

Abstract
Fatty acid amide hydrolase (FAAH) is responsible for regulating concentrations of the endocannabinoid arachidonoyl ethanolamide. Multiple FAAH inhibitors have been developed for clinical trials and have failed to demonstrate efficacy at treating pain, despite promising preclinical data. One approach toward increasing the efficacy of FAAH inhibitors is to concurrently inhibit other targets responsible for regulating pain. Here, we designed dual inhibitors targeting the enzymes FAAH and soluble epoxide hydrolase (sEH), which are targets previously shown to synergize at reducing inflammatory and neuropathic pain. Exploration of the sEH/FAAH inhibitor structure–activity relationship started with PF-750, a FAAH inhibitor (IC50 = 19 nM) that weakly inhibited sEH (IC50 = 640 nM). Potency was optimized resulting in an inhibitor with improved potency on both targets (11, sEH IC50 = 5 nM, FAAH IC50 = 8 nM). This inhibitor demonstrated good target selectivity, pharmacokinetic properties (AUC = 1200 h nM, t1/2 = 4.9 h in mice), and in vivo target engagement.
Introduction
The endocannabinoid system is an attractive target for anti-inflammatory and analgesic therapeutics. It maintains physiologic homeostasis through two receptors, the cannabinoid receptor 1 (CB1) and 2 (CB2), that are activated primarily by two endocannabinoids, arachidonoyl ethanolamide (AEA) and 2-arachidonoyl glycerol. Agonists directly targeting these receptors, including Δ9-tetrahydrocannabinol, may be useful as analgesics but can cause motor impairment and prevent daily function.1 By comparison, increasing concentrations of endocannabinoids by targeting their hydrolytic metabolism is an alternative approach for producing the same therapeutic effect with reduced side effects. Inhibition or genetic deletion of fatty acid amide hydrolase (FAAH), the enzyme primarily responsible for regulating AEA, has been efficacious in numerous experimental rodent models of inflammatory and neuropathic pain with no functional impairment.2−4
Several types of FAAH inhibitors have been designed. Most inhibit either by interacting with catalytic serine by forming reversible transition-state mimics, such as α-ketoheterocycles,5,6 or through irreversible carbamoylation, such as carbamates7,8 or trisubstituted ureas.4,9−11 Both mechanisms are general for serine hydrolases, and thus, the medicinal chemistry of FAAH inhibitors has focused on optimizing target selectivity in addition to potency. In the case of the FAAH inhibitor BIA 10-2474, off-target inhibition coupled with relatively low potency was the potential cause of a human fatality during phase I clinical trials.12,13 By comparison, PF-04457845, a highly potent FAAH inhibitor with an excellent selectivity profile had no observed side effects but was unable to produce efficacy in phase II osteoarthritic pain trials.14,15 Although there may be therapeutic potential for FAAH inhibitors for other indications, no other clinical studies studying FAAH inhibition on pain have demonstrated biological efficacy in human.16
One approach toward improving biological efficacy, and thus enhancing their therapeutic potential, is to combine inhibitors of multiple targets into a single therapy. Several multitarget inhibitors/modulators have been proposed for FAAH inhibition, including cyclooxygenase (COX),17,18 monoacylglycerol lipase (MAGL),19 cytosolic phospholipase A2,20,21 and the dopamine 3 receptor.22 When FAAH and COX inhibition were combined, the resulting drug not only improves the potency relative to targeting a single molecule but also reduces gastrointestinal side effects associated with nonsteroidal anti-inflammatory drugs.17 In addition to these targets, soluble epoxide hydrolase (sEH) similarly synergizes with FAAH to improve potency on both inflammatory and neuropathic models of pain.23 Soluble epoxide hydrolase regulates biologically active epoxy-fatty acids, including epoxyeicosatrienoic acids, through conversion to their less active diols.24,25 Although the mechanism of synergy between sEH and FAAH is poorly understood, this efficacy is possibly mediated through epoxy-fatty ethanolamides (EpFEAs), CB2 receptor agonists that are likely metabolized by both FAAH and sEH.26,27 In support of this hypothesis, the analgesic effect of sEH inhibitors alone is partially blocked by CB2 but not CB1 antagonists.28
Previously, we reported dual sEH/FAAH inhibitors that were potent on human forms of either enzymes but were unsuitable for use in experimental rodent models.29 Here, we sought to design new dual sEH/FAAH inhibitors that could be used in experimental in vivo models. These inhibitors were designed by integrating a urea pharmacophore common to the medicinal chemistry of both sEH and FAAH inhibitors.
Chemistry and Structure Activity Relationship
The design for potent FAAH and sEH inhibitors is well characterized and described in several recent reviews.30−32 Both FAAH and sEH inhibitors utilize urea pharmacophores to obtain compounds with nM potencies (Figure 1A).4,9−11,33−35 Between these two targets, the urea functional group plays a different role in the mechanism of inhibition. Urea-based sEH inhibitors satisfy hydrogen bonds between H-donating tyrosine residues and the catalytic aspartate residue, resulting in reversible transition-state mimics.36,37 Disubstituted urea inhibitors generally have the highest potency, but highly potent amide, trisubstituted urea, and carbamate inhibitors have been described also.31 The amide-based GSK2256294A has sub-nM potency and has been developed through phase I clinical trials.38 By comparison, urea-based FAAH inhibitors form a covalent intermediate by carbamoylating the catalytic serine residue. Two criteria are critical for this activity: (1) one of the urea nitrogens must carry an aromatic group that becomes a leaving group after nucleophilic attack, and (2) the other nitrogen must bear a heterocycle group (such as a piperidine or piperazine) that provides strain on the molecule.9,39,40 This carbamoylating reaction is relatively unique to FAAH over other serine hydrolases; thus, these inhibitors have high selectivity for FAAH.
Figure 1.

(A) Previously reported sEH, FAAH, and dual sEH/FAAH inhibitors that were used to optimize newly reported dual sEH/FAAH inhibitors. FAAH enzyme preincubation times are reported next to IC50 values. Publications with reported values indicated next to compound name. (B) General approach for the design of dual sEH/FAAH inhibitors. (Ahn et al., 2007,1 Johnson et al., 2010,2 Ahn et al., 2009,3 Keith et al., 2008,4 Kodani et al., 2018,5 Hwang et al., 2007,6 Rose et al., 2010,7 Podolin et al., 2013,8).
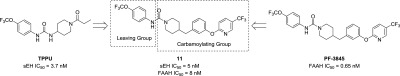
Given the structural overlap between FAAH and sEH inhibitors, we speculated that optimization of current FAAH inhibitors could be used to develop dual sEH/FAAH inhibitors. To investigate this possibility, the potency of several commercially available FAAH inhibitors was measured on human sEH and FAAH enzymes (Table 1). These inhibitors have been well described in the literature with nM potency, and some have been used in rodent models of pain and other disease.4,9,10,41PF-750 had the highest potency toward sEH with an IC50 of 360 nM. From this initial structure, we sought to rationally design an inhibitor with improved potency toward both enzymes by modifying the portion of the molecule that is removed by FAAH (here termed the “leaving group”) and the portion of the molecule that covalently forms a carbamate intermediate with the FAAH active site (here termed the “carbomoylating group”) (Figure 1B).
Table 1. Potency of Several Commercially Available FAAH Inhibitors on Recombinant Human sEH and FAAH.
| name | sEH IC50 (nM)a (5 min preincubation) | FAAH IC50 (nM)b (5 min preincubation) |
|---|---|---|
| PF-750 | 360 | 6.4 |
| PF-622 | 15 000 | 4.0 |
| PF-3845 | 12 000 | 0.65 |
| PF-04457845 | 7800 | 1.1 |
| JNJ-1661010 | 27 000 | 14 |
sEH IC50 was measured using CMNPC ([S]final = 5 μM) in sodium phosphate buffer (0.1 M, pH = 7.4, 0.1 mg/mL BSA).
FAAH IC50 was measured using OMP ([S]final = 100 μM) in sodium phosphate buffer (0.1 M, pH = 8.0, 0.1 mg/mL BSA).
On the basis of previous SAR studies of sEH and FAAH inhibition, we observed the effect of various substitutions on this aromatic “leaving group” has opposite effects on improving the potency of these two targets.11,35 To test the hypothesis that sEH and FAAH potency could be balanced by modifying the carbamoylating and leaving groups, a series of compounds were synthesized where the carbamoylating group from PF-750 was kept the same and the aromatic leaving group was modified. All molecules were synthesized based on a modification of previously described methods (Scheme 1).9,42 The boc-protected 4-substituted piperidines were synthesized by cross-coupling using a Pd catalyst and deprotected under acidic conditions. The final urea was generated by reacting the secondary amine with the commercially available aromatic isocyanate.
Scheme 1. Reagents and Conditions: (a) t-Butyl–Methylenepiperidine-1-carboxylate, 9-BBN, Pd(dppf)Cl2, K2CO3, 5:1 DMF/H2O; (b) K2CO3, Phenylacyl Bromide, DMF, 110 °C, Overnight; (c) 2 N HCl in MeOH, 60 °C, 2 h; (d) R4-CNO, THF, Overnight; and (e) R4-Phenyl Carbamate, DIPEA, DMSO, 55 °C.

Substitution of the leaving group resulted in opposite effects on potency toward sEH and FAAH comparable to previously reported SAR (Table 2).11,35 The unsubstituted ring was modified to add fluorine (1) and chlorine (2) in the 4 position. This increased activity toward sEH 7- and 30-fold, respectively, and decreased activity toward FAAH 5- and 3-fold. Addition of fluorine to the 2 position (3) decreased potency 3-fold toward FAAH while maintaining low potency of sEH compared to PF-750. Altering the aromatic group to have a 4-trifluoromethoxy (4) improved sEH potency 80-fold, whereas also reducing FAAH potency by 21-fold. In comparison, 4-methoxy (5) had a 5-fold increase in potency toward sEH without any change in the potency on FAAH. The 3-pyridine-substituted inhibitor (6) had very little potency toward sEH, but maintained high potency toward FAAH.
Table 2. Effect of Modification of the “Leaving Group” from PF-750 on sEH and FAAH Potency.
![]()

To investigate the contribution of the carbamoylating group to the sEH SAR, we tested several carbamoylating groups from previously described FAAH inhibitors while keeping the 4-methoxy leaving group constant (Table 3). The synthesis of most of these used the same cross-coupling reactions as 1–6 with the exception of 10, in which the piperidine was generated by cyclization between 2-bromoacetophenone and N-boc-4-aminothiocarbonylpiperidine to form the corresponding thiazole.43 Modification of 3-quinoline (5) to 3-phenyl-thiazole (10) resulted in a loss of potency toward both targets, and modification to 2-quinoline (7) resulted in a loss of potency toward sEH with no change in potency toward FAAH. Although the thiazole was used instead of the thiadiazole, previous studies indicate that this change does not impact potency toward FAAH.43 Modification to the 2-naphthylene (8) or 2-phenoxy-5-(trifluoromethyl)pyridine (9) increased activity toward FAAH (4-fold and 30-fold, respectively) without sacrificing potency toward sEH. Because of its high FAAH potency, the 2-phenoxy-5-(trifluoromethyl)pyridine group was used to further optimize the leaving group.
Table 3. Effect of Modification of the “Carbamoylating Group” on sEH and FAAH Potency.
![]()

Combining the sEH potent 4-trifluoromethoxy aniline with the 2-(3-(piperidin-4-ylmethyl)phenoxy)-5-(trifluoromethyl)pyridine leaving group (11) resulted in a potent inhibitor for both enzymes (sEH IC50 = 5 nM, FAAH IC50 = 8 nM). We further tested whether additional substitutions in the leaving group could improve potency (Table 4). For 14–18, the corresponding isocyanate was not commercially available, so a phenyl carbamate generated using phenyl chloroformate was used instead. Using the 4-chloro (12) resulted in a slightly less potent inhibitor on sEH (4-fold reduction). The 4-trifluoromethyl (13) was not substantially different from 11. Adding nitrogen in either the 2 position (15) or the 3 position (14) of the ring reduced sEH potency 12- to 14-fold while either reducing (15) or improving (14) FAAH potency 3-fold. Modification to the pyridazine (16) or the 3,4-dimethyl-isoxazole (17), both potent substitutions on FAAH,10 dramatically decreased potency toward sEH. Additionally, adding fluorine in the 2 position (18) had minor changes on potency in both enzymes (<3-fold difference). It should be noted that the large difference in potencies on sEH with minor modifications in the leaving groups (2400-fold between 11 and PF-3845) is consistent with previous SARs and is likely due to an inability for the sEH active site to accommodate hydrophilic inhibitors.35
Table 4. Effect of Modification of the “Leaving Group” from PF-3845 on sEH and FAAH Potency.


Species Selectivity
Although human recombinant enzymes were used for understanding the structure activity relationships on both FAAH and sEH, most experimental models for testing biological efficacy are in rodents. Because PF-3845 is less potent on rodent FAAH than on human FAAH,4 we expected the potency of all of the new inhibitors on rat and mouse FAAH to be reduced relative to their potency on human FAAH (Table 5). All of the dual inhibitors had IC50 values >10 000 nM on rat enzyme. On the mouse enzyme, 11 and 14 had IC50 values of 1400 and 560 nM, respectively, whereas 13 and 18 had IC50 values >10 000 nM. As expected, increasing the preincubation time increased the potency of all inhibitors in both species while maintaining the relative rank in potency between inhibitors.
Table 5. Comparison of Kinetic Parameters for 11, 13, 14, and 18 with TPPU and PF-3845a.
| TPPU | PF-3845 | 11 | 13 | 14 | 18 | |
|---|---|---|---|---|---|---|
| Human | ||||||
| FAAH IC50 (nM) | - | 0.65 | 8 | 8 | 3 | 3 |
| Ki (nM) | - | 3.5 | 77 | 37 | 14 | 24 |
| kinact (min–1) | - | 0.012 | 0.022 | 0.019 | 0.020 | 0.017 |
| kinact/Ki (nM–1 s–1) | - | 0.0034 | 0.0003 | 0.0005 | 0.0014 | 0.0007 |
| sEH IC50 (nM) | 3.7 | 12 000 | 5 | 7 | 60 | 9 |
| koff × 10–3 (s–1) | 1.4 | - | 0.99 | 1.2 | 6.0 | 2.7 |
| t1/2 (min) | 8.3 | - | 11.8 | 9.9 | 2.0 | 4.4 |
| Mouse | ||||||
| FAAH IC50 (nM) (5 min pre-incubation) | - | 15 | 1400 | >10 000 | 560 | >10 000 |
| FAAH IC50 (nM) (60 min pre-incubation) | - | 0.4 | 66 | 290 | 28 | 340 |
| sEH IC50 (nM) | 43 | 1200 | 27 | 290 | 550 | 360 |
| Rat | ||||||
| FAAH IC50 (nM) (5 min pre-incubation) | - | 29 | >10 000 | >10 000 | >10 000 | >10 000 |
| FAAH IC50 (nM) (60 min pre-incubation) | - | 0.2 | 300 | 710 | 110 | 1100 |
| sEH IC50 (nM) | 70 | 780 | 18 | 40 | 210 | 240 |
| solubility (DI water) (nM) | - | - | 0.13 | 1.6 | 36.0 | 2.8 |
All IC50 values were determined with a 5 min preincubation unless stated otherwise.
The potency on sEH is similarly reduced in both rat and mouse for all inhibitors tested. However, compared to the >100-fold difference between species on FAAH, the species difference on sEH ranged from 4-fold to 40-fold. The best compound in this series, 11, had a better rodent potency than the reference sEH inhibitor TPPU; thus, it is unlikely that differences in potency on sEH would limit the use of this compound for sEH inhibition.
Kinetics of Inhibition
IC50s with a short (5 min) preincubation are a relatively close approximation of affinity for characterizing the SAR of dual sEH/FAAH inhibitors. However, many groups have argued that defining enzyme kinetics more accurately predicts the in vivo pharmacodynamics.10,41 For irreversible FAAH inhibition, these values include the inhibitor association constant (Ki), the rate constant for covalent inactivation (kinact), and the rate constant for enzyme reactivation (kreact). To define kinact and Ki constants, an experimental design was used that was originally described by Main44 and subsequently used for the characterization of FAAH inhibitors (Figure S1A, Table 5).10,41 This approach concurrently adds the enzyme, substrate, and inhibitor and measures the rate of change of substrate hydrolysis. The observed rate of enzyme inactivation (kobs) is plotted against multiple inhibitor concentrations to derive the Ki and kinact. Between PF-3845, 11, 13, 14, and 18, there was relatively little variation between the kinact values (within 1.8-fold between PF-3845 and 11) and most of the variation was in the Ki (22-fold between PF-3845 and 11). This indicates that the difference between inhibitors is mostly in their affinity for the active site rather than the ability to form a covalent intermediate.
In addition to the rate of reactivation (kreact), the rate of dissociation (koff) and the corresponding length of target occupancy on the enzyme (t1/2) contribute to the overall in vivo potency of the inhibitor. Previous experiments with PF-3845, which result in the same carbamate intermediate as 11, demonstrate that the kreact of the catalytic serine residue on the FAAH enzyme is such that the target occupancy is at least several hours.4 To determine the rate of reversible dissociation on the sEH enzyme, a FRET-based assay was used in which 100-fold excess of a high affinity fluorescent reporter was added to a solution of the enzyme–inhibitor complex (1:1 molar equivalent) and the rate of inhibitor dissociation was measured as the rate of FRET generated by the formation of the enzyme–reporter complex.34,45 Compared to TPPU, which had a half-life on the enzyme (t1/2) of 8.3 min in this study, 11 and 13 had a similar half-life at 11.8 and 9.9 min, respectively, and 14 and 18 had lower half-lives at 2.0 and 4.4 min, respectively (Table 5). Because the target occupancy of these compounds on both sEH and FAAH is comparable to their respective reference inhibitors (TPPU and PF-3845), a similar pharmacodynamic profile is expected in vivo.
A poorly explored challenge to the design of dual modulators is the relative preference between the two targets in vivo. In an isolated system such as the recombinant enzyme assays used in this study, the two targets are treated equally and independently. In vivo, these two targets are unlikely to be equivalent because of differences in the target abundance of competing substrates or differences in the relative occupancy on the target. We speculate that the kinetics of inhibition for 11 on sEH and FAAH targets avoids these issues with intertarget dynamics. The target occupancy of 11 on both sEH and FAAH enzymes is comparable to the corresponding single-target inhibitors but is substantially longer on FAAH than on sEH (>10×). By comparison, the relative potency of 11 on mouse sEH (IC50 = 27 nM) is 52-fold greater than FAAH (IC50 = 1400 nM). This trade-off between target occupancy and potency could likely result in good in vivo activity on both targets that could be compromised if target occupancy was longer on sEH or if potency was higher on FAAH.
Pharmacokinetics
To test whether these dual inhibitors could be viable tools to use in experimental rodent models, pharmacokinetic analysis was performed in mice and rats. Cassette dosing has been used previously to characterize and compare the pharmacokinetics of multiple similar sEH inhibitors.46 Cassette dosing was used here to compare the pharmacokinetics of 11, 13, 14, and 18 and to identify the best inhibitor for further studies (1 mg/kg, p.o. in PEG400). All four inhibitors had relatively comparable PK profiles in both species (Figure 2, Table 6). The tmax for all inhibitors in both animals was 4 h and the Cmax ranged from 37 to 140 nM. The t1/2 ranged from 4.9 to 8.9 h and the AUC ranged from 410 to 2200 nM h. Between the two species, 14 had the best AUC, 18 had the worst AUC, and 11 and 13 had intermediate values. On the basis of this profile, 11 was the only inhibitor to reach a blood concentration within the IC50 of sEH inhibition in both species by 2 h postdosing and last within this range at least 8 h postdosing.
Figure 2.

Pharmacokinetic analysis of inhibitors by cassette dosing. Rats (n = 4) or mice (n = 4) were dosed with a cocktail of inhibitors (1 mg/kg each inhibitor, p.o., in PEG300) and sampled at given intervals by tail vein collection. Results are represented as averages of the group.
Table 6. Pharmacokinetic Parameters of Several Dual sEH/FAAH Inhibitorsa.
| species | AUC (h nM) | Cmax (nM) | t1/2 (h) | |
|---|---|---|---|---|
| rat | 11 | 530 | 44 | 7.8 |
| 13 | 740 | 66 | 7.2 | |
| 14 | 1200 | 95 | 7.4 | |
| 18 | 600 | 58 | 5.8 | |
| mouse | 11 | 1200 | 98 | 4.9 |
| 13 | 750 | 65 | 5.1 | |
| 14 | 2200 | 140 | 8.9 | |
| 18 | 410 | 37 | 6.0 | |
Mice or rats were dosed with 1 mg/kg of a cocktail of inhibitors by oral gavage (formulated in PEG300), and blood was collected at intervals indicated in the methods. Pharmacokinetic parameters were determined using PKSolver. Curves were fit to a noncompartmental extravascular model using a linear trapezoidal method for quantifying the area under the curve (AUC). Values were calculated using data up to 24 h postdosing.
Solubility is a major limitation to the described compounds that likely accounts for the long absorption time (tmax ≈ 4 h). Between the inhibitors, 11 had the worst water solubility (0.13 nM) (Table 5) which made it near impossible to dose by any route of administration in any water-based solution. Solubility of 11 was also tested in a variety of vehicles for in vivo dosing (Table S3). Because of the high solubility, a minimal volume of dimethyl sulfoxide (DMSO) dosed by i.p. administration was used for further in vivo experiments.
In Vivo Target Engagement
On the basis of the balance of sEH/FAAH potency and pharmacokinetics, compound 11 was chosen for further exploration for in vivo target engagement. On the basis of its potency toward FAAH (Table 5), 11 should have low in vivo activity on FAAH inhibition in rodents. However, the irreversible mechanism of inhibition may result in greater inhibition of the enzyme than would be predicted by in vitro assays. Thus, the residual enzyme activities of FAAH and sEH were determined 4 h after dosing mice with 11 and compared to TPPU and PF-3845 (Figure 3). Activity was quantified in liver to determine target engagement in normal tissues and in the brain to determine ability to penetrate the blood–brain barrier. To measure FAAH activity, the same fluorescent substrate from the SAR [N-(6-methoxypyridin-3-yl)octanamide (OMP)] was used. Because of the reversible nature of sEH inhibitors, we used [3H]-JHIII as a low activity substrate47 to reduce the dilution of enzyme, and thus of the inhibitor, required for measuring sEH activity in the steady-state range. On the basis of the large dilution required (>20-fold) in the liver due to the high abundance of sEH, this sEH assay was only tested in the brain tissue of dosed animals. Dosing mice with a relatively high dose of TPPU (10 mg/kg) did not alter residual FAAH activity, as expected, but did reduce brain sEH activity to 22 ± 8% of the vehicle-treated activity. PF-3845 (1 mg/kg) reduced liver and brain FAAH activity to 22 ± 2 and 0.2 ± 0.1% of the control values, respectively. Interestingly, PF-3845 also reduced apparent sEH activity to 63 ± 14% of the control. On the basis of the low potency of PF-3845 on murine sEH (IC50 = 1200 nM), it is unlikely that this reduction is due to direct action on sEH but through some other unknown mechanism. By comparison, 11 (10 mg/kg) appeared to reduce liver FAAH to 22 ± 2% of control, similar to PF-3845, but was not as efficient at inhibiting brain FAAH with 22 ± 15% of the activity remaining. 11 appeared to marginally reduce brain sEH to 61 ± 12% of control. A dose response of 11 ranging from 1 to 100 mg/kg demonstrated >60% FAAH inhibition even at the lowest dose tested (Figure S2).
Figure 3.

In vivo target engagement of 11 on sEH and FAAH enzyme in liver and brain. Mice were dosed with either vehicle (20 μL DMSO), TPPU (10 mg/kg), PF-3845 (1 mg/kg), or 11 (10 mg/kg) by i.p. injection and sacrificed 4 h after dosing. Residual FAAH activity was measured fluorescently with a OMP ([S] = 100 μM), and sEH activity was measured radiometrically using [3H]JHIII ([S] = 5 μM). Results are represented as averages ± standard deviation. *p < 0.05 from vehicle control (n = 4).
Off-Target Selectivity
Many serine hydrolase inhibitors suffer from poor target selectivity because of their common mechanisms of action.48 Thus, to test whether 11 broadly inhibited serine hydrolases or whether the inhibition is selective to FAAH, activity-based protein profiling (ABPP) was used on both mouse brain and liver tissue homogenate (Figure S4).9,48 This technique uses a rhodamine-labeled fluorophosphonate probe that tags serine hydrolase enzymes, which are then separated by SDS-PAGE and visualized using a Cy3 filter.4911, 13, 14, and 18 were compared with two commonly used inhibitors, URB597 and PF-3845. URB597 is known to target a number of other hydrolases including carboxylesterase 2.4,9,50 By comparison, PF-3845 is considered as a highly selective inhibitor of FAAH.4 This selectivity is based on the relatively unique ability of FAAH to hydrolyze urea inhibitors because of a distorted amide bond when in complex with FAAH that increases the reactivity of the urea.9,39 In mouse brain tissue, the intensity of the FAAH band is reduced by URB597 and PF-3845 and no other bands were reduced by any of the inhibitors. Although 11, 13, 14, and 18 does not appear to fully inhibit the band corresponding to FAAH, this may be due to the low apparent potency of these inhibitors on the mouse enzyme. In the mouse liver tissue, URB597 reduced the intensity of a band around 62.5 kDa (corresponding to carboxylesterase enzyme), whereas neither 11, 13, 14, 18, nor PF-3845 had any effect on the intensity of other bands. In addition to using ABPP to compare selectivity, the IC50 in several recombinant human enzyme preparations was compared between 11, URB597, and PF-3845 (Table S3). Both 11 and PF-3845 weakly inhibited human CES2 (IC50 = 560 and 1100 nM, respectively, 5 min IC50) and did not inhibit any other tested enzyme. By comparison, URB597 inhibited human CES1, CES2, and AADAC with IC50’s ranging from 39 to 190 nM. Thus, compared to URB597, the series of inhibitors described herein are highly selective for FAAH over other serine hydrolases.
Conclusions
Here, we described a series of dual sEH/FAAH inhibitors with 11 as the optimized structure (human sEH IC50 = 5 nM, human FAAH IC50 = 8 nM). Our previous attempt to design dual sEH/FAAH inhibitors (A-24, Figure 1A)29 resulted in compounds that were potent on both enzymes in human (sEH IC50 = 3.5 nM, FAAH IC50 = 24 nM) but only potent on sEH in rodent species (mouse: sEH IC50 = 5.7, FAAH IC50 = 350 nM; rat: sEH IC50 = 54 nM, FAAH IC50 = 1700 nM). 11 similarly has reduced potency on rodent FAAH (5 min mouse IC50 = 1400 nM), but the irreversible nature of this inhibition results in a higher in vitro potency with longer incubation times (60 min mouse IC50 = 66 nM) which results in effective in vivo target engagement. Furthermore, on the basis of the high selectivity for FAAH over other serine hydrolase inhibitors and excellent pharmacokinetic properties, we expect 11 to be a suitable tool for studying dual sEH/FAAH inhibition in experimental rodent models.
The inhibitors described here will be useful for exploring therapeutic benefits of dual sEH/FAAH inhibition. Given that dual sEH/FAAH inhibition likely modulates EpFEAs that activate the CB2 receptor, 11 may be useful in multiple indications where the CB2 receptor is a major target, including in the regulation of energy homeostasis51−53 and the regulation of organ damage response and fibrosis.54
Methods
General Synthetic Procedures and Methods
Solvents and reagents were used without purification from commercial sources. For 1H and 13C NMR analysis, either a 300 MHz Varian Mercury, 400 Bruker AVANCE, or 600 MHz Varian VNMRS spectrometer was used. Analysis of the high-resolution mass spectra was performed using a Thermo Fisher Scientific LTQ Orbitrap XL with the following settings: centroid mode, spray voltage: 4.5 kV, capillary temperature: 275 °C, sheath gas: 15. Melting point was determined using the Optimelt Automated Melting Point System. Purity was checked by measuring the quenching of green fluorescence indicator (λabs = 254 nm) on thin-layer chromatography and confirmed using high-performance liquid chromatography (HPLC) with an Agilent 1200 Infinity LC system on a Luna C18 column (150 mm × 2.1 mm, 5 μm particle size, 100 Å pore size). The solvent gradient for HPLC is as follows (A = 0.1% formic acid in water; B = 0.1% formic acid in ACN): 0–2 min—90:10 A/B, 2–15—90:10 to 2:98 A/B, 15–21—2:98 A/B, 21–22—2:98 to 90:10 A/B, 22–25—90:10 A/B. Percent purity is reported based on UV absorption at λabs = 250 nm. The general procedures for the synthesis of the described inhibitors are described below, and detailed procedures and characterization are described in the Supporting Information.
General Procedure for the Synthesis of Boc-Protected Piperidines (General Procedure A)
To a solution of tert-butyl 4-methylenepiperidine-1-carboxylate (1 equiv) in THF (10 mL) was added a 0.5 M solution of 9-BBN in THF (1.05 equiv). After stirring an hour, the solution was added to a preprepared solution of the corresponding bromines (1.2 equiv), potassium carbonate (1 equiv), and Pd(dppf)Cl2 complexed with dichloromethane (0.05 equiv) in a 5:1 solution of DMF/H2O (50 mL). The solution was heated to 60 °C and allowed to stir overnight. The reaction was quenched by the addition of 1 M NaOH, extracted with EtOAc, and dried over NaSO4. The product was run on a column of hexane/EtOAc with a gradient from 8:2 to 6:4, yielding the product.
General Procedure for Boc Deprotection (General Procedure B)
The corresponding boc-protected piperidine was dissolved in 2 N HCl in MeOH (30 mL) and was heated to 60 °C for 2 h. The excess MeOH was removed by a rotary evaporator and diluted with H2O (30 mL), and the solution was treated with NaOH pellets to reach pH = 11. The product was extracted with EtOAc, dried over NaSO4, and evaporated to afford the product.
General Procedure for Urea Synthesis through an Isocyanate (1–13) (General Procedure C)
The corresponding isocyanate (1.2 equiv) was added to a solution of the corresponding amine (1 equiv) dissolved in dry THF (10 mL) and stirred overnight. The reaction was quenched with 1 N HCl (10 mL) and stirred for 5 min. The reaction was neutralized by addition of Na2CO3 and extracted three times with EtOAc. The product was further purified by flash chromatography to afford the product.
General Procedure for Urea Synthesis through a Carbamate (14–18) (General Procedure D)
The corresponding amine (1 equiv), phenyl carbamate (1.2 equiv), and N,N′-diisopropylethylamine (1.2 equiv) were dissolved in DMSO (15 mL). After stirring overnight at 55 °C, the reaction was quenched with water and stirred for 5 min. The crude mixture was extracted with EtOAc, dried with Na2CO3, and evaporated. The product was purified by flash chromatography.
Preparation of Soluble Epoxide Hydrolase (sEH) and FAAH Enzyme Extracts
Recombinant mouse, rat, and human sEH were prepared as previously described29 using an insect cell/baculovirus system and affinity-based purification (purity > 95% as judged by SDS-PAGE) that removes all measurable esterase or glutathione-S-transferase activity.55
Recombinant FAAH enzyme from baculovirus was prepared as previously described.29,56 Briefly, homogenized (3 × 15 s) baculovirus-infected high five cells were centrifuged (9000g, 20 min) to collect the S9 fraction which was resuspended in buffer (50 mM tris/HCl buffer, pH = 8.0) containing 1 mM CHAPS and 10% glycerol and stored frozen (−80 °C) until use. For measuring rat FAAH inhibition, brain microsomes were prepared by homogenizing tissue in phosphate buffer [20 mM, pH = 7.4, 5 mM ethylenediaminetetraacetic acid (EDTA)], collecting the S9 fraction by centrifugation (9000g, 20 min), and further centrifuging the S9 fraction at 100 000g for 1 h. The subsequent microsomes were resuspended in phosphate buffer (10 mM, pH = 7.4, 2.5 mM EDTA) containing 20% glycerol and stored frozen (−80 °C) until use. For measuring mouse FAAH inhibition, crude tissue homogenate was prepared in phosphate buffer (20 mM, pH = 7.4, 5 mM EDTA) and stored frozen (−80 °C) until use.
Measurement of Inhibitor Potency Using Fluorescent Assays
Methods for the quantification of inhibitor potencies have been previously published for sEH,35,57,58 FAAH,29,59 and other esterases,55,60 and the individual details are described below. Generally, fluorescence generated by enzymatic hydrolysis was quantified every 30 s for 10 min, and the reaction velocity (vinhibitor) was determined from the linear portion of the curve. Values were subtracted from background using wells containing no enzyme. The IC50 values were derived using simple linear regression of the log [I] versus % remaining activity (vinhibitor/vDMSO) and determining x when y = 0.50. All measurements were the average of triplicates. For all assays, the final DMSO concentration was 2%.
sEH Assay
The substrate cyano(6-methoxynaphthalen-2-yl)methyl((3-phenyloxiran-2-yl)methyl)carbonate (CMNPC) ([S]final = 5 μM) was added to wells containing sEH in sodium phosphate buffer [0.1 M, pH = 7.4 and 0.1 mg/mL bovine serum albumin (BSA)], and formation of the fluorescent 6-methoxynaphthaldehyde (λexcitation = 330 nm, λemission = 465 nm, 30 °C) was measured.
FAAH Assay
The substrate OMP ([S]final = 100 μM) was added to wells containing FAAH in sodium phosphate buffer (0.1 M, pH = 8, 0.1 mg/mL BSA), and formation of the fluorescent 6-methoxypyridine (λexcitation = 303 nm, λemission = 394 nm, 37 °C) was measured.
For measuring Ki and kinact, OMP ([S]final = 100 μM) was added to various inhibitor concentrations and the mixture was incubated at 37 °C for 5 min. Crude enzyme was then added to the well, and the generation of fluorescent signal was immediately monitored over 5 min. The value of kobs was calculated by the following equation
and kobs was fit to
to calculate Ki and kobs.
Esterase Assay
CES1, CES2, and AADAC crude recombinant enzyme extracts were prepared as previously described.29 To measure esterase activity, the substrate cyano(6-methoxynaphthalen-2-yl)methyl acetate ([S]final = 50 μM) was added to wells containing either CES1, CES2, or AADAC in sodium phosphate buffer (0.1 M, pH = 8, 0.1 mg/mL BSA) and formation of the fluorescent 6-methoxynaphthaldehyde (λexcitation = 330 nm, λemission = 465 nm, 37 °C) was measured.
Purified recombinant MAGL enzyme was kindly provided by Dr. Christian Krintel. To measure MAGL activity, the substrate 4-nitrophenyl acetate ([S]final = 50 μM) was added to wells containing MAGL in sodium phosphate buffer (0.1 M, pH = 8, 0.1 mg/mL BSA) and formation of the colorimetric 4-nitrophenol (λabs = 412 nm, 37 °C) was measured.
Solubility
For measuring water solubility, 2–5 mg of each material was added to a 1.7 mL polypropylene tube and was shaken for 2 days at 37 °C. The resulting water solution was then filtered through a 0.22 μM PTFE filter and diluted twofold in MeOH containing 15 nM B-12. The concentration was quantified by LC/MS/MS.
Activity-Based Protein Profiling for Determining Enzyme Selectivity
Activity-based protein profiling (ABPP) was performed by incubating lysates with an inhibitor, then with the fluorophosphonate probe, followed by separation by SDS-PAGE and visualization. Inhibitor (in 1 μL of DMSO) was added to a tube containing either brain or liver tissue homogenate (2 mg/mL, 50 μL) and incubated for 20 min at 30 °C. ActivX TAMRA-FP serine hydrolase probe (1 μL, Thermo Scientific, Waltham, MA) ([TAMRA-FP]final = 2 μM) was added, and the mixture was incubated another 20 min at 30 °C. The reaction was quenched with 4× loading buffer, boiled at 90 °C for 5 min, and loaded on a Bolt 4–12% Bis–Tris SDS-PAGE gel (Thermo Scientific, Waltham, MA). The proteins were separated at 200 V for 35 min run in MOPS SDS buffer (Thermo Scientific, Waltham, MA). The bands were visualized using Bio-Rad ChemiDoc MP (Bio-Rad, Hercules, CA) using a Cy3 filter and analyzed using ImageLab 5.0 (Bio-Rad, Hercules, CA).
In Vivo FAAH and sEH Target Engagement
Animal experiments were performed according to established protocols approved by the University of California Institutional Animal Care and Use Committee. Male Swiss Webster mice were dosed with the corresponding compound in DMSO (20 μL, i.p.) and sacrificed 4 h after dosing. Tissues were homogenized in phosphate buffer (20 mM, pH = 7.4, 5 mM EDTA) with an approximate fivefold dilution by weight. After homogenizing, the samples were either used as is (sEH assay) or were diluted to the appropriate linear range for the assay (BCA protein assay and FAAH assay). sEH assay: to a solution of 100 μL tissue homogenate was added 1 μL of [3H]JHIII in MeOH ([S]final = 5 μM, approximately 10 000 CPM) and the reaction was incubated for 20 min. To quench the reaction, 100 μL of MeOH and 250 μL of iso-octane were added and the mixture was vortexed, centrifuged for 5 min at 2000 rpm, and 50 μL of the aqueous solution was collected, placed in 1 mL of liquid scintillation fluid, and counted by liquid scintillation. The values were normalized by the protein concentration in each sample and compared to vehicle-treated animals such that values represent % remaining [3H]JHIII hydrolysis activity. FAAH assay: samples were diluted 1000-fold in phosphate buffer (100 mM, pH = 8.0, 0.1% BSA) for liver and 100-fold for brain. OMP in 2 μL of DMSO ([S]final = 100 μM) was added to each well, and fluorescence was measured at λexc = 303 nm and λemm = 394 nm for 10 min. The values were normalized by the protein concentration in each sample and compared to vehicle-treated animals such that values represent % remaining OMP hydrolysis activity. Protein assay: the BCA protein assay was performed as specified by the manufacturer’s instructions, using BSA as standard.
Acknowledgments
This work was supported by the National Institute of Environmental Health Sciences grant R01 ES002710 and the National Institute of Environmental Health Sciences Superfund Research Program grant P42 ES004699. S.D.K. was supported by a NIGMS-funded Pharmacology Training Program (T32GM099608). We would also like to thank Dr. Christian Krintel for providing MAGL enzyme and Drs. Jun Yang and Kin Sing Stephen Lee for technical assistance.
Glossary
Abbreviations
- AADAC
arylacetamide deacetylase
- ABPP
activity-based protein profiling
- AEA
arachidonoyl ethanolamide
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- CES1
carboxylesterase 1
- CES2
carboxylesterase 2
- CMNPC
cyano(6-methoxynaphthalen-2-yl)methyl((3-phenyloxiran-2-yl)methyl) carbonate
- COX
cyclooxygenase
- EpFEA
epoxy fatty ethanolamides
- FAAH
fatty acid amide hydrolase
- MAGL
monoacylglycerol lipase
- OMP
N-(6-methoxypyridin-3-yl)octanamide
- SAR
structure–activity relationship
- sEH
soluble epoxide hydrolase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01625.
Synthesis; pharmacokinetics; HPLC solvent gradient to separate dual sEH/FAAH inhibitors for PK analysis; optimized parameters for multiple-reaction-monitoring (MRM) analysis of inhibitors for PK analysis; solubility of 11 in a variety of vehicles for in vivo dosing; potency of URB597, PF-3845, and 11 on several human recombinant serine hydrolase enzymes; analysis of kinact and Ki on FAAH and koff on sEH for 11; dose response of 11 on residual FAAH and sEH tissue activity; and activity-based protein profiling of dual inhibitors to identify off-target inhibition of serine hydrolases (PDF)
The authors declare the following competing financial interest(s): SDK and BDH are authors on a patent own by the University of California for compounds presented in this text (WO2017160861). BDH, KMW and SSH are part of Eicosis L.L.C., a company developing sEH inhibitors for clinical trials.
Supplementary Material
References
- Pertwee R. G. The Central Neuropharmacology of Psychotropic Cannabinoids. Pharmacol. Ther. 1988, 36, 189–261. 10.1016/0163-7258(88)90106-4. [DOI] [PubMed] [Google Scholar]
- Cravatt B. F.; Saghatelian A.; Hawkins E. G.; Clement A. B.; Bracey M. H.; Lichtman A. H. Functional dissociation of the central and peripheral fatty acid amide signaling systems. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 10821–10826. 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley D.; Gaetani S.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005, 313, 352–358. 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Ahn K.; Johnson D. S.; Mileni M.; Beidler D.; Long J. Z.; McKinney M. K.; Weerapana E.; Sadagopan N.; Liimatta M.; Smith S. E.; Lazerwith S.; Stiff C.; Kamtekar S.; Bhattacharya K.; Zhang Y.; Swaney S.; Van Becelaere K.; Stevens R. C.; Cravatt B. F. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 2009, 16, 411–420. 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D. L.; Sato H.; Lerner A. E.; Hedrick M. P.; Fecik R. A.; Miyauchi H.; Wilkie G. D.; Austin B. J.; Patricelli M. P.; Cravatt B. F. Exceptionally potent inhibitors of fatty acid amide hydrolase: The enzyme responsible for degradation of endogenous oleamide and anandamide. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 5044–5049. 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman A. H.; Leung D.; Shelton C. C.; Saghatelian A.; Hardouin C.; Boger D. L.; Cravatt B. F. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J. Pharmacol. Exp. Ther. 2004, 311, 441–448. 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- Mor M.; Rivara S.; Lodola A.; Plazzi P. V.; Tarzia G.; Duranti A.; Tontini A.; Piersanti G.; Kathuria S.; Piomelli D. Cyclohexylcarbamic Acid 3′- or 4′-Substituted Biphenyl-3-yl Esters as Fatty Acid Amide Hydrolase Inhibitors: Synthesis, Quantative Structure-Activity Relationships, and Molecular Modeling Studies. J. Med. Chem. 2004, 47, 4998–5008. 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- Moreno-Sanz G.; Duranti A.; Melzig L.; Fiorelli C.; Ruda G. F.; Colombano G.; Mestichelli P.; Sanchini S.; Tontini A.; Mor M.; Bandiera T.; Scarpelli R.; Tarzia G.; Piomelli D. Synthesis and structure-activity relationship studies of O-biphenyl-3-yl carbamates as peripherally restricted fatty acid amide hydrolase inhibitors. J. Med. Chem. 2013, 56, 5917–5930. 10.1021/jm4007017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K.; Johnson D. S.; Fitzgerald L. R.; Liimatta M.; Arendse A.; Stevenson T.; Lund E. T.; Nugent R. A.; Nomanbhoy T. K.; Alexander J. P.; Cravatt B. F. Novel Mechanistic Class of Fatty Acid Amide Hydrolase Inhibitors with Remarkable Selectivity†. Biochemistry 2007, 46, 13019–13030. 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- Johnson D. S.; Stiff C.; Lazerwith S. E.; Kesten S. R.; Fay L. K.; Morris M.; Beidler D.; Liimatta M. B.; Smith S. E.; Dudley D. T.; Sadagopan N.; Bhattachar S. N.; Kesten S. J.; Nomanbhoy T. K.; Cravatt B. F.; Ahn K. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med. Chem. Lett. 2011, 2, 91–96. 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith J. M.; Apodaca R.; Xiao W.; Seierstad M.; Pattabiraman K.; Wu J.; Webb M.; Karbarz M. J.; Brown S.; Wilson S.; Scott B.; Tham C.-S.; Luo L.; Palmer J.; Wennerholm M.; Chaplan S.; Breitenbucher J. G. Thiadiazolopiperazinyl ureas as inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2008, 18, 4838–4843. 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]
- Kerbrat A.; Ferré J.-C.; Fillatre P.; Ronzière T.; Vannier S.; Carsin-Nicol B.; Lavoué S.; Vérin M.; Gauvrit J.-Y.; Le Tulzo Y.; Edan G. Acute Neurologic Disorder from an Inhibitor of Fatty Acid Amide Hydrolase. N. Engl. J. Med. 2016, 375, 1717–1725. 10.1056/nejmoa1604221. [DOI] [PubMed] [Google Scholar]
- van Esbroeck A. C. M.; Janssen A. P. A.; Cognetta A. B.; Ogasawara D.; Shpak G.; van der Kroeg M.; Kantae V.; Baggelaar M. P.; de Vrij F. M. S.; Deng H.; Allarà M.; Fezza F.; Lin Z.; van der Wel T.; Soethoudt M.; Mock E. D.; den Dulk H.; Baak I. L.; Florea B. I.; Hendriks G.; De Petrocellis L.; Overkleeft H. S.; Hankemeier T.; De Zeeuw C. I.; Di Marzo V.; Maccarrone M.; Cravatt B. F.; Kushner S. A.; van der Stelt M. Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10-2474. Science 2017, 356, 1084–1087. 10.1126/science.aaf7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins J. P.; Smart T. S.; Langman S.; Taylor L.; Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 2012, 153, 1837–1846. 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Li G. L.; Winter H.; Arends R.; Jay G. W.; Le V.; Young T.; Huggins J. P. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br. J. Clin. Pharmacol. 2012, 73, 706–716. 10.1111/j.1365-2125.2011.04137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenlehner F. M. E.; van Till J. W. O.; Houbiers J. G. A.; Martina R. V.; Cerneus D. P.; Melis J. H. J. M.; Majek A.; Vjaters E.; Urban M.; Ramonas H.; Shoskes D. A.; Nickel J. C. Fatty Acid Amide Hydrolase Inhibitor Treatment in Men With Chronic Prostatitis/Chronic Pelvic Pain Syndrome: An Adaptive Double-blind, Randomized Controlled Trial. Urology 2017, 103, 191–197. 10.1016/j.urology.2017.02.029. [DOI] [PubMed] [Google Scholar]
- Sasso O.; Migliore M.; Habrant D.; Armirotti A.; Albani C.; Summa M.; Moreno-Sanz G.; Scarpelli R.; Piomelli D. Multitarget fatty acid amide hydrolase/cyclooxygenase blockade suppresses intestinal inflammation and protects against nonsteroidal anti-inflammatory drug-dependent gastrointestinal damage. FASEB J. 2015, 29, 2616–2627. 10.1096/fj.15-270637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore M.; Habrant D.; Sasso O.; Albani C.; Bertozzi S. M.; Armirotti A.; Piomelli D.; Scarpelli R. Potent multitarget FAAH-COX inhibitors: Design and structure-activity relationship studies. Eur. J. Med. Chem. 2016, 109, 216–237. 10.1016/j.ejmech.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J. Z.; Nomura D. K.; Vann R. E.; Walentiny D. M.; Booker L.; Jin X.; Burston J. J.; Sim-Selley L. J.; Lichtman A. H.; Wiley J. L.; Cravatt B. F. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 20270–20275. 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirer K.; Steinhilber D.; Proschak E. Inhibitors of the Arachidonic Acid Cascade: Interfering with Multiple Pathways. Basic Clin. Pharmacol. Toxicol. 2014, 114, 83–91. 10.1111/bcpt.12134. [DOI] [PubMed] [Google Scholar]
- Forster L.; Ludwig J.; Kaptur M.; Bovens S.; Elfringhoff A. S.; Holtfrerich A.; Lehr M. 1-Indol-1-yl-propan-2-ones and related heterocyclic compounds as dual inhibitors of cytosolic phospholipase A(2)alpha and fatty acid amide hydrolase. Bioorg. Med. Chem. 2010, 18, 945–952. 10.1016/j.bmc.2009.11.028. [DOI] [PubMed] [Google Scholar]
- De Simone A.; Russo D.; Ruda G. F.; Micoli A.; Ferraro M.; Di Martino R. M. C.; Ottonello G.; Summa M.; Armirotti A.; Bandiera T.; Cavalli A.; Bottegoni G. Design, Synthesis, Structure-Activity Relationship Studies, and Three-Dimensional Quantitative Structure-Activity Relationship (3D-QSAR) Modeling of a Series of O-Biphenyl Carbamates as Dual Modulators of Dopamine D3 Receptor and Fatty Acid Amide Hydrolase. J. Med. Chem. 2017, 60, 2287–2304. 10.1021/acs.jmedchem.6b01578. [DOI] [PubMed] [Google Scholar]
- Sasso O.; Wagner K.; Morisseau C.; Inceoglu B.; Hammock B. D.; Piomelli D. Peripheral FAAH and soluble epoxide hydrolase inhibitors are synergistically antinociceptive. Pharmacol. Res. 2015, 97, 7–15. 10.1016/j.phrs.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C.; Hammock B. D. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 37–58. 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodani S. D.; Hammock B. D. The 2014 Bernard B. Brodie award lecture-epoxide hydrolases: drug metabolism to therapeutics for chronic pain. Drug Metab. Dispos. 2015, 43, 788–802. 10.1124/dmd.115.063339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider N. T.; Nast J. A.; Tesmer L. A.; Hollenberg P. F. A cytochrome P450-derived epoxygenated metabolite of anandamide is a potent cannabinoid receptor 2-selective agonist. Mol. Pharmacol. 2009, 75, 965–972. 10.1124/mol.108.053439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougle D. R.; Watson J. E.; Abdeen A. A.; Adili R.; Caputo M. P.; Krapf J. E.; Johnson R. W.; Kilian K. A.; Holinstat M.; Das A. Anti-inflammatory omega-3 endocannabinoid epoxides. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E6034–E6043. 10.1073/pnas.1610325114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K.; Inceoglu B.; Hammock B. D. Soluble epoxide hydrolase inhibition, epoxygenated fatty acids and nociception. Prostaglandins Other Lipid Mediators 2011, 96, 76–83. 10.1016/j.prostaglandins.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodani S. D.; Bhakta S.; Hwang S. H.; Pakhomova S.; Newcomer M. E.; Morisseau C.; Hammock B. D. Identification and optimization of soluble epoxide hydrolase inhibitors with dual potency towards fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2018, 28, 762–768. 10.1016/j.bmcl.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otrubova K.; Ezzili C.; Boger D. L. The discovery and development of inhibitors of fatty acid amide hydrolase (FAAH). Bioorg. Med. Chem. Lett. 2011, 21, 4674–4685. 10.1016/j.bmcl.2011.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H. C.; Hammock B. D. Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J. Med. Chem. 2012, 55, 1789–1808. 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodola A.; Castelli R.; Mor M.; Rivara S. Fatty acid amide hydrolase inhibitors: a patent review (2009-2014). Expert Opin. Ther. Pat. 2015, 25, 1247–1266. 10.1517/13543776.2015.1067683. [DOI] [PubMed] [Google Scholar]
- Hwang S. H.; Tsai H.-J.; Liu J.-Y.; Morisseau C.; Hammock B. D. Orally Bioavailable Potent Soluble Epoxide Hydrolase Inhibitors. J. Med. Chem. 2007, 50, 3825–3840. 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. S. S.; Liu J.-Y.; Wagner K. M.; Pakhomova S.; Dong H.; Morisseau C.; Fu S. H.; Yang J.; Wang P.; Ulu A.; Mate C. A.; Nguyen L. V.; Hwang S. H.; Edin M. L.; Mara A. A.; Wulff H.; Newcomer M. E.; Zeldin D. C.; Hammock B. D. Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. J. Med. Chem. 2014, 57, 7016–7030. 10.1021/jm500694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose T. E.; Morisseau C.; Liu J.-Y.; Inceoglu B.; Jones P. D.; Sanborn J. R.; Hammock B. D. 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J. Med. Chem. 2010, 53, 7067–7075. 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argiriadi M. A.; Morisseau C.; Goodrow M. H.; Dowdy D. L.; Hammock B. D.; Christianson D. W. Binding of alkylurea inhibitors to epoxide hydrolase implicates active site tyrosines in substrate activation. J. Biol. Chem. 2000, 275, 15265–15270. 10.1074/jbc.m000278200. [DOI] [PubMed] [Google Scholar]
- Argiriadi M. A.; Morisseau C.; Hammock B. D.; Christianson D. W. Detoxification of environmental mutagens and carcinogens: Structure, mechanism and evolution of liver epoxide hydrolase. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 10637–10642. 10.1073/pnas.96.19.10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolin P. L.; Bolognese B. J.; Foley J. F.; Long E. III; Peck B.; Umbrecht S.; Zhang X.; Zhu P.; Schwartz B.; Xie W.; Quinn C.; Qi H.; Sweitzer S.; Chen S.; Galop M.; Ding Y.; Belyanskaya S. L.; Israel D. I.; Morgan B. A.; Behm D. J.; Marino J. P. Jr.; Kurali E.; Barnette M. S.; Mayer R. J.; Booth-Genthe C. L.; Callahan J. F. In vitro and in vivo characterization of a novel soluble epoxide hydrolase inhibitor. Prostaglandins Other Lipid Mediators 2013, 104–105, 25–31. 10.1016/j.prostaglandins.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Palermo G.; Branduardi D.; Masetti M.; Lodola A.; Mor M.; Piomelli D.; Cavalli A.; De Vivo M. Covalent inhibitors of fatty acid amide hydrolase: a rationale for the activity of piperidine and piperazine aryl ureas. J. Med. Chem. 2011, 54, 6612–6623. 10.1021/jm2004283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodola A.; Capoferri L.; Rivara S.; Tarzia G.; Piomelli D.; Mulholland A.; Mor M. Quantum mechanics/molecular mechanics modeling of fatty acid amide hydrolase reactivation distinguishes substrate from irreversible covalent inhibitors. J. Med. Chem. 2013, 56, 2500–2512. 10.1021/jm301867x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileni M.; Johnson D. S.; Wang Z.; Everdeen D. S.; Liimatta M.; Pabst B.; Bhattacharya K.; Nugent R. A.; Kamtekar S.; Cravatt B. F.; Ahn K.; Stevens R. C. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 12820–12824. 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenbucher J. G.; Keith J. M.; Tichenor M. S.; Chambers A. L.; Jones W. M.; Hawryluk N. A.; Timmons A. K.; Merit J. E.; Seierstad M.. Heteroaryl-substituted urea modulators of fatty acid amide hydrolase. WO2010068453 A1, 2010.
- Kono M.; Matsumoto T.; Kawamura T.; Nishimura A.; Kiyota Y.; Oki H.; Miyazaki J.; Igaki S.; Behnke C. A.; Shimojo M.; Kori M. Synthesis, SAR study, and biological evaluation of a series of piperazine ureas as fatty acid amide hydrolase (FAAH) inhibitors. Bioorg. Med. Chem. 2013, 21, 28–41. 10.1016/j.bmc.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Main A. R.; Dauterman W. C. Determination of the biomolecular rate constant for the reaction between organophosphorous inhibitors and esterases in the presence of substrate. Nature 1963, 198, 551–553. 10.1038/198551a0. [DOI] [Google Scholar]
- Lee K. S. S.; Morisseau C.; Yang J.; Wang P.; Hwang S. H.; Hammock B. D. Forster resonance energy transfer competitive displacement assay for human soluble epoxide hydrolase. Anal. Biochem. 2013, 434, 259–268. 10.1016/j.ab.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T.; Schulz D.; Morisseau C.; Hammock B. D. High-throughput pharmacokinetic method: Cassette dosing in mice associated with minuscule serial bleedings and LC/MS/MS analysis. Anal. Chim. Acta 2006, 559, 37–44. 10.1016/j.aca.2005.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T.; Morisseau C.; Maxwell J. E.; Argiriadi M. A.; Christianson D. W.; Hammock B. D. Biochemical evidence for the involvement of tyrosine in epoxide activation during the catalytic cycle of epoxide hydrolase. J. Biol. Chem. 2000, 275, 23082–23088. 10.1074/jbc.m001464200. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Saraf A.; Kolasa T.; Bhatia P.; Zheng G. Z.; Patel M.; Lannoye G. S.; Richardson P.; Stewart A.; Rogers J. C.; Brioni J. D.; Surowy C. S. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology 2007, 52, 1095–1105. 10.1016/j.neuropharm.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Patricelli M. P.; Cravatt B. F. Activity-based protein profiling: The serine hydrolases. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 14694–14699. 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow J. A.; Bittles V.; Borazjani A.; Potter P. M.; Ross M. K. Covalent inhibition of recombinant human carboxylesterase 1 and 2 and monoacylglycerol lipase by the carbamates JZL184 and URB597. Biochem. Pharmacol. 2012, 84, 1215–1222. 10.1016/j.bcp.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz K.; Mangels N.; Häussler A.; Ferreirós N.; Fleming I.; Tegeder I. Pro-inflammatory obesity in aged cannabinoid-2 receptor-deficient mice. Int. J. Obes. 2016, 40, 366–379. 10.1038/ijo.2015.169. [DOI] [PubMed] [Google Scholar]
- Verty A. N. A.; Stefanidis A.; McAinch A. J.; Hryciw D. H.; Oldfield B. Anti-Obesity Effect of the CB2 Receptor Agonist JWH-015 in Diet-Induced Obese Mice. PLoS One 2015, 10, e0140592. 10.1371/journal.pone.0140592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F.; Bellini G.; Luongo L.; Manzo I.; Tolone S.; Tortora C.; Bernardo M. E.; Grandone A.; Conforti A.; Docimo L.; Nobili B.; Perrone L.; Locatelli F.; Maione S.; del Giudice E. M. Cannabinoid Receptor 2 as Antiobesity Target: Inflammation, Fat Storage, and Browning Modulation. J. Clin. Endocrinol. Metab. 2016, 101, 3469–3478. 10.1210/jc.2015-4381. [DOI] [PubMed] [Google Scholar]
- Mallat A.; Teixeira-Clerc F.; Lotersztajn S. Cannabinoid signaling and liver therapeutics. J. Hepatol. 2013, 59, 891–896. 10.1016/j.jhep.2013.03.032. [DOI] [PubMed] [Google Scholar]
- Morisseau C.; Merzlikin O.; Lin A.; He G.; Feng W.; Padilla I.; Denison M. S.; Pessah I. N.; Hammock B. D. Toxicology in the fast lane: application of high-throughput bioassays to detect modulation of key enzymes and receptors. Environ. Health Perspect. 2009, 117, 1867–1872. 10.1289/ehp.0900834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodani S. D.; Overby H. B.; Morisseau C.; Chen J.; Zhao L.; Hammock B. D. Parabens inhibit fatty acid amide hydrolase: A potential role in paraben-enhanced 3T3-L1 adipocyte differentiation. Toxicol. Lett. 2016, 262, 92–99. 10.1016/j.toxlet.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. D.; Wolf N. M.; Morisseau C.; Whetstone P.; Hock B.; Hammock B. D. Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal. Biochem. 2005, 343, 66–75. 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf N. M.; Morisseau C.; Jones P. D.; Hock B.; Hammock B. D. Development of a high-throughput screen for soluble epoxide hydrolase inhibition. Anal. Biochem. 2006, 355, 71–80. 10.1016/j.ab.2006.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Nishi K.; Tsai H.-J.; Hammock B. D. Development of highly sensitive fluorescent assays for fatty acid amide hydrolase. Anal. Biochem. 2007, 363, 12–21. 10.1016/j.ab.2006.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodani S. D.; Barthélemy M.; Kamita S. G.; Hammock B.; Morisseau C. Development of amide-based fluorescent probes for selective measurement of carboxylesterase 1 activity in tissue extracts. Anal. Biochem. 2017, 539, 81–89. 10.1016/j.ab.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.