizures are highly reproducible and electrophysiologically consistent with human seizures. Indeed, numerous ASDs work in the MES test10 (Table 1), including sodium channel‐blocking agents, like phenytoin and carbamazepine. This test thus represents a commonly used, clinically validated assay for the initial identification of potential anticonvulsant activity. It should be cautioned, however, that some clinically effective ASDs (e.g., levetiracetam, vigabatrin, and others) do not show efficacy in this test, so drug testing by the MES test alone is not sufficient to identify relevant antiseizure efficacy.
The 6 Hz seizure model in mice was first described by Brown and colleagues in 1953,25 but was abandoned for more than 40 years due to the lack of anticonvulsant activity of phenytoin in this assay. Barton and colleagues defined the pharmacologic profile of the 6 Hz assay in CF‐1 mice in 2001,16 demonstrating that this assay could effectively differentiate ASDs. This test relies on first determining a median convulsant current (CC50, that is, the current needed to evoke a seizure in 50% of a population), which is generally considered the seizure threshold for this and other electrical stimulation models. It is important to note that the pharmacosensitivity of this model is inversely correlated to the stimulation intensity in use; for example, many ASDs are effective against stimulation at the CC97 (e.g. the threshold to evoke a seizure in 97% of the population), whereas most lose their efficacy at 1.5–2.0‐fold greater current intensities.16 The 6 Hz test‐induced seizures are believed to model focal seizures in humans,25 and as the stimulation intensity increases, those seizures become more and more pharmacoresistant. Moreover, this test is highly differentiating relative to other acute seizure models, such as MES. Levetiracetam can block seizures evoked by a stimulus 1.5‐fold greater than the CC97 in the 6 Hz assay (e.g., 32 mA for male CF‐1 mice), whereas it is ineffective in the MES test and subcutaneous (s.c.) PTZ assays.16 Conversely, sodium channel blockers, such as phenytoin, are ineffective in the 6 Hz assay at doses that do not produce overt motor impairments (e.g., protective index [PI; the ratio of TD50/ED50] is less than 1.5), thereby suggesting that the 6 Hz assay may be a pharmacoresistant model for some categories of ASDs, such as sodium channel modulators.16 The 6 Hz test thus now serves as a primary screening platform in the NINDS ETSP and is frequently used in the discovery of ASDs.10 Moreover, the 6 Hz assay may identify compounds with potentially novel mechanisms of action for the treatment of focal seizures in humans, including anti‐inflammatory agents like minocycline,26 serotonin receptor agonists,10 and adenosine receptor agonists.10 Variations on the protocol of Barton and colleagues16 certainly exist; for example, Walrave and colleagues27 report seizure duration as a reliable outcome measure in the 6 Hz assay and provide an approach for target validation using intracerebroventricular (i.c.v.) delivery of promising test compounds with unknown or limited blood–brain barrier permeability. Finally, genetic background can strongly influence treatment resistance in the 6 Hz seizure model.28 Thus, any attempt to implement the 6 Hz assay in other strains or models with variation in genetic background should first define the CC97 of the test population, and then conduct subsequent pharmacologic studies at 1.5‐ to 2‐fold greater current intensities where pharmacoresistance begins to occur, dependent on the degree of pharmacologic differentiation desired.
Background for electroconvulsive seizure testing
Regardless of intensity, electroconvulsant seizures are induced through corneal or ear clip electrodes. When using corneal electrodes, the animal should be lightly restrained while the electrodes and stimulus are applied to open eyes. A 0.9% saline and local anesthetic (e.g., tetracaine/lidocaine) solution is applied to the eyes to improve electrical conductance and tolerability of the electrode placement. The concentration and timing of local anesthetic administration should be noted in the CRF. Time interval between local anesthesia and stimulation depends on activity of the local anesthetic. Alternatively, electrodes can be attached tightly to the ears of the animal and the stimulus can be applied in the freely moving animal. Conductance through the ear clips can be improved by using topical electrode gels. Care should be taken to ensure that the same electrode location and pressure (for corneal electrodes) is applied to every animal to minimize variation in current conductance (i.e., if stimulation is applied via an open or closed eye or via the edge or the center of the pinna). A specialized device for electrical stimulation of rodents is suitable to deliver the electrical stimulus, as long as the instrument is capable of delivering sufficient electrical current necessary to evoke a seizure (e.g., 150 mA for rats).29 It is important to note the device manufacturer information (e.g., the ECT [electroconvulsive therapy] unit from Ugo Basile30), as this can affect the wave form shape used to deliver the electrical stimulation.28 Finally, electrical wave shape should also be noted (e.g., square wave) because it could also influence the outcome.
For the MES and 6 Hz tests, pharmacologic studies are conducted by administering the same current intensity (e.g., CC97 in the 6 Hz test) to all animals and determining the extent of protection or other seizure outcome following ASD treatment. For the seizure threshold tests, the effect of an investigational agent on the response of a population (recommended minimum n = 20 subjects) can be defined using escalating current intensities until at least 3 points are defined between the limits of 0% and 100% of animals with seizure. As an example, a dose‐response could resemble the following outcomes following administration of compound X (Fig. 1): at 12.5 mg/kg, 0/8 mice are protected from seizures; at a dose of 25 mg/kg, 2/8 mice are protected from seizures; at a dose of 35 mg/kg, 4/8 mice are protected from seizures; at 50 mg/kg, 7/8 mice are protected from seizures; and at 100 mg/kg, 8/8 mice are protected from seizures. With these doses, an ED50 calculated by the Probit method would be 33.46 mg/kg (95% CIs, 25.42–43.03), with a line slope of 6.316 and standard error of 2.085. The suggestions presented below may assist with development and implementation of any electroconvulsive test for pharmacologic studies of anticonvulsant efficacy in rodents. Specifically, these should accompany the Maximal Electroshock Seizure Test (MES)/Maximal Electroshock Seizure Threshold Test (MEST) and Acute 6 Hz Model CRFs.
Seizure Threshold Test (minimal clonic, maximal electroshock threshold, or 6 Hz): This test defines the effect of a drug on the seizure threshold of a population of animals using a staircase procedure.14, 16, 31, 32 The reaction of one animal to the stimulus dictates the current intensity for the following animal. The initial intensity should be the threshold intensity known for the rodent strain, age, or gender, based on whichever animal characteristics (strain, age, etc.) are known; for example, if a genetically modified mouse population on a C57Bl/6 background is going to be tested, we suggest that any initial/pilot studies with that genetic model use information from age‐ and gender‐matched wild‐type C57Bl/6 as a guide until appropriate study parameters can be defined. If the first animal displays the endpoint to be measured (for example, a seizure, such as a tonic hind limb extension seizure for MEST), the current intensity is decreased for the next animal by a predefined amount; if the animal does not show the endpoint, the current intensity is step‐wise increased by a predefined amount, using staircase procedures. A CC50 is defined using this staircase procedure and calculated by Probit,18 as for an ED50. A median threshold can thus be calculated to define the CC50, but the thresholds for individual animals cannot be determined. In addition to a median threshold for a group of animals, drug‐induced increases (anticonvulsant) or decreases (proconvulsant) in seizure threshold can also be determined using the seizure threshold test.33
Model‐specific use of the CRF and CDE The below‐detailed sections should be referenced when completing each CRF for an MES/MES threshold (MEST) or 6 Hz/6 Hz threshold test.
CRF: MES and MEST test
CRF Filenames: 2 MES and MEST.docx
CDE chart Filename: 2 MES and MEST CDE Chart.docx
Stimulation protocol
A suprathreshold stimulus that will reliably induce hind limb tonus in 100% of naive animals is used; this stimulus should be low enough that ASDs (e.g., phenytoin or phenobarbital) dose‐dependently suppress seizures. For example, 50 or 60 Hz of alternating current (i.e., 50 mA in CF‐1 mice and 150 mA in Sprague‐Dawley rats19) is most often delivered for 0.2 s by corneal electrodes. It is not possible to determine the seizure susceptibility of an individual animal in this assay. Therefore, the type of test used (MES) or MES threshold (MEST) should be selected a priori in the CRF module. As detailed in the preceding general section for electroconvulsant models, general parameters of stimulation duration, frequency, intensity, waveform characteristics, and so on, are essential to record in this CRF module. Surgical preparation and local anesthetic application: Stress can affect seizure threshold and potentially confound any study results; thus any surgical preparations should be recorded on the CRF.34 However, the surgical specifications should be recorded in the General Core CRF. Local anesthetic information should also be noted in this section. Outcome measures: The specific seizure endpoint measures, for example, hindlimb extension and/or duration of extension, should also be clearly noted in the CRF. The use of binary protection criteria (e.g., protected/not protected) allows for determination of an ED50 by the Probit method18 and thus, if a dose‐response study is used, this criteria should be clearly defined and recorded. Recordings of electrographic seizures via EEG are possible and, and if used, should be clearly indicated on the CRF.
CRF: acute 6 Hz test
CRF Filenames: 3 Acute 6 Hz CRF.docx
CDE chart Filename: 3 Acute 6 Hz CDE Chart.xlsx
Stimulation protocol
Limbic psychomotor seizures are induced via corneal stimulation according to the following specifications: 6 Hz frequency, 0.2 msec rectangular pulse width, 3 s duration.16 The stimulation current should be defined for the animal strain in use and be sufficient to elicit seizures in all animals (i.e., CC97; this would be 22 mA for male CF‐1 mice derived from Charles River10, 16). As the stimulation intensity increases, the pharmacosensitivity of the 6 Hz assay is reduced.10, 16 Thus it is imperative that the stimulation intensity be defined a priori for the strain and species in use.35 Surgical preparation and local anesthetic application: As with the MES/MEST CRF, basic latency between surgical intervention and seizure testing should be noted to minimize the potential for stress‐induced confounding effects. Outcome measures: Seizures following 6 Hz stimulation in mice and rats are characterized by a stunned or fixed posture, forelimb clonus, and elevated Straub tail16 (for detailed images and description of the Straub tail, see Ref. 36). Although the 6 Hz approach is widely used in mice, there is no consensus on the endpoints to define anticonvulsant protection: some groups describe protection as the presence or absence of seizures,16 whereas others consider an animal to be protected if it resumes normal exploratory behavior within 7–10 s after stimulation such that total seizure duration (in seconds) is an additional endpoint measure.27, 28 For these reasons, the various endpoint measures to be included in the study should be defined clearly in the 6 Hz CRF module. As with MES, binary endpoint measurements (protected/not protected) based on the specific seizure endpoints will accommodate an ED50 determination.18
Chemoconvulsant models
CRF Filename: 4 Subcutaneous chemoconvulsant CRF.docx; 5 Timed intravenous chemoconvulsant infusion test CRF.docx
CDE chart Filename: 4 Subcutaneous chemoconvulsant CDE Chart.docx; 5 Timed intravenous chemoconvulsant infusion test CDE Chart.docx
Background for chemoconvulsant seizure testing
Chemoconvulsant models have been at the forefront of ASD discovery for decades. These models can discriminate ASDs, due, in part, to the differential route of chemoconvulsant administration (e.g., s.c. vs. i.v.), presumed pharmacologic mechanism of action, and seizure pattern observed in response to each chemoconvulsant agent. For example, Goodman and colleagues defined the differential presentation of maximal seizures induced by the intravenous PTZ infusion versus the maximal electroshock (or MES) test in 1953, providing some of the first information to differentiate phenytoin from other ASDs.37 Of note, the i.v. PTZ test was found to produce maximal (generalized clonic and tonic) seizures similar to the MES test, but the patterns of seizure presentation differed markedly between the 2 tests; for example, i.v. PTZ results in first a clonic phase and then a tonic phase (i.e., clonic‐tonic seizures), whereas MES results in tonic‐clonic seizures,37 that is, the tonic phase precedes the clonic phase. This work further confirmed that phenytoin does not raise seizure threshold, but rather prevents seizure spread,38 setting the stage for ASD differentiation studies for decades.15 Moreover, the subcutaneous (s.c.) PTZ test has been validated clinically with the identification of ethosuximide (and trimethadione) for absence seizures.39, 40 This is likely because ethosuximide and trimethadione exhibit a mechanism of action that is substantially different from other ASDs, for example, they block thalamic “T” type calcium channels implicated in the spike‐wave discharges of absence epilepsy.41 An added advantage for the use of chemoconvulsant models is that they are also relatively simple and easily implemented for early pharmacologic evaluations in large cohorts of animals. Thus, the chemoconvulsant models are useful to differentiate ASDs in acute seizures. However, they also have limitations. For instance, the anti‐absence activity of levetiracetam and lamotrigine or the absence‐aggravating effect of vigabatrin were not determined by the s.c. PTZ test, but instead in genetic models of absence epilepsy.42 Thus, genetic rodent models with spontaneous spike‐wave discharges are more predictive for clinical anti‐absence efficacy.41
Model‐specific use of the CRF and CDE
The following sections should facilitate completion of the Subcutaneous Chemoconvulsants and Timed Intravenous Chemoconvulsant Infusion CRFs. Of note, the CDEs of these models require a baseline determination of the effective convulsant dose (e.g., CD95) of any chemoconvulsant at a fixed concentration in the preclinical species of interest. The sections below provide doses of PTZ, bicuculline (Bic), or picrotoxin (Pic), as historically defined for use in male CF‐1 mice derived from Charles River Laboratories weighing between 18 and 35 g.43 The infusion rates and concentrations, where relevant, should also be defined by an investigator a priori for the animal model of interest, and this information should also be included as a core component of this model and recorded in the CRF.
CRF: subcutaneous chemoconvulsant
CRF Filename: 4 Subcutaneous chemoconvulsant CRF.docx
CDE chart Filename: 4 Subcutaneous chemoconvulsant CDE Chart.docx
Animal history
As with the electroconvulsive seizure models, animal history that could affect seizure susceptibility should be recorded. Test parameters. This section details essential information to conduct a subcutaneous chemoconvulsant study. For each type of subcutaneous chemoconvulsant most frequently used, we provide model‐specific information to facilitate such a study. Regardless of the chemoconvulsant used, the essential information to be recorded include the chemoconvulsant dose, concentration, solvent, and duration of monitoring.
Subcutaneous pentylenetetrazol
Administration of subcutaneous PTZ produces clonic and, later, infrequent tonic seizures in rodents. At times that are dependent on the compound under study, the dose of PTZ that will induce convulsions in >97% of animals (e.g., 85 mg/kg in a 0.85% solution in saline in male CF‐1 mice; 68 mg/kg in a 3.4% solution in saline in male Sprague‐Dawley rats44) is injected into a loose fold of skin in the midline of the neck. The animals are placed in isolation cages to minimize stress43 and observed for an extended time frame (typically 30 min). Outcome measures in this assay include the presence or absence of a generalized clonic seizure as measured according the Racine rating scale45 or other appropriate scale, latency to first clonic twitch, latency to tonic seizure, number of clonic events, and so on. Other measures can also be made, such as electrographic seizures or mortality, depending on the goals of the study.
Subcutaneous bicuculline or picrotoxin
Subcutaneous administration of the chemoconvulsants bicuculline (Bic) and picrotoxin (Pic), can induce clonic and tonic seizures in a manner behaviorally similar to that of s.c. PTZ.46 These models can demonstrate a candidate compound's ability to confer protection against effects of a subcutaneous injection of the chemoconvulsant (e.g., CD97 of Bic and Pic, respectively: 2.7 or 2.5 mg/kg in male CF‐1 mice). After administration of the chemoconvulsant, we recommend a period of behavioral seizure observation, typically at minimum 30 min for Bic, and 45 min for Pic because of the slower absorption of this agent.46, 47 Bic‐ and Pic‐induced seizures typically consist of an episode of clonic seizures of the fore‐ and hind limbs, jaws, and vibrissae.46, 47 Bic‐induced clonic seizures are generally followed by tonic extension of the hind limbs and, in some cases, death.46, 47 Outcome measures (e.g., presence/absence of seizure) and associated considerations are similar to those described for s.c. PTZ.
CRF: timed intravenous chemoconvulsant infusion test
CRF Filename: 5 Timed intravenous chemoconvulsant infusion test CRF.docx
CDE chart Filename: 5 Timed intravenous chemoconvulsant infusion test CDE Chart.docx
Test parameters
The i.v. PTZ test is used to determine whether a compound affects seizure threshold; for example, whether a compound increases or decreases the threshold at which a seizure can be induced. Mice are pretreated with the investigational agent prior to being injected with i.v. PTZ.48 Infusion protocol. At the time of desired testing (e.g., time of peak effect of the ASD), PTZ is infused at a constant rate (for CF‐1 mice, 0.34 ml/min) into the lateral tail vein. Restriction of animals during infusion may lead to stress‐induced threshold alterations, so infusion without restriction (via a flexible tubing) is preferred. At the start of the PTZ infusion, a hemostat clamped to the guide tubing to prevent backflow is removed, the infusion started, and recording of latency to each outcome measure is started. The latency (in seconds) from the start of the infusion to the appearance of the “first twitch” and/or the onset of sustained clonus is a primary outcome measure of this model. Outcome measures and parameters determined during infusion. Other behavioral outcome measures can be included as supplemental elements, for example, mortality rate, duration of clonus, etc. Because subtle changes in environmental conditions may impact seizure threshold, a vehicle treatment group should always be run in parallel. The mean latency for each of the groups, and the significant difference between the test groups and the vehicle‐treated group is calculated to define the impact of a test compound on seizure threshold. An increase in mg/kg to first twitch or to clonus indicates that the test substance increases seizure threshold.
The times to each endpoint are converted to mg/kg of PTZ for each animal as follows:
CRF: kindling models for antiseizure testing
CRF Filenames: 6 Intracerebral electrical kindling CRF.docx; 7 Corneal kindling CRF.docx; 8 Chemoconvulsant kindling CRF.docx;
CDE Chart Filenames: 6 Intracerebral electrical kindling CDE chart.xlsx 7 Corneal kindling CDE chart.xlsx; 8 Chemoconvulsant kindling CDE chart.xlsx;
Background for kindling models
Kindling models mimic, at least in part, the epileptogenesis process and continue to be used widely because of the ability of these models to predict clinically useful drugs with anticonvulsant efficacy against focal and secondarily generalized seizures.49 As such, electrical amygdala kindling in rats has been clinically validated because this model correctly predicted the efficacy of levetiracetam against focal seizures in patients. The phenomenon of kindling is defined as a process in which repetitive administration of an initial subconvulsive stimulus leads to the progressive development of first, focal seizures, followed by recruitment of more brain areas and secondary generalized seizures. Kindling, typically in mice or rats, is suited to both evaluation of chronic drug intervention to impact epileptogenesis,50 as well as to evaluation of acute anticonvulsant efficacy once seizures are established.10, 51, 52, 53, 54, 55 For this reason, the model description and CRFs/CDEs are closely linked. Because of the diversity of kindling models (chemical, electrical; implanted, corneal) and intended outcomes for these studies (e.g., acute anticonvulsant vs. antiepileptogenesis studies), the CRFs and CDEs developed herein have been limited. Furthermore, we have elected to broadly define kindling models in this guideline document, but specific aspects of each type of kindling are including in the respective CRFs/CDEs.
Model‐specific use of the CRFs and CDEs
The first and best characterized kindling model is kindling using an electrical stimulus of the basolateral amygdala in rats, thus this manuscript guideline will primarily focus on electrical kindling models. The reader is referred to “CRF module 6: Intracerebral electrical kindling models – antiseizure” to guide implementation of electrical kindling for acute anticonvulsant efficacy studies. Animal history. Kindled rodents can be repeatedly used for acute drug studies after acquisition of the fully kindled state, thus an animal's history should be expressly recorded in all CRFs for antiseizure kindling studies. Surgery. Electrical kindling can be achieved by stimulus of numerous brain areas, such as hippocampus or perirhinal cortex. Kindling parameters. In completing the CRF, the stimulation location and stimulation parameters should be noted carefully as the kindling rates, the exact sequence, and manifestations of the behavioral stages in the progression of kindling and the recruited network differ when kindling is performed in different brain sites,56 or by different stimulation parameters.57, 58 Stimulation parameters. The stimulation parameters should be noted (e.g., current and intensity of stimulations, frequency of stimulation trains, behavioral vs. electrographic seizure scoring, etc.), including information concerning outcome measures to establish a fully kindled experimental animal. Rodents implanted are amenable to the recording of afterdischarge threshold (ADT) and afterdischarge duration (ADD), which can provide an additional outcome measure for seizure testing.
There are numerous electrical kindling protocols in use for acute anticonvulsant pharmacologic studies, including single‐day kindling in rats57, 59 and corneal kindling in mice.51 The following guidance should be used to complete the “CRF module 8: Corneal kindling.” Stimulation protocol: Much like electrical kindling in rats, the corneal kindled mouse has various stimulation parameters. Therefore, the stimulator equipment should be recorded as well as the stimulation parameters themselves (e.g., frequency, current intensity, number of stimulation events/day, and delay between stimulation events). Relative to the traditional 60 Hz corneal kindling model, the 6 Hz kindling procedure demonstrates a more pharmacoresistant profile.30 Differences in mouse strain may also promote accelerated kindling acquisition60 and this is one reason why mouse strain is a CDE to be recorded in the Core CDE file4 before commencing use of any model‐specific CRF, including Corneal Kindling. As with the electrical seizure models, the corneal kindling model requires a topical anesthetic applied to the corneas. Furthermore, the scoring system to define a seizure and determine kindling criterion should be clearly noted. Drug administration studies: Although the corneal kindled mouse was originally described nearly 3 decades prior to robust incorporation for drug discovery for epilepsy,61 this model has since gained popularity because of the noninvasive, less labor‐intensive, and more cost‐effective process51, 62 relative to the classical electrical kindling paradigms. This is further supported by the fact that the corneal kindled mouse is now one front‐line assay used for preclinical ASD screening by the NINDS ETSP.10 The animal is typically stimulated for a baseline seizure and then tested for presentation of a seizure at some protracted time point post‐drug administration; the separation between baseline seizure testing and post‐drug administration testing should be clearly noted. There is typically a 24‐h delay between baseline seizure testing and drug administration,62 albeit modifications certainly exist (e.g., same day protocol63). Therefore, the latency between baseline and drug testing should be noted.
It is also feasible to kindle rodents by repetitive chemoconvulsant administration. Animal history. As for the electrical and corneal kindling model CRFs, the prior animal history should be noted. Chemoconvulsant kindling parameters. The chemoconvulsant information (dose, purity, and formulation information) is essential to be recorded. The frequency of administration and route of administration also can dramatically affect the kindling acquisition rate and is thus a core element of the chemoconvulsant kindling CRF/CDEs. With the exception of the kindling stimulus, the use of chemoconvulsant‐kindled rodents does not differ significantly from electrically kindled rodents in the outcome measures for antiseizure studies.
For all antiseizure studies in kindled rodents, it is noteworthy that appropriate and similar handling of all experimental animals before the experiments (i.e., handling habituation) is also recommended to reduce bias because acute stress mostly exerts anticonvulsant effects, whereas chronic stress conditions might worsen seizures.34 This is one reason that several aspects of housing are included in the Core CDE (see Harte‐Hargrove et al.4). To alleviate any potential confounds of stress‐induced effects on seizure susceptibility, control groups of rodents should receive adequate mock stimulations and similar handling, as well.
Background of kindling for antiepileptogenesis/disease modification studies
Kindling models can be broadly applied to antiepileptogenesis or disease modification studies; for example, a drug can be administered to determine whether there could be a delay or modification of the kindling process.64 For example, levetiracetam displayed disease‐modifying effects in the amygdala‐kindled rat.52, 65 Interpretations of these models have been also applied to models of pharmacoresistant seizures, including the selection of phenytoin‐resistant and phenytoin‐sensitive amygdala‐kindled rats,66 and lamotrigine‐resistant amygdala‐kindled rats67 and corneal kindled mice,68 which are kindled in the presence of therapeutic doses of lamotrigine and are subsequently resistant to lamotrigine and carbamazepine. Thus, kindling is a useful platform on which to evaluate acute anticonvulsant effects with established chronic seizures, as well as to evaluate the impact of therapeutic intervention on the epileptogenesis process and behavioral comorbidities associated with epilepsy. It should be noted, however, that the antiepileptogenic or disease‐modifying effects identified in this way have not been validated in human trials to date, which is to say that no treatment has been identified for clinical use as an antiepileptogenic agent. The same is true for other animal models of epileptogenesis (e.g., post‐status epilepticus models discussed below).
In addition to spontaneous seizures, kindled rodents exhibit a number of behavioral comorbidities of epilepsy,60, 63, 69, 70, 71, 72 a finding consistent with clinical reports that seizures in humans are just one symptom of epilepsy diagnosis.73 It is important to note that kindled rodents consistently demonstrate increased anxiety‐like behaviors and cognitive deficits in a diversity of approaches performed by numerous independent investigators,60, 63, 67, 68 further supporting the utility of kindling for studies of disease‐modification and/or epileptogenesis and demonstrating that these behavioral comorbidities are relevant biomarkers in kindling models, as well as models with spontaneous recurrent seizures (SRS). Kindled models offer the capability to tightly control the epileptogenesis process in a manner that is distinct from other models of epilepsy.74, 75 Kindled rodents do not exhibit spontaneous seizures at the time of acquisition of kindling criterion, but spontaneous seizures do occur in kindling models76 (i.e., “overkindling”). The lack of spontaneous seizures early in the kindling process is thus a potential limitation of kindled rodent models, which may only reflect partial epileptogenesis at this stage. Nonetheless, the use of kindled rodents for pharmacologic evaluations may help identify drugs that improve comorbidities, and kindled rodent models provide numerous avenues to evaluate both acute anticonvulsant effects of investigational therapies, as well as antiepileptogenic or disease‐modifying effects of such agents.
Model‐specific use of the CRFs and CDEs
Antiepileptogenesis studies in kindling models requires similar test parameters as included for antiseizure testing (e.g., surgical procedures, kindling parameters, and so on). Antiepileptogenesis/disease modification studies. Antiepileptogenesis studies differ from acute antiseizure studies in that treatment should be administered prior to or during kindling acquisition, rather than after establishment of the fully kindled state. Therefore, the timing of the drug administration and criteria for treatment should be recorded. For antiepileptogenesis studies in the kindling model, it is important to continue with kindling stimulations after drug washout to exclude that any effect on kindling acquisition is only secondary to an anticonvulsant effect of the compound.52
Status epilepticus models
CRF Filename: 9 CS_SE CRF.docx; 10 CS_ESE CRF.docx; 11 CS_fKASE CRF.docx;
CDE chart Filename: 9 CS_SE CDE Chart.xlsx; 10 CS_ESE CDE Chart.xlsx; 11 CS_fKASE CDE Chart.xlsx
Background of status epilepticus models
The most widely used experimental approach to study epileptogenesis and/or the effects of drugs on SRS is the induction of status epilepticus (SE) in rodents (whether anesthetized or conscious) either by the systemic or focal injection of chemoconvulsants (i.e., kainic acid or pilocarpine) or by electrical stimulation of selected brain regions (e.g., self‐sustained limbic SE). Chemoconvulsant‐induced SE in rodents serves as an important surrogate to that observed in humans and is often employed in the search for novel therapies that can halt refractory SE and prevent the negative behavioral and neuroanatomic consequences associated with uncontrolled SE.77 These models are amenable to EEG recordings to confirm acute SE, and also for long‐term video‐EEG monitoring, as these animals develop SRS, neuropathologic damage, and cognitive deficits that reproduce the facets of temporal lobe epilepsy (TLE).77, 78, 79, 80, 81 Chemoconvulsant models are routinely used in mice82, 83, 84, 85 and rats80, 81, 86; the self‐sustaining limbic status epilepticus (SSLSE) model is also applicable to both mice87, 88 and rats.89 With the exception of the induction of the precipitating insult (either chemoconvulsant or electrical stimulation) that initiates epileptogenesis (e.g., SE), these models share many characteristics and can thus be generally described for the use of pharmacologic studies. These models all lead to an acute SE period, and subsequent onset of SRS days to weeks later.81, 82, 83, 86, 90, 91 One important limitation of the post‐SE models is the high mortality rate associated with the neurologic insult, as well as the need to intervene during the SE insult period to improve survival rates. This is in stark contrast to other models described earlier (e.g., kindling models), wherein pharmacologic intervention is not needed to improve long‐term survival and rodents generally do not exhibit significant mortality during the kindling acquisition process.87 Another limitation of post‐SE models is the need for laborious video‐EEG monitoring over prolonged periods to quantitate the occurrence and frequency of SRS. Video recording alone is not sufficient, because most focal (nonconvulsive) seizures are missed without EEG, recorded via single or multiple skull or depth electrodes. In most post‐SE models, the frequency of spontaneous electroclinical seizures is low and SRS often occur in clusters with long intercluster intervals, thus it is recommended that continuous (24/7) video‐EEG monitoring is performed over several weeks,92 rather than intermittent recordings. Although several automated or semi‐automated methods for seizure detection in the EEG have been described, a generally accepted and validated algorithm is lacking, so visual video‐EEG analysis of seizures is still a gold standard. The location and number of recording electrodes will depend on the specific animal model, to what extent each model has been previously characterized with regard to EEG features, and on the specific research objectives.
As an alternative to continuous 24/7 video‐EEG monitoring for SRS in pharmacologic studies in epileptic rodents, Blanco et al. suggested that the acute induction of MES or PTZ seizures in such epileptic rodents might constitute an effective and valuable method to screen ASDs and to study mechanisms involved in pharmacoresistant TLE.93 This idea has been explored by other groups, demonstrating that determination of electrical or chemical seizure thresholds in epileptic rodents (as is produced by pilocarpine) provides an interesting tool for drug testing in chronically epileptic rodents in a moderate‐throughput testing approach suitable for pharmacologic studies.94
The sections below are to be used when referring to the Chronic Seizures Following Chemoconvulsant‐Induced Status Epilepticus (CS/SE) CRF.
CRF: chronic seizures following chemoconvulsant‐induced status epilepticus (CS/SE)
CRF Filename: 9 CS_SE CRF.docx
CDE chart Filename: 9 CS_SE CDE Chart.xlsx
Model‐specific use of the CRFs and CDEs
Electrographic Seizure Electrode Implantation: Animals subjected to video‐EEG monitoring allow investigators to record the seizures that occur during the electrographic component of SE (electrographic SE; ESE) and chronic seizures. The recordings can also be made so that the onset of chronic seizures is defined. Electrodes should be implanted at least 7 days before SE to allow for sufficient post‐procedural recovery and metabolism of any accompanying anesthetic. The specific locations of the electrodes should be decided according to study objectives. In addition, information concerning the recording electrode specifications and materials should be recorded, if applicable.
Convulsive and Electrographic Seizure Induction: Kainic acid (KA) and pilocarpine are 2 common methods for SE induction in mice and rats and are provided in this section to serve as specific examples for usage of this CRF. This portion of the Chronic seizure induced by chemoconvulsant‐induced status epilepticus (CS/SE) CRF details the explicit study endpoints used to define the seizure severity and onset of SE. Acute SE Onset and Acute Behavioral Observations. If a study is relying solely on behavioral seizures and not EEG activity, the Racine scale is useful,95 but modifications of this scale, or alternative observational strategies are also appropriate as they align with the study goals.
Study design should be used to identify the study goals (e.g., acute antiseizure or antiepileptogenesis). The SE models are amenable to acute pharmacologic interventions to halt SE at various time points (e.g., pretreatment before the administration of the chemoconvulsant or post‐treatment after the administration of the chemoconvulsant). The specific timing of pharmacologic intervention should be carefully designed to align with investigator objectives; for example, a study might be designed to test the hypothesis that a drug will prevent SE, whereas a disease‐modification/antiepileptogenesis study could be designed to test the hypothesis that treatment will influence outcomes after SE96. Drug testing and outcome measures. If a pharmacologic intervention is used to arrest or attenuate SE activity, the specific drug information, dose(s), route, formulation vehicle, and timing of administration should also be recorded. Often, in the case of post‐SE studies, the SE insult is terminated after a predetermined duration (e.g., 90 min in chemoconvulsant models and 3–4 h in electrical stimulation models) to reduce mortality (however, SE may recur during subsequent hours because of the rapid elimination of most ASDs used to halt SE). This also needs to be recorded in the CRFs because it is likely to influence later outcome measures if animals are to be used for studies of longer duration than acute SE intervention studies (e.g., antiepileptogenesis). Investigational compounds can be administered at relevant time points of interest (pre‐SE onset, during active SE, or into the latent period, or in the chronic phase of SRS), with doses, source, route recorded in the CRF. Thus, use of the CRF can assist investigators in comparing their data to those in the literature and making consistent evaluations in their own laboratories.
CRF: electrical stimulation model of status epilepticus: SSLSE induced by continuous hippocampal stimulation
CRF Filename: 10 CS_ESE CRF.docx
CDE chart Filename: 10 CS_ESE CDE Chart.xlsx
Model‐specific use of the CRFs and CDEs
Amygdala (lateral and basolateral nuclei), hippocampus, and perforant path are the brain structures of the limbic system in which area‐specific stimulation paradigms are most commonly applied to induce SE.79, 89, 97 The advantage of using electrical stimulation models of SE versus chemoconvulsant‐based models of SE is that it is possible to stop the stimulation as soon as SE becomes self‐sustained, that is, when SE continues after the epileptogenic stimulus is withdrawn. This feature allows tight control of the severity of SE, thereby reducing the mortality (~10%), despite a high percentage of animals (>90%) that develop self‐sustained SE and TLE.95 Moreover, this procedure allows a high‐degree of consistency in the response of individual animals, providing a drug‐testing platform that requires far fewer animals than other, more variable models of post‐insult‐acquired epilepsy.98 Nevertheless, stimulation models require dedicated equipment to deliver electrical stimulation, surgical preparation, and are more time consuming.
The protocols below are suggested based on the continuous hippocampal stimulation (CHS) paradigm originally described by Lothman and colleagues.97 It should be noted also that the model described below is for CSE induced by electrical stimulation of the hippocampus, but the reader is also referred to other examples wherein nonconvulsive ESE is induced by continuous stimulation of the perforant path to also subsequently lead to a TLE‐like condition without hippocampal sclerosis.99
CHS implant surgery
Experimental subjects are implanted bilaterally in the temporal pole of the hippocampus (recommend coordinate from bregma in male Sprague Dawley rats: AP −4.7 mm, ML ±5.0 mm, −5.0 mm below dura). The electrode coordinates, metal type, formation (twisted/concentric), length, and diameter are all essential to include in the CRF documents. The stimulation of the temporal aspect of the hippocampus promotes a higher rate of inducing SE versus the septal pole,97 but the investigator is encouraged to determine the specific location prior to embarking on a pharmacologic study. Electrodes should be tested with an Ohm‐meter before use to identify possible breaks in the insulation as a consequence of the manufacturer process (especially with twisted wire electrodes), which can result in stimulation failure. As described originally, “the tips of the twisted electrodes are cut with scissors at a 45‐degree angle, with the tips separated approximately 1 mm.”100
Electrical stimulation
As with any electrode implant surgery, animals should be given several days (typically 7 days) to recover from surgical electrode implantation, and then at least 30 min of baseline electroencephalographic (EEG) activity is recorded before the afterdischarge threshold (ADT) is determined in each rat using constant current stimulus (1 msec monopolar square waves, 50 Hz for 2 s). Determination of the ADT is done using an ascending stepwise procedure,99 which is similar to that for CC50 or seizure threshold determination described earlier.97 As an example, the initial current intensity could be set at 50 μA; then the current is increased by 10 μA up to a maximal value of 250 μA by intervals of 1 min until one afterdischarge (AD) of at least 3 s duration is elicited. Only animals with threshold values ≤250 μA (in our experience approximately 90% of the implanted rats) are used; the lack of AD induction in rats exposed up to 250 μA reduces the chances to successfully evoke SE.97 The duration of stimulation and other recording parameters should thus be recorded on this CRF.
Before electrical stimulation, EEG baseline hippocampal activity in freely moving rats should be recorded for a recommended period for 24 h. Then, rats are unilaterally stimulated according to the strain and gender in use. As an example of stimulation procedure in male Sprague Dawley rats, unilateral stimulation (same hemisphere of ADT) in the hippocampus is introduced at 400 μA (50 Hz, 1 msec biphasic square waves in 10 s trains delivered every 11 s) to override postictal refractoriness.97 This stimulation paradigm is applied for 9 min, followed by 1 min of “stimulus‐off” period, during which EEG and behavior seizure score (according to Racine rating scale) are recorded.97, 101, 102 Together, the stimulation and the “stimulus‐off” periods constitute a 10 min epoch. In our experience, at least 6 epochs of stimulation will reliably induce self‐sustained SE; if this endpoint is not reached following 9 epochs of stimulation, SE is unlikely to occur; thus the stimulation should be stopped and the rat removed from further study.97 However, the study goals will dictate the recording and stimulation paradigm necessary. EEG SE has to occur for at least 30–120 min after the end of CHS for the rats to be considered positive for the development of self‐sustained limbic SE (SSLSE). However, as with all other models described for pharmacologic studies, the investigator is encouraged to determine the duration, frequency of stimulation, and other stimulation induction parameters necessary to elicit SSLSE in the strain in use. SE‐induced mortality is not observed in this model, unlike chemoconvulsant SE models. In this regard, the electrical stimulation SE model is amenable to pharmacologic studies.
CRF: focal kainic acid seizure induction
CRF Filename: 11 CS_fKASE CRF.docx
CDE chart Filename: 11 CS_fKASE CDE Chart.xlsx
Background of the focal kainic acid model
Although similar to other SE models due to the induction of SE, the focal KA SE model is becoming increasingly popular because it is associated with reduced mortality and leads to frequent spontaneous electrographic discharges, although convulsive seizures vary in frequency.103, 104, 105, 106 Linking the CRF about SE induction to other CRFs about the EEG will allow investigators to document the variables associated with SE induction, as well as long‐term recordings of spontaneous seizures (see Ono et al. in this special issue).
Model‐specific use of the CRF and CDEs
In addition to systemic KA administration, KA can be focally injected in hippocampus, amygdala, or overlying cortical sites to produce an ipsilateral epileptic focus in mice or rats.94, 104 Information concerning surgical procedures should thus be recorded in the Surgery section. Kainic Acid (KA). This diversity of methods is the reason that the CRF needs to be clear about the use of KA. Information concerning the focal injection site coordinates, KA source, KA concentration, and infusion volume, and rate are all critical components to include in this CRF. SE Induction and SE Assessment include important parameters concerning how KA is administered to induce SE and the endpoints to define SE onset itself. Once the SE induction procedure has terminated and an animal is a candidate for pharmacologic study, the study type should be accordingly noted in Study Type (e.g., antiseizure or antiepileptogenesis). Drug Testing and Outcome Measures. As with the previously described CRF/CDE modules, this section should contain the relevant drug administration parameters (e.g., pre‐ or post‐treatment, route of administration, and other outcome measures of the fKASE model. It is important to note that the acute and long‐term animal disposition should also be recorded, for example, frequency and duration of EEG recording and/or duration of behavioral monitoring, as well as housing conditions (single‐housed, paired, etc.). Drug Administration During Chronic Period (Antiseizure Study). Long‐term monitoring. Because the fKASE model is amenable to both antiseizure and antiepileptogenesis studies, the long‐term monitoring parameters should be accordingly noted. This includes whether animals are housed singly or socially, the duration of monitoring, and whether behavioral comorbidities are assessed.
Conclusions
We presently provide the background and rationale for designing CRFs and CDEs for pharmacologic approaches to preclinical epilepsy research, focusing on animal models that have made significant contributions to preclinical development of clinically approved ASDs. The reader is referred to the reference list for a more exhaustive description of the models presently detailed. The use of CDEs in preclinical epilepsy research is gaining increasing priority to harmonize approaches to data collection and scientific methodology of these studies,5 as well as limit interinvestigator variability. It is thus our intention that the CRFs and CDEs developed and presented herein may provide a means to further standardize laboratory approaches to preclinical pharmacologic interventions in models of acute and chronic seizures and epilepsy.
Disclosure of Conflicts of Interest
MBH, LCHH, and CB have received reimbursement by the AES, ILAE, and NINDS for attending the meetings of the ILAE/AES Joint Translational Task Force. BX, TR, IS, and WL declare no conflicts of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Appendix S1. Core CRF and CDE files. The CDE and CRF modules linked to this article can be found and downloaded as a zip folder.
Acknowledgments
This report was written by experts selected by the International League Against Epilepsy (ILAE) and the American Epilepsy Society (AES) and was approved for publication by the ILAE and the AES. Opinions expressed by the authors, however, do not necessarily represent the policy or position of the ILAE or the AES. We are also grateful to the AES, ILAE, and the National Institutes of Health (NIH)/NINDS for partial sponsoring of the activities of the ILAE/AES Joint Translational Task Force. This report is a product of the Pharmacology working group of the TASK3 of the ILAE/AES Joint Translational Task Force. We gratefully acknowledge the editorial review of numerous versions of this manuscript by Drs. Jacqueline French, Aristea S Galanopoulou, and Helen Scharfman. MBH has received support from the National Institute of Neurological Disorders and Stroke Epilepsy Therapy Screening Program contracts HHSN 271201100029C and 271201600048C, the University of Washington, and the Institute for Translational Health Sciences supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1 TR002319. LCH received travel reimbursement for meetings for the work done through the TASK3 of the ILAE/AES Joint Translational Task Force. LCH is currently Associate Director of Research at Citizens United for Research in Epilepsy (CURE), but this position has created no conflict of interest for the content of this manuscript. TR has received support from the European Union Seventh Framework Program (FP7/2007‐2013) under grant agreement no. 602102 (Targets and biomarkers for antiepileptogenesis, EPITARGET) and Citizen United for Research in Epilepsy (CURE). IS has received grant support from Vrije Universiteit Brussel (OZR2102, SRP40), the Fund for Scientific Research Flanders (G.028716N) and the Queen Elisabeth Medical Foundation (Prize ING). BX has received support from the National Natural Science Fund of China, no. 81671299 and 81371435. CB and WL acknowledge support from the German Research Foundation (DFG, Bonn, Germany; grant # LO 274/1–LO 274/16), the National Institutes of Health (NIH; Bethesda, MD, USA: grant R21 NS049592), the Niedersachsen‐Research Network on Neuroinfectiology (N‐RENNT) of the Ministry of Science and Culture of Lower Saxony in Germany, and the European Union's Seventh Framework Programme (FP7) under grant agreement 602102 (EPITARGET). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding agencies listed.
Biography
Melissa Barker‐Haliski is research assistant professor of pharmacy at the University of Washington.

References
- 1. Arrowsmith J. Trial watch: phase II failures: 2008‐2010. Nat Rev Drug Discov 2011;10:328–329. [DOI] [PubMed] [Google Scholar]
- 2. Perrin S. Preclinical research: make mouse studies work. Nature 2014;507:423–425. [DOI] [PubMed] [Google Scholar]
- 3. Loring DW, Lowenstein DH, Barbaro NM, et al. Common data elements in epilepsy research: development and implementation of the NINDS epilepsy CDE project. Epilepsia 2011;52:1186–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harte‐Hargrove LC, French JA, Pitkanen A, et al. Common data elements for preclinical epilepsy research: standards for data collection and reporting. A TASK3 report of the AES/ILAE Translational Task Force of the ILAE. Epilepsia 2017;58(Suppl 4):78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lapinlampi N, Melin E, Aronica E, et al. Common data elements and data management: remedy to cure underpowered preclinical studies. Epilepsy Res 2017;129:87–90. [DOI] [PubMed] [Google Scholar]
- 6. Loscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 2011;20:359–368. [DOI] [PubMed] [Google Scholar]
- 7. Putnam TJ, Merritt HH. Experimental determination of the anticonvulsant properties of some phenyl derivatives. Science 1937;85:525–526. [DOI] [PubMed] [Google Scholar]
- 8. Oakley JC, Kalume F, Yu FH, et al. Temperature‐ and age‐dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc Natl Acad Sci USA 2009;106:3994–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. J Physiol 2010;588:1849–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barker‐Haliski ML, Johnson K, Billingsley P, et al. Validation of a preclinical drug screening platform for pharmacoresistant epilepsy. Neurochem Res 2017;42:1904–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kehne JH, Klein BD, Raeissi S, et al. The National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP). Neurochem Res 2017;42:1894–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Libbey JE, Doty DJ, Sim JT, et al. The effects of diet on the severity of central nervous system disease: one part of lab‐to‐lab variability. Nutrition 2016;32:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Loscher W, Ferland RJ, Ferraro TN. The relevance of inter‐ and intrastrain differences in mice and rats and their implications for models of seizures and epilepsy. Epilepsy Behav 2017;73:214–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frankel WN, Taylor L, Beyer B, et al. Electroconvulsive thresholds of inbred mouse strains. Genomics 2001;74:306–312. [DOI] [PubMed] [Google Scholar]
- 15. Swinyard EA, Woodhead JH, White HS, et al. General principles: experimental selection, quantification, and evaluation of anticonvulsants In Levy R, Mattson R, Meldrum B, et al. (Eds) Antiepileptic drugs. 3rd Ed New York: Raven Press Ltd, 1989:85–102. [Google Scholar]
- 16. Barton ME, Klein BD, Wolf HH, et al. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res 2001;47:217–227. [DOI] [PubMed] [Google Scholar]
- 17. Loscher W. (Ed). Valproate. Basel: Springer; 1999. [Google Scholar]
- 18. Finney DJ. Probit analysis. A statistical treatment of the sigmoid response curve. Cambridge: University Press; 1952. [Google Scholar]
- 19. Barker‐Haliski ML, Loscher W, White HS, et al. Neuroinflammation in epileptogenesis: insights and translational perspectives from new models of epilepsies. Epilepsia 2017;58(Suppl 3):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Loscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 2011;52:657–678. [DOI] [PubMed] [Google Scholar]
- 21. Otto JF, Yang Y, Frankel WN, et al. Mice carrying the szt1 mutation exhibit increased seizure susceptibility and altered sensitivity to compounds acting at the m‐channel. Epilepsia 2004;45:1009–1016. [DOI] [PubMed] [Google Scholar]
- 22. White HS, Barker‐Haliski M. Antiepileptic drug discovery In Shorvon SD, Perucca E, Engel J., Jr (Eds) The treatment of epilepsy. Chichester, West Sussex: John Wiley & Sons, Ltd., 2016:52–60. [Google Scholar]
- 23. Barker‐Haliski M, White HS. Antiepileptic drug development and experimental models In Wyllie E, Gidal BE, Goodkin HP. (Eds) Wyllie's treatment of epilepsy. Philadelphia, PA: Lippencott, Williams & Wilkins, 2015:516–521. [Google Scholar]
- 24. Smith M, Wilcox KS, White HS. Discovery of antiepileptic drugs. Neurotherapeutics 2007;4:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brown WC, Schiffman DO, Swinyard EA, et al. Comparative assay of antiepileptic drugs by ‘psychomotor’ seizure test and minimal electroshock threshold test. J Pharmacol Exp Ther 1953;107:273–283. [PubMed] [Google Scholar]
- 26. Wang DD, Englot DJ, Garcia PA, et al. Minocycline‐ and tetracycline‐class antibiotics are protective against partial seizures in vivo. Epilepsy Behav 2012;24:314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walrave L, Maes K, Coppens J, et al. Validation of the 6 Hz refractory seizure mouse model for intracerebroventricularly administered compounds. Epilepsy Res 2015;115:67–72. [DOI] [PubMed] [Google Scholar]
- 28. Leclercq K, Kaminski RM. Genetic background of mice strongly influences treatment resistance in the 6 Hz seizure model. Epilepsia 2015;56:310–318. [DOI] [PubMed] [Google Scholar]
- 29. Loscher W, Fassbender CP, Nolting B. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. II. Maximal electroshock seizure models. Epilepsy Res 1991;8:79–94. [DOI] [PubMed] [Google Scholar]
- 30. Leclercq K, Matagne A, Kaminski RM. Low potency and limited efficacy of antiepileptic drugs in the mouse 6 Hz corneal kindling model. Epilepsy Res 2014;108:675–683. [DOI] [PubMed] [Google Scholar]
- 31. Singh NA, Pappas C, Dahle EJ, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet 2009;5:e1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singh NA, Otto JF, Dahle EJ, et al. Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J Physiol 2008;586:3405–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Loscher W, Nau H. Pharmacological evaluation of various metabolites and analogues of valproic acid. Anticonvulsant and toxic potencies in mice. Neuropharmacology 1985;24:427–435. [DOI] [PubMed] [Google Scholar]
- 34. Swinyard EA, Miyahara JT, Clark LD, et al. The effect of experimentally‐induced stress on pentylenetetrazol seizure threshold in mice. Psychopharmacologia 1963;4:343–353. [DOI] [PubMed] [Google Scholar]
- 35. Metcalf CS, West PJ, Thomson KE, et al. Development and pharmacologic characterization of the rat 6 Hz model of partial seizures. Epilepsia 2017;58:1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bilbey DL, Salem H, Grossman MH. The anatomical basis of the straub phenomenon. Br J Pharmacol Chemother 1960;15:540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goodman LS, Grewal MS, Brown WC, et al. Comparison of maximal seizures evoked by pentylenetetrazol (metrazol) and electroshock in mice, and their modification by anticonvulsants. J Pharmacol Exp Ther 1953;108:168–176. [PubMed] [Google Scholar]
- 38. Toman JE, Goodman LS. Anticonvulsants. Physiol Rev 1948;28:409–432. [DOI] [PubMed] [Google Scholar]
- 39. Snead OC 3rd. Pharmacological models of generalized absence seizures in rodents. J Neural Transm Suppl 1992;35:7–19. [DOI] [PubMed] [Google Scholar]
- 40. Loscher W, Honack D, Fassbender CP, et al. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. III. Pentylenetetrazole seizure models. Epilepsy Res 1991;8:171–189. [DOI] [PubMed] [Google Scholar]
- 41. Tringham E, Powell KL, Cain SM, et al. T‐type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci Transl Med 2012;4:121ra119. [DOI] [PubMed] [Google Scholar]
- 42. Hosford DA, Wang Y. Utility of the lethargic (lh/lh) mouse model of absence seizures in predicting the effects of lamotrigine, vigabatrin, tiagabine, gabapentin, and topiramate against human absence seizures. Epilepsia 1997;38:408–414. [DOI] [PubMed] [Google Scholar]
- 43. White HS, Woodhead JH, Franklin MR, et al. General principles: experimental selection, quantification, and evaluation of antiepileptic drugs In Levy RH, Mattson RH, Meldrum BS. (Eds) Antiepileptic drugs. 4th Ed New York: Raven Press Ltd, 1995:99–110. [Google Scholar]
- 44. Sobol E, Yagen B, Steve White H, et al. Preclinical evaluation of 2,2,3,3‐tetramethylcyclopropanecarbonyl‐urea, a novel, second generation to valproic acid, antiepileptic drug. Neuropharmacology 2006;51:933–946. [DOI] [PubMed] [Google Scholar]
- 45. Auvin S, Shin D, Mazarati A, et al. The utility of testing pentylenetetrazol threshold. Epilepsia 2006;47:662–663; author reply 663‐664. [DOI] [PubMed] [Google Scholar]
- 46. Engstrom FL, Woodbury DM. Seizure susceptibility in DBA and C57 mice: the effects of various convulsants. Epilepsia 1988;29:389–395. [DOI] [PubMed] [Google Scholar]
- 47. Swinyard EA, Woodhead JH, White HS, et al. General principles: experimental selection, quantification, and evaluation of anticonvulsants In Levy RH, Mattson RH, Meldrum BS, Penri JK, Dreyfuss FE. (Eds) Antiepileptic drugs. New York: Raven Press, 1989:85–102. [Google Scholar]
- 48. Orlof MJ, Williams HL, Pfeiffer CC. Timed intravenous infusion of Metrazol and strychnine for testing anticonvulsant drugs. Proc Soc Exp Biol Med 1949;70:254–257. [DOI] [PubMed] [Google Scholar]
- 49. Klitgaard H. Levetiracetam: the preclinical profile of a new class of antiepileptic drugs? Epilepsia 2001;42(Suppl 4):13–18. [PubMed] [Google Scholar]
- 50. Loscher W, Brandt C. Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol Rev 2010;62:668–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matagne A, Klitgaard H. Validation of corneally kindled mice: a sensitive screening model for partial epilepsy in man. Epilepsy Res 1998;31:59–71. [DOI] [PubMed] [Google Scholar]
- 52. Loscher W, Honack D, Rundfeldt C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J Pharmacol Exp Ther 1998;284:474–479. [PubMed] [Google Scholar]
- 53. Otsuki K, Morimoto K, Sato K, et al. Effects of lamotrigine and conventional antiepileptic drugs on amygdala‐ and hippocampal‐kindled seizures in rats. Epilepsy Res 1998;31:101–112. [DOI] [PubMed] [Google Scholar]
- 54. Wauquier A, Zhou S. Topiramate: a potent anticonvulsant in the amygdala‐kindled rat. Epilepsy Res 1996;24:73–77. [DOI] [PubMed] [Google Scholar]
- 55. Loscher W, Honack D. Profile of ucb L059, a novel anticonvulsant drug, in models of partial and generalized epilepsy in mice and rats. Eur J Pharmacol 1993;232:147–158. [DOI] [PubMed] [Google Scholar]
- 56. McIntyre DC, Poulter MO, Gilby K. Kindling: some old and some new. Epilepsy Res 2002;50:79–92. [DOI] [PubMed] [Google Scholar]
- 57. Lothman EW, Hatlelid JM, Zorumski CF, et al. Kindling with rapidly recurring hippocampal seizures. Brain Res 1985;360:83–91. [DOI] [PubMed] [Google Scholar]
- 58. Krug M, Koch M, Grecksch G, et al. Pentylenetetrazol kindling changes the ability to induce potentiation phenomena in the hippocampal CA1 region. Physiol Behav 1997;62:721–727. [DOI] [PubMed] [Google Scholar]
- 59. Mazarati A, Shin D, Auvin S, et al. Age‐dependent effects of topiramate on the acquisition and the retention of rapid kindling. Epilepsia 2007;48:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Remigio GJ, Loewen JL, Heuston S, et al. Corneal kindled C57BL/6 mice exhibit saturated dentate gyrus long‐term potentiation and associated memory deficits in the absence of overt neuron loss. Neurobiol Dis 2017;105:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sangdee P, Turkanis SA, Karler R. Kindling‐like effect induced by repeated corneal electroshock in mice. Epilepsia 1982;23:471–479. [DOI] [PubMed] [Google Scholar]
- 62. Rowley NM, White HS. Comparative anticonvulsant efficacy in the corneal kindled mouse model of partial epilepsy: correlation with other seizure and epilepsy models. Epilepsy Res 2010;92:163–169. [DOI] [PubMed] [Google Scholar]
- 63. Albertini G, Walrave L, Demuyser T, et al. 6 Hz corneal kindling in mice triggers neurobehavioral comorbidities accompanied by relevant changes in c‐Fos immunoreactivity throughout the brain. Epilepsia 2017;59:67–78. [DOI] [PubMed] [Google Scholar]
- 64. Loscher W. Strategies for antiepileptogenesis: antiepileptic drugs versus novel approaches evaluated in post‐status epilepticus models of temporal lobe epilepsy In Noebels JL, Avoli M, Rogawski MA, et al. (Eds) Jasper's basic mechanisms of the epilepsies. Bethesda, MD: National Center for Biotechnology Information (US), 2012:1563–1580. [PubMed] [Google Scholar]
- 65. Stratton SC, Large CH, Cox B, et al. Effects of lamotrigine and levetiracetam on seizure development in a rat amygdala kindling model. Epilepsy Res 2003;53:95–106. [DOI] [PubMed] [Google Scholar]
- 66. Loscher W, Cramer S, Ebert U. Selection of phenytoin responders and nonresponders in male and female amygdala‐kindled Sprague–Dawley rats. Epilepsia 1998;39:1138–1147. [DOI] [PubMed] [Google Scholar]
- 67. Srivastava AK, White HS. Carbamazepine, but not valproate, displays pharmacoresistance in lamotrigine‐resistant amygdala kindled rats. Epilepsy Res 2013;104:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Koneval Z, Knox KM, White HS, et al. Lamotrigine‐resistant corneal‐kindled mice: a model of pharmacoresistant partial epilepsy for moderate‐throughput drug discovery. Epilepsia 2018;59:1245–1256. [DOI] [PubMed] [Google Scholar]
- 69. Hannesson DK, Howland JG, Pollock M, et al. Anterior perirhinal cortex kindling produces long‐lasting effects on anxiety and object recognition memory. Eur J Neurosci 2005;21:1081–1090. [DOI] [PubMed] [Google Scholar]
- 70. Hannesson DK, Mohapel P, Corcoran ME. Dorsal hippocampal kindling selectively impairs spatial learning/short‐term memory. Hippocampus 2001;11:275–286. [DOI] [PubMed] [Google Scholar]
- 71. Hannesson DK, Howland J, Pollock M, et al. Dorsal hippocampal kindling produces a selective and enduring disruption of hippocampally mediated behavior. J Neurosci 2001;21:4443–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Barker‐Haliski ML, Vanegas F, Mau MJ, et al. Acute cognitive impact of antiseizure drugs in naive rodents and corneal‐kindled mice. Epilepsia 2016;57:1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Brooks‐Kayal AR, Bath KG, Berg AT, et al. Issues related to symptomatic and disease‐modifying treatments affecting cognitive and neuropsychiatric comorbidities of epilepsy. Epilepsia 2013;54(Suppl 4):44–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sutula TP, Kotloski RJ. Kindling: a model and phenomenon of epilepsy In Pitkanen A, Buckmaster PS, Galanopoulou AS, et al. (Eds) Models of seizure and epilepsy. London: Academic Press, 2017:813–825. [Google Scholar]
- 75. Sutula TP. Experimental models of temporal lobe epilepsy: new insights from the study of kindling and synaptic reorganization. Epilepsia 1990;31(Suppl 3):S45–S54. [DOI] [PubMed] [Google Scholar]
- 76. Brandt C, Tollner K, Klee R, et al. Effective termination of status epilepticus by rational polypharmacy in the lithium‐pilocarpine model in rats: window of opportunity to prevent epilepsy and prediction of epilepsy by biomarkers. Neurobiol Dis 2015;75:78–90. [DOI] [PubMed] [Google Scholar]
- 77. White HS, Alex AB, Pollock A, et al. A new derivative of valproic acid amide possesses a broad‐spectrum antiseizure profile and unique activity against status epilepticus and organophosphate neuronal damage. Epilepsia 2012;53:134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sharma AK, Reams RY, Jordan WH, et al. Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol Pathol 2007;35:984–999. [DOI] [PubMed] [Google Scholar]
- 79. Brandt C, Glien M, Potschka H, et al. Epileptogenesis and neuropathology after different types of status epilepticus induced by prolonged electrical stimulation of the basolateral amygdala in rats. Epilepsy Res 2003;55:83–103. [DOI] [PubMed] [Google Scholar]
- 80. Hellier JL, Dudek FE. Spontaneous motor seizures of rats with kainate‐induced epilepsy: effect of time of day and activity state. Epilepsy Res 1999;35:47–57. [DOI] [PubMed] [Google Scholar]
- 81. Hellier JL, Patrylo PR, Buckmaster PS, et al. Recurrent spontaneous motor seizures after repeated low‐dose systemic treatment with kainate: assessment of a rat model of temporal lobe epilepsy. Epilepsy Res 1998;31:73–84. [DOI] [PubMed] [Google Scholar]
- 82. Puttachary S, Sharma S, Tse K, et al. Immediate epileptogenesis after kainate‐induced status epilepticus in C57BL/6J mice: evidence from long term continuous video‐EEG telemetry. PLoS ONE 2015;10:e0131705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tse K, Puttachary S, Beamer E, et al. Advantages of repeated low dose against single high dose of kainate in C57BL/6J mouse model of status epilepticus: behavioral and electroencephalographic studies. PLoS ONE 2014;9:e96622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Umpierre AD, Bennett IV, Nebeker LD, et al. Repeated low‐dose kainate administration in C57BL/6J mice produces temporal lobe epilepsy pathology but infrequent spontaneous seizures. Exp Neurol 2016;279:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cavalheiro EA, Santos NF, Priel MR. The pilocarpine model of epilepsy in mice. Epilepsia 1996;37:1015–1019. [DOI] [PubMed] [Google Scholar]
- 86. Turski WA, Cavalheiro EA, Schwarz M, et al. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res 1983;9:315–335. [DOI] [PubMed] [Google Scholar]
- 87. Mazarati A, Lu X, Shinmei S, et al. Patterns of seizures, hippocampal injury and neurogenesis in three models of status epilepticus in galanin receptor type 1 (GalR1) knockout mice. Neuroscience 2004;128:431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kienzler F, Norwood BA, Sloviter RS. Hippocampal injury, atrophy, synaptic reorganization, and epileptogenesis after perforant pathway stimulation‐induced status epilepticus in the mouse. J Comp Neurol 2009;515:181–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mazarati A, Bragin A, Baldwin R, et al. Epileptogenesis after self‐sustaining status epilepticus. Epilepsia 2002;43(Suppl 5):74–80. [DOI] [PubMed] [Google Scholar]
- 90. Liu Z, Gatt A, Werner SJ, et al. Long‐term behavioral deficits following pilocarpine seizures in immature rats. Epilepsy Res 1994;19:191–204. [DOI] [PubMed] [Google Scholar]
- 91. Curia G, Longo D, Biagini G, et al. The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods 2008;172:143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mazarati AM, Wasterlain CG, Sankar R, et al. Self‐sustaining status epilepticus after brief electrical stimulation of the perforant path. Brain Res 1998;801:251–253. [DOI] [PubMed] [Google Scholar]
- 93. Blanco MM, dos Santos JG Jr, Perez‐Mendes P, et al. Assessment of seizure susceptibility in pilocarpine epileptic and nonepileptic Wistar rats and of seizure reinduction with pentylenetetrazole and electroshock models. Epilepsia 2009;50:824–831. [DOI] [PubMed] [Google Scholar]
- 94. Loscher W. Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res 2016;126:157–184. [DOI] [PubMed] [Google Scholar]
- 95. Trandafir CC, Pouliot WA, Dudek FE, et al. Co‐administration of subtherapeutic diazepam enhances neuroprotective effect of COX‐2 inhibitor, NS‐398, after lithium pilocarpine‐induced status epilepticus. Neuroscience 2015;284:601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Loscher W. Single versus combinatorial therapies in status epilepticus: novel data from preclinical models. Epilepsy Behav 2015;49:20–25. [DOI] [PubMed] [Google Scholar]
- 97. Lothman EW, Bertram EH, Bekenstein JW, et al. Self‐sustaining limbic status epilepticus induced by ‘continuous’ hippocampal stimulation: electrographic and behavioral characteristics. Epilepsy Res 1989;3:107–119. [DOI] [PubMed] [Google Scholar]
- 98. Gorter JA, van Vliet EA. Post‐status epilepticus models: electrical stimulation In Pitkanen A, Buckmaster PS, Galanopoulou AS, et al. (Eds) Models of seizures and epielspy. London: Academic Press, 2017:637–650. [Google Scholar]
- 99. Sloviter RS. “Epileptic” brain damage in rats induced by sustained electrical stimulation of the perforant path. I. Acute electrophysiological and light microscopic studies. Brain Res Bull 1983;10:675–697. [DOI] [PubMed] [Google Scholar]
- 100. Mazarati A, Thompson KW, Suchomelova S, et al. Status epilepticus models: electrical stimulation In Pitkanen A, Schwartzkroin PA, Moshe SL. (Eds) Models of seizures and epilepsy. Amsterdam: Academic Press, 2005:449–464. [Google Scholar]
- 101. Pauletti A, Terrone G, Shekh‐Ahmad T, et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 2017;140:1885–1899. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 102. Walker LE, Frigerio F, Ravizza T, et al. Molecular isoforms of high‐mobility group box 1 are mechanistic biomarkers for epilepsy. J Clin Invest 2017;127:2118–2132. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 103. Riban V, Bouilleret V, Pham‐Le BT, et al. Evolution of hippocampal epileptic activity during the development of hippocampal sclerosis in a mouse model of temporal lobe epilepsy. Neuroscience 2002;112:101–111. [DOI] [PubMed] [Google Scholar]
- 104. Bouilleret V, Ridoux V, Depaulis A, et al. Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience 1999;89:717–729. [DOI] [PubMed] [Google Scholar]
- 105. Duveau V, Pouyatos B, Bressand K, et al. Differential effects of antiepileptic drugs on focal seizures in the intrahippocampal kainate mouse model of mesial temporal lobe epilepsy. CNS Neurosci Ther 2016;22:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Venceslas D, Corinne R. A mesiotemporal lobe epilepsy mouse model. Neurochem Res 2017;42:1919–1925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Core CRF and CDE files. The CDE and CRF modules linked to this article can be found and downloaded as a zip folder.