

Abstract
While screening off-target effects of rigid (N)-methanocarba-adenosine 5′-methylamides as A3 adenosine receptor (AR) agonists, we discovered μM binding hits at the δ-opioid receptor (DOR) and translocator protein (TSPO). In an effort to increase OR and decrease AR affinity by structure activity analysis of this series, antagonist activity at κ-(K)OR appeared in 5′-esters (ethyl 24 and propyl 30), which retained TSPO interaction (μM). 7-Deaza modification of C2-(arylethynyl)-5′-esters but not 4′-truncation enhanced KOR affinity (MRS7299 28 and 29, Ki ≈ 40 nM), revealed μ-OR and DOR binding, and reduced AR affinity. Molecular docking and dynamics simulations located a putative KOR binding mode consistent with the observed affinities, placing C7 in a hydrophobic region. 3-Deaza modification permitted TSPO but not OR binding, and 1-deaza was permissive to both; ribose-restored analogues were inactive at both. Thus, we have repurposed a known AR nucleoside scaffold for OR antagonism, with a detailed hypothesis for KOR recognition.
Introduction
Nucleoside analogues containing a bicyclo[3.1.0]hexane ring system (termed methanocarba, Chart 1) in place of ribose are being developed as highly selective A3 adenosine receptor (AR) agonists, which have potential in treatment of chronic neuropathic pain and other conditions.1,2 The position of cyclopropyl and cyclopentyl ring fusion enforces a South (S) or a North (N) envelope conformation, which we previously showed to promote A3AR interaction.2,3 This rigid scaffold shows promise as a privileged but not promiscuous structural class for interaction with diverse protein targets.4 Varied off-target effects of these nucleosides at biogenic amine receptors, including as 5HT2B and 5HT2C serotonin receptor antagonists, and as allosteric modulators of the dopamine transporter (DAT), were detected at higher concentrations than their nM affinities at the A3AR. Moreover, it was possible to enhance these novel activities, while at the same time deselecting for AR affinity.5,6
Chart 1. Rigid (N)-Methanocarba Nucleosides with Reported Off-Target Activity at rTSPO24a.

a All three nucleosides lack significant binding affinity at the hKOR. N6-(3-Chlorobenzyl) derivative 4 also activates the human (h) A3AR (Ki = 3 nM), and corresponding 4′-truncated derivative 3 is an A3AR antagonist (Ki = 100 nM).
We report here that certain (N)-methanocarba nucleosides can also antagonize opioid receptors (ORs): δ- (DOR), κ- (KOR), and μ- (MOR), with moderate preference for KOR. We probed the structure–activity relationship (SAR) of rigid bicyclic nucleosides to enhance KOR affinity and modeled their receptor interactions. We have also proposed a structural basis for the unexpected interaction of rigid nucleosides with the KOR, especially 7-deaza analogues,7,8 utilizing modeling approaches that complement published reports on KOR modeling.9−11 We also observed a partial SAR convergence with the translocator protein (TSPO), but this was not the target of the present study. The affinity at TSPO and DOR was noted previously for a few (N)-methanocarba adenosine derivatives.4 For example, the N6-dicyclopropylmethyl 5′-ester derivatives 1 and 2 displayed roughly μM affinity at TSPO, while also binding to A1AR and A3AR.5 Other rigid bicyclic nucleosides, 3 and 4, with enlarged substituents at the N6 and C2 positions were shown to bind at TSPO as well, and compound 4 also bound weakly at DOR.4
High-affinity antagonists of KOR have been reported, including both morphinans and non-morphinans.10,12−15 Centrally active KOR antagonists, including those containing novel scaffolds and having shorter half-lives in vivo, are sought for the treatment of mood disorders, drug addiction, pain, and depression resulting from chronic stress.16,17 KOR activation by the native peptide dynorphin in the amygdala inhibits presynaptic glutamate release, and its deletion or blockade with selective antagonists produces an anxiolytic phenotype.18 KOR activation impeded lung cancer cell growth through activation of glycogen synthase kinase 3β.19 MOR antagonists may be applied to addiction treatment and to reducing side effects of opioid pain killers when their action is peripherally restricted.20−22 TSPO, an off-target interaction of many of nucleosides in this chemical series, is a component of the permeability transition pore on the outer mitochondrial membrane in microglial and other cells, and its ligands provide neuroprotective and anticancer properties.23−29 Structurally diverse TSPO ligands are being explored for treatment of chronic pain, inflammation, anxiety, mood disorders, neurodegeneration, and diseases of cellular proliferation.30,31 Thus, both of these membrane-bound protein families are important therapeutic targets.
Results
Here, we extend the polypharmacology of rigid nucleosides to enhance affinity at KOR by functional group or heterocyclic replacement on the same scaffold, in some cases with accompanying moderate affinity at TSPO. Furthermore, we discovered modifications of the adenine moiety and its substituents that allowed MOR and/or DOR interactions as antagonists. The receptor interactions of representative nucleosides, based on a KOR X-ray crystallographic structure, were modeled to provide a self-consistent hypothesis for recognition. In this study, we did not model the TSPO interaction or probe the effects of the methanocarba ring isomer having the opposite South (S) conformation.32 Thus, we have repurposed a nucleoside scaffold from ARs to bind at another rhodopsin-like G protein-coupled receptor (GPCR) family.5,33
Compound Design
The previously noted binding of (N)-methanocarba nucleosides 1 and 2 at the rat kidney TSPO was dependent on the presence of a 5′-ethyl or 5′-n-propyl ester.7 These esters bound to TSPO with greater affinity than the corresponding 5′-methyl ester or 5′-methylamide, which had Ki values >10 μM (data not shown). Removing the alkyloxycarbonyl group of 1, that is, to give an N6-dicyclopropylmethyl 4′-truncated analogue (structure not shown), also prevented TSPO binding.4 Thus, a medium-sized 5′-alkyl ester was conducive to TSPO interaction. However, with enlarged substituents at both N6 and C2 positions, 5′-methylamide 4 bound with greater affinity at the TSPO than 5′-esters 1 and 2 or its 4′ truncated equivalent 3. Thus, we explored here the interplay between the 5′, N6 and C2 positions and adenine ring nitrogens in the SAR of rigid nucleosides initially at TSPO and subsequently at the ORs.
The current study began with a focus on N6-3-chlorobenzyl 5′-methylamide derivatives (Table 1, 4–9), but that scope was subsequently expanded to include a range of other N6-alkyl or N6-alkylaryl derivatives with 5′-carbonyl groups (Tables 2 and 3, 10–43). To explore the SAR around the detected OR and TSPO interactions of nucleosides, we chose to modify the rigid adenosine derivatives at the 5′ position, from amides to esters, and at the N6 and C2 positions. The default C2 substitution was 5-chloro-thien-2-yl-ethynyl, upon which halogen was substituted, or other arylethynyl groups were introduced. We included various 4′-truncated (Tables 4 and 5, 47–58) and other derivatives that were synthesized previously in the context of ARs or DAT.2,3,5,6,34,35 In addition, N6-propyl (19 and 20) and 1-, 3-, or 7-deaza-adenine (17, 26–29, 39–44 and 54–58) analogues were prepared for this study.
Table 1. Structures and Modulation of Binding and Activity at hORs, rTSPO and A3AR of (N)-Methanocarba Adenosine Derivatives (4–21) Containing a 5′-Carbonyl Group; (N)-Methanocarba-5′-amides; X, Z = N and R1 = CH3, Unless Noted.

| compound | R2=, other changes | R3= | DOR,a,bKi, nM | TSPO,bKi, nM | DAT,b % of control at 10 μM (%) | A3AR binding Ki, nM (species),d or % of control at 10 μM |
|---|---|---|---|---|---|---|
| 4d | 3,4-F2-phenylethynyl | CH2-(3-Cl-Ph) | 2480 ± 790 | 340 ± 72 | 16 | 3.49 ± 1.84 (h), 3.08 ± 0.23 (m) |
| 5d | Cl | CH2-(3-Cl-Ph) | e | f | 3 | 0.29 ± 0.04 (h) |
| 6 | phenyl-ethynyl | CH2-(3-Cl-Ph) | e | 2650 ± 290 | 7 | 1.35 ± 0.30 (h) |
| 7d | 4-F-phenylethynyl | CH2-(3-Cl-Ph) | 6620 ± 1700 | 253 ± 57 | 21 | 2.16 ± 0.34 (h) |
| 8d | 2-Cl-phenylethynyl | CH2-(3-Cl-Ph) | f | 344 ± 149 | 50 | 1.92 ± 0.57 (h) |
| 9 | 3-Cl-phenylethynyl | CH2-(3-Cl-Ph) | 5870 ± 2120 | 1770 ± 910 | 26 | 4.45 ± 1.39 (h) |
| 10 | phenylethynyl | trans-cPr-Ph | e | f | 2310c | 6.16 ± 0.22 (h) |
| 11d | 5-Cl-thienyl-ethynyl | CH3 | e | e | –322 | 1.65 ± 0.08 (h), 86 ± 6 (m) |
| 12 | phenylethynyl | CH3 | e | e | –211 | 0.85 ± 0.22 (h) |
| 13 | 2-Cl-phenyl-ethynyl | CH3 | e | e | –484 | 0.58 ± 0.04 (h), 110 ± 5 (m) |
| 14d | 5-Cl-thienyl-ethynyl | CH3 | e | 684 ± 175 | –556 | 0.70 ± 0.11 (h), 36 ± 5 (m) |
| 15d | 5-Br-thienyl-ethynyl | CH3 | e | f | –235 | 0.44 ± 0.12 (h), 43.7 ± 2.1 (m) |
| 16d | 5-Cl-thienyl-ethynyl, X = CH | CH3 | e | e | 1 | 3.0 ± 0.8 (h), 31 ± 2 (m) |
| 17 | H, Z = CH | CH3 | e | e | –14 | 498 ± 46 (h), 38 ± 1% (m) |
| 18d | 5-Cl-thienyl-ethynyl | (CH2)2CH3 | e | 1310 ± 210 | –159 | 1.1 ± 0.3 (h), 6.8 ± 0.3 (m) |
| 19 | 5-Cl-thienyl-ethynyl | (CH2)2CF3 | e | 1300 ± 150 | 15 | 3.7 ± 1.0 (h), 71 ± 2 (m) |
| 20 | 5-Cl-thienyl-ethynyl | (CH2)3OH | e | 6160 ± 50 | –107 ± 4 | 2.04 ± 1.46 (h), 105 ± 2 (m) |
| 21a | 5-Cl-thienylethynyl, R1 = (CH2)2NH2 | CH3 | f | >10 000 | –136 | 158 ± 8 (h), 0% (m) |
KOR and MOR binding inhibition is <50% at 10 μM, unless noted. 21: KOR, Ki = 2.79 ± 0.77 μM.
Modulation of inhibition of binding of opioid agonist radioligands [3H]Tyr-d-Ala-Gly-Phe-d-Leu ([3H]DADLE, 97, 0.2 nM) for DOR, [3H]N-methyl-2-phenyl-N-[(5R,7S,8S)-7-(pyrrolidin-1-yl)-1-oxaspiro[4.5]dec-8-yl]acetamide ([3H]U69593, 98, 0.3 nM) for KOR, [3H]Ala2-MePhe4-Glyol5-Enkephalin ([3H]DAMGO, 99, 0.3 nM) for MOR; and [3H]nociceptin 100 (0.5–2.0 nM) for NOP. [3H]N-Butan-2-yl-1-(2-chlorophenyl)-N-methylisoquinoline-3-carboxamide ([3H]PK11195, 101, 1.0 nM) was used for rat TSPO, and [3H]methyl(1R,2S,3S)-3-(4-fluorophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([3H]WIN35428 103, 0.5 nM) was used for DAT. NOP binding Ki values (μM, n = 1) were found to be 4 (3.90) and 9 (>10). Reference ligands and their Ki values (nM) were DOR, natrindole 108, 0.81; KOR, salvinorin A 109, 1.93; MOR, morphine 110, 3.29; NOP, 7-[[4-(2,6-dichlorophenyl)-1-piperidinyl]methyl]-6,7,8,9-tetrahydro-1-methyl-5H-benzocyclohepten-5-ol (SB612111) 111, 6.58; TSPO, 4′-chlorodiazepam (Ro5-4864) 112, 27.6; DAT, 1-[2-[bis-(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine (GBR12909) 114, 3.04. Values are expressed as the mean ± SEM of N = 2–4 assays performed in duplicate, unless indicated.
Ki (μM), inhibition of binding of [3H]103 at hDAT: 10, 2.31.5
A3AR and other AR binding data and procedures from Tosh et al.2,3,6,34,35,37 [125I]N6-(4-Amino-3-iodobenzyl)adenosine-5′-N-methyl-uronamide ([125I]AB-MECA, 102, 0.5 nM) was used for hA3AR. Reference ligand [adenosine-5′-N ethyluronamide (NECA, 113)], and its Ki value (nM) was hA3AR, 35; mA3AR, 0.45. Representative binding inhibition at hA1AR is (% at 10 μM) 7, 22%; 18, 22%; 19, 46%; 26, 39%; 40, 35%; 41, 44%; 42, 34%; 43, 26%. Representative binding inhibition at hA2AAR (% at 10 μM): 18, 34%; 19, 21%; 26, 16%; 40, 22%; 41, 25%; 42, 20%; 43, <10%. Ki values (hA1AR, nM): 55, 1300 ± 290; 56, 650 ± 71; 73, 1110 ± 470; determined as reported.37 mA1AR and mA2AAR binding data, % inhibition at 10 μM, respectively: 24, 43 ± 2, 10 ± 3; 26, 54 ± 1, 5 ± 1; 28, 32 ± 2, 10 ± 3; 30, 53 ± 2, 13 ± 2; 46, 62 ± 2, 12 ± 4. Values are expressed as the mean ± SEM of N = 3 assays performed in duplicate.
30–50% inhibition of radioligand binding at 10 μM.
<30% inhibition of radioligand binding at 10 μM.
Table 2. Structures and Modulation of Binding and Activity at hORs, rTSPO and A3AR of (N)-Methanocarba Adenosine Derivatives (22–43) and Iodo Derivative (82) Containing a 5′-Carbonyl Group; (N)-Methanocarba 5′-Esters and Carboxylate; X, Y and Z = N, R3 = CH3 and R4 = Cl, Unless Noted.

| Compound | R1=, other changes | DOR,aKi, nM | KOR,aKi, nM | MOR,aKi, nM | TSPO,aKi, nM | DAT,a inhib., % of control at 10 μM, % | A3AR binding Ki, nM (species),b or % at 10 μM |
|---|---|---|---|---|---|---|---|
| 22b | CH3 | d | 3130 ± 300 | c | c | –352 | 5.38 ± 0.03 (h), 36 ± 1% (m) |
| 23b | CH3, R4 = Br | d | 2090 ± 330 | c | 8120 ± 670 | –370 | 8.56 ± 0.10 (h), 57 ± 1% (m) |
| 24b | CH2CH3 | c | 396 ± 29 | d | 1290 ± 70 | –539 | 14.5 ± 2.3 (h), 45 ± 5% (m) |
| 25 | CH2CH3, R4 = Br | c | 975 ± 198 | c | 1520 ± 250 | –430 | 6.42 ± 0.35 (h), 1800 ± 90 (m) |
| 26 | CH2CH3, X = CH | c | 806 ± 263 | c | 3810 ± 100 | <10 | 29.4 ± 13.8 (h), 828 ± 51 (m) |
| 27 | CH2CH3, Y = CH | d | d | d | 3390 ± 640 | –354 | 47.3 ± 18.9 (h), 18 ± 4% (m) |
| 28 | CH2CH3, Z = CH | 786 ± 210 | 42 ± 1 | 637 ± 367 | 869 ± 160 | –172 ± 57 (3) | 448 ± 13 (h), 15 ± 1% (m) |
| 77 | CH2CH3, R2 = Cl-thienyl-C≡C | 1990 ± 1020 | 52 ± 23 | 1530 ± 560 | d | –20 | 1650 ± 330 (h) |
| 29 | CH2CH3, Z = CH, R4 = Br | 437 ± 94 | 39 ± 1 | 368 ± 135 | 765 ± 118 | –102 ± 8 (3) | 466 ± 20 (h), 24 ± 2% (m) |
| 30 | (CH2)2CH3 | d | 437 ± 167 | d | 1160 ± 560 | –426 | 5.78 ± 1.45 (h), 2810 ± 150 (m) |
| 31 | CH(CH3)2 | d | d | d | d | <20 | 42.9 ± 22.8 (h), 30 ± 1% (m) |
| 32 | (CH2)3CH3 | d | 1210 ± 100 | d | >10 000 | –285 | 17.5 ± 1.6 (h), 54 ± 1% (m) |
| 33 | (CH2)2–CH(CH3)2 | d | 1090 ± 320 | d | >10 000 | –196 ± 58 | 24.4 ± 2.8 (h), 32 ± 2% (m) |
| 34 | (CH2)2–cHex | c | >10 000 | d | c | <20 | 334 ± 132 (h), 32 ± 1% (m) |
| 35 | CH2–Ph | d | 629 ± 183 | c | 4050 ± 740 | <20 | 7.81 ± 2.40 (h), 891 ± 105 (m) |
| 36 | (CH2)2–Ph | d | 3670 ± 1640 | d | d | <20 | 114 ± 64 (h), 31 ± 1% (m) |
| 37 | (CH2)3–Ph | d | 8920 ± 1080 | d | 1090 ± 290 | <20 | 132 ± 68 (h), 40 ± 1% (m) |
| 38 | H | d | d | d | d | –50 | 684 ± 195 (h), 0% (m) |
| 39 | CH2CH3, R2 = I | 1590 ± 280 | 104 ± 35 | c | d | <10 | 390 ± 139 (h) |
| 73 | CH2CH3, R2 = I | >10 000 | 276 ± 65 | 7600 | >10 000 | –39 | 4050 ± 740 (h) |
| 40 | CH2CH3, R2 = phenylethynyl | 1780 ± 410 | 91.5 ± 23.7 | 1480 ± 450 | >10 000 | –66 | 344 ± 40 (h), 15 ± 1% (m) |
| 41 | CH2CH3, R3 = cPr, R2 = phenylethynyl | 2400 ± 90 | 852 ± 137 | 2110 ± 590 | c | –312 | 228 ± 115 (h), 41 ± 1% (m) |
| 42 | CH2CH3, R3 = CH2-cPr, R2 = phenylethynyl | c | 1400 ± 210 | 1300 ± 530 | 4470 ± 1330 | –13 | 791 ± 433 (h), 30 ± 1% (m) |
| 43 | CH2CH3, R3 = (CH2)2Ph, R2 = phenylethynyl | 627 ± 178 | 207 ± 21 | 1770 ± 540 | 480 ± 200 | 21 | 483 ± 62 (h), 51 ± 1% (m) |
| 82 | CH2CH3, R3 = (CH2)2Ph, R2 = phenylethynyl | 881 ± 158 | 217 ± 100 | >10 000 | >10 000 | –24 | 905 ± 144 (h) |
Binding assays performed as specified in Table 1, unless noted. NOP binding Ki values (μM, n = 1) were found to be 24 (>10), 25 (9.40), 28 (3.45), 29 (2.09), 35 (>10), and 40 (>10). Values are expressed as the mean ± SEM of N = 2–4 assays performed in duplicate, unless indicated.
A3AR binding data from Tosh et al.2,3,6,34,35,37 Representative binding inhibition at hA1AR is (% at 10 μM) 26, 39%; 40, 35%; 41, 44%; 42, 34%; 43, 26%. Representative binding inhibition at hA2AAR is (% at 10 μM) 26, 16%; 40, 22%; 41, 25%; 42, 20%; 43, <10%; determined as reported.37 mA1AR and mA2AAR binding data, % inhibition at 10 μM (mean ± SEM, n = 3), respectively: 24, 43 ± 2, 10 ± 3; 26, 54 ± 1, 5 ± 1; 28, 32 ± 2, 10 ± 3; 30, 53 ± 2, 13 ± 2. Values are expressed as the mean ± SEM of N = 3 assays performed in duplicate.
30–50% inhibition of radioligand binding at 10 μM.
<30% inhibition of radioligand binding at 10 μM.
Table 3. Structures and Modulation of Binding and Activity at hORs, rTSPO and A3AR of (N)-Methanocarba Adenosine Derivatives (44 and 45) and Ribose Derivative (46) Containing a 5′-Carbonyl Group; (N)-Methanocarba-5′-ketone and 9-Riboside Derivativesa.

| compound | R1=, other change | KOR,bKi, nM | DAT,b inhib., % of control at 10 μM, % | A3AR binding Ki, nM (species)b or % of control at 10 μm |
|---|---|---|---|---|
| 44 | 2610 ± 660 | –9 | 98 ± 30 (h), 41 ± 1% (m) | |
| 45c | NHCH3 | e | –321 | 1.55 ± 0.03 (h), 1170 ± 40 (m) |
| 46c | OCH3 | d | –220 | 11.5 ± 0.9 (h), 34 ± 2 (m) |
DOR, MOR, and TSPO binding inhibition is <50% at 10 μM. NOP binding Ki value (μM, n = 1): 44 (8.65).
Binding assays performed as specified in Table 1. Values are expressed as the mean ± SEM of N = 2–4 assays performed in duplicate, unless indicated.
A3AR binding data from Tosh et al.2,3,6,34,35,37 Representative binding inhibition at mA1AR and mA2AAR, % inhibition at 10 μM, respectively: 46, 62 ± 2, 12 ± 4. Values are expressed as the mean ± SEM of N = 3 assays performed in duplicate.
30–50% inhibition of radioligand binding at 10 μM.
<30% inhibition of radioligand binding at 10 μM.
Table 4. Binding Activity of 4′-Truncated (N)-Methanocarba Derivatives at hORs, hA3AR and TSPO and Transporters; 4′-Truncated (N)-Methanocarba Adenosine Derivatives.

| compound | R2= | R3= | DOR,a,bKi, nM | TSPO,bKi, nM | DAT,b inhibition, % of control at 10 μM, % | NET,b inhibition, % of control at 10 μM, % | A3AR binding Ki, nM (species)b,e |
|---|---|---|---|---|---|---|---|
| 3b | 3,4-F2-phenyl-ethynyl | CH2-(3-Cl-Ph) | g | 1750 ± 360 | 16 | 51 | 100 ± 30 (h) |
| 47 | phenyl-ethynyl | CH2-(3-Cl-Ph) | 8000 ± 1000 | 1640 ± 160 | 37 | 34 | 39.0 ± 20.0 (h), 299 ± 3 (m) |
| 48b | H | CH2-(3-Cl-Ph) | g | g | –2 | 35 | 4.9 ± 0.7 (h) |
| 49b | phenyl-ethynyl | CH3 | g | f | –213 | c | 5.48 ± 1.23 (h), 1530 ± 240 (m) |
| 50b | phenyl-ethynyl | CH2CH3 | g | f | –154 | d | 5.02 ± 2.19 (h), 1480 ± 170 (m) |
| 51b | phenyl-ethynyl | (CH2)2-Ph | g | g | 24 | d | 20 ± 6 (h), 480 ± 90 (m) |
| 52b | 2-Cl-phenyl-ethynyl | (CH2)2-Ph | f | 877 ± 368 | 7 | 24 | 37.0 ± 7.0 (h) |
| 53b | phenyl-ethynyl | CH2CH-(Ph)2 | g | 3570 ± 1820 | 17 | 5780d | 200 ± 20 (h) |
KOR and MOR binding inhibition is <50% at 10 μM. NOP binding Ki for compound 47 was found to be >10 μM.
Binding assays performed as specified in Table 1, unless noted. Values are expressed as the mean ± SEM of N = 2–4 assays performed in duplicate, unless indicated.
61% inhibition of radioligand binding at 10 μM.
Ki (μM), inhibition of binding of [3H]104 at NET: 50, 8.66; 51, 2.8.6
A3AR binding data from Tosh et al.3,34,35 Representative binding inhibition at hA1AR is (% at 10 μM) 49, 18%; 50, 36%; 51, 30%; representative binding inhibition at hA2AAR is (% at 10 μM, or Ki) 47, 670 nM; 49, 18%; 50, 42%; 51, 22%; determined as reported.37 Values are expressed as the mean ± SEM of N = 3 assays performed in duplicate.
30–50% inhibition of radioligand binding at 10 μM.
<30% inhibition of radioligand binding at 10 μM.
Table 5. Binding Activity of 4′-Truncated (N)-Methanocarba Derivatives at hORs, hA3AR and TSPO and Transporters; 4′-Truncated (N)-Methanocarba 7-Deaza-adenosine Derivativesa.

| compound | R2= | R3= | DOR,bKi, nM | KOR,bKi, nM | MOR,bKi, nM | TSPO,bKi, nM | A3AR binding Ki, nM (species),c or % of control at 10 μM |
|---|---|---|---|---|---|---|---|
| 54a | phenyl-ethynyl | CH3 | d | 1120 ± 220 | 2670 ± 280 | e | 85.6 ± 12.0 (h), 11 ± 1% (m) |
| 55a | phenyl-ethynyl | (CH2)2-Ph | 3660 ± 1330 | 1370 ± 180 | 4020 ± 890 | 2120 ± 230 | 217 ± 65 (h), 29 ± 2% (m) |
| 56 | phenyl-ethynyl | CH(cPr)2 | 2550 ± 660 | d | 1440 ± 750 | 4210 ± 1840 | 178 ± 32 (h) |
| 57 | 5-Br-thienyl-ethynyl | CH(cPr)2 | >10 000 | 3020 ± 90 | >10 000 | >10 000 | 2440 ± 430 (h) |
| 58 | I | CH(cPr)2 | e | e | e | e | 5310 ± 1310 (h) |
DAT and NET binding inhibition is <50% at 10 μM, unless noted. 54: DAT, −58%; 55: NET, Ki = 5.56 μM.
Binding assays performed as specified in Table 1, unless noted. Values are expressed as the mean ± SEM (n = 2–4). Values are expressed as the mean ± SEM of N = 2–4 assays performed in duplicate.
A3AR binding data from Tosh et al.2,3,6,34,35,37 Representative binding Ki values at hA1AR (nM): 55, 1300 ± 290; 56, 650 ± 71; determined as reported.37 Values are expressed as the mean ± SEM of N = 3 assays performed in duplicate.
30–50% inhibition of radioligand binding at 10 μM.
<30% inhibition of radioligand binding at 10 μM.
Chemical Synthesis
We previously reported the synthesis of numerous 5′-methylamide and 5′-ester derivatives (and truncated derivatives) of N6-alkyl C2-arylalkynyl (N)-methanocarba nucleosides similar to 1–4.2,3,6,36,37 The synthesis of analogues with C2–H (17, Scheme S1), N6-propyl-modified (19, 20, Scheme S2), and 1-deaza (26, Scheme S3) modifications is described in the Supporting Information.
The synthesis of target 7-deaza-2-arylalkynyl nucleosides began with a C2-iodo derivative 72 of the nucleobase (Scheme 1). Initially, compound 60 (Supporting Information, Scheme S4) was subjected to a lithiation reaction with n-BuLi in the presence of Bu3SnCl. However, instead of the desired product, it gave an unanticipated butyl keto derivative 63, which was further transformed to 2-iodo compound 64. C6 amination of compound 64 with methylamine followed by a Sonogashira coupling with 5-chloro-thienylacetylene and subsequent acid hydrolysis provided the 5′-(butyl-keto) nucleoside 44. To overcome this barrier to the synthesis of 7-deaza nucleosides, a convergent approach was designed. 7-Deaza-6-chloro purine 67 was silylated with TIPSCl in the presence of NaH to yield the protected pyrrolo[2,3-d]pyrimidine 68. Stannylation of 68 with Bu3SnCl in the presence of n-BuLi provided the tin derivative 69, which upon treatment with iodine and subsequent silyl deprotection afforded 7-deaza-2-iodo-purine 71. Mitsunobu condensation of 71 with glycosyl donor 59(38) gave the nucleoside precursor 72, which was aminated at the 6 position with various amines to provide compounds 73–76 (Scheme 1). Sonogashira coupling of iodo derivatives (73–76) with various alkynes using PdCl2(Ph3P)2 as a catalyst followed by acid hydrolysis of the respective derivatives (77–82) with 10% trifluoroacetic acid (TFA) afforded the final hypermodified nucleosides (28–29, 40–43). Acid hydrolysis of 2-iodo derivative 73 yielded the nucleoside 5′-ester 39.
Scheme 1. Synthesis of 7-Deaza (N)-Methanocarba 5′-Esters.

Reagents and conditions: (i) TIPSCl, NaH, THF, 0 °C; (ii) 2,2,6,6-tetramethylpiperdine, n-BuLi, Bu3SnCl, THF, −78 °C; (iii) I2, THF, rt; (iv) TBAF, THF, 0 °C to rt; (v) 7-deaza-2-iodo-6-chloro-purine, Ph3P, DIAD, THF, rt; (vi) MeNH2·HCl, Et3N, MeOH, rt; (vii) 2-chloro or 2-bromo-5-ethynylthiophene, Pd(Ph3P)2Cl2, CuI, Et3N, DMF, rt; and (viii) 10% TFA, MeOH, 70 °C.
4′-Truncated 7-deaza nucleosides (54–58) were synthesized in a similar fashion starting from the truncated pseudosugar 83(39) (Scheme 2). Intermediate 83 was coupled with 7-deaza-2-iodo-purine 71 under Mitsunobu conditions to give the nucleoside derivative 84, which was aminated with various amines to yield compounds 85–87. A Sonogashira coupling of compounds 85–87 with various alkynes gave C2-functionalized derivatives 88–91. Acid hydrolysis of compounds 88 and 89 with 10% TFA afforded final nucleosides 54 and 55. However, attempted acid hydrolysis of compounds 90 and 91 in the presence of 10% TFA gave complex mixtures because of the high acid sensitivity of the dicyclopropylmethylamino group;7 Dowex 50 was used as an alternative. Thus, isopropylidene deprotection of compound 87 with Dowex 50 followed by a Sonogashira coupling with phenylacetylene or 5-bromo-thien-2-ylacetylene afforded nucleosides 56 and 57, respectively.
Scheme 2. Synthesis of 4′-Truncated 7-Deaza (N)-Methanocarba Nucleosides.

Reagents and Conditions: (i) 7-Deaza-2-iodo-6-chloro-purine, Ph3P, DIAD, THF, rt; (ii) R3NH2, Et3N, MeOH, rt; (iii) alkynes, Pd(Ph3P)2Cl2, CuI, Et3N, DMF, rt; (iv) 10% TFA, MeOH, 70 °C; and (v) Dowex 50, MeOH–H2O, 50 °C
Pharmacological Testing
Assays of ORs and TSPO
Screening of the nucleoside derivatives at the human (h) ORs (Tables 1–5, Figures S1–S3, Supporting Information), including selected analogues at the nociceptin receptor (NOP), and rat kidney TSPO (Figure S4) and at other off-target sites, was performed by the Psychoactive Drug Screening Program (PDSP). Initially, radioligand binding assays were performed using membranes of mammalian cells overexpressing the receptor of interest.40 The following opioid agonist radioligands were used: [3H]Tyr-d-Ala-Gly-Phe-d-Leu ([3H]DADLE, 97) for DOR; [3H]N-methyl-2-phenyl-N-[(5R,7S,8S)-7-(pyrrolidin-1-yl)-1-oxaspiro[4.5]dec-8-yl]acetamide ([3H]U69593, 98) for KOR; [3H]Ala2-MePhe4-Glyol5-Enkephalin ([3H]DAMGO, 99) for MOR; and [3H]nociceptin 100 for NOP. [3H]N-Butan-2-yl-1-(2-chlorophenyl)-N-methylisoquinoline-3-carboxamide ([3H]PK11195, 101) was used for TSPO. Other reagents are found in the Supporting Information. Binding data for three neurotransmitter transporters, that is, DAT, the norepinephrine transporter (NET) and the serotonin transporter (SERT), as well as the A3AR, using methods previously reported,3,33 were also included for comparison. A primary radioligand binding screen was performed at each target protein using a fixed nucleoside concentration of 10 μM, and those compounds showing >50% inhibition were assayed with full concentration-dependent curves. Binding data determined by PDSP for these compounds at other diverse receptors and channels are described below and detailed in Figure S5 (Supporting Information) and in some cases in previous publications.2,5,8 To further explore the potential off-target activities in the (N)-methanocarba nucleoside series, we examined A3AR agonists 14 and 18 in broad screens of 240 GPCRs and 486 kinases, and there were no additional outstanding interactions at 10 μM at any of these sites (Supporting Information). Thus, the specificity for a few interactions was high, and these nucleosides are not promiscuous binders.
3,4-Difluoro 5′-methylamide derivative 4 has been studied as an A3AR agonist for reducing chronic neuropathic pain, and some of its weak off-target activities, including DOR, were already reported.4,36 Compound 4 bound to DOR with a Ki value of 2.48 ± 0.79 μM (n = 4), while its 4-fluoro-7 and 3-chloro-phenylethynyl 9 analogues bound more weakly. Substitution of the terminal 3,4-difluorophenyl group of 4 with 2-Cl-Ph 8 prevented binding affinity at DOR, but this and other compounds in this series (4 and 7) bound to TSPO with Ki values ∼300 nM.4 Thus, the DOR affinity was sensitive to the C2-terminal aryl group, whereas the TSPO affinity was less sensitive. A N6-(trans-phenyl-cyclopropyl) group in 10 (diastereomeric mixture) was not associated with measurable binding affinity at TSPO or ORs. None of the subsequent 5′-methylamides 11–20 at 10 μM significantly inhibited DOR binding (by >50%).
In the series of N6-methyl 5′-methylamides, a C2-(5-chlorothien-2-yl-ethynyl) 14 but not a 5-bromothien-2-yl-ethynyl 15 group was tolerated in TSPO binding, but only one 5-chlorothien-2-yl-ethynyl derivative in the 5′-methylamide series, 21, displayed OR binding affinity. Thus, a 5′-amide extension as a 2-aminoethylamide in 21 introduced μM affinity at KOR (Ki 2.9 μM) while reducing TSPO binding affinity (Figure S2). A 1-deaza substitution of 14 in compound 16 prevented TSPO binding, but its effect on KOR could not be probed in the 5′-amide series, because the reference compound 14 was inactive. N6-Alkyl extensions to substituted n-propyl moieties in 18–20 maintained μM affinity at TSPO with no significant KOR binding.
Unlike 5′-methylamide 14, the corresponding 5′-methyl ester 22 and its 5-bromothien-2-yl-ethynyl analogue 23 displayed μM affinity at KOR but weaker affinity than 14 at TSPO. Furthermore, an increased affinity of homologated 5′-alkyl esters at KOR was revealed, and TSPO affinity of <1 μM was determined (Figure S2). Specifically, the ethyl 24 and n-propyl 31 esters displayed Ki values at KOR of ∼400 nM. The 5-halo-thiophene substitution in the ethyl ester series indicated an order of affinity of Cl ≥ Br (25) at KOR. The 5′-i-propyl ester 31 and the corresponding carboxylic acid 38 were inactive at both KOR and TSPO. Further 5′-ester elongation to n-Bu in compound 32 and i-pentyl in compound 33 decreased the KOR affinity ∼threefold compared with 24 but greatly reduced TSPO affinity. Inclusion of cyclohexyl and phenyl groups on the 5′-ester moiety in 34–37 led to variable moderate (μM) KOR affinity or inactivity, with O-benzyl derivative 35 having the highest affinity (Ki 629 nM).
Compound 26 allowed the effects of the 1-deaza modification to be examined in the 5′-ester series, demonstrating small affinity decreases at KOR and TSPO compared with 24. A 3-deaza modification in 27 was permissive for TSPO binding but prevented KOR binding. In contrast to the requirement for the N3 in KOR binding, a 7-deaza modification in esters 28 and 29 both enhanced KOR affinity (∼40 nM) and allowed DOR and MOR affinity to appear (Figure S3). The preference for KOR in comparison to DOR and MOR was 19- and 15-fold for 28 and 11- and 9-fold for 29, respectively. The preference for KOR in comparison to TSPO was ∼20-fold for both 28 and 29.
In the 7-deaza-5′-ester series, modified N6 and C2 substituents were explored. C2-phenylethynyl analogue 40 had 2–3-fold lower affinity at ORs than the corresponding C2-(5-chlorothien-2-yl-ethynyl) analogue 28 and was inactive at TSPO. N6-Cyclopropyl 41 and cyclopropylmethyl 42 groups reduced the OR affinity compared with N6-methyl 40. N6-2-Phenylethyl-substituted analogue 43 was comparable to N6-methyl analogue 40 in OR affinity, but with enhanced TSPO affinity (Ki 480 nM). Compound 77, a 2′,3′-isopropylidene-protected precursor of 28 had the same binding affinity at KOR, but was weaker in TSPO binding. Compound 82, the isopropylidene-protected precursor of 43, maintained affinity at both DOR and KOR, but not MOR.
The inactivity of the ribose derivatives 45 and 46 compared with their corresponding (N)-methanocarba derivatives 14 and 22, respectively, showed that the rigid (N)-methanocarba ring is required for interaction with both KOR and TSPO. In the methanocarba series, 2-iodo 5′-ethyl ester 39 was included to test the requirement for an extended C2 substituent; in comparison to 28, there was only a ∼2-fold affinity decrease at DOR and KOR and a large decrease at MOR and TSPO.
We extended the SAR analysis to a set of 4′-truncated nucleosides in the same methanocarba series (Tables 4 and 5), to determine if a 7-deaza modification was also OR affinity-enhancing without a 5′-ester and to analyze the effects of substitution at N6 and C2 positions (Tables 4 and 5, 3, 47–58). Starting with the lead of DOR and TSPO binding reported for compound 4, the truncated N6-(3-chlorobenzyl) analogues 3 and 47 with extended C2 groups, and compound 48 that had no C2 substitution, were weaker or inactive in OR binding, but 3 and 47 bound weakly to TSPO. Small N6-methyl 49 and ethyl 50 groups or an N6-2-phenylethyl 51 group did not produce appreciable OR or TSPO binding. C2-modified analogue 52 and N6-(2,2-diphenylethyl) derivative 53 bound measurably at TSPO but not ORs.
7-Deaza-modified truncated analogues were compared: the N6-methyl 54, N6-(2-phenylethyl) 55 and N6-(dicyclopropylmethyl) 56 analogues (all C2-phenylethynyl) bound to various ORs in the micromolar range, while 5-bromothien-2-yl-ethynyl derivative 57 bound only to KOR. However, lack of an extended C2 substituent in 58 prevented interaction with both KOR and TSPO. Therefore, as in the 5′-amide series, the 4′-truncated (N)-methanocarba nucleosides bound with moderate affinity at ORs.
Binding assays at NOP41 indicated moderate-to-weak affinity (Ki, μM) for 4 (4.56 ± 0.45), 9 (>10), 24 (>10), 25 (>10), 28 (3.71 ± 0.78), 29 (2.76 ± 0.67), 35 (>10), 40 (>10), 44 (>10), and 47 (>10). Single-point functional assays at the three ORs, using a TANGO assay of β-arrestin mobilization,42 demonstrated that 5′-amide and 5′-ester nucleosides 4, 24, 25, 28, 29, 35, and 45 were antagonists at all three ORs, and no agonist effect was observed at 10 μM at DOR or MOR (Table 6). Agonist activity at KOR was not determined in the TANGO assay. An additional functional output was determined by conducting dose–response curves at KOR using an enzyme complementation assay to measure β-arrestin2 recruitment to the receptor in U2OS cells (Figure 1). Compounds 28, 39, and 40 inhibited the agonist response of 1 μM N-methyl-2-phenyl-N-[(5R,7S,8S)-7-(pyrrolidin-1-yl)-1-oxaspiro[4.5]dec-8-yl]acetamide (U69593, 98). Compound 28 was the most potent antagonist with an IC50 value equal to 794 ± 50 nM (Table 7). Compounds 43 and 54 did not inhibit agonist 98 in the assay. The compounds were also tested as agonists, and only 43 stimulated β-arrestin2 recruitment and only at the highest concentrations (30 ± 4.6% relative to full agonist 98). These compounds were further tested as agonists and antagonists in the KOR-induced Gαi-dependent inhibition of forskolin-stimulated cAMP accumulation (Figure 2). Compounds 28, 39, and 40 inhibited the agonist response of 100 nM 98, with compound 28 again being the most potent antagonist (IC50 = 2220 ± 830 nM, Table 7). Compounds 43 and 54 did not inhibit agonist 98 in the assay, and 43 was a weak agonist for KOR-mediated cAMP inhibition (28 ± 11% relative to 98).
Table 6. Functional Activity at 10 μM (Percent Activity in β-Arrestin-Recruitment TANGO Assaya Compared with Standard Agonist or Antagonist) at Human ORsa.
| DOR | KOR | MOR | |||
|---|---|---|---|---|---|
| no. | Ag (%) | Antag (%) | Antag (%) | Ag (%) | Antag (%) |
| 4 | –10.3 | 25.3 | 125 | –1.6 | 18.8 |
| 24 | 0.7 | 35.3 | 58.2 | 0.3 | –13.4 |
| 25 | –1.7 | –6.0 | 88.1 | –1.2 | 22.6 |
| 28 | –0.6 | 85.4 | 99.6 | 0.4 | 96.4 |
| 29 | –1.1 | 93.2 | 104.5 | 1.6 | 96.9 |
| 35 | –1.9 | 14.1 | 112.9 | 3.1 | 82.1 |
| 44 | 6.7 | 20.3 | 65.5 | 6.0 | –44.4 |
Determined by PDSP, using the Tango GPCR assays as described:42 the principle of the assay: receptor activation recruits a β-arrestin fusion protein connected to tobacco etch virus (TEV) protease to the activated OR. The cleavage by TEV protease releases the hybrid factor for transcription GAL4-VP16 from its position fused to the OR. The liberation of the transcription factor induces expression of the β-lactamase reporter gene.49 KOR agonist activity was not determined. Values are expressed as the mean ± SEM of one assay performed in duplicate. The following standard DOR, KOR, and MOR ligands were used for comparison: agonists (set as 100% activation at 10 μM) DAMGO 99, salvinorin A 109, and morphine 110, respectively. Reference antagonist used was naloxone 121 (set as 100% inhibition, at 10 μM), of the effects of corresponding agonist (nM): DOR, DALDE 97; KOR, 109 (3); MOR, 99 (300).
Figure 1.

Ligand effects on βarrestin2 recruitment to hKOR using the DiscoveRx PathHunter enzyme fragment complementation assay. (A) Antagonism of stimulated βarrestin2 recruitment by 1 μM agonist U69593 98. Norbinaltorphimine (NorBNI, 120) is the reference KOR antagonist. (B) Agonist activity; EC50 for 98 = 75 ± 9 nM; not derived for 43 due to lack of plateau. N = 3 assays performed in duplicate; mean ± SEM presented.
Table 7. Antagonism of KOR in the βarrestin2-KOR Enzyme Fragment Complementation Assay (DiscoveRx) and cAMP Cisbio Assaya,b.
| βarrestin2
recruitment |
inhibition of cAMP accumulation |
|||
|---|---|---|---|---|
| compound | IC50 (nM) | Imax (% 120) | IC50 (nM) | Imax (% 120) |
| 120 | 2.0 ± 0.3 | 100 | 4.0 ± 0.8 | 100 |
| 28 | 794 ± 50 | 92 ± 1 | 2220 ± 830 | 122 ± 13 |
| 39 | 4200 ± 940 | 77 ± 3 | 13100 ± 6000 | 105 ±3 |
| 40 | 2220 ± 540 | 76 ± 2 | 12100 ± 4400 | 100 ± 1 |
| 43 | NC | (10* ± 15) | NC | (23 ± 5) |
| 54 | NC | (35* ± 4) | NC | (16 ± 6) |
Figure 2.

Ligand effects on hKOR-induced inhibition of forskolin-stimulated cAMP accumulation using the Cisbio cAMP Dynamic 2 assay. (A) Antagonism of 100 nM agonist U69593 98-stimulated KOR. NorBNI (120) is the reference KOR antagonist. (B) Agonist activity; EC50 for 98 = 3.3 ± 1.8 nM. Not derived for 43 due to lack of plateau. N = 3, performed in duplicate; mean ± SEM presented.
Activity at the ARs, Transporters, and Other Off-Target Proteins
Some of these nucleoside derivatives, particularly 5′-methylamides, were previously characterized as AR ligands. For example, compound 14 is also a fully efficacious hA3AR agonist of high affinity (Ki = 0.70 nM).2 The hA3AR binding affinities in the series of 5′-esters, such as 23, were consistently in the μM range or weaker, and the affinity at mA3AR was generally much weaker than at hA3AR. The hA1AR and hA2AAR affinities of the nucleosides in this study were generally >10 μM. As reported earlier,6 5′-ester 22 displayed eightfold lower affinity than corresponding 5′-methylamide 14 in hA3AR binding; thus, the substitution of ester for amide tended to increase both affinity and preference for KOR. The 7-deaza modification of 5′-ethyl ester 28 reduced hA3AR binding affinity by 31-fold compared with 24, which is greater than the affinity loss in 1-deaza 26 and 3-deaza 27 analogues. Furthermore, 27 and 28 were inactive in mA3AR binding. 7-Deaza 28 was inactive as an agonist in a functional assay of hA3AR-mediated inhibition of cAMP formation (n = 3),43 with maximal efficacy at 10 μM of 0.59 ± 6.62%, compared with full agonist 5′-N-ethyluronamidoadenosine (100%). Thus, the combination of 7-deaza modification with a 5′-small alkyl ester in place of the 5′-amide appears to turn A3AR agonists into weakly binding A3AR antagonists or non-A3AR binding compounds. The functional activity of 5′-esters 22 and 27 as hA3AR partial agonists was reported.6 Therefore, the 5′-ester modification alone reduces A3AR efficacy, while increasing OR antagonist affinity. A 5′-n-propyl ester 30 had higher affinity at both mouse (m) and hA3ARs than the corresponding 5′-ethyl ester 24, and among the extended esters, 5′-benzyl ester 35 displayed the highest affinity at hA3AR (Ki 7.81 nM) and at mA3AR (Ki 891 nM). 4′-Truncated (N)-methanocarba-adenosine derivatives displayed varied affinities at hA3AR, as we previously reported.35 The 7-deaza modification reduced hA3AR affinity by 16-fold (in a comparison of 54 and 49) or 11-fold (in a comparison of 55 and 51), and the 7-deaza analogues were inactive at the mA3AR.
Another recently discovered target of certain C2-extended (N)-methanocarba-adenosine derivatives was DAT, as 5′-methyl and ethyl esters in this series were potent allosteric binding enhancers of tropane radioligand affinity (resulting in enhanced level of binding in the primary screen) and inhibitors of dopamine uptake (Figure 3).6 Enhancement of radioligand binding at NET was also observed in this chemical series but less potently than at DAT. Thus, it was important to establish effects of the 7-deaza and other modifications on transporters, such as radioligand binding enhancement in the DAT primary screen (Tables 1–3, shown as a negative percent inhibition at 10 μM) and inhibition at NET (Tables 4 and 5). Various 1-, 3- or 7-deaza derivatives (26–29) were compared to probe the participation of these H-bond acceptor nitrogens of adenine for DAT recognition. The 1-deaza modification in the 5′-methylamide series prevented enhancement of tropane radioligand binding at DAT, but not A3AR binding.5,37 Similarly, the conversion of an N6-methyl to N6-propyl group in 18 greatly reduced DAT interaction, and its 3,3,3-trifluoropropyl equivalent in 19 eliminated it. The corresponding 3-deaza 5′-ethyl ester 27 was nearly as efficacious as 24 in enhancing binding at DAT, whereas the 7-deaza 5′-ester 28 was much less efficacious in enhancing DAT binding. EC50 values for binding enhancement at hDAT determined by the PDSP for 28 and 29 were >10–6 M. Using an alternate tropane radioligand [125I]methyl (1R,2S,3S)-3-(4-iodophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate (RTI-55, 118) as described,628 enhanced hDAT binding by 320 ± 23% at 10 μM with an EC50 value of 1410 ± 340 nM (Figure 3, Table S1, Supporting Information). Using a nontropane radioligand [3H](±)-5-(4-chlorophenyl)-3,5-dihydro-2H-imidazo[2,1-a]isoindol-5-ol (mazindol, 119) as described,828 enhanced hDAT binding by a maximal 267 ± 17% with an EC50 value of 2160 ± 550 nM. The IC50 for inhibition of dopamine uptake by 28 was >9800 nM. The potencies in enhancement of binding at hNET and hSERT and in inhibition of uptake were >10 μM. Thus, in the 5′-ester series, the N1 nitrogen atom, but not N3, was essential for interaction with neurotransmitter symporters, and the 7-deaza modification greatly reduced these interactions. Byproducts of the synthesis in the 7-deaza series were inactive at DAT, that is, C2-truncated 5′-methylamide 17 and 5′-butylketone 44. In the 4′-truncated series, two compounds with C2-phenylethynyl and small N6 groups, 49 and 50, displayed weak-to-moderate DAT interaction, but this binding enhancement disappeared with extension of the N6 group in 51.
Figure 3.

Interaction of selected nucleosides 5′-methylamide 14, 5′-ethyl ester 24, and 7-deaza 5′-ethyl ester 28 as allosteric modulators of hDAT expressed in HEK cells, as characterized using a tropane, methyl (1R,2S,3S)-3-(4-iodophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate (RTI-55, 118) (A), and a nontropane, (±)-5-(4-chlorophenyl)-3,5-dihydro-2H-imidazo[2,1-a]isoindol-5-ol (mazindol, 119). (B) Radioligand binding and inhibition of [3H]dopamine ([3H]DA) uptake (C). Comparison with known binding and uptake inhibitors, cocaine and mazindol, is shown. Methods and data for 14 and 24 were described in Tosh et al.6 The 7-deaza modification in 28 greatly reduces the binding enhancement and inhibition of dopamine uptake compared to 24. Compound 30 enhanced DAT binding with an EC50 value of 294 ± 82 nM (not shown). EC50 and IC50 values for the curves shown and archival compounds for comparison are provided in Supporting Information.
The nucleoside derivatives were screened at other off-target interactions by the PDSP (Supporting Information, all values Ki, μM). In addition to those off-target interactions already reported for the archival compounds,2,4,6,37 affinity at 5HT2A (27, 1.30 ± 0.29; 57, 4.34 ± 1.74), 5HT2B (6, 2.96 ± 0.60; 27, 1.78 ± 0.28; 77, 4.18; 55, 3.06 ± 0.17; 58, 1.33), and 5HT2C (21, 0.51; 23, 2.33±; 27, 0.40; 31, 0.43; 46, 3.24 ± 1.05; 77, 0.016) serotonin receptors was detected. Thus, compound 27 bound to all three 5HT2Rs. Curiously, 2′,3′-isopropylidene protected compound 77 displayed a high affinity and selectivity (260-fold compared with 5HT2BR) at 5HT2CR. Compounds 6 and 43 bound to the human α2C adrenergic (6.3 and 0.51, respectively) and compound 6 to the β3 adrenergic (2.6) receptor. Compound 10 bound to the human 5HT1A serotonin (6.3) and β3 (2.5) receptors. Weak affinity was noted at the muscarinic M1 (55, 6.97) and M5 (27, 3.85) receptors. Compounds 35 and 73 bound to the human SERT (5.56 and 6.58, respectively). Intermediate affinity was found in inhibition of NET binding (6, 3.16; 10, 2.31; 43, 2.82; 51, 2.80; 53, 5.78; 55, 5.65). Compound 57 bound to the rat brain benzodiazepine receptor with a Ki of 118 nM. Widespread affinity at the rat σ2 receptor was detected: 6, 1.54; 10, 2.51; 26, 1.76; 36, 5.87; 42, 1.11; 47, 2.73; 51, 1.48; 52, 1.82; 56, 0.67; 57, 6.92. There were fewer and weaker interactions at the σ1 receptor: 9, 7.55; 12, 7.11; 22, 8.73; 27, 1.04; 47, 7.66.
Molecular Modeling at KOR
Docking and molecular dynamics (MD) were exploited to rationalize the observed experimental data by applying the knowledge gained from the analysis of X-ray structures of KOR, MOR, and DOR with bound antagonists.43−45 Although a TSPO X-ray structure was recently reported,52 we did not propose a binding mode at this protein and therefore focused our attention on the molecular signatures determining OR affinity and selectivity. In particular, we generated an initial hypothesis for the KOR binding mode of the 7-deaza 5′-ethyl ester derivative 28 (Ki = 42 nM) and used it to rationalize the SAR of a subset of structurally related analogues (22, 24, 30–37). Compound 28 was initially docked by Induced Fit Docking (IFD) at a hKOR homology model built upon an available X-ray structure with a bound 4-phenylpiperidine antagonist43 (PDB ID: 4DJH, see Supporting Information). In the resulting docking pose (Figure 4), the ligand resided in the upper portion of the TM (transmembrane) bundle with the (N)-methanocarba ring pointing toward the EC side and the N6-methyl group buried in the binding cleft. The C2 extension was directed toward a Tyr-rich region at the interface of TM2, TM1, and TM7. The ligand established interactions with residues located mainly in TM3, TM6, and TM7, consisting of two H-bond interactions of the C3′ and the C2′ hydroxyl groups with the side chains of Tyr312 (7.35) and Tyr139 (3.33), respectively, and π–π stacking interaction between the 5-chlorothienyl moiety and the side chain of Tyr320 (7.43). Surprisingly, the ligand did not form a H-bond with the conserved Asp138 (3.32) residue.
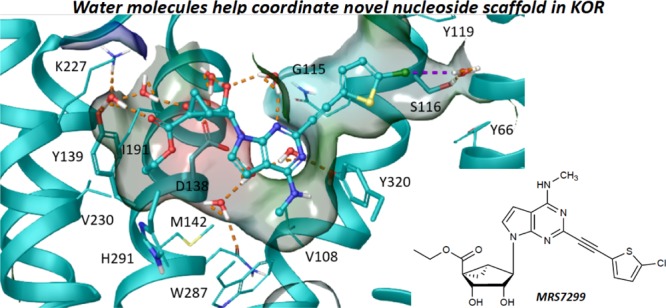
Figure 4.
Binding mode predicted by IFD N6-methyl 5′-ethylester 7-deaza (N)-methanocarba derivative 28 (pink carbon atoms, sticks representation) at hKOR. Side chains of residues important for ligand recognition (gray carbon atoms) are reported as sticks. Residues in close contact with the ligand are depicted as transparent surfaces color-coded according to the residue type (red: negatively charged; blue: positively charged; cyan: polar, green: hydrophobic). H-bonds and π–π stacking interactions are pictured as dashed orange and cyan lines, respectively. Nonpolar hydrogen atoms are omitted. The PDB ID of the X-ray structure used as starting points for molecular modeling was 4DJH.
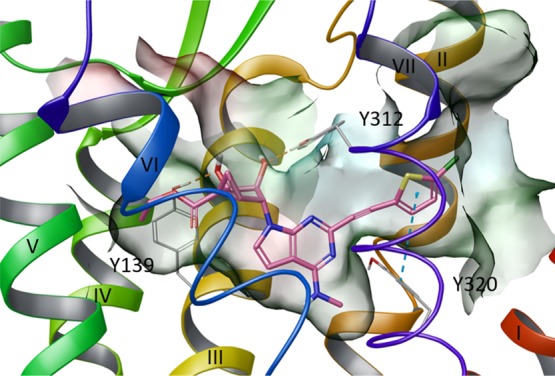
We subjected the initial docking pose of 28 to 30 ns of membrane MD simulation (run in triplicate). Replicas were run by using different initial randomly assigned atom velocities, and ligand–receptor interactions were analyzed qualitatively (ligand and protein root-mean-square deviation (RMSD) values in Table S2). We considered a key His residue surrounding the binding site, that is, His291 (6.52), in two possible alternative protonation states and compared the two result sets (Video S1). A preliminary analysis of the His(6.52) environment in the available antagonist-bound KOR,43 MOR (PDB ID: 4DKL),44 and DOR (PDB ID: 4N6H)45 X-ray structures unequivocally determined it as protonated on the Nε atom (hereby denoted as HSE), as the residue established a H-bond with the aromatic hydroxyl moiety of the co-crystallized antagonists mediated by two highly conserved water molecules. As we did not retain those water molecules during the IFD and the binding mode of 28 (Figure 4) did not predict the presence of a H-bond donor moiety in proximity to His291 (6.52), we also considered the alternative protonation state featuring a hydrogen atom on the Nδ (hereby denoted as HSD).
Notably, in all the simulations run with both HSE and HSD models, the ligand moved deeper in the binding site with respect to the initial docking pose to establish a persistent H-bond between the 3′-OH group and the conserved Asp138 (3.32) side chain. However, as reported in Table S2 and visualized in Video S1 (right panel), in the HSD model, the ligand was characterized by a higher average RMSD value and was less stable. By superimposing the lowest interaction energy (IE) complexes extracted for each replica of the two models (Figure 5A,B), it was evident that TM1, which was considerably displaced with respect to the TM bundle in the initial X-ray structure,43 maintained its displaced position in the HSD model and approached the TM bundle in the HSE model. To investigate the reason for the different placement of TM1 in the two models, we analyzed in more detail a selected trajectory for each model (Table S6 and Video S1) and focused our attention on the ligand-protein complexes characterized by the lowest IE value extracted from each trajectory. As depicted in Figure 5C, in the HSD model, His291 (6.52) rotated and pointed toward TM3. This rotation was accompanied by the diffusion of water molecules into the binding cavity that connected His291 (6.52) to the conserved Asp138 (3.32) (Video S1, right panel). The presence of this network of water molecules pushed the ligand slightly higher in the binding site. In the HSE model, His291 (6.52) maintained its initial conformation facing TM5, thus allowing the ligand to be accommodated deeper in the binding site (Video S1, left panel). Concerning the placement of TM1 with respect to the TM bundle, in the HSE model, the movement of TM1 occurred early during the equilibration phase (data not shown) and was triggered by favorable hydrophobic interactions established by 5-chlorothienyl moiety of the ligand with residues at the interface between TM1 (Ile62) and TM2 (Tyr119), which persisted during the production phase (Video S1, left panel).
Figure 5.
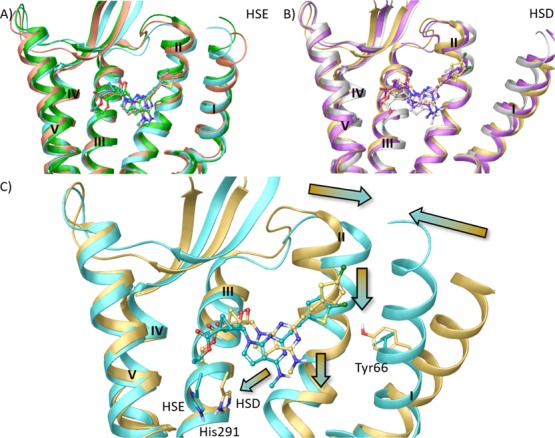
Superimposition of ligand–protein complexes characterized by lowest IE values during the MD simulation (replicas represented with different colors) of (A) HSE and (B) HSD hKOR models in complex with N6-methyl 5′-ethylester 7-deaza (N)-methanocarba derivative 28 (ball and sticks representation). (C) Superimposition of ligand–protein complexes characterized by lowest IE values among three replicas (see Table S2) for the HSE (cyan ribbon, and ligand in cyan ball and sticks) and the HSD (yellow ribbons, ligand in yellow ball and sticks) hKOR models in complex with N6-methyl 5′-ethylester 7-deaza (N)-methanocarba derivative 28. Side chains of residues undergoing considerable conformational changes during the MD simulation are highlighted (sticks representation with carbon atoms matching the color of the model). Arrows indicate the shift of ligand position and TMs between the two models. TM6, EL3, and TM7 were omitted to aid visualization. The PDB coordinates of both complexes are available as separate Supporting Information files. The PDB ID of the X-ray structure used as starting points for molecular modeling was 4DJH.
Considering both higher ligand stability in the MD simulations and the analysis of the X-ray structures,43−45 we considered the HSE model more reliable and inspected the ligand environment in the selected trajectory more carefully. In the ligand–protein complex characterized by the lowest IE values (Figure 6), the ligand was almost completely buried and shielded from the aqueous environment, except for the 2′,3′-OH groups and the 5′-carbonyl group (Figure 6B). Consequently, the interactions involving those groups were mainly direct and water-mediated H-bonds established with polar residues (Figure 6A,C). The C3′ group established an H-bond with the conserved Asp138 (3.32) and a water-mediated H-bond with Tyr139 (3.33). The C2′ hydroxyl was H-bonded to the conserved Asp138 (3.32) through the interplay of another water molecule. Additional water molecules connected the N3 nitrogen to the C2′ hydroxyl group and the side chain of Tyr312 (7.37) and the 5′-carbonyl group to the side chains of Tyr139 (3.33) and Lys227 (5.39). The 5′-ethyl ester moiety was accommodated in a small hydrophobic pocket delimited by Met142 (3.36), Tyr139 (3.33), and Val230 (5.42). The N6-methyl group was hosted in a hydrophobic pocket delimited by the conserved Trp187 (6.48), and the fused cyclopropyl ring of the (N)-methanocarba system established hydrophobic contacts with Ile194 (6.55). The 5-chlorothienyl moiety lay in a hydrophobic pocket delimited by Tyr312 (7.35), Tyr119 (2.64), Tyr66 (1.39), Met112 (2.57), and Tyr320 (7.43) with a water-mediated halogen bond interaction connecting the chlorine atom to the side chain of Ser116 (2.61).
Figure 6.
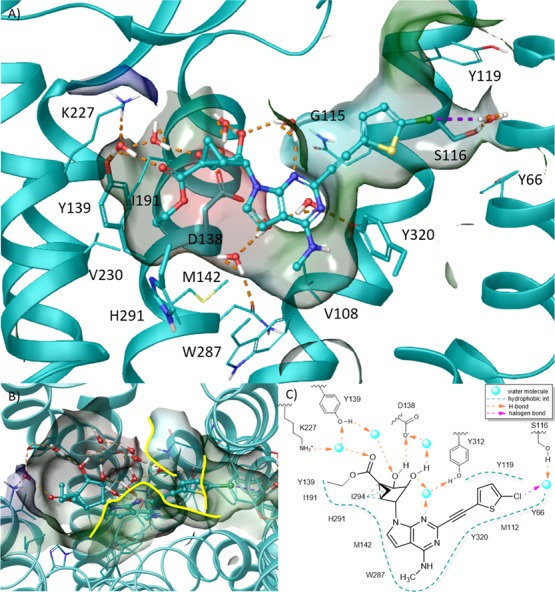
(A) Side view, (B) top view, and (C) schematic representation of the hypothetical binding mode of N6-methyl 5′-ethylester 7-deaza (N)-methanocarba derivative 28 (cyan carbon atoms, ball and sticks representation) at hKOR as predicted by MD simulation. Side chains of residues important for ligand recognition (cyan carbon atoms) and water molecules are represented as sticks. Residues in close contact with the ligand are depicted as transparent surfaces color-coded according to the residue type (red: negatively charged; blue: positively charged; cyan: polar, green: hydrophobic) with boundaries to solvent exposure highlighted with yellow solid lines. H-bonds and halogen bonds are pictured as dashed orange and purple lines, respectively. Nonpolar hydrogen atoms are omitted. TM6, EL3, and TM7 were omitted to aid visualization. The PDB ID of the X-ray structure used as starting points for molecular modeling was 4DJH.
We finally tested the ability of the binding hypothesis suggested by MD simulation (Figure 6) in rationalizing the SAR at KOR of a subset of structurally related ligands, namely, 7-aza 22, 24, and 30–37. In the corresponding docking poses (Figure 7), all ligands maintained the above described pattern of H-bond interactions while differing in the 5′-ester group placement. The docking analysis suggested that the hKOR affinity modulation arises from the ability of this group to fit the narrow hydrophobic pocket delimited by Met142 (3.36), Val230 (5.42), and Tyr139 (3.33). In particular, the hydrophobic contact of the ester with Tyr139 (3.33) seems to favorably modulate the KOR binding affinity, as the 7-aza methyl-ester derivative 22 (Ki = 3130 nM) binds in a pose that superimposes well with the one predicted for 28 (Ki = 42 nM) and 24 (Ki = 396 nM) but lacks this interaction (Figure 7A). Slight elongation of the alkyl ester chain is moderately tolerated, whereas further alkyl elongation/branching or the introduction of aliphatic/aromatic cycles requires the position of the bulkier groups to be either outside the above-mentioned hydrophobic pocket (Figure 7B) or completely solvent exposed (Figure 7C), respectively.
Figure 7.
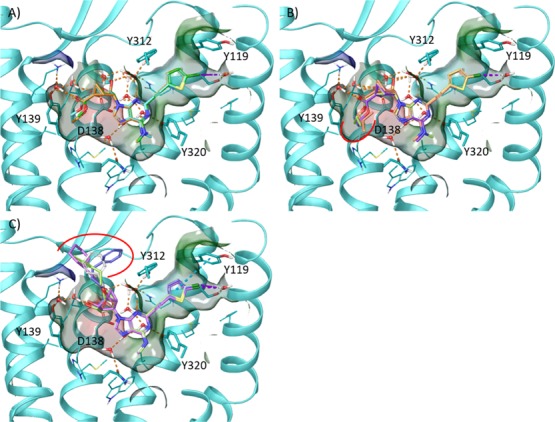
Docking poses of 7-aza 5′-ester (N)-methanocarba derivatives at the hKOR. (A) Predicted docking poses of 22 (green carbon atoms), 24 (cyan carbon atoms), and 30 (orange carbon atoms). (B) Predicted docking poses of 30 (purple carbon atoms), 31 (green carbon atoms), 32 (yellow carbon atoms), and 33 (orange carbon atoms). (C) Predicted docking poses of 34 (magenta carbon atoms), 35 (gray carbon atoms), 36 (green carbon atoms), and 37 (blue carbon atoms). Side chains of residues important for ligand recognition (cyan carbon atoms) and water molecules are represented as sticks. Residues in close contact with the ligand are depicted as transparent surfaces color-coded according to the residue type (red: negatively charged; blue: positively charged; cyan: polar, green: hydrophobic). H-bonds, π–π stacking interactions and halogen bonds are pictured as dashed orange, cyan, and purple lines, respectively. Nonpolar hydrogen atoms are omitted. Red solid lines highlight the changes with respect to the binding mode predicted for the 7-deaza derivative 28 (Figure 3). TM6, EL3, and TM7 were omitted to aid visualization. The PDB ID of the X-ray structure used as starting points for molecular modeling was 4DJH.
Concerning the hKOR affinity increase and the display of MOR and DOR affinity in 7-deaza derivatives, we speculate that the presence of a 7-nitrogen atom would trap unstable water molecules in the small pocket delimited by the conserved Trp287 (6.48), thus disrupting the hydrophobic contacts with the N6-methyl and the C7 atom. Although we were not able to confirm this hypothesis, the MD simulation of the HSD model clearly suggested the presence of ligand-destabilizing water molecules in this binding cavity region. Moreover, the superimposition between the proposed hKOR binding mode and the WaterMap computed by Goldfeld et al. on the KOR X-ray structure9 (Figure S7) highlighted that the hydrophobic moieties of 28, such as the 5′-ethyl ester, the 7-deaza position, the N6-methyl group and the C2-(5-chlorothien-2-yl-ethynyl) moiety, overlapped well with regions predicted to be occupied by “unhappy” water molecules. On the other hand, the N1 and N3 adenine nitrogen atoms, the 5′-carbonyl group, and the C2′ and C3′ hydroxyl groups lie close to a region rich in “happy” water molecules.
Finally, our modeling analysis also suggested that the moderate hKOR preference might arise from a water-mediated H-bond between the N3 nitrogen and the non-conserved Tyr312 (7.35). Binding data suggested that a 3-deaza modification might be incompatible with the ORs as it clearly caused a loss of hKOR affinity (e.g., 24 vs its 3-deaza analogue 27, Ki = 396 nM and % of control at 10 μM < 10%, respectively). However, further derivatives need to be synthetized and tested to fully support this speculation.
ADME-Tox Testing
The in vivo pharmacokinetics of 28 was studied in the Sprague-Dawley rat (Figure 8 and Table S3), and in vitro preclinical testing was performed using described methods (Supporting Information).5 Upon oral administration of 1, 5, and 10 mg/kg doses, the bioavailability was determined to be 58, 92, and 60 % F, respectively. Furthermore, in vitro preclinical testing (Tables S4–S7, Figures 8 and 9) demonstrated that 28 was not an inhibitor of hERG (IC50 > 30 μM, using a fluorescence polarization assay) or CYP450 enzymes (IC50 ≥ 10 μM at 1A2, 2C9, 2C19, 2D6 and 3A4 isoforms) or cytotoxic to Hep-G2 cells (CC50 > 30 μM). It was stable in human plasma (98% remaining after 2 h) and in simulated gastric and intestinal fluids (100% remaining after 240 min). The CACO-2 cell permeability (Papp, apical to basal) was 1.42 × 10–6 cm/s, with efflux indicated (ratio 15.6).
Figure 8.
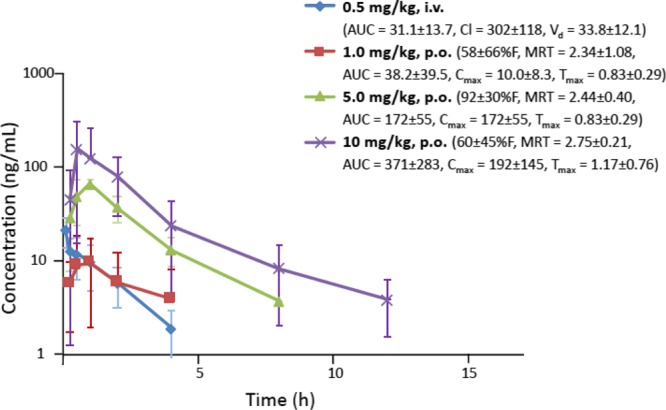
Pharmacokinetics of 28 in male SD rats. Rats were fasted overnight for three oral doses, but fed for the i.v. dose. The in vivo half-life (t1/2, h, p.o.) of 28 was 1 mg/kg, 2.16 ± 0.45; 3 mg/kg, 1.55 ± 0.42; 10 mg/kg, 1.91 ± 0.28. The t1/2 for the i.v. dose was 1.31 ± 0.06 h. Oral bioavailability (% F) and other pharmacokinetic parameters (units) are indicated: MRT (mean residence time, h); AUC (area under the curve, time 0 to ∞, ng·h/mL); Cl (clearance, mL/min/kg); Vd (volume of distribution, L/kg); Cmax = 10.0 ± 8.3 (max. concentration, ng/mL); Tmax (time at max. concentration, h).
Figure 9.

SAR summary at ORs, TSPO, and A3AR of rigid (N)-methanocarba nucleosides from binding data.
Discussion
The affinities of the present nucleoside derivatives are not comparable to the most potent indolyl or tetrahydroquinolyl antagonists of KOR43 or sub nM antagonists of MOR or to known ligands of TSPO,25,28,29 but nucleosides represent a novel scaffold for the ORs and possibly TSPO. However, to effectively repurpose this versatile scaffold, it was desired to reduce the A3AR affinity, and in some cases A3AR agonist efficacy was also reduced, while enhancing KOR affinity. Furthermore, activity at other sites, such as inhibition of binding at NET or enhancement of binding at DAT was observed in some derivatives. Thus, a major challenge in this study was to identify principles of preference for KOR, which was largely accomplished in the 7-deaza 5′-ethyl ester series.
In general, affinity at both KOR and TSPO was evident in this series with extended C2 substituents, with weaker activity at DOR and MOR (Figure 9). Nevertheless, an affinity correlation plot (Figure S6) showed no direct correlation between KOR and TSPO, and we have no reason to expect a binding site structural similarity. One of the most potent ligands in KOR binding 28 (ligand efficiency = 0.25) was consistently the most potent KOR antagonist tested in two functional assays, that is, to block the activity of a reference agonist (98, U69593, nor-BNI) in βarrestin2-dependent and Gi-protein-dependent signaling. Among 5′-methylamides, the TSPO ligands with highest binding affinity (Ki 200–300 nM) were the 3,4-diF-Ph 4, 4-F-Ph 7, and 2-Cl-Ph 8 analogues,7 but not 3-chloro 9. However, we also functionalized the molecules in a manner to distinguish between the two receptor classes. Compound 14 bound with moderate affinity at TSPO and was inactive at ORs. Among the more potent KOR antagonists, 7-deaza compounds 28 and 29 bound to both ORs and TSPO, but ester group extension greatly reduced the TSPO affinity. Also, compounds 39 and 40 with a modified C2 substitution and the unusual 2′,3′-isopropylidene intermediate 82 bound with a preference for KOR compared with TSPO. Binding at the ORs was sensitive to the terminal ring of the C2 substituent. The C2-phenylethynyl group provided a slightly greater peference than C2-5-chlorothienylethynyl for KOR with respect to TSPO.
Ribose analogues 45 and 46 were inactive at ORs and TSPO, although they corresponded to (N)-methanocarba analogues that bound to TSPO (14) or KOR (22). The equivalent 9-unsubstituted adenine nucleobase moiety of 3 and 4 did not bind appreciably to TSPO or ORs (Ki > 10 μM).6 Thus, the pseudoribose moiety is needed for interaction at these sites. The rigid (N)-methanocarba ring promoted interaction with these receptors, possibly by maintaining a specific receptor-preferred conformation of this pseudoribose moiety.
There is an overlap in the nucleoside structures that act allosterically at DAT (e.g., 14 and 24) and those that inhibit KOR binding. As for the KOR binders, a 5′-ester group favored DAT interaction. However, changing the size of the 5′-ester group was a means of separating those activities. While both 5′-methyl and ethyl esters acted at DAT, the 5′-methyl ester was inactive at KOR. The larger (5′-propyl and beyond) esters were less potent and efficacious than smaller esters in enhancing tropane radioligand binding at DAT but retained some KOR affinity. The 5′-propyl ester 30 enhanced human DAT binding with an EC50 value of 294 ± 82 nM, which is less potent than the 5′-ethyl ester 24 (EC50 34 ± 13 nM).33 Thus, even in the 7-aza series, we have partially separated the activity at KOR from interactions with other receptors and transporters.
We have shown that a 3-deaza modification removed OR affinity, but TSPO interaction is still present. Compound 28 had an 11-fold preference for binding at KOR versus TSPO. The 1-deaza (26) and to some extent the 7-deaza (28 and 29) modifications are a means of eliminating DAT interaction in this series. The most effective means of separating OR binding affinity relative to other sites of action, that is, DAT and ARs, is the 7-deaza modification. However, many 7-deaza nucleosides here also bound to TSPO. A 5-bromothien-2-yl-ethynyl group prevented TSPO binding in the 5′-amide but not 5′-ester or 4′-truncated series. Curiously, various nucleosides with large N6 substituents (e.g., N6-2-phenylethyl in 43, 51, and 55) weakly inhibited NET binding, rather than enhancing it as seen in radioligand assays with reference compounds 14 and 24.
Truncation of the 5′-amide or ester group was compatible with moderate binding at TSPO and weaker binding at ORs. The most potent truncated ligands (Ki, nM) at TSPO were 52 (877) and 57 (353). The most potent truncated ligands (Ki, nM) were 54 (1120) and 55 (1130) at KOR and 56 (1440) at MOR (all C2-phenylethynyl 7-deaza analogues). We expect the 4′-truncated analogues to bind to ORs in a similar orientation as the full nucleosides, but without the 5′-group (fitting a narrow hydrophobic pocket) the ligands are likely not well anchored in the binding site.
There is a possibility that a compound acting centrally as both KOR antagonist and dopamine uptake inhibitor could have a beneficial, synergistic antidepressant effect based on these two mechanisms. KOR activation reduces the availability of dopamine in the brain, and KOR agonists induce depression.16,46 However, we have not evaluated the benefit of additivity of activities of these nucleosides at receptors, channels, and transporters.
We have demonstrated oral bioavailability of the KOR antagonist 28. The presence of an ester group that is conducive to high OR affinity in the present chemical series might be predictive of a short in vivo half-life. However, the half-life (1.6–2.2 h) or mean residence time (2.3–2.8 h) of 28 (p.o.) in rats did not indicate rapid hydrolysis.17 A similar result was obtained with a different series of (N)-methanocarba nucleosides containing 5′-esters and 5′-amides;25 thus, the ester functionality in this (N)-methanocarba nucleoside series appears to be sterically protected from hydrolysis by the adjacent bicyclic ring system.
Furthermore, we have not evaluated the ability of these relatively complex nucleosides to cross the blood–brain barrier. However, except for carrier-mediated transport of simple adenosine derivatives by the equilibrative nucleoside tranporter1, nucleoside derivatives that are derivatized at the N6 and C2 positions for potent interaction with GPCRs tend not to readily cross the blood–brain barrier, as indicated by a study of A1AR agonists.47 The potential use of OR antagonists acting peripherally for cancer treatment has been proposed, based on experiments in MOR-knockout mice and an unplanned post hoc analysis of patients receiving MOR antagonist methylnaltrexone parenterally for OIC.48 Thus, there are applications for selective OR antagonists in the periphery. Further study of these OR antagonists is needed to determine if their action is predominantly in the periphery when administered orally or i.p.
Conclusions
In conclusion, as with our previous reports on repurposing the (N)-methanocarba scaffold from ARs to 5HT2B and 5HT2C receptors and the DAT,6,7 we now show that moderate affinity at KOR within the same chemical series is achievable. The current findings expand our hypothesis that these rigid nucleosides are indeed privileged structures.4 The highest affinity as KOR antagonist achieved was ∼40 nM for 7-deaza ethyl esters 28 and 29, which displayed at least an order of magnitude preference at KOR, including in comparison to the A3AR and TSPO. Moreover, leads for ligands that bind at DOR, MOR, and even NOP have been identified, and the KOR affinity and selectivity of this repurposed scaffold could potentially be enhanced in future studies. Unlike many of the known OR antagonists, these conformationally locked nucleosides interact potently with KOR in the absence of a free basic amino group, which is present in the native ligands. The rigidity of this chemical series was exploited in the molecular modeling of the KOR interaction of 28 using MD simulations. Thus, we have repurposed a ligand class designed originally for one GPCR to a different class of GPCRs and analyzed its KOR interactions structurally in a coherent manner.
Experimental Procedures
Chemical Synthesis
Materials and Instrumentation
Similar syntheses of (N)-methancarba nucleosides have been reported.2,3,5,6,33,35,38 Sigma-Aldrich (St. Louis, MO) was the source for most of the reagents and solvents. Other reagents were purchased from Small Molecules, Inc. (Hoboken, NJ), Anichem (North Brunswick, NJ), PharmaBlock, Inc. (Sunnyvale, CA), Frontier Scientific (Logan, UT), and Tractus (Perrineville, NJ). 1H NMR spectra were measured on a Bruker 400 spectrometer with CDCl3, CD3OD, or dimethyl sulfoxide as solvent. Chemical shifts are given as δ values in ppm from the standard tetramethylsilane signal set at 0.00 when CDCl3 was used as solvent and the water peak at δ 3.30 when CD3OD was used as a solvent. A Bruker AV spectrometer equipped with a z-gradient [1H, 13C, 15N]-cryoprobe was used. Analytical thin layer chromatography analysis was performed using silica gel F254 (0.2 mm) coated on glass plates (Sigma-Aldrich, St. Louis, MO). The purity of the terminal nucleoside derivatives was analyzed by high-performance liquid chromatography (HPLC) (1100 series, Agilent Technologies Inc., Palo Alto, CA) using as a column Agilent Zorbax SB, 50 × 4.6 mm, with a mobile phase consisting of a linear gradient (80:20 to 0:100 in 13 min) of 5 mM TBAP (tetrabutylammonium dihydrogenphosphate) to CH3CN with a 0.5 mL/min flow. UV diode array detection was followed at 230, 254, and 280 nm. The final product nucleosides tested biologically displayed >95% HPLC purity (254 nm). Routine mass spectrometry (MS) was performed using a JEOL SX102 spectrometer with 6 kV Xe atoms following desorption from a glycerol matrix or LC/MS 1100 MSD (Agilent), equipped with an Atlantis C18 column (Waters, Milford, MA). High-resolution MS (HRMS) was performed using a Q-TOF-2 (Micromass-Waters), calibrated (external polyalanine calibration) for proteomics was used. Observed accurate masses (uncorrected for temporal drift) are as expected, consistent with instrumental performance and standard compounds’ mass trends maintained over time.
Ethyl(1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-4-(methylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (28)
TFA (10%, 3 mL) was added to a solution of compound 77 (74 mg, 0.144 mmol) in MeOH (3 mL) and heated at 70 °C for 3 h. The solvent was evaporated, and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH = 30:1) to give the compound 28 (60 mg, 89%) as a syrup. 1H NMR (CD3OD, 400 MHz): δ 7.30 (d, J = 4.4 Hz, 1H), 7.03 (d, J = 4.4 Hz, 1H), 7.01 (d, J = 4.0 Hz, 1H), 6.61 (d, J = 3.6 Hz, 1H), 5.12 (d, J = 6.4 Hz, 1H), 5.04 (s, 1H), 4.29–4.21 (m, 2H), 3.92 (d, J = 6.4 Hz, 1H), 3.09 (s, 3H), 2.10–2.07 (m, 1H), 1.96 (t, J = 5.2 Hz, 1H), 1.66–1.62 (m, 1H), 1.30 (t, J = 7.2 Hz, 3H). HRMS calcd for C22H22N4O4SCl (M + H)+: 473.1050; found, 473.1043.
Ethyl(1S,2R,3S,4R,5S)-4-(2-((5-bromothiophen-2-yl)ethynyl)-4-(methylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (29)
Compound 29 (85%) was prepared from compound 78 by adapting the method provided for compound 28. 1H NMR (CD3OD, 400 MHz): δ 7.26 (d, J = 4.0 Hz, 1H), 7.14 (d, J = 4.0 Hz, 1H), 7.03 (d, J = 3.6 Hz, 1H), 6.61 (d, J = 3.6 Hz, 1H), 5.11 (d, J = 5.2 Hz, 1H), 5.02 (s, 1H), 4.29–4.21 (m, 2H), 3.92 (d, J = 5.2 Hz, 1H), 3.09 (s, 3H), 2.10–2.07 (m, 1H), 1.96 (t, J = 5.2 Hz, 1H), 1.66–1.62 (m, 1H), 1.30 (t, J = 7.2 Hz, 3H). HRMS calcd for C22H22N4O4SBr (M + H)+: 517.0545; found, 517.0548.
Ethyl(1S,2R,3S,4R,5S)-2,3-dihydroxy-4-(2-iodo-4-(methylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)bicyclo[3.1.0]hexane-1-carboxylate (39)
Compound 39 (90%) was prepared from compound 73 by adapting the method provided for compound 28. 1H NMR (CD3OD, 400 MHz): δ 6.8 (d, J = 3.6 Hz, 1H), 6.52 (br s, 1H), 5.15 (d, J = 6.8 Hz, 1H), 4.92 (s, 1H), 4.29–4.21 (m, 2H), 3.92 (d, J = 6.8 Hz, 1H), 3.03 (br s, 3H), 2.04–2.01 (m, 1H), 1.89 (t, J = 5.2 Hz, 1H), 1.62–1.59 (m, 1H), 1.31 (t, J = 7.2 Hz, 3H). HRMS calcd for C16H20N4O4I (M + H)+: 459.0529; found, 459.0521.
Ethyl(1S,2R,3S,4R,5S)-2,3-dihydroxy-4-(4-(methylamino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)bicyclo[3.1.0]hexane-1-carboxylate (40)
Compound 40 (87%) was prepared from compound 79 by adapting the method provided for compound 28. 1H NMR (CD3OD, 400 MHz): δ 7.68–7.66 (m, 2H), 7.45–7.43 (m, 3H), 7.05 (d, J = 3.6 Hz, 1H), 6.64 (d, J = 3.6 Hz, 1H), 5.13 (d, J = 5.6 Hz, 1H), 5.05 (s, 1H), 4.29–4.21 (m, 2H), 3.83 (d, J = 5.6 Hz, 1H), 3.12 (s, 3H), 2.11–2.08 (m, 1H), 1.97 (t, J = 5.2 Hz, 1H), 1.66–1.63 (m, 1H), 1.30 (t, J = 7.2 Hz, 3H). HRMS calcd for C24H25N4O4 (M + H)+: 433.1876; found, 433.1879.
Ethyl(1S,2R,3S,4R,5S)-4-(4-(cyclopropylamino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (41)
Compound 41 (85%) was prepared from compound 80 by adapting the method provided for compound 28. 1H NMR (CD3OD, 400 MHz): δ 7.68–7.66 (m, 2H), 7.45–7.43 (m, 3H), 7.07 (d, J = 3.6 Hz, 1H), 6.78 (br s, 1H), 5.14 (d, J = 7.2 Hz, 1H), 5.06 (s, 1H), 4.29–4.19 (m, 2H), 3.94 (d, J = 6.8 Hz, 1H), 3.02–2.97 (m, 1H), 2.11–2.08 (m, 1H) 1.97 (t, J = 5.2 Hz, 1H), 1.67–1.63 (m, 1H), 1.30 (t, J = 7.2 Hz, 3H), 0.95–0.90 (m, 2H), 0.71–0.67 (m, 2H). HRMS calcd for C26H27N4O4 (M + H)+: 459.2032; found, 459.2027.
Ethyl(1S,2R,3S,4R,5S)-4-(4-((cyclopropylmethyl)amino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (42)
Compound 42 (84%) was prepared from compound 81 by adapting the method provided for compound 28. 1H NMR (CD3OD, 400 MHz): δ 7.67–7.65 (m, 2H), 7.44–7.43 (m, 3H), 7.04 (d, J = 3.6 Hz, 1H), 6.73 (d, J = 3.6 Hz, 1H), 5.13 (d, J = 6.8 Hz, 1H), 5.05 (s, 1H), 4.29–4.21 (m, 2H), 3.93 (d, J = 6.4 Hz, 1H), 3.47 (d, J = 6.8 Hz, 2H), 2.12–2.08 (m, 1H), 1.97 (t, J = 5.2 Hz, 1H), 1.66–1.62 (m, 1H), 1.30 (t, J = 7.2 Hz, 3H), 1.25–1.16 (m, 1H), 0.60–0.55 (m, 2H), 0.36–0.33 (m, 2H). HRMS calcd for C27H29N4O4 (M + H)+: 473.2189; found, 473.2189.
Ethyl(1S,2R,3S,4R,5S)-2,3-dihydroxy-4-(4-(phenethylamino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)bicyclo[3.1.0]hexane-1-carboxylate (43)
Compound 43 (87%) was prepared from compound 82 by adapting the method provided for compound 28. 1H NMR (CD3OD, 400 MHz): δ 7.68–7.67 (m, 2H), 7.46–7.44 (m, 3H), 7.30–7.29 (m, 4H), 7.22–7.19 (m, 4H), 7.09 (d, J = 3.6 Hz, 1H), 6.69 (d, J = 3.6 Hz, 1H), 5.14 (d, J = 6.8 Hz, 1H), 5.05 (s, 1H), 4.26–4.21 (m, 2H), 3.96 (d, J = 6.8 Hz, 1H), 3.85 (t, J = 7.2 Hz, 2H), 3.02 (t, J = 7.2 Hz, 2H), 2.12–2.08 (m, 1H), 1.96 (t, J = 5.2 Hz, 1H), 1.67–1.63 (m, 1H), 1.29 (t, J = 7.2 Hz, 3H). HRMS calcd for C31H31N4O4 (M + H)+: 523.2345; found, 523.2348.
(1R,2R,3S,4R,5S)-4-(4-(Methylamino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)bicyclo[3.1.0]hexane-2,3-diol (54)
TFA (10%, 2 mL) was added to a solution of compound 88 (20 mg, 0.05 mmol) in MeOH (2 mL) and heated at 70 °C for 3 h. The solvent was evaporated, and the residue was purified on flash silica gel column chromatography (ethylacetate/MeOH = 50:1) to give the compound 54 (16 mg, 90%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz): δ 7.68–7.66 (m, 2H), 7.47–7.43 (m, 3H), 7.36 (d, J = 3.6 Hz, 1H), 6.68 (s, 1H), 5.07 (s, 1H), 4.69 (br s, 1H), 3.77 (d, J = 6.4 Hz, 1H), 3.15 (s, 3H), 1.99–1.96 (m, 1H), 1.63–1.59 (m, 1H), 1.38–1.35 (m, 1H), 0.77–0.72 (m, 1H). HRMS calcd for C21H21N4O2 (M + H)+: 361.1665; found, 361.1672.
(1R,2R,3S,4R,5S)-4-(4-(Phenethylamino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)bicyclo[3.1.0]hexane-2,3-diol (55)
Compound 55 (92%) was prepared from compound 89 by adapting the method provided for compound 54. 1H NMR (CD3OD, 400 MHz): δ 7.70 (d, J = 3.6 Hz, 2H), 7.50–7.43 (m, 4H), 7.30–7.27 (m, 4H), 7.23–7.20 (m, 1H), 6.79 (m, 1H), 5.09 (s, 1H), 4.69 (br s, 1H), 3.89 (t, J = 7.2 Hz, 2H), 3.78 (d, J = 6.4 Hz, 1H), 3.05 (t, J = 7.2 Hz, 2H), 2.00–1.96 (m, 1H), 1.63–1.58 (m, 1H), 1.36–1.34 (m, 1H), 0.79–0.73 (m, 1H). HRMS calcd for C28H27N4O2 (M + H)+: 451.2134; found, 451.2133.
(1R,2R,3S,4R,5S)-4-(4-((Dicyclopropylmethyl)amino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)bicyclo[3.1.0]hexane-2,3-diol (56)
PdCl2(PPh3)2 (1.68 mg, 0.0024 mmol), CuI (1.0 mg, 0.0012 mmol), phenylacetylene (8 μL, 0.072 mmol), and triethylamine (16 μL, 0.12 mmol) were added to a solution of compound 58 (5.6 mg, 0.012 mmol) in anhydrous dimethylformamide (DMF) (0.6 mL) and stirred at room temperature for overnight. The solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (hexane/ethyl acetate = 1:1) to give the compound 56 (4 mg, 80%) as a brownish syrup. 1H NMR (CD3OD, 400 MHz): δ 7.66–7.63 (m, 2H), 7.44–7.42 (m, 3H), 7.28 (d, J = 3.6 Hz, 1H), 6.72 (d, J = 3.6 Hz, 1H), 5.03 (s, 1H), 4.65 (t, J = 5.6 Hz, 1H), 3.74 (d, J = 6.4 Hz, 1H), 3.59 (t, J = 7.6 Hz, 1H), 2.00–1.94 (m, 1H), 1.65–1.60 (m, 1H), 1.38–1.35 (m, 1H), 1.21–1.12 (m, 2H), 0.76–0.71 (m, 1H), 0.59–0.54 (m, 2H), 0.47–0.42 (m, 6H). HRMS calcd for C27H29N4O2 (M + H)+: 441.2291; found, 441.2290.
(1R,2R,3S,4R,5S)-4-(2-((5-Bromothiophen-2-yl)ethynyl)-4-((dicyclopropylmethyl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)bicyclo[3.1.0]hexane-2,3-diol (57)
Compound 57 (78%) was prepared from compound 58 by adapting the method provided for compound 56. 1H NMR (CD3OD, 400 MHz): δ 7.28 (d, J = 3.6 Hz, 1H), 7.25 (d, J = 3.6 Hz, 1H), 7.14 (d, J = 4.0 Hz, 1H), 7.20 (d, J = 3.6 Hz, 1H), 5.01 (s, 1H), 4.68–4.63 (m, 1H), 3.73 (d, J = 6.0 Hz, 1H), 3.56 (t, J = 8.0 Hz, 1H), 2.01–1.94 (m, 1H), 1.63–1.59 (m, 1H), 1.37–1.34 (m, 1H), 1.20–1.09 (m, 2H), 0.76–0.67 (m, 1H), 0.59–0.54 (m, 2H), 0.45–0.39 (m, 6H). HRMS calcd for C25H26N4O2SBr (M + H)+: 525.0960; found, 525.0953.
(1R,2R,3S,4R,5S)-4-(4-((Dicyclopropylmethyl)amino)-2-iodo-7H-pyrrolo[2,3-d]pyrimidin-7-yl)bicyclo[3.1.0]hexane-2,3-diol (58)
Dowex 50 (40 mg) was added to a solution of compound 87 (67 mg, 0.13 mmol) in MeOH (1 mL)–water (1 mL) and heated at 50 °C for 1.5 h. The reaction mixture was filtered and filtrate was evaporated under vacuum. The residue was purified on flash silica gel column chromatography (hexane/ethylacetate = 1:1) to give the compound 58 (38 mg, 63%). 1H NMR (CD3OD, 400 MHz): δ 7.05 (d, J = 3.6 Hz, 1H), 6.62 (d, J = 3.6 Hz, 1H), 4.93 (s, 1H), 4.67 (t, J = 5.6 Hz, 1H), 3.72 (d, J = 6.4 Hz, 1H), 1.95–1.91 (m, 1H), 1.57–1.52 (m, 1H), 1.31–1.28 (m, 1H), 1.15–1.07 (m, 2H), 0.74–0.68 (m, 1H), 0.58–0.52 (m, 2H), 0.45–0.34 (m, 6H). HRMS calcd for C19H24N4O2I (M + H)+: 467.0944; found, 467.0947.
Ethyl(3aR,3bS,4aS,5R,5aS)-5-(4-chloro-2-iodo-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (72)
Diisopropyl-azodicarboxylate (DIAD) (0.15 mL, 0.76 mmol) was added to a solution of triphenylphosphine (199 mg, 0.76 mmol) and 7-deaza-2-iodo-6-chloro-purine (212 mg, 0.76 mmol) in dry THF (4 mL) at 0 °C and stirred at room temperature for 10 min. A solution of compound 59 (92 mg, 1.0 mmol) in THF (1 mL) was added to the reaction mixture and stirred overnight at room temperature. The solvent was evaporated, and the residue was purified on flash silica gel column chromatography (hexane/ethyl acetate = 5:1) to give the compound 72 (131 mg, 69%) as a colorless foamy solid. 1H NMR (CD3OD, 400 MHz): δ 7.46 (d, J = 3.6 Hz, 1H), 6.66 (d, J = 3.6 Hz, 1H), 5.83 (d, J = 6.8 Hz, 1H), 5.0 (s, 1H), 4.83 (d, J = 6.8 Hz, 1H), 4.32–4.24 (m, 2H), 2.28–2.24 (m, 1H), 1.68–1.64 (m, 1H), 1.54–1.51 (m, 4H), 1.33 (d, J = 7.2 Hz, 3H), 1.2 (s, 3H). HRMS calculated for C18H20N3O4ClI (M + H)+: 504.0187; found, 504.0191.
Ethyl(3aR,3bS,4aS,5R,5aS)-5-(2-iodo-4-(methylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (73)
Methylamine hydrochloride (164 mg, 2.43 mmol) and triethylamine (0.67 mL, 4.87 mmol) were added to a solution of compound 72 (245 mg, 0.48 mmol) in methanol (5 mL) and stirred at room temperature for overnight. The reaction mixture was evaporated under vacuum, and the residue was purified on flash column chromatography (hexane/ethyl acetate = 1:1) to give the desired product 73 (198 mg, 82%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz): δ 6.91 (d, J = 3.6 Hz, 1H), 6.49 (d, J = 3.6 Hz, 1H), 5.78 (d, J = 6.4 Hz, 1H), 4.93 (s, 1H), 4.74 (d, J = 6.8 Hz, 1H), 4.30–4.24 (m, 2H), 3.02 (br s, 3H), 2.19–2.14 (m, 1H), 1.64–1.60 (m, 1H), 1.53 (s, 3H), 1.50 (t, J = 5.2 Hz, 1H), 1.32 (t, J = 7.2 Hz, 3H), 1.28 (s, 3H). HRMS calcd for C19H24N4O4I (M + H)+: 499.0842; found, 499.0844.
Ethyl(3aR,3bS,4aS,5R,5aS)-5-(4-(cyclopropylamino)-2-iodo-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (74)
Compound 74 (85%) was prepared from compound 72 by adapting the method provided for compound 73. 1H NMR (CD3OD, 400 MHz): δ 6.94 (d, J = 3.6 Hz, 1H), 6.67 (br s, 1H), 5.81 (d, J = 7.2 Hz, 1H), 4.76 (d, J = 6.0 Hz, 1H), 4.30–4.22 (m, 2H), 2.96–2.91 (m, 1H), 2.19–2.15 (m, 1H), 1.54–1.60 (m, 1H), 1.53 (s, 3H), 1.50 (t, J = 5.6 Hz, 1H), 1.32 (t, J = 7.2 Hz, 3H), 1.26 (s, 3H), 0.89–0.85 (m, 2H), 0.64–0.63 (m, 2H). HRMS calcd for C21H26N4O4I (M + H)+: 525.0999; found, 525.0994.
Ethyl(3aR,3bS,4aS,5R,5aS)-5-(4-((cyclopropylmethyl)amino)-2-iodo-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (75)
Compound 75 (83%) was prepared from compound 72 by adapting the method provided for compound 73. 1H NMR (CD3OD, 400 MHz): δ 6.89 (d, J = 3.6 Hz, 1H), 6.56 (d, J = 3.6 Hz, 1H), 5.80 (d, J = 6.8 Hz, 1H), 4.74 (d, J = 7.2 Hz, 1H), 4.30–4.21 (m, 2H), 3.36–3.34 (m, 2H), 2.18–2.14 (m, 1H), 1.63–1.59 (m, 1H), 1.52 (s, 3H), 1.48 (t, J = 5.6 Hz, 1H), 1.30 (t, J = 7.2 Hz, 3H), 1.27 (s, 3H), 1.18–1.08 (m, 1H), 0.54–0.50 (m, 2H), 0.32–0.29 (m, 2H). HRMS calcd for C22H28N4O4I (M + H)+: 539.1155; found, 539.1150.
Ethyl(3aR,3bS,4aS,5R,5aS)-5-(2-iodo-4-(phenethylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (76)
Compound 76 (85%) was prepared from compound 72 by adapting the method provided for compound 73. 1H NMR (CD3OD, 400 MHz): δ 7.28–7.27 (m, 4H), 7.21–7.17 (m, 1H), 6.90 (d, J = 3.6 Hz, 1H), 6.48 (d, J = 3.6 Hz, 1H), 5.81 (d, J = 3.6 Hz, 1H), 5.81 (d, J = 6.4 Hz, 1H), 4.75 (d, J = 7.2 Hz, 1H), 4.31–4.22 (m, 2H), 3.71 (t, J = 7.2 Hz, 2H), 2.94 (d, J = 7.2 Hz, 2H), 2.19–2.15 (m, 1H), 1.64–1.60 (m, 1H), 1.53 (s, 3H), 1.50 (t, J = 5.2 Hz, 1H), 1.33 (t, J = 7.2 Hz, 3H), 1.28 (s, 3H). HRMS calcd for C26H30N4O4I (M + H)+: 589.1312; found, 589.1309.
Ethyl(3aR,3bS,4aS,5R,5aS)-5-(2-((5-chlorothiophen-2-yl)ethynyl)-4-(methylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (77)
PdCl2(PPh3)2 (19.7 mg, 0.02 mmol), CuI (2.6 mg, 0.01 mmol), 2-chloro-5-ethynylthiophene (100 mg, 0.70 mmol), and triethylamine (0.2 mL, 1.4 mmol) were added to a solution of compound 73 (70 mg, 0.14 mmol) in anhydrous DMF (1.5 mL) and stirred at room temperature overnight. The solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (hexane/ethyl acetate = 2:1) to give the compound 77 (55 mg, 76%) as a yellow syrup. 1H NMR (CD3OD, 400 MHz): δ 7.34 (d, J = 4.0 Hz, 1H), 7.09 (d, J = 3.6 Hz, 1H), 7.01 (d, J = 4.0 Hz, 1H), 6.59 (d, J = 3.6 Hz, 1H), 5.82 (d, J = 6.4 Hz, 1H), 5.04 (s, 1H), 4.71 (d, J = 6.4 Hz, 1H), 4.26–4.16 (m, 2H), 3.08 (s, 3H), 2.25–2.20 (m, 1H), 1.71–1.67 (m, 1H), 1.57–1.64 (m, 4H), 1.28 (s, 3H), 1.24 (t, J = 7.2 Hz, 3H). HRMS calcd for C25H26N4O4SCl (M + H)+: 513.1363; found, 513.1362.
Ethyl(3aR,3bS,4aS,5R,5aS)-5-(2-((5-bromothiophen-2-yl)ethynyl)-4-(methylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (78)
Compound 78 (72%) was prepared from compound 73 by adapting the method provided for compound 77. 1H NMR (CD3OD, 400 MHz): δ 7.32 (d, J = 4.0 Hz, 1H), 7.14 (d, J = 4.0 Hz, 1H), 7.09 (d, J = 3.6 Hz, 1H), 6.59 (d, J = 3.6 Hz, 1H), 5.82 (d, J = 6.4 Hz, 1H), 5.04 (s, 1H), 4.71 (d, J = 6.4 Hz, 1H), 4.26–4.18 (m, 2H), 3.08 (s, 3H), 2.25–2.21 (m, 1H), 1.71–1.67 (m, 1H), 1.57–1.54 (m, 4H), 1.28 (s, 3H), 1.24 (t, J = 7.2 Hz, 3H). HRMS calcd for C25H26N4O4SBr (M + H)+: 557.0858; found, 557.0853.
Ethyl(3aR,3bS,4aS,5R,5aS)-2,2-dimethyl-5-(4-(methylamino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)tetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (79)
Compound 79 (82%) was prepared from compound 73 by adapting the method provided for compound 77. 1H NMR (CD3OD, 400 MHz): δ 7.72–7.70 (m, 2H), 7.45–7.43 (m, 3H), 7.08 (d, J = 3.6 Hz, 1H), 6.61 (d, J = 3.6 Hz, 1H), 5.86 (d, J = 7.2 Hz, 1H), 5.07 (s, 1H), 4.74 (d, J = 7.2 Hz, 1H), 4.24–4.15 (m, 2H), 3.10 (s, 3H), 2.25–2.21 (m, 1H), 1.71–1.67 (m, 1H), 1.56 (t, J = 5.2 Hz, 1H), 1.54 (s, 3H), 1.29 (s, 3H), 1.21 (t, J = 7.2 Hz, 3H). HRMS calcd for C27H29N4O4 (M + H)+: 473.2189; found, 473.2191.
Ethyl(3aR,3bS,4aS,5R,5aS)-5-(4-(cyclopropylamino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (80)
Compound 80 (84%) was prepared from compound 74 by adapting the method provided for compound 77. 1H NMR (CD3OD, 400 MHz): δ 7.72–7.70 (m, 2H), 7.45–7.43 (m, 3H), 7.11 (d, J = 3.2 Hz, 1H), 6.76 (br s, 1H), 5.86 (d, J = 6.4 Hz, 1H), 5.07 (s, 1H), 4.75 (d, J = 7.2 Hz, 1H), 4.24–4.15 (m, 2H), 3.02–2.96 (m, 1H), 2.25–2.21 (m, 1H), 1.71–1.67 (m, 1H), 1.56 (t, J = 5.6 Hz, 1H), 1.54 (s, 3H), 1.29 (s, 3H), 1.21 (t, J = 7.2 Hz, 3H), 0.94–0.89 (m, 2H), 0.70–0.66 (m, 2H). HRMS calcd for C29H31N4O4 (M + H)+: 499.2345; found, 499.2341.
Ethyl(3aR,3bS,4aS,5R,5aS)-5-(4-((cyclopropylmethyl)amino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (81)
Compound 81 (82%) was prepared from compound 75 by adapting the method provided for compound 77. 1H NMR (CD3OD, 400 MHz): δ 7.72–7.69 (m, 2H), 7.44–7.42 (m, 3H), 7.07 (d, J = 3.6 Hz, 1H), 6.70 (d, J = 3.6 Hz, 1H), 5.85 (d, J = 6.4 Hz, 1H), 5.08 (s, 1H), 4.74 (d, J = 7.2 Hz, 1H), 4.26–4.12 (m, 2H), 3.46 (d, J = 6.8 Hz, 2H), 2.25–2.21 (m, 1H), 1.71–1.67 (m, 1H), 1.56 (t, J = 5.6 Hz, 1H), 1.54 (s, 3H), 1.28 (m, 4H), 1.22 (d, J = 7.2 Hz, 3H), 0.58–0.54 (m, 2H), 0.35–0.31 (m, 2H). HRMS calcd for C30H33N4O4 (M + H)+: 513.2502; found, 513.2507.
Ethyl(3aR,3bS,4aS,5R,5aS)-2,2-dimethyl-5-(4-(phenethylamino)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)tetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (82)
Compound 82 (83%) was prepared from compound 76 by adapting the method provided for compound 77. 1H NMR (CD3OD, 400 MHz): δ 7.72–7.71 (m, 2H), 7.45–7.43 (m, 4H), 7.30–7.29 (m, 4H), 7.21–7.18 (m, 1H), 7.08 (d, J = 3.6 Hz, 1H), 6.61 (d, J = 3.6 Hz, 1H), 5.86 (d, J = 7.2 Hz, 1H), 5.08 (s, 1H), 4.74 (d, J = 6.4 Hz, 1H), 4.22–4.14 (m, 2H), 3.82 (t, J = 7.2 Hz, 2H), 3.02 (d, J = 7.2 Hz, 2H), 2.26–2.22 (m, 1H), 1.72–1.68 (m, 1H), 1.57 (t, J = 5.2 Hz, 1H), 1.54 (s, 3H), 1.29 (s, 3H), 1.22 (t, J = 7.2 Hz, 3H). HRMS calcd for C34H35N4O4 (M + H)+: 563.2658; found, 563.2654.
4-Chloro-7-((3aR,3bR,4aS,5R,5aS)-2,2-dimethylhexahydrocyclopropa[3,4]cyclo-penta[1,2-d][1,3]dioxol-5-yl)-2-iodo-7H-pyrrolo[2,3-d]pyrimidine (84)
DIAD (0.27 mL, 1.4 mmol) was added to a solution of triphenylphosphine (369 mg, 1.4 mmol) and 7-deaza-2-iodo-6-chloro-purine (295 mg, 1.4 mmol) in dry THF (2 mL) at 0 °C and stirred at room temperature for 10 min. A solution of compound 83 (120 mg, 0.7 mmol) in THF (1.5 mL) was added to the reaction mixture and stirred overnight at room temperature. The solvent was evaporated, and the residue was purified on flash silica gel column chromatography (hexane/ethyl acetate = 5:1) to give the compound 84 (206 mg, 68%) as a colorless foamy solid. 1H NMR (CD3OD, 400 MHz): δ 7.55 (d, J = 3.6 Hz, 1H), 6.66 (d, J = 3.6 Hz, 1H), 5.36–5.33 (m, 1H), 5.13 (s, 1H), 4.66 (d, J = 7.2 Hz, 1H), 2.09–2.02 (m, 1H), 1.71–1.65 (m, 1H), 1.52 (s, 3H), 1.24 (s, 3H), 0.96–0.88 (m, 2H). HRMS calcd for C15H16N3O2ClI (M + H)+: 431.9976; found, 431.9969.
7-((3aR,3bR,4aS,5R,5aS)-2,2-Dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-iodo-N-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (85)
Methylamine hydrochloride (63 mg, 0.93 mmol) and triethylamine (0.33 mL, 1.8 mmol) were added to a solution of compound 84 (81 mg, 0.18 mmol) in methanol (2 mL) and stirred at room temperature for overnight. The reaction mixture was evaporated under vacuum, and the residue was purified on flash column chromatography (hexane/ethyl acetate = 2:1) to give the desired product 85 (66 mg, 83%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz): δ 7.03 (d, J = 3.6 Hz, 1H), 6.53 (s, 1H), 5.30 (t, J = 6.0 Hz, 1H), 5.04 (s, 1H), 4.56 (d, J = 7.2 Hz, 1H), 3.03 (br s, 3H), 2.03–1.97 (m, 1H), 1.65–1.60 (m, 1H), 1.51 (s, 3H), 1.24 (s, 3H), 0.93–0.84 (m, 2H). HRMS calcd for C16H20N4O2I (M + H)+: 427.0631; found, 427.0635.
7-((3aR,3bR,4aS,5R,5aS)-2,2-Dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-iodo-N-phenethyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (86)
Compound 86 (80%) was prepared from compound 84 by adapting the method provided for compound 85. 1H NMR (CD3OD, 400 MHz): δ 7.31–7.20 (m, 4H), 7.21–7.19 (m, 1H), 7.02 (d, J = 3.6 Hz, 1H), 6.51 (d, J = 3.6 Hz, 1H), 5.30 (t, J = 6.4 Hz, 1H), 5.05 (s, 1H), 4.55 (d, J = 7.2 Hz, 1H), 3.72 (t, J = 7.2 Hz, 2H), 2.95 (t, J = 7.2 Hz, 2H), 2.03–1.97 (m, 1H), 1.65–1.60 (m, 1H), 1.51 (s, 3H), 1.24 (s, 3H), 0.93–0.84 (m, 2H). HRMS calcd for C23H26N4O2I (M + H)+: 517.1101; found, 517.1093.
N-(Dicyclopropylmethyl)-7-((3aR,3bR,4aS,5R,5aS)-2,2-dimethylhexahydrocyclopropa [3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-iodo-7H-pyrrolo[2,3-d]pyrimidin-4-amine (87)
Dicyclopropylmethyl amine hydrochloride (160 mg, 1.05 mmol) and DIPEA (0.37 mL, 2.1 mmol) were added to a solution of compound 84 (93 mg, 0.21 mmol) in 2-propanol (2 mL) and heated at 100 °C for 2 h under microwave condition. The reaction mixture was evaporated under vacuum, and the residue was purified on flash column chromatography (hexane/ethyl acetate = 2:1) to give the desired product 87 (86 mg, 79%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz): δ 7.00 (d, J = 3.6 Hz, 1H), 6.63 (d, J = 6.4 Hz, 1H), 5.03 (t, J = 3.6 Hz, 1H), 5.03 (s, 1H), 4.56 (d, J = 7.2 Hz, 1H), 2.01–1.97 (m, 1H), 1.64–1.59 (m, 1H), 1.50 (s, 3H), 1.30 (t, J = 5.2 Hz, 1H), 1.23 (s, 3H), 1.14–1.06 (m, 1H), 0.92–0.84 (m, 1H), 0.58–0.52 (m, 2H), 0.45–0.35 (m, 6H). HRMS calcd for C22H28N4O2I (M + H)+: 507.1257; found, 507.1264.
7-((3aR,3bR,4aS,5R,5aS)-2,2-Dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-N-methyl-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (88)
PdCl2(PPh3)2 (9.8 mg, 0.014 mmol), CuI (1.3 mg, 0.07 mmol), phenylethyne (46 μL, 0.42 mmol), and triethylamine (98 μL, 0.7 mmol) were added to a solution of compound 85 (30 mg, 0.07 mmol) in anhydrous DMF (1.2 mL) and stirred at room temperature overnight. The solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (hexane/ethyl acetate = 3:1) to give the compound 88 (20 mg, 71%) as a brownish syrup. 1H NMR (CD3OD, 400 MHz): δ 7.69–7.66 (m, 2H), 7.44–7.42 (m, 4H), 7.24 (d, J = 3.6 Hz, 1H), 6.64 (d, J = 3.6 Hz, 1H), 5.30–5.25 (m, 2H), 4.54 (d, J = 7.2 Hz, 1H), 3.11 (s, 3H), 2.05–2.01 (m, 1H), 1.72–1.67 (m, 1H), 1.52 (s, 3H), 1.24 (s, 3H), 0.99–0.96 (m, 1H), 0.92–0.88 (m, 1H). HRMS calcd for C24H25N4O2 (M + H)+: 401.1978; found, 401.1983.
7-((3aR,3bR,4aS,5R,5aS)-2,2-Dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-N-phenethyl-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (89)
Compound 89 (74%) was prepared from compound 86 by adapting the method provided for compound 88. 1H NMR (CD3OD, 400 MHz): δ 7.69–7.67 (m, 2H), 7.44–7.42 (m, 3H), 7.30–7.26 (m, 4H), 7.21 (d, J = 3.6 Hz, 1H), 7.20–7.19 (m, 1H), 6.64 (d, J = 3.6 Hz, 1H), 5.30–5.26 (m, 2H), 4.54 (d, J = 7.2 Hz, 1H),3.83 (t, J = 7.2 Hz, 2H), 3.03–2.99 (t, J = 7.2 Hz, 2H), 2.06–2.01 (m, 1H), 1.72–1.67 (m, 1H), 1.52 (s, 3H), 1.24 (s, 3H), 1.00–0.96 (m, 1H), 0.92–0.87 (m, 1H). HRMS calcd for C31H31N4O2 (M + H)+: 491.2447; found, 491.2449.
N-(Dicyclopropylmethyl)-7-((3aR,3bR,4aS,5R,5aS)-2,2-dimethylhexahydrocyclopropa [3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-2-(phenylethynyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (90)
Compound 90 (73%) was prepared from compound 87 by adapting the method provided for compound 88. 1H NMR (CD3OD, 400 MHz): δ 7.65–7.66 (m, 2H), 7.43–7.42 (m, 3H), 7.23 (d, J = 3.6 Hz, 1H), 6.76 (d, J = 3.6 Hz, 1H), 5.30–5.25 (m, 2H), 4.54 (d, J = 7.2 Hz, 1H), 3.59 (t, J = 3.6 Hz, 1H), 2.07–2.01 (m, 1H), 1.73–1.68 (m, 1H), 1.52 (s, 3H), 1.24 (s, 3H), 1.21–1.12 (m, 2H), 1.00–0.97 (m, 1H), 0.92–0.87 (m, 1H), 0.59–0.54 (m, 2H), 0.46–0.41 (m, 6H). HRMS calcd for C30H33N4O2 (M + H)+: 481.2604; found, 481.2609.
2-((5-Bromothiophen-2-yl)ethynyl)-N-(dicyclopropylmethyl)-7-((3aR,3bR,4aS,5R,5aS)-2,2-dimethylhexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (91)
Compound 91 (71%) was prepared from compound 87 by adapting the method provided for compound 88. 1H NMR (CD3OD, 400 MHz): δ 7.26 (d, J = 4.0 Hz, 1H), 7.24 (d, J = 3.6 Hz, 1H), 7.13 (d, J = 4.0 Hz, 1H), 6.75 (d, J = 3.6 Hz, 1H), 5.29 (t, J = 6.0 Hz, 1H), 5.22 (s, 1H), 4.53 (d, J = 7.2 Hz, 1H), 3.58 (t, J = 8.0 Hz, 1H), 2.06–2.00 (m, 1H), 1.72–1.67 (m, 1H), 1.52 (s, 3H), 1.30 (t, J = 5.2 Hz, 1H), 1.24 (s, 3H), 1.20–1.11 (m, 1H), 0.99–0.95 (m, 1H), 0.92–0.86 (m, 1H), 0.58–0.53 (m, 2H), 0.45–0.39 (m, 6H). HRMS calcd for C28H30N4O2SBr (M + H)+: 565.1095; found, 565.1089.
Pharmacological Procedures
Radioligand Binding and Uptake Assays
Initial screening at ORs and other drug targets was performed by the PDSP. Ki determinations using 96-well plates and binding profiles in a broad screen of receptors and channels were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. For experimental details please refer to the PDSP web site at: https://pdspdb.unc.edu/pdspWeb/content/PDSP%20Protocols%20II%202013-03-28.pdf. The cell line used for preparing membranes for binding in human KOR, DOR, and MOR is HEK-293s cells stably expressing the receptors, grown in DMEM medium containing 200 (DOR and MOR) or 500 (KOR) μg/mL geneticin, 10% fetal bovine serum, and penicillin/streptomycin (1 U/mL). The buffer used for binding was 50 mM Tris·HCl, 10 mM MgCl2, 0.1 mM EDTA, pH 7.4, room temperature. The buffer used for washing was 50 mM Tris·HCl, pH 7.4, 4° to 8 °C. The radioligand Kd values (and concentration range used, nM) were [3H]Tyr-d-Ala-Gly-Phe-d-Leu ([3H]DADLE, 97) for DOR, 1.85 ± 0.15 (1.0–2.0) nM; [3H]N-methyl-2-phenyl-N-[(5R,7S,8S)-7-(pyrrolidin-1-yl)-1-oxaspiro[4.5]dec-8-yl]acetamide ([3H]U69593, 98) for KOR, 1.07 ± 0.10 (0.6–1.2) nM; and [3H]Ala2-MePhe4-Glyol5-Enkephalin ([3H]DAMGO, 99) for MOR, 1.73 ± 0.14 (1.0–2.0) nM. The hDOR, hKOR, and hMOR reference ligands had Ki values of natrindole 108, 0.81 ± 0.08 nM; salvinorin A 109, 1.93 ± 0.45 nM; and morphine 110, 2.62 ± 0.22 nM, respectively.
The radioligand for binding in membranes of HEKT cells expressing hNOP was [3H]nociceptin 100 (pKi 9.11 ± 0.12, 0.5–2.0 nM used), and the reference ligand was 7-[[4-(2,6-dichlorophenyl)-1-piperidinyl]methyl]-6,7,8,9-tetrahydro-1-methyl-5H-benzocyclohepten-5-ol (SB612111), 111 (Ki = 6.58 ± 1.42 nM).
The buffer used for TSPO binding was 50 mM Tris·acetate, pH 7.4, room temperature. The reference ligand 4′-chlorodiazepam for TSPO binding had a Ki value of 27.6 ± 2.3 nM.
Binding assays at DAT, NET, and SERT were performed as described.5,8,40 The protein content and Ki values were determined, as reported.50,51 The radioligands used by PDSP for NET and SERT binding were [3H](R,S)-3-(2-methoxyphenoxy)-N-methyl-3-phenylpropan-1-amine 104 and [3H](R,S)-1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile 105, respectively.
βArrestin2 Recruitment Assay
The DiscoveRx PathHunter enzyme complementation assay (PathHunter U2OS OPRK1 β-Arrestin Cell Line) cell line was used to assess βarrestin2 recruitment to KOR according to the manufacturer’s instruction and as described in detail previously.53 Nonlinear regression analysis was used to generate IMAX, EC50 and IC50 values in GraphPad Prism, v. 7.0 (San Diego, CA).
cAMP Accumulation Assay
Cyclic AMP accumulation was measured by treating CHO cells stably, expressing the hKOR with test compounds in the presence of 20 μM forskolin and 25 μM 4-(3-butoxy-4-methoxybenzyl)imidazolidin-2-one for 30 min at room temperature. Detection of cAMP accumulation was done using Cisbio’s dynamic 2 kit (Cisbio Bioassays, Bedford, MA) according to the manufacturer’s instructions. Data were analyzed using GraphPad Prism, v. 7.0 (San Diego, CA).
Acknowledgments
We acknowledge the support from the NIH Intramural Research Program (NIDDK, ZIA DK031117-28). We thank John Lloyd (NIDDK) for mass spectral determinations and Robert O’Connor (NIDDK) for NMR spectra. We thank Dr. Bryan L. Roth (University of North Carolina at Chapel Hill) and National Institute of Mental Health’s Psychoactive Drug Screening Program (contract # HHSN-271-2008-00025-C) for screening data. We also thank the NIH National Institute on Drug Abuse (NIDA)/VA Interagency Agreement #ADA12013; the Methamphetamine Abuse Research Center (P50 DA018165-06), and the Department of Veterans Affairs Research Career Scientist and Merit Review Programs, and NIDA (R01DA031297) to L.M.B.
Glossary
Abbreviations
- AR
adenosine receptor
- HEK 293
human embryonic kidney 293
- DAT
the dopamine transporter
- DMF
dimethylformamide
- DIAD
diisopropyl-azodicarboxylate
- DIPEA
diisopropylethylamine
- DOR
δ-opioid receptor
- GPCR
G protein-coupled receptor
- HRMS
high resolution mass spectrometry
- HSD
His protonated on the Nδ atom
- HSE
His protonated on the Nε atom
- IE
interaction energy
- IFD
Induced Fit Docking
- KOR
κ-opioid receptor
- MD
molecular dynamics
- MOR
μ-opioid receptor
- NET
the norepinephrine transporter
- NOP
nociceptin receptor
- PDSP
Psychoactive Drug Screening Program
- RSMD
root mean square deviation
- SAR
structure–activity relationship
- SERT
the serotonin transporter
- Tango
transcriptional activation following arrestin translocation
- TBAP
tetrabutylammonium dihydrogenphosphate
- TFA
trifluoroacetic acid
- THF
Tetrahydrofuran
- TIPSCl
tri-isopropylsilyl chloride
- TM
transmembrane
- TSPO
the translocator protein.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01237.
PDB file of the following ligand–protein complex: minimized lowest IE structure returned during the MD simulation of 28 in the hKOR HSE model (PDB)
PDB file of the following ligand–protein complex: minimized lowest IE structure returned during the MD simulation of 28 in the hKOR HSD model (complexes superimposed in Figure 5C) (PDB)
MD simulation of 28 (Video S1) (AVI)
Chemical strings file (CSV).
Chemical synthesis (with Schemes S1–S4); reagents for diverse and off-target binding activities; binding inhibition curves (KOR and TSPO, Figures S1–S4); binding inhibition curves (other off-targets, Figure S5); parameters for interaction with DAT and NET (Table S1); lack of correlation of binding at KOR and TSPO (Figure S6); molecular modeling procedures and results (Table S2 and Figure S7); and pharmacokinetic methods and ADME-toxicity data for 28 (Tables S3–S7 and Figures 8 and 9) (PDF)
Author Present Address
# Queen’s University Belfast, School of Pharmacy, 96 Lisburn Rd, Belfast BT9 7BL, UK.
Author Contributions
⊥ D.K.T. and A.C. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Little J. W.; Ford A.; Symons-Liguori A. M.; Chen Z.; Janes K.; Doyle T.; Xie J.; Luongo L.; Tosh D. K.; Maione S.; Bannister K.; Dickenson A. H.; Vanderah T. W.; Porreca F.; Jacobson K. A.; Salvemini D. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 2015, 138, 28–35. 10.1093/brain/awu330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Finley A.; Paoletta S.; Moss S. M.; Gao Z.-G.; Gizewski E. T.; Auchampach J. A.; Salvemini D.; Jacobson K. A. In Vivo Phenotypic Screening for Treating Chronic Neuropathic Pain: Modification of C2-Arylethynyl Group of Conformationally Constrained A3 Adenosine Receptor Agonists. J. Med. Chem. 2014, 57, 9901–9914. 10.1021/jm501021n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Deflorian F.; Phan K.; Gao Z.-G.; Wan T. C.; Gizewski E.; Auchampach J. A.; Jacobson K. A. Structure-Guided Design of A3 Adenosine Receptor-Selective Nucleosides: Combination of 2-Arylethynyl and Bicyclo[3.1.0]hexane Substitutions. J. Med. Chem. 2012, 55, 4847–4860. 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Tosh D. K.; Toti K. S.; Ciancetta A. Polypharmacology of conformationally locked methanocarba nucleosides. Drug Discovery Today 2017, 22, 1782–1791. 10.1016/j.drudis.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Ciancetta A.; Warnick E.; Crane S.; Gao Z.-G.; Jacobson K. A. Structure-Based Scaffold Repurposing for G Protein-Coupled Receptors: Transformation of Adenosine Derivatives into 5HT2B/5HT2C Serotonin Receptor Antagonists. J. Med. Chem. 2016, 59, 11006–11026. 10.1021/acs.jmedchem.6b01183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Janowsky A.; Eshleman A. J.; Warnick E.; Gao Z.-G.; Chen Z.; Gizewski E.; Auchampach J. A.; Salvemini D.; Jacobson K. A. Scaffold repurposing of nucleosides (adenosine receptor agonists): enhanced activity at the human dopamine and norepinephrine sodium symporters. J. Med. Chem. 2017, 60, 3109–3123. 10.1021/acs.jmedchem.7b00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coen L. M.; Heugebaert T. S. A.; García D.; Stevens C. V. Synthetic entries to and biological activity of pyrrolopyrimidines. Chem. Rev. 2016, 116, 80–139. 10.1021/acs.chemrev.5b00483. [DOI] [PubMed] [Google Scholar]
- Perlíková P.; Hocek M. Pyrrolo[2,3-d ]pyrimidine (7-deazapurine) as a privileged scaffold in design of antitumor and antiviral nucleosides. Med. Res. Rev. 2017, 37, 1429–1460. 10.1002/med.21465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfeld D. A.; Murphy R.; Kim B.; Wang L.; Beuming T.; Abel R.; Friesner R. A. Docking and free energy perturbation studies of ligand binding in the kappa opioid receptor. J. Phys. Chem. B 2015, 119, 824–835. 10.1021/jp5053612. [DOI] [PubMed] [Google Scholar]
- Zheng Z.; Huang X.-P.; Mangano T. J.; Zou R.; Chen X.; Zaidi S. A.; Roth B. L.; Stevens R. C.; Katritch V. Structure-Based Discovery of New Antagonist and Biased Agonist Chemotypes for the Kappa Opioid Receptor. J. Med. Chem. 2017, 60, 3070–3081. 10.1021/acs.jmedchem.7b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino K. A.; Shang Y.; Filizola M. Insights into the function of opioid receptors from molecular dynamics simulations of available crystal structures. Br. J. Pharmacol. 2017, 175, 2834–2845. 10.1111/bph.13774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens W. C.; Jones R. M.; Subramanian G.; Metzger T. G.; Ferguson D. M.; Portoghese P. S. Potent and selective indolomorphinan antagonists of the kappa-opioid receptor. J. Med. Chem. 2000, 43, 2759–2769. 10.1021/jm0000665. [DOI] [PubMed] [Google Scholar]
- Thomas J. B.; Atkinson R. N.; Vinson N. A.; Catanzaro J. L.; Perretta C. L.; Fix S. E.; Mascarella S. W.; Rothman R. B.; Xu H.; Dersch C. M.; Cantrell B. E.; Zimmerman D. M.; Carroll F. I. Identification of (3R)-7-Hydroxy-N-((1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)- 3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro- 3-isoquinolinecarboxamide as a Novel Potent and Selective Opioid κ Receptor Antagonist. J. Med. Chem. 2003, 46, 3127–3137. 10.1021/jm030094y. [DOI] [PubMed] [Google Scholar]
- Urbano M.; Guerrero M.; Rosen H.; Roberts E. Antagonists of the kappa opioid receptor. Bioorg. Med. Chem. Lett. 2014, 24, 2021–2032. 10.1016/j.bmcl.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Rorick-Kehn L. M.; Witkin J. M.; Statnick M. A.; Eberle E. L.; McKinzie J. H.; Kahl S. D.; Forster B. M.; Wong C. J.; Li X.; Crile R. S.; Shaw D. B.; Sahr A. E.; Adams B. L.; Quimby S. J.; Diaz N.; Jimenez A.; Pedregal C.; Mitch C. H.; Knopp K. L.; Anderson W. H.; Cramer J. W.; McKinzie D. L. LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology 2014, 77, 131–144. 10.1016/j.neuropharm.2013.09.021. [DOI] [PubMed] [Google Scholar]
- Dale E.; Bang-Andersen B.; Sánchez C. Emerging mechanisms and treatments for depression beyond SSRIs and SNRIs. Biochem. Pharmacol. 2015, 95, 81–97. 10.1016/j.bcp.2015.03.011. [DOI] [PubMed] [Google Scholar]
- Carroll F. I.; Carlezon W. A. Development of κ Opioid Receptor Antagonists. J. Med. Chem. 2013, 56, 2178–2195. 10.1021/jm301783x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley N. A.; Bloodgood D. W.; Hardaway J. A.; Kendra A. M.; McCall J. G.; Al-Hasani R.; McCall N. M.; Yu W.; Schools Z. L.; Krashes M. J.; Lowell B. B.; Whistler J. L.; Bruchas M. R.; Kash T. L. Dynorphin controls the gain of an amygdalar anxiety circuit. Cell Rep. 2016, 14, 2774–2783. 10.1016/j.celrep.2016.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzumaki N.; Suzuki A.; Narita M.; Hosoya T.; Nagasawa A.; Imai S.; Yamamizu K.; Morita H.; Nagase H.; Okada Y.; Okano H. J.; Yamashita J. K.; Okano H.; Suzuki T.; Narita M. Effect of κ-opioid receptor agonist on the growth of non-small cell lung cancer (NSCLC) cells. Br. J. Cancer 2012, 106, 1148–1152. 10.1038/bjc.2011.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnaturi R.; Aricò G.; Ronsisvalle G.; Parenti C.; Pasquinucci L. Multitarget opioid ligands in pain relief: New players in an old game. Eur. J. Med. Chem. 2016, 108, 211–228. 10.1016/j.ejmech.2015.11.028. [DOI] [PubMed] [Google Scholar]
- Leppert W. Emerging therapies for patients with symptoms of opioid-induced bowel dysfunction. Drug Des., Dev. Ther. 2015, 2015, 2215–2231. 10.2147/dddt.s32684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Holzer P. Non-analgesic effects of opioids: Management of opioid-induced constipation by peripheral opioid receptor antagonists: Prevention or withdrawal?. Curr. Pharm. Des. 2012, 18, 6010–6020. 10.2174/138161212803582388. [DOI] [PubMed] [Google Scholar]; b Anantharamu T.; Sharma S.; Gupta A.; Dahiya N.; Singh Brashier D.; Sharma A. Naloxegol: First oral peripherally acting mu opioid receptor antagonists for opioid-induced constipation. J. Pharmacol. Pharmacother. 2015, 6, 188–192. 10.4103/0976-500x.162015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Pozzo E.; Giacomelli C.; Costa B.; Cavallini C.; Taliani S.; Barresi E.; Da Settimo F.; Martini C. TSPO PIGA ligands promote neurosteroidogenesis and human astrocyte well-being. Int. J. Mol. Sci. 2016, 17, 1028. 10.3390/ijms17071028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa B.; Da Pozzo E.; Giacomelli C.; Barresi E.; Taliani S.; Da Settimo F.; Martini C. TSPO ligand residence time: a new parameter to predict compound neurosteroidogenic efficacy. Sci. Rep. 2016, 6, 18164. 10.1038/srep18164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barresi E.; Bruno A.; Taliani S.; Cosconati S.; Da Pozzo E.; Salerno S.; Simorini F.; Daniele S.; Giacomelli C.; Marini A. M.; La Motta C.; Marinelli L.; Cosimelli B.; Novellino E.; Greco G.; Da Settimo F.; Martini C. Deepening the topology of the translocator protein binding site by novel N,N-dialkyl-2-arylindol-3-ylglyoxylamides. J. Med. Chem. 2015, 58, 6081–6092. 10.1021/acs.jmedchem.5b00689. [DOI] [PubMed] [Google Scholar]
- Lenglet T.; Lacomblez L.; Abitbol J. L.; Ludolph A.; Mora J. S.; Robberecht W.; Shaw P. J.; Pruss R. M.; Cuvier V.; Meininger V. A phase II–III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur. J. Neurol. 2014, 21, 529–536. 10.1111/ene.12344. [DOI] [PubMed] [Google Scholar]
- Daniele S.; Barresi E.; Zappelli E.; Marinelli L.; Novellino E.; Da Settimo F.; Taliani S.; Trincavelli M. L.; Martini C. Long lasting MDM2/Translocator protein modulator: a new strategy for irreversible apoptosis of human glioblastoma cells. Oncotarget 2016, 7, 7866–7884. 10.18632/oncotarget.6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damont A.; Médran-Navarrete V.; Cacheux F.; Kuhnast B.; Pottier G.; Bernards N.; Marguet F.; Puech F.; Boisgard R.; Dollé F. Novel Pyrazolo[1,5-a]pyrimidines as Translocator Protein 18 kDa (TSPO) Ligands: Synthesis, in Vitro Biological Evaluation, [18F]-Labeling, and in Vivo Neuroinflammation PET Images. J. Med. Chem. 2015, 58, 7449–7464. 10.1021/acs.jmedchem.5b00932. [DOI] [PubMed] [Google Scholar]
- Li F.; Liu J.; Liu N.; Kuhn L. A.; Garavito R. M.; Ferguson-Miller S. Translocator protein 18 kDa (TSPO): An old protein with new functions?. Biochemistry 2016, 55, 2821–2831. 10.1021/acs.biochem.6b00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavish M.; Bachman I.; Shoukrun R.; Katz Y.; Veenman L.; Weisinger G.; Weizman A. Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 1999, 51, 629–650. [PubMed] [Google Scholar]
- Daugherty D. J.; Selvaraj V.; Chechneva O. V.; Liu X.-B.; Pleasure D. E.; Deng W. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol. Med. 2013, 5, 891–903. 10.1002/emmm.201202124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruoka H.; Barrett M. O.; Ko H.; Tosh D. K.; Melman A.; Burianek L. E.; Balasubramanian R.; Berk B.; Costanzi S.; Harden T. K.; Jacobson K. A. Pyrimidine Ribonucleotides with Enhanced Selectivity as P2Y6Receptor Agonists: Novel 4-Alkyloxyimino, (S)-Methanocarba, and 5′-Triphosphate γ-Ester Modifications†. J. Med. Chem. 2010, 53, 4488–4501. 10.1021/jm100287t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletta S.; Tosh D. K.; Finley A.; Gizewski E. T.; Moss S. M.; Gao Z.-G.; Auchampach J. A.; Salvemini D.; Jacobson K. A. Rational Design of Sulfonated A3 Adenosine Receptor-Selective Nucleosides as Pharmacological Tools To Study Chronic Neuropathic Pain. J. Med. Chem. 2013, 56, 5949–5963. 10.1021/jm4007966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Paoletta S.; Chen Z.; Moss S. M.; Gao Z.-G.; Salvemini D.; Jacobson K. A. Extended N6 substitution of rigid C2-arylethynyl nucleosides for exploring the role of extracellular loops in ligand recognition at the A3 adenosine receptor. Bioorg. Med. Chem. Lett. 2014, 24, 3302–3306. 10.1016/j.bmcl.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Paoletta S.; Phan K.; Gao Z.-G.; Jacobson K. A. Truncated Nucleosides as A3 Adenosine Receptor Ligands: Combined 2-Arylethynyl and Bicyclohexane Substitutions. ACS Med. Chem. Lett. 2012, 3, 596–601. 10.1021/ml300107e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Padia J.; Salvemini D.; Jacobson K. A. Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain. Purinergic Signalling 2015, 11, 371–387. 10.1007/s11302-015-9459-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Crane S.; Chen Z.; Paoletta S.; Gao Z.-G.; Gizewski E.; Auchampach J. A.; Salvemini D.; Jacobson K. A. Rigidified A3 Adenosine Receptor Agonists: 1-Deazaadenine Modification Maintains High in Vivo Efficacy. ACS Med. Chem. Lett. 2015, 6, 804–808. 10.1021/acsmedchemlett.5b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi B. V.; Melman A.; Mackman R. L.; Jacobson K. A. Synthesis of ethyl (1S,2R,3S,4S,5S)-2,3-O-(isopropylidene)-4-hydroxy-bicyclo[3.1.0]hexane-carboxylate from L-ribose: A versatile chiral synthon for preparation of adenosine and P2 receptor ligands. Nucleosides, Nucleotides Nucleic Acids 2008, 27, 279–291. 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Chinn M.; Ivanov A. A.; Klutz A. M.; Gao Z.-G.; Jacobson K. A. Functionalized Congeners of A3Adenosine Receptor-Selective Nucleosides Containing a Bicyclo[3.1.0]hexane Ring System†. J. Med. Chem. 2009, 52, 7580–7592. 10.1021/jm900426g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X.-P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R. C.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri N. T. Nociceptin opioid receptor (NOP) as a therapeutic target: progress in translation from preclinical research to clinical utility. J. Med. Chem. 2016, 59, 7011–7028. 10.1021/acs.jmedchem.5b01499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze W. K.; Sassano M. F.; Huang X.-P.; Lansu K.; McCorvy J. D.; Giguère P. M.; Sciaky N.; Roth B. L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015, 22, 362–369. 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Wacker D.; Mileni M.; Katritch V.; Han G. W.; Vardy E.; Liu W.; Thompson A. A.; Huang X.-P.; Carroll F. I.; Mascarella S. W.; Westkaemper R. B.; Mosier P. D.; Roth B. L.; Cherezov V.; Stevens R. C. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S.; Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Weis W. I.; Kobilka B. K. Structure of the δ-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van’t Veer A.; Carlezon W. A. Jr. Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology 2013, 229, 435–452. 10.1007/s00213-013-3195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaddelee M. P.; Read K. D.; Cleypool C. G. J.; IJzerman A. P.; Danhof M.; de Boer A. G. Brain penetration of synthetic adenosine A1 receptor agonists in situ: role of the rENT1 nucleoside transporter and binding to blood constituents. Eur. J. Pharm. Sci. 2005, 24, 59–66. 10.1016/j.ejps.2004.09.010. [DOI] [PubMed] [Google Scholar]
- a Mathew B.; Lennon F. E.; Siegler J.; Mirzapoiazova T.; Mambetsariev N.; Sammani S.; Gerhold L. M.; LaRiviere P. J.; Chen C.-T.; Garcia J. G. N.; Salgia R.; Moss J.; Singleton P. A. The Novel Role of the Mu Opioid Receptor in Lung Cancer Progression. Anesth. Analg. 2011, 112, 558–567. 10.1213/ane.0b013e31820568af. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Janku F.; Johnson L. K.; Karp D. D.; Atkins J. T.; Singleton P. A.; Moss J. Treatment with methylnaltrexone is associated with increased survival in patients with advanced cancer. Ann. Oncol. 2016, 27, 2032–2038. 10.1093/annonc/mdw317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski I.; Ma J.; Triezenberg S.; Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature 1988, 335, 563–564. 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- Yung-Chi C.; Prusoff W. H. Relationship between inhibition constant (K1) and concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic-reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Li F.; Liu J.; Zheng Y.; Garavito R. M.; Ferguson-Miller S. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 2015, 347, 555–558. 10.1126/science.1260590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L.; Lovell K. M.; Frankowski K. J.; Slauson S. R.; Phillips A. M.; Streicher J. M.; Stahl E.; Schmid C. L.; Hodder P.; Madoux F.; Cameron M. D.; Prisinzano T. E.; Aubé J.; Bohn L. M. Development of functionally selective, small molecule agonists at kappa opioid receptors. J. Biol. Chem. 2013, 288, 36703–36716. 10.1074/jbc.m113.504381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.