

Magnetotactic bacteria (MTB) are a group of organisms that form intracellular nanometer-scale magnetic crystals though a complex process involving lipid and protein scaffolds. These magnetic crystals and their lipid membranes, termed magnetosomes, are model systems for studying bacterial cell biology and biomineralization and are potential platforms for biotechnological applications. Due to a lack of genetic tools and unculturable representatives, the mechanisms of magnetosome formation in phylogenetically deeply branching MTB remain unknown. These MTB contain elongated bullet-/tooth-shaped magnetite and greigite crystals that likely form in a manner distinct from that of the cubooctahedral-shaped magnetite crystals of the genetically tractable MTB within the Alphaproteobacteria. Here, we present a method for genome editing in Desulfovibrio magneticus RS-1, a cultured representative of the deeply branching MTB of the class Deltaproteobacteria. This marks a crucial step in developing D. magneticus as a model for studying diverse mechanisms of magnetic particle formation by MTB.
KEYWORDS: Desulfovibrio, biomineralization, genome editing, iron, magnetosomes, magnetotactic bacteria, organelles
ABSTRACT
Magnetosomes are complex bacterial organelles that serve as model systems for studying bacterial cell biology, biomineralization, and global iron cycling. Magnetosome biogenesis is primarily studied in two closely related Alphaproteobacteria of the genus Magnetospirillum that form cubooctahedral-shaped magnetite crystals within a lipid membrane. However, chemically and structurally distinct magnetic particles have been found in physiologically and phylogenetically diverse bacteria. Due to a lack of molecular genetic tools, the mechanistic diversity of magnetosome formation remains poorly understood. Desulfovibrio magneticus RS-1 is an anaerobic sulfate-reducing deltaproteobacterium that forms bullet-shaped magnetite crystals. A recent forward genetic screen identified 10 genes in the conserved magnetosome gene island of D. magneticus that are essential for its magnetic phenotype. However, this screen likely missed mutants with defects in crystal size, shape, and arrangement. Reverse genetics to target the remaining putative magnetosome genes using standard genetic methods of suicide vector integration have not been feasible due to the low transconjugation efficiency. Here, we present a reverse genetic method for targeted mutagenesis in D. magneticus using a replicative plasmid. To test this method, we generated a mutant resistant to 5-fluorouracil by making a markerless deletion of the upp gene that encodes uracil phosphoribosyltransferase. We also used this method for targeted marker exchange mutagenesis by replacing kupM, a gene identified in our previous screen as a magnetosome formation factor, with a streptomycin resistance cassette. Overall, our results show that targeted mutagenesis using a replicative plasmid is effective in D. magneticus and may also be applied to other genetically recalcitrant bacteria.
IMPORTANCE Magnetotactic bacteria (MTB) are a group of organisms that form intracellular nanometer-scale magnetic crystals though a complex process involving lipid and protein scaffolds. These magnetic crystals and their lipid membranes, termed magnetosomes, are model systems for studying bacterial cell biology and biomineralization and are potential platforms for biotechnological applications. Due to a lack of genetic tools and unculturable representatives, the mechanisms of magnetosome formation in phylogenetically deeply branching MTB remain unknown. These MTB contain elongated bullet-/tooth-shaped magnetite and greigite crystals that likely form in a manner distinct from that of the cubooctahedral-shaped magnetite crystals of the genetically tractable MTB within the Alphaproteobacteria. Here, we present a method for genome editing in Desulfovibrio magneticus RS-1, a cultured representative of the deeply branching MTB of the class Deltaproteobacteria. This marks a crucial step in developing D. magneticus as a model for studying diverse mechanisms of magnetic particle formation by MTB.
INTRODUCTION
Magnetotactic bacteria (MTB) are a group of diverse microorganisms that align along magnetic fields via their intracellular chains of magnetic crystals (1, 2). Each magnetic crystal consists of either magnetite (Fe3O4) or greigite (Fe3S4) and is synthesized within a complex organelle called a magnetosome (3). The first cultured MTB were microaerophilic members of the Alphaproteobacteria, which form cubooctahedral-shaped magnetite crystals and have served as model organisms for understanding magnetosome formation (4–7). Early studies on Magnetospirillum spp. revealed a lipid-bilayer membrane, with a unique suite of proteins, surrounding each magnetite crystal (8–10). The development of genetic tools in Magnetospirillum magneticum AMB-1 and Magnetospirillum gryphiswaldense MSR-1 revealed a conserved magnetosome gene island (MAI) that contains the factors necessary and sufficient for the formation of the magnetosome membrane, magnetite biomineralization within the lumen of the magnetosome, and alignment of the magnetosomes in a chain along the length of the cell (3, 11). These molecular advances, along with the magnetic properties of magnetosomes, have made MTB ideal models for the study of compartmentalization and biomineralization in bacteria as well as a target for the development of biomedical and industrial applications.
Improvements in isolation techniques and sequencing have revealed that MTB are ubiquitous in many aquatic environments. On the basis of phylogeny and magnetosome morphology, MTB can be categorized into two subgroups. The first subgroup includes members of the Alphaproteobacteria and Gammaproteobacteria, such as Magnetospirillum spp., that synthesize cubooctahedral, elongated octahedral, or elongated prisms of magnetite (12). The second subgroup comprises MTB from more deep-branching lineages, including members of the Deltaproteobacteria class and the Nitrospirae and Omnitrophica phyla, which synthesize elongated bullet-/tooth-shaped magnetite and/or greigite crystals (13, 14). While all MTB sequenced to date have their putative magnetosome genes arranged in distinct regions of their genomes (3, 15–17), many of the genes essential for magnetosome biogenesis in Magnetospirillum spp. are missing from the genomes of deep-branching MTB (14). Likewise, a conserved set of mad (magnetosome-associated Deltaproteobacteria) genes are only found in deep-branching MTB (14, 18–20). This suggests a genetic diversity underpinning the control of magnetosome morphology and physiology in nonmodel MTB that is distinct from that of the well-characterized Magnetospirillum spp.
Desulfovibrio magneticus RS-1, one of the few cultured MTB outside the Alphaproteobacteria, is an anaerobic sulfate-reducing member of the Deltaproteobacteria that forms irregular bullet-shaped crystals of magnetite (21, 22). As with the Magnetospirillum spp., the magnetosome genes of D. magneticus are located within a MAI and include homologs to some mam genes as well as mad genes (14, 18, 23). Recently, we used a forward genetic screen combining random chemical and UV mutagenesis with whole-genome resequencing to identify mutations that resulted in nonmagnetic phenotypes. These included many mutants that had the entire MAI deleted (ΔMAI) as well as mutants with point mutations, frameshift mutations, and transposon insertions in 10 mam and mad genes of the D. magneticus MAI that resulted in nonmagnetic phenotypes (20). However, this screen relied on a strict selection scheme for nonmagnetic mutants. As such, we likely missed magnetosome genes that are important for regulating the shape, size, and arrangement of magnetosomes. To elucidate the degree of conservation between mam genes and determine the function of the proteins encoded by mad genes in D. magneticus, a reverse genetic method for targeted mutagenesis is necessary.
D. magneticus and other Desulfovibrio spp. have gained much attention for their importance in the global cycling of numerous elements, in biocorrosion, and in the bioremediation of toxic metal ions (24, 25). The development of genetic tools, such as expression vectors, transposons, and targeted genome-editing systems, has enabled a more detailed examination of the important activities of a few Desulfovibrio spp. (26, 27). Targeted mutagenesis using a one-step double recombination method was first achieved in Desulfovibrio fructosivorans and, more recently, in Desulfovibrio gigas and Desulfovibrio desulfuricans ND132 (28–30). With this method, plasmids that are electroporated into the cell are thought to be rapidly linearized by endogenous restriction modification systems (30–32). The linearized plasmid DNA, carrying a selectable marker flanked by upstream and downstream regions of homology to a target gene, can then undergo double recombination into the chromosome in one step (Fig. 1A). This efficient one-step method, which is dependent on electroporation of the plasmid (28–30), is unlikely to be applicable for D. magneticus, because plasmid uptake has only been demonstrated using conjugal transfer (20). The second targeted mutagenesis method, used in Desulfovibrio vulgaris Hildenborough, is a two-step double recombination that makes use of a nonreplicative, or suicide, vector (31, 32). In the first step of this method, a suicide vector, with sequences upstream and downstream of the target gene, integrates into the genome upon the first homologous recombination event (Fig. 1B). Next, a second recombination event occurs whereby the vector is excised from the genome, and cells with the desired genotype are selected with an antibiotic marker and/or a counterselection marker (31, 32) (Fig. 1B). For many bacteria, including D. magneticus, plasmid uptake and integration occur at frequencies that are too low for genetic manipulation via suicide vectors (20).
FIG 1.

Schematic of deletion methods used in Desulfovibrio spp. Plasmids (black lines) were designed to replace a target gene (X, aqua arrows) in the chromosome (blue lines) with a streptomycin resistance cassette (strAB, purple arrows). Regions upstream (*) and downstream (**) of the target gene (blue boxes) on the chromosome undergo recombination (red lines) with homologous regions that are cloned into the deletion plasmid. Key steps, such as recombination events (red crosses), are indicated in the boxes, and the selection steps are labeled in red. (A) Double recombination can occur in one step after plasmids are linearized (dashed lines) by endogenous restriction enzymes. Mutants are selected using the marker (e.g., strAB) that was exchanged with the target gene. (B) Two-step double recombination is possible when suicide vectors integrate into the chromosome in the first homologous recombination event and then recombine out after the second homologous recombination event. The first step and second step are selected for with antibiotic resistance markers (e.g., npt) and counterselectable markers (e.g., sacB), respectively. (C) A replicative deletion plasmid designed to target genes for deletion may undergo double recombination in one or two steps as shown in panels A and B, respectively. After passaging the cells without antibiotic, the mutants are selected with an antibiotic resistance cassette (e.g., strAB) and a counterselectable marker (e.g., sacB). mob, mobilization genes (mobA′, mobB, mobC); npt, kanamycin-resistance gene; oriDm, origin of replication for D. magneticus; oriEc, origin of replication for E. coli.
Here, we describe the method we developed for targeted gene deletion using a replicative plasmid, thereby bypassing the need for suicide vector integration (Fig. 1C). We generated a mutant resistant to 5-fluorouracil by making a markerless deletion of the upp gene, which encodes an enzyme in the pyrimidine salvage pathway that is nonessential under standard laboratory conditions. Additionally, we deleted kupM, a gene encoding a potassium transporter that acts as a magnetosome formation factor (20), via marker exchange with a streptomycin resistance cassette. The deletion of both upp and kupM conferred the expected phenotypes, which were subsequently complemented in trans. Overall, our results show that targeted mutagenesis using a replicative plasmid is possible in D. magneticus. It may also be suitable for other bacteria for which replicative plasmid uptake is possible but at a rate too low for suicide vector integration.
RESULTS
Design of a replicative deletion plasmid using sacB counterselection.
Targeted genetic manipulation in most bacteria requires a method to efficiently deliver foreign DNA destined for integration into the chromosome. One commonly used method involves suicide vector uptake and integration prior to the first selection step (Fig. 1B). In D. magneticus, plasmid transfer has only been achieved via conjugation at low efficiencies, making the uptake and subsequent integration of suicide vectors into its chromosome an unlikely event (20). As such, we attempted to bypass the use of suicide vectors and use a stable replicative plasmid designed to delete specific genes via homologous recombination (Fig. 1C). Two features of this method enable the isolation of desired mutants: (i) a selectable marker is used to identify double recombination events at the targeted site and (ii) a counterselectable marker distinguishes the desired mutant cells, which have lost all remaining copies of the plasmid.
sacB is a common counterselection marker that is effective in many bacteria. The sacB gene from Bacillus subtilis encodes levansucrase, which converts sucrose to levans that are lethal to many Gram-negative bacteria, including D. vulgaris Hildenborough (31, 33, 34). To test its functionality in D. magneticus, we inserted sacB under the expression of the mamA promoter of D. magneticus (described in reference 20) in a plasmid that replicates in both Escherichia coli and D. magneticus (Fig. 2A). This plasmid (pAK914) and a control plasmid were then conjugated into D. magneticus. We found no growth inhibition for D. magneticus cells with the control plasmid in the presence of sucrose and kanamycin. In contrast, cells expressing sacB were unable to grow with kanamycin and sucrose concentrations of 1% (wt/vol) or higher (data not shown). To test if the plasmids could be cured, D. magneticus with pAK914 was passaged two times in liquid medium containing no antibiotic and plated on 1% sucrose. Individual sucrose-resistant (Sucr) colonies were inoculated and screened for kanamycin sensitivity (Kans). All isolated colonies (n = 16) were Kans, suggesting that the cells had lost the plasmid. These experiments demonstrate that sacB is a suitable counterselection marker in D. magneticus.
FIG 2.

Plasmids constructed for the present study. (A) Expression plasmid pAK914 expresses sacB from the mamA promoter and is the parent vector for the deletion plasmids and upp expression plasmid described below. (B) Replicative deletion plasmid to target upp for markerless deletion. The upp deletion cassette was cloned into XbaI-SacI of pAK914. (C) Expression plasmid used for upp complementation. The upp gene and its promoter were cloned into BamHI-SacI of pAK914. (D) Replicative deletion plasmid to target kupM for marker exchange mutagenesis with strAB. The kupM::strAB deletion cassette was cloned into XbaI of pAK914. Labeling and colors correspond to those in Fig. 1.
Construction of a Δupp strain by markerless deletion.
To test our replicative deletion method, we chose to target the upp gene, the mutation of which has a selectable phenotype. The upp gene encodes uracil phosphoribosyltransferase (UPRTase), a key enzyme in the pyrimidine salvage pathway that catalyzes the reaction of uracil with 5-phosphoribosyl-α-1-pyrophosphate (PRPP) to UMP and PPi (35) (Fig. 3A). When given the pyrimidine analog 5-fluorouracil (5-FU), UPRTase catalyzes the production of 5-fluoroxyuridine monophosphate (5-FUMP). 5-FUMP is further metabolized and incorporated into DNA, RNA, and sugar nucleotides resulting in eventual cell death (Fig. 3A) (36, 37). Previous studies have shown that Δupp mutants of D. vulgaris Hildenborough are resistant to 5-FU, while wild-type (WT) cells are effectively killed by the pyrimidine analog (32, 38). The D. magneticus genome has a homolog (DMR_08390) to the D. vulgaris Hildenborough upp gene that is likely functional, as detected by the sensitivity of D. magneticus to 5-FU (Fig. 3B and 4A). To show that the upp gene product confers 5-FU sensitivity and to validate our replicative deletion system, we chose to target the D. magneticus upp gene for markerless deletion.
FIG 3.

(A) The upp gene encodes UPRTase, which is a key enzyme in the uracil salvage pathway. The product of the UPRTase reaction, UMP, is processed by downstream enzymes in pathways for RNA, DNA, and sugar nucleotide synthesis. 5-FU causes cell death by incorporating into this pathway via UPRTase. (B) Schematic of genomic regions of upp in the WT or the ΔMAI mutant (top) and the Δupp mutant (bottom). (C) Genomic region of kup in WT (top) and kup::strAB (bottom) strains. Primers used to screen for the correct genotype are indicated with half arrows. (D) Δupp mutants in WT and ΔMAI backgrounds were confirmed by PCR using primers P19/P20 and agarose gel electrophoresis. WT and ΔMAI strains show a band corresponding to the upp gene (2,691 bp), while the Δupp mutants have a smaller band corresponding to a markerless deletion of the upp gene (2,079 bp). The lower bands are likely nonspecific PCR products. (E) kupM::strAB genotype confirmation by PCR and agarose gel electrophoresis using primers P21/P22 (WT, 3,069 bp; kupM::strAB, 3,263 bp; ΔMAI, not applicable [NA]).
FIG 4.
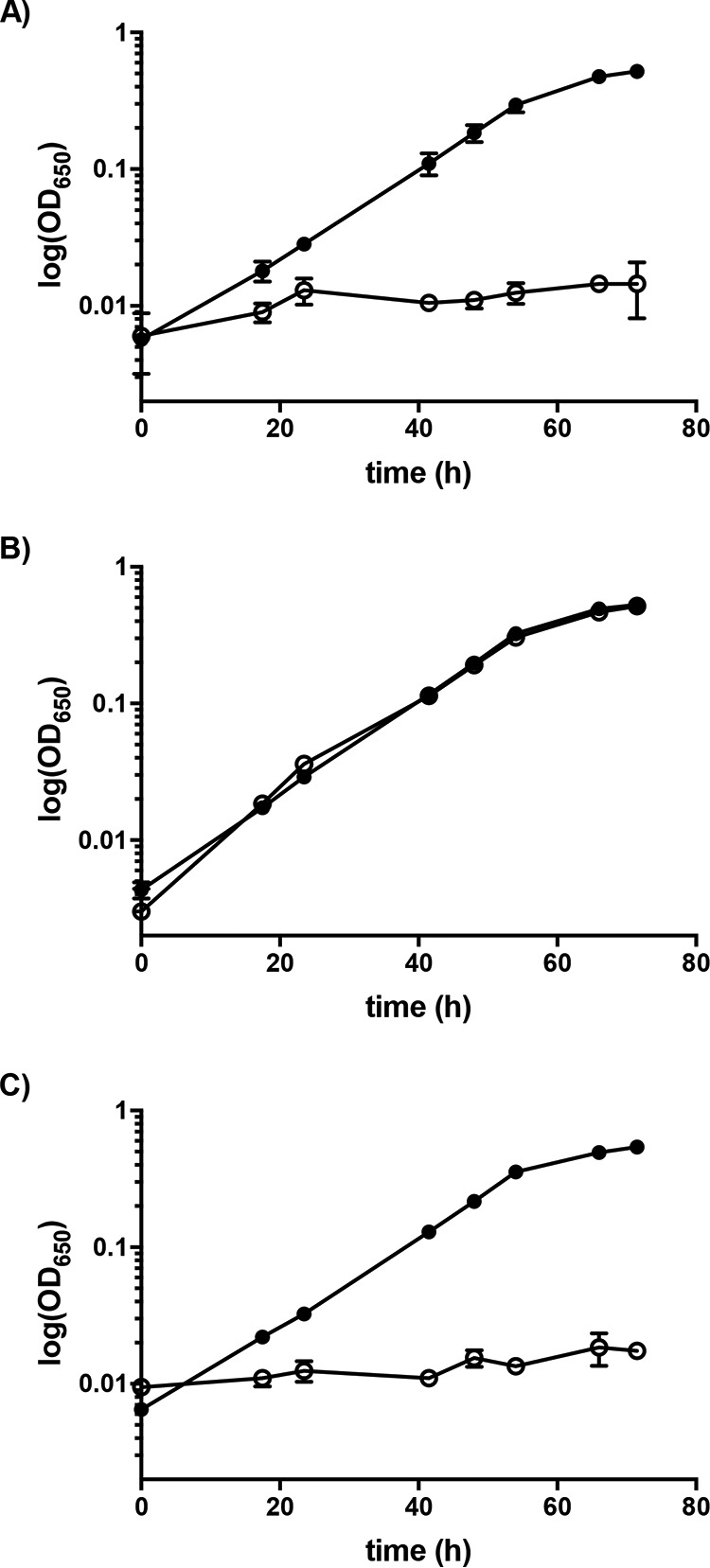
upp mutant and complementation phenotype. Growth of the parent strain (ΔMAI) (A), upp deletion (ΔMAI Δupp) (B), and complementation of the upp deletion (ΔMAI Δupp/upp+) (C) when grown with 1.25 μg/ml 5-FU (○) or without 5-FU (●). Data presented are averages from 2 to 3 independent cultures; error bars indicate the standard deviations.
To construct a upp deletion vector, a markerless cassette containing the regions upstream and downstream of the upp gene were inserted into plasmid pAK914 (Fig. 2B). The resulting plasmid (pAK1126) was transferred to WT D. magneticus by conjugation and single kanamycin-resistant (Kanr) colonies were isolated and passaged in growth medium containing no antibiotic. Since D. magneticus has interesting features independent of its magnetosomes, the same deletion procedure was also carried out in a nonmagnetic strain (ΔMAI) isolated in our previous genetic studies (20). After the third passage, upp mutants that had lost the vector backbone were selected for with 5-FU and sucrose. Compared with those obtained using a control plasmid (pAK914), >20-fold more 5-FU-resistant (5-FUr) mutants were generated using pAK1126 at a frequency of approximately 10−6. PCR of the region flanking the upp gene confirmed that the 5-FUr colonies harboring pAK1126 resulted from a markerless deletion of upp (Δupp), while 5-FUr colonies harboring pAK914 were likely the result of point mutations (Fig. 3B and D). Similar to the results obtained for D. vulgaris Hildenborough (32), the Δupp mutant of D. magneticus grew in the presence of 5-FU (Fig. 4B and Table 1). Complementation of the upp gene in trans restored UPRTase function, and the cells no longer grew with 5-FU (Fig. 2C and 4C and Table 1). These experiments demonstrate that a replicative plasmid can be used to directly edit the D. magneticus genome.
TABLE 1.
Growth rates and generation times of the parent strain (ΔMAI), Δupp mutant, and upp complementation in trans with and without treatment with 5-FU
| Strain | Growth rate (h−1) |
Generation time (h) |
||
|---|---|---|---|---|
| Without 5-FU | With 5-FU | Without 5-FU | With 5-FU | |
| ΔMAI | 0.077 ± 0.0017 | NAa | 9.1 ± 0.2 | NA |
| ΔMAI Δupp | 0.079 ± 0.0017 | 0.070 ± 0.0040 | 8.8 ± 0.2 | 10.0 ± 0.6 |
| ΔMAI Δupp/upp+ | 0.076 ± 0.0041 | NA | 9.1 ± 0.5 | NA |
NA, not applicable.
Construction of a Δkup strain by marker exchange mutagenesis.
Because many genetic mutations do not confer a selectable phenotype, we sought to develop our replicative deletion plasmid for marker exchange mutagenesis. To test this system, we chose to replace a gene with a known phenotype, kupM (DMR_40800), with a streptomycin-resistance gene cassette (strAB). kupM is located in the D. magneticus MAI and encodes a functional potassium transporter (20). Mutant alleles in kupM, including missense, nonsense, and frameshift mutations, were previously identified in our screen for nonmagnetic mutants (20). These kupM mutations resulted in cells that rarely contained electron-dense particles and were unable to turn in a magnetic field, as measured by the coefficient of magnetism (Cmag) (20).
To mutate kupM, we inserted a marker exchange cassette, with regions upstream and downstream of kupM flanking strAB, into pAK914 (Fig. 2D) to create the deletion plasmid pAK941. Following conjugation, single colonies of D. magneticus harboring pAK941 were isolated by kanamycin selection. After three passages in growth medium without selection, potential mutants were isolated at a frequency of approximately 10−6 on plates containing streptomycin and sucrose. Single colonies that were streptomycin resistant (Strr) and Sucr were inoculated in liquid medium and screened for Kans. Of the isolates screened (n = 48), 20% were Kans and 4% had the correct genotype (ΔkupM::strAB) as confirmed by PCR and sequencing (Fig. 3C and E).
Similar to the phenotypes previously observed in kupM mutants (20), ΔkupM::strAB cells were severely defective in magnetosome synthesis and turning in a magnetic field (Fig. 5). Although a slight Cmag was measured, few cells contained electron-dense particles or magnetosomes. Importantly, the WT phenotype was rescued by expressing kupM from a plasmid in the ΔkupM::strAB mutant (Fig. 5). These results confirm that the replicative deletion plasmid method described here can be used successfully for marker exchange mutagenesis.
FIG 5.

kupM mutant and complementation phenotype. Cmag values (A) and electron micrographs of WT (B), kupM::strAB (C), and ΔkupM::strAB/kup+ (D) strains. Scale bars, 200 nm. Data presented are averages from 4 independent cultures; error bars indicate the standard deviations.
DISCUSSION
In this study, we expand the genetic toolbox for D. magneticus to include a replicative plasmid method for targeted mutagenesis (Fig. 1C). We show the utility of this method for markerless deletion of genes with a selectable phenotype and for marker exchange mutagenesis. Some of the earliest examples of targeted mutagenesis in Gram-negative bacteria used replicative plasmids, similar to the method described here (34, 39). These studies, which predated the application of suicide vectors, relied on plasmid instability by introducing a second plasmid of the same incompatibility group or by limiting nutrients in the growth medium (34, 39).
Because the D. magneticus genetic toolbox has a limited number of plasmids, antibiotic markers, and narrow growth constraints, we used a replicative plasmid and established sacB as a counterselection marker to generate and isolate mutants. While sacB counterselection was ultimately successful, a large number of false positives were also isolated at the sucrose selection step. Mutations in sacB have been found to occur at a high frequency in many bacteria (31, 40–43). Indeed, we found that deletions and mutations in PmamA-sacB are abundant in the false-positive Sucr Strr isolates (data not shown). Alternative counterselection markers, including upp, have been shown to select for fewer false positives (32, 43–45). Since D. magneticus is sensitive to 5-FU only when the upp gene is present (Fig. 4), the upp mutants generated in this study may be used as the parent strains for future targeted mutagenesis with upp, rather than sacB, serving as a counterselectable marker. Additionally, the combined use of upp and sacB for counterselection might reduce the false-positive background that results from the accumulation of mutations in these markers.
The replicative deletion plasmid described here was designed to replace a target gene with an antibiotic resistance marker. As such, the construction of strains with multiple directed mutations will be complicated by the need for additional antibiotic-resistance markers, which are limited in D. magneticus. These limitations may be overcome by removing the chromosomal antibiotic marker in subsequent steps (34, 46, 47). Ultimately, improvements in conjugation efficiency or methods for electroporation with high transformation efficiency are desired. Similar to the ongoing development of genetics in D. vulgaris Hildenborough, the establishment of a suicide vector delivery system in D. magneticus will enable more high-throughput targeted mutagenesis and even the construction of markerless deletion mutants (26, 32).
Overall, we demonstrated the utility of a replicative deletion plasmid to generate targeted mutants of D. magneticus. This method marks a crucial step in developing D. magneticus as a model for the study of anaerobic sulfate reduction and diverse mechanisms of magnetic particle formation by MTB. Both MTB and sulfate-reducing bacteria have been singled out for their role in the global cycling of numerous elements and for potential applications, such as bioremediation (24, 25, 48, 49). D. magneticus, in particular, may be useful in the bioremediation of heavy metals and in the global cycling of iron, since it can form both magnetosomes and other iron-containing organelles (50, 51). Through genetic manipulation of D. magneticus, pathways of elemental cycling and heavy-metal turnover may now be explored. Additionally, genetic manipulation of D. magneticus will further our understanding of magnetosome formation and provide answers to many longstanding questions for the deeply branching MTB. Which proteins regulate and control magnetosome formation? To what extent are lipid membranes involved in forming these crystals? How is the elongated and irregular crystal shape achieved? Finally, in addition to D. magneticus, the method described here may extend to other bacteria that are not amenable to targeted mutagenesis with suicide vectors but are able to accommodate replicative plasmids.
MATERIALS AND METHODS
Strains, media, and growth conditions.
The bacterial strains used in this study are listed in Table 2. All E. coli strains were cultured aerobically with continuous shaking at 250 rpm at 37°C in lysogeny broth (LB). D. magneticus strains were grown anaerobically at 30°C in sealed Balch tubes with a N2 headspace containing RS-1 growth medium (RGM) that was degassed with N2, unless otherwise stated (51). Sodium pyruvate (10 mM) was used as an electron donor with fumaric acid disodium (10 mM) as the terminal electron acceptor. RGM was buffered with HEPES, and the pH was adjusted to 6.7 with NaOH (20). Before inoculating with cells, RGM was supplemented with 0.8% (vol/vol) Wolfe's vitamins, 100 μM ferric malate, and 285 μM cysteine-HCl (51). Solid agar plates were prepared by adding 1.5% agar (wt/vol) to LB and 1% agar (wt/vol) to RGM. Vitamins (0.8% [vol/vol]), ferric malate (20 μM), and cysteine (285 μM), as well as antibiotics and selective agents, were added to the molten RGM agar as needed. For D. magneticus, all plating steps were carried out aerobically, and the bacteria were transferred to an anaerobic jar and incubated at 30°C for 10 to 14 days, as described previously (20). The antibiotics and selective agents used are as follows: kanamycin (50 μg/ml for E. coli strains, 125 μg/ml for D. magneticus strains), streptomycin (50 μg/ml for E. coli and D. magneticus strains), diaminopimelic acid (300 μM for E. coli WM3064), 5-FU (2.5 μg/ml for D. magneticus strains), and sucrose (1% for D. magneticus strains).
TABLE 2.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristics | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α λpir | Cloning strain | Lab strain |
| WM3064 | Conjugation strain; DAP auxotroph used for plasmid transfer | Lab strain |
| D. magneticus | ||
| AK80 | Nonmotile mutant of D. magneticus strain RS-1, referred to as wild type | 51 |
| AK201 | ΔMAI | 20 |
| AK267 | ΔMAI Δupp | This study |
| AK268 | Δupp | This study |
| AK270 | ΔkupM::strAB | This study |
| Plasmids | ||
| pBMK7 | Conjugative vector with pBG1 and pMB1 replicons; Kanr | 53 |
| pBMC7 | Conjugative vector with pBG1 and pMB1 replicons; Cmr | 53 |
| pBMS6 | Cloning vector; source of strAB; Strr | 53 |
| pLR6 | pBMK7 with PmamA in HindIII-SalI; source of Pnpt; Kanr | 20 |
| pLR41 | pLR6 with PmamA-kupM in SalI; Kanr | 20 |
| pAK0 | Cloning vector, source of sacB; Kanr | 10 |
| pAK914 | pLR6 with sacB in SalI-XbaI; Kanr | This study |
| pAK920 | pBMC7 with Pnpt-strAB inserted into SacI site; Cmr Strr | This study |
| pAK941 | pAK914 with cassette of 1,064 bp upstream and 1,057 bp downstream of kupM flanking Pnpt-strAB in XbaI; Kanr Strr | This study |
| pAK1126 | pAK914 with cassette of 991 bp upstream and 1,012 bp downstream of upp in XbaI-SacI; Kanr | This study |
| pAK1127 | pAK914 with Pupp-upp in BamHI-SacI; Kanr | This study |
Plasmids and cloning.
All plasmids used in this work are listed in Table 2. All cloning was performed in E. coli DH5α λpir using the Gibson method (52) or restriction enzyme ligation. For PCR amplification, KOD (EMD Millipore, Germany) and GoTaq (Promega, USA) DNA polymerases were used with the primers listed in Table 3. All upstream and downstream homology regions were amplified from D. magneticus genomic DNA. strAB and Pnpt were amplified from pBMS6 and pLR6, respectively, and subcloned into pBMC7 to make pAK920, which served as the template for amplifying Pnpt-strAB for the deletion vectors. sacB was amplified from pAK0 and inserted into pLR6 digested with SalI and XbaI to create pAK914. To construct a plasmid for the targeted deletion of upp (DMR_08390), 991 bp upstream and 1,012 bp downstream of upp were amplified and inserted into pAK914 digested with XbaI and SacI using a 3-piece Gibson assembly. To create the upp complementation plasmid, pAK914 was digested with BamHI and SacI, and the upp gene, with its promoter, was PCR amplified from D. magneticus genomic DNA. To construct pAK941 for marker exchange mutagenesis of kupM, a cassette of a 1,064-bp upstream region and 1,057-bp downstream region flanking Pnpt-strAB was assembled using Gibson cloning. The cassette was amplified and inserted into pAK914 digested with XbaI using a two-piece Gibson assembly.
TABLE 3.
Primers used in this study
| Name | Sequence from 5′ end | Descriptiona |
|---|---|---|
| P1 | AAGCCAAGAAAAACGTCGCCAACGTCGACATGAACATCAAAAAGTTTGCA | F sacB for pAK914 |
| P2 | GCTCGGTACCCGGGGATCCTCTAGAGGCCAATAGGATATCGGCATTT | R sacB for pAK914 |
| P3 | CGACTCTAGAGGATCCCCGGGTACCGTAGCTTCACGCTGCCGCAAG | F Pnpt for pAK920 |
| P4 | CCCGAATGTGCATGCGAAACGATCCTCATCCTGTC | R Pnpt for pAK920 |
| P5 | AGGATCGTTTCGCATGCACATTCGGGATATTTCTCTA | F strAB for pAK920 |
| P6 | TAATACGACTCACTATAGGGAATTCGCCCAGGGGATAGGAGAAGTC | R strAB for pAK920 |
| P7 | AAATGCCGATATCCTATTGGCCTCTAGAGAGATCGCGAAGCAGAGC | F kupM upstream for pAK941 |
| P8 | TGCGGCAGCGTGAAGCTACGGTACCGCCGTAATGCGTCAGAAAGT | R kupM upstream for pAK941 |
| P9 | CTTCTCCTATCCCCTGGGCGAATTCAGCCGGGTCATGGAAGTC | F kupM downstream for pAK941 |
| P10 | CGAGCTCGGTACCCGGGGATCCTCTAGAGGCCAGGGAATGGAGTTT | R kupM downstream for pAK941 |
| P11 | GGTACCGTAGCTTCACGCTGCCGCA | F Pnpt-strAB for pAK941 |
| P12 | GAATTCGCCCAGGGGATAGGAGAAGTCGCT | R Pnpt-strAB for pAK941 |
| P13 | GCCGATATCCTATTGGCCTCTAGAGCCTCCCAGATCGACCAGTC | F upp upstream for pAK1126 |
| P14 | CTATTTGGTGCCGGATCCCATGGACGCGCTCCTGGG | R upp upstream for pAK1126 |
| P15 | AGCGCGTCCATGGGATCCGGCACCAAATAGGGGG | F upp downstream for pAK1126 |
| P16 | CGACTCACTATAGGGAATTCGAGCTCGCCAGGCAGACGGCGGTG | R upp downstream for pAK1126 |
| P17 | GCCGATATCCTATTGGCCTCTAGAGAAGCTCGCCGAAAAGACC | F Pupp-upp for pAK1127 |
| P18 | CGACTCACTATAGGGAATTCGATGAAGGCGAACGAGGAAC | R Pupp-upp for pAK1127 |
| P19 | GCCCGCATTGAGGACGTG | To check upp deletion |
| P20 | CAGCGCCCCGAGCTTGCC | To check upp deletion |
| P21 | CGTCAGCAGGCAAACGG | To check kupM deletion |
| P22 | ACCGTTGTCTCCCATGTCTC | To check kupM deletion |
F, forward; R, reverse.
upp and kup mutant generation and complementation.
Replicative deletion plasmids were transformed into E. coli WM3064 by heat shock and transferred to D. magneticus by conjugation, as described previously (20). Single colonies of Kanr D. magneticus were isolated and inoculated in RGM containing no antibiotic. Cultures were passaged and, after the third passage, approximately 2 × 108 cells were spread on 1% agar RGM plates containing either 50 μg/ml streptomycin and 1% sucrose or 2.5 μg/ml 5-FU and 1% sucrose. 5-FUr Sucr and Strr Sucr colonies harboring plasmids pAK1126 and pAK941, respectively, were recovered at a frequency of approximately 10−6. Single colonies were screened for Kans and by PCR using the primers listed in Table 3. Successful upp and kup mutants were confirmed by Sanger sequencing. The expression plasmids for the complementation of Δkup::strAB and Δupp, as well as empty vectors for controls, were transferred to D. magneticus strains as described above. Transconjugants were inoculated in RGM containing kanamycin to maintain the plasmids.
Mutant phenotype and complementation analyses.
The growth and coefficient of magnetism (Cmag) of D. magneticus strains were measured in a Spec20 spectrophotometer at an optical density of 650 nm (OD650), as described previously (10, 51). For upp mutant and complementation analyses, RGM was supplemented with 5-FU (1.25 μg/ml in 0.01% dimethyl sulfoxide [DMSO]) or DMSO (0.01%), and the growth was measured for WT and Δupp strains with an empty vector (pAK914) and for the Δupp strain with the complementation plasmid pAK1127. For kup mutant and complementation analyses, the Cmag was measured by placing a large bar magnet parallel or perpendicular to the sample to measure the maximum or minimum absorbance, respectively, as the D. magneticus strains rotate 90° with the magnetic field. The ratio of maximum to minimum absorbances was calculated as the Cmag (10). Whole-cell transmission electron microscopy (TEM) was performed as previously described (51). The Cmag calculations and TEM were performed for WT D. magneticus with an empty vector (pBMK7) and the Δkup::strAB strain with an empty vector (pBMK7) or complementation plasmid (pLR41). For all growth measurements, Cmag measurements, and TEM, the cells harboring the plasmids were maintained with 125 μg/ml kanamycin.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (R01GM84122 and R35GM127114), the National Science Foundation (1504681), and the Office of Naval Research (N000141310421).
REFERENCES
- 1.Bellini S. 2009. On a unique behavior of freshwater bacteria. Chin J Oceanol Limnol 27:3. doi: 10.1007/s00343-009-0003-5. [DOI] [Google Scholar]
- 2.Blakemore R. 1975. Magnetotactic bacteria. Science 190:377–379. doi: 10.1126/science.170679. [DOI] [PubMed] [Google Scholar]
- 3.Uebe R, Schüler D. 2016. Magnetosome biogenesis in magnetotactic bacteria. Nat Rev Microbiol 14:621–637. doi: 10.1038/nrmicro.2016.99. [DOI] [PubMed] [Google Scholar]
- 4.Bazylinski DA, Frankel RB, Jannasch HW. 1988. Anaerobic magnetite production by a marine, magnetotactic bacterium. Nature 334:518–519. doi: 10.1038/334518a0. [DOI] [Google Scholar]
- 5.Blakemore RP, Maratea D, Wolfe RS. 1979. Isolation and pure culture of a freshwater magnetic spirillum in chemically defined medium. J Bacteriol 140:720–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsunaga T, Sakaguchi T, Tadakoro F. 1991. Magnetite formation by a magnetic bacterium capable of growing aerobically. Appl Microbiol Biotechnol 35:651–655. doi: 10.1007/BF00169632. [DOI] [Google Scholar]
- 7.Schüler D, Köhler M. 1992. The isolation of a new magnetic spirillum. Zentralbl Mikrobiol 147:150–151. doi: 10.1016/S0232-4393(11)80377-X. [DOI] [Google Scholar]
- 8.Balkwill DL, Maratea D, Blakemore RP. 1980. Ultrastructure of a magnetotactic spirillum. J Bacteriol 141:1399–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorby YA, Beveridge TJ, Blakemore RP. 1988. Characterization of the bacterial magnetosome membrane. J Bacteriol 170:834–841. doi: 10.1128/jb.170.2.834-841.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komeili A, Vali H, Beveridge TJ, Newman DK. 2004. Magnetosome vesicles are present before magnetite formation, and MamA is required for their activation. Proc Natl Acad Sci U S A 101:3839–3844. doi: 10.1073/pnas.0400391101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komeili A. 2012. Molecular mechanisms of compartmentalization and biomineralization in magnetotactic bacteria. FEMS Microbiol Rev 36:232–255. doi: 10.1111/j.1574-6976.2011.00315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pósfai M, Lefèvre C, Trubitsyn D, Bazylinski DA, Frankel R. 2013. Phylogenetic significance of composition and crystal morphology of magnetosome minerals. Front Microbiol 4:344. doi: 10.3389/fmicb.2013.00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lefèvre CT, Bazylinski DA. 2013. Ecology, diversity, and evolution of magnetotactic bacteria. Microbiol Mol Biol Rev 77:497–526. doi: 10.1128/MMBR.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin W, Zhang W, Zhao X, Roberts AP, Paterson GA, Bazylinski DA, Pan Y. 2018. Genomic expansion of magnetotactic bacteria reveals an early common origin of magnetotaxis with lineage-specific evolution. ISME J 12:1508–1519. doi: 10.1038/s41396-018-0098-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolinko I, Lohße A, Borg S, Raschdorf O, Jogler C, Tu Q, Pósfai M, Tompa É, Plitzko JM, Brachmann A, Wanner G, Müller R, Zhang Y, Schüler D. 2014. Biosynthesis of magnetic nanostructures in a foreign organism by transfer of bacterial magnetosome gene clusters. Nat Nanotechnol 9:193–197. doi: 10.1038/nnano.2014.13. [DOI] [PubMed] [Google Scholar]
- 16.Murat D, Quinlan A, Vali H, Komeili A. 2010. Comprehensive genetic dissection of the magnetosome gene island reveals the step-wise assembly of a prokaryotic organelle. Proc Natl Acad Sci U S A 107:5593–5598. doi: 10.1073/pnas.0914439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murat D, Falahati V, Bertinetti L, Csencsits R, Körnig A, Downing K, Faivre D, Komeili A. 2012. The magnetosome membrane protein, MmsF, is a major regulator of magnetite biomineralization in Magnetospirillum magneticum AMB-1. Mol Microbiol 85:684–699. doi: 10.1111/j.1365-2958.2012.08132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lefèvre CT, Trubitsyn D, Abreu F, Kolinko S, Jogler C, de Almeida LGP, de Vasconcelos ATR, Kube M, Reinhardt R, Lins U, Pignol D, Schüler D, Bazylinski DA, Ginet N. 2013. Comparative genomic analysis of magnetotactic bacteria from the Deltaproteobacteria provides new insights into magnetite and greigite magnetosome genes required for magnetotaxis. Environ Microbiol 15:2712–2735. doi: 10.1111/1462-2920.12128. [DOI] [PubMed] [Google Scholar]
- 19.Lin W, Deng A, Wang Z, Li Y, Wen T, Wu L-F, Wu M, Pan Y. 2014. Genomic insights into the uncultured genus “Candidatus Magnetobacterium” in the phylum Nitrospirae. ISME J 8:2463–2477. doi: 10.1038/ismej.2014.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahn-Lee L, Byrne ME, Zhang M, Sage DL, Glenn DR, Milbourne T, Walsworth RL, Vali H, Komeili A. 2015. A genetic strategy for probing the functional diversity of magnetosome formation. PLoS Genet 11:e1004811. doi: 10.1371/journal.pgen.1004811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakaguchi T, Arakaki A, Matsunaga T. 2002. Desulfovibrio magneticus sp. nov., a novel sulfate-reducing bacterium that produces intracellular single-domain-sized magnetite particles. Int J Syst Evol Microbiol 52:215–221. doi: 10.1099/00207713-52-1-215. [DOI] [PubMed] [Google Scholar]
- 22.Sakaguchi T, Burgess JG, Matsunaga T. 1993. Magnetite formation by a sulphate-reducing bacterium. Nature 365:47–49. doi: 10.1038/365047a0. [DOI] [Google Scholar]
- 23.Nakazawa H, Arakaki A, Narita-Yamada S, Yashiro I, Jinno K, Aoki N, Tsuruyama A, Okamura Y, Tanikawa S, Fujita N, Takeyama H, Matsunaga T. 2009. Whole genome sequence of Desulfovibrio magneticus strain RS-1 revealed common gene clusters in magnetotactic bacteria. Genome Res 19:1801–1808. doi: 10.1101/gr.088906.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barton LL, Fauque GD. 2009. Chapter 2. Biochemistry, physiology and biotechnology of sulfate-reducing bacteria, p 41–98. In Laskin A, Gadd G, Sariaslani S (ed), Advances in applied microbiology, vol 68 Academic Press, Cambridge, MA. [DOI] [PubMed] [Google Scholar]
- 25.Heidelberg JF, Seshadri R, Haveman SA, Hemme CL, Paulsen IT, Kolonay JF, Eisen JA, Ward N, Methe B, Brinkac LM, Daugherty SC, Deboy RT, Dodson RJ, Durkin AS, Madupu R, Nelson WC, Sullivan SA, Fouts D, Haft DH, Selengut J, Peterson JD, Davidsen TM, Zafar N, Zhou L, Radune D, Dimitrov G, Hance M, Tran K, Khouri H, Gill J, Utterback TR, Feldblyum TV, Wall JD, Voordouw G, Fraser CM. 2004. The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat Biotechnol 22:554–559. doi: 10.1038/nbt959. [DOI] [PubMed] [Google Scholar]
- 26.Keller KL, Wall JD. 2011. Genetics and molecular biology of the electron flow for sulfate respiration in Desulfovibrio. Front Microbiol 2:135. doi: 10.3389/fmicb.2011.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wall JD, Hemme CL, Rapp-Giles B, Ringbauer JA, Casalot L, Giblin T. 2003. Genes and genetic manipulations of Desulfovibrio, p 85–98. In Ljungdahl LG, Adams MW, Barton LL, Ferry JG, Johnson MK (ed), Biochemistry and physiology of anaerobic bacteria. Springer, New York, NY. [Google Scholar]
- 28.Broco M, Rousset M, Oliveira S, Rodrigues-Pousada C. 2005. Deletion of flavoredoxin gene in Desulfovibrio gigas reveals its participation in thiosulfate reduction. FEBS Lett 579:4803–4807. doi: 10.1016/j.febslet.2005.07.044. [DOI] [PubMed] [Google Scholar]
- 29.Parks JM, Johs A, Podar M, Bridou R, Hurt RA, Smith SD, Tomanicek SJ, Qian Y, Brown SD, Brandt CC, Palumbo AV, Smith JC, Wall JD, Elias DA, Liang L. 2013. The genetic basis for bacterial mercury methylation. Science 339:1332–1335. doi: 10.1126/science.1230667. [DOI] [PubMed] [Google Scholar]
- 30.Rousset M, Dermoun Z, Chippaux M, Bélaich JP. 1991. Marker exchange mutagenesis of the hydN genes in Desulfovibrio fructosovorans. Mol Microbiol 5:1735–1740. doi: 10.1111/j.1365-2958.1991.tb01922.x. [DOI] [PubMed] [Google Scholar]
- 31.Fu R, Voordouw G. 1997. Targeted gene-replacement mutagenesis of dcrA, encoding an oxygen sensor of the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Microbiology 143:1815–1826. doi: 10.1099/00221287-143-6-1815. [DOI] [PubMed] [Google Scholar]
- 32.Keller KL, Bender KS, Wall JD. 2009. Development of a markerless genetic exchange system for Desulfovibrio vulgaris Hildenborough and its use in generating a strain with increased transformation efficiency. Appl Environ Microbiol 75:7682–7691. doi: 10.1128/AEM.01839-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gay P, Coq DL, Steinmetz M, Ferrari E, Hoch JA. 1983. Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli. J Bacteriol 153:1424–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ried JL, Collmer A. 1987. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in Gram-negative bacteria by marker exchange-eviction mutagenesis. Gene 57:239–246. doi: 10.1016/0378-1119(87)90127-2. [DOI] [PubMed] [Google Scholar]
- 35.Neuhard J. 1983. Utilization of preformed pyrimidine bases and nucleosides, p 95–148. In Munch-Petersen A. (ed), Metabolism of nucleotides, nucleosides and nucleobases in microorganisms. Academic Press, New York, NY. [Google Scholar]
- 36.Singh V, Brecik M, Mukherjee R, Evans JC, Svetlíková Z, Blaško J, Surade S, Blackburn J, Warner DF, Mikušová K, Mizrahi V. 2015. The complex mechanism of antimycobacterial action of 5-fluorouracil. Chem Biol 22:63–75. doi: 10.1016/j.chembiol.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 37.Cohen SS, Flaks JG, Barner HD, Loeb MR, Lichtenstein J. 1958. The mode of action of 5-fluorouracil and its derivatives. Proc Natl Acad Sci U S A 44:1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bender KS, Cheyen H, Wall JD. 2006. Analysing the metabolic capabilities of Desulfovibrio species through genetic manipulation. Biotechnol Genet Eng Rev 23:157–174. doi: 10.1080/02648725.2006.10648083. [DOI] [PubMed] [Google Scholar]
- 39.Ruvkun GB, Ausubel FM. 1981. A general method for site-directed mutagenesis in prokaryotes. Nature 289:85–88. doi: 10.1038/289085a0. [DOI] [PubMed] [Google Scholar]
- 40.Bloor AE, Cranenburgh RM. 2006. An efficient method of selectable marker gene excision by Xer recombination for gene replacement in bacterial chromosomes. Appl Environ Microbiol 72:2520–2525. doi: 10.1128/AEM.72.4.2520-2525.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai YP, Wolk CP. 1990. Use of a conditionally lethal gene in Anabaena sp. strain PCC 7120 to select for double recombinants and to entrap insertion sequences. J Bacteriol 172:3138–3145. doi: 10.1128/jb.172.6.3138-3145.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaniga K, Delor I, Cornelis GR. 1991. A wide-host-range suicide vector for improving reverse genetics in Gram-negative bacteria: inactivation of the blaA gene of Yersinia enterocolitica. Gene 109:137–141. doi: 10.1016/0378-1119(91)90599-7. [DOI] [PubMed] [Google Scholar]
- 43.Ma W, Wang X, Mao Y, Wang Z, Chen T, Zhao X. 2015. Development of a markerless gene replacement system in Corynebacterium glutamicum using upp as a counter-selection marker. Biotechnol Lett 37:609–617. doi: 10.1007/s10529-014-1718-8. [DOI] [PubMed] [Google Scholar]
- 44.Graf N, Altenbuchner J. 2011. Development of a method for markerless gene deletion in Pseudomonas putida. Appl Environ Microbiol 77:5549–5552. doi: 10.1128/AEM.05055-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Zhang C, Gong T, Zuo Z, Zhao F, Fan X, Yang C, Song C. 2015. An upp-based markerless gene replacement method for genome reduction and metabolic pathway engineering in Pseudomonas mendocina NK-01 and Pseudomonas putida KT2440. J Microbiol Methods 113:27–33. doi: 10.1016/j.mimet.2015.03.022. [DOI] [PubMed] [Google Scholar]
- 46.Fabret C, Dusko Ehrlich S, Noirot P. 2002. A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol Microbiol 46:25–36. doi: 10.1046/j.1365-2958.2002.03140.x. [DOI] [PubMed] [Google Scholar]
- 47.Huang LC, Wood EA, Cox MM. 1997. Convenient and reversible site-specific targeting of exogenous DNA into a bacterial chromosome by use of the FLP recombinase: the FLIRT system. J Bacteriol 179:6076–6083. doi: 10.1128/jb.179.19.6076-6083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen AP, Berounsky VM, Chan MK, Blackford MG, Cady C, Moskowitz BM, Kraal P, Lima EA, Kopp RE, Lumpkin GR, Weiss BP, Hesse P, Vella NGF. 2014. Magnetic properties of uncultivated magnetotactic bacteria and their contribution to a stratified estuary iron cycle. Nat Commun 5:4797. doi: 10.1038/ncomms5797. [DOI] [PubMed] [Google Scholar]
- 49.Lin W, Bazylinski DA, Xiao T, Wu L-F, Pan Y. 2014. Life with compass: diversity and biogeography of magnetotactic bacteria. Environ Microbiol 16:2646–2658. doi: 10.1111/1462-2920.12313. [DOI] [PubMed] [Google Scholar]
- 50.Arakaki A, Takeyama H, Tanaka T, Matsunaga T. 2002. Cadmium recovery by a sulfate-reducing magnetotactic bacterium, Desulfovibrio magneticus RS-1, using magnetic separation. Appl Biochem Biotechnol 98–100:833–840. [DOI] [PubMed] [Google Scholar]
- 51.Byrne ME, Ball DA, Guerquin-Kern J-L, Rouiller I, Wu T-D, Downing KH, Vali H, Komeili A. 2010. Desulfovibrio magneticus RS-1 contains an iron- and phosphorus-rich organelle distinct from its bullet-shaped magnetosomes. Proc Natl Acad Sci U S A 107:12263–12268. doi: 10.1073/pnas.1001290107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 53.Rousset M, Casalot L, Rapp-Giles BJ, Dermoun Z, de Philip P, Bélaich J-P, Wall JD. 1998. New shuttle vectors for the introduction of cloned DNA in Desulfovibrio. Plasmid 39:114–122. doi: 10.1006/plas.1997.1321. [DOI] [PubMed] [Google Scholar]