

Abstract
Interferon lambda (IFN-λ) is a relatively unexplored, yet promising antiviral agent. IFN-λ has recently been tested in clinical trials of chronic hepatitis B virus infection (CHB), with the advantage that side effects may be limited compared with IFN-α, as IFN-λ receptors are found only in epithelial cells. To date, IFN-λ's downstream signaling pathway remains largely unelucidated, particularly via proteomics methods. Here, we report that IFN-λ3 inhibits HBV replication in HepG2.2.15 cells, reducing levels of both HBV transcripts and intracellular HBV DNA. Quantitative proteomic analysis of HBV-transfected cells was performed following 24-hour IFN-λ3 treatment, with parallel IFN-α2a and PBS treatments for comparison using a dimethyl labeling method. The depth of the study allowed us to map the induction of antiviral proteins to multiple points of the viral life cycle, as well as facilitating the identification of antiviral proteins not previously known to be elicited upon HBV infection (e.g. IFITM3, XRN2, and NT5C3A). This study also shows up-regulation of many effectors involved in antigen processing/presentation indicating that this cytokine exerted immunomodulatory effects through several essential molecules for these processes. Interestingly, the 2 subunits of the immunoproteasome cap (PSME1 and PSME2) were up-regulated whereas cap components of the constitutive proteasome were down-regulated upon both IFN treatments, suggesting coordinated modulation toward the antigen processing/presentation mode. Furthermore, in addition to confirming canonical activation of interferon-stimulated gene (ISG) transcription through the JAK-STAT pathway, we reveal that IFN-λ3 restored levels of RIG-I and RIG-G, proteins known to be suppressed by HBV. Enrichment analysis demonstrated that several biological processes including RNA metabolism, translation, and ER-targeting were differentially regulated upon treatment with IFN-λ3 versus IFN-α2a. Our proteomic data suggests that IFN-λ3 regulates an array of cellular processes to control HBV replication.
Keywords: Quantification, Immunology, Mass Spectrometry, Systems biology, Viruses, dimethyl labeling, immunoproteasome, quantitative proteomics, RIG-I, type III IFN
Chronic hepatitis B (CHB)1 is a major health problem worldwide, affecting 240 million people throughout the world with a prevalence in Africa and South-East Asia (1). Chronic HBV-infected individuals mostly acquire the virus at a young age through vertical transmission or contact with the blood or other body fluids of an infected person. CHB eventually progresses to severe and high-mortality liver diseases including liver cirrhosis and hepatocellular carcinoma (HCC) resulting in 600,000 deaths annually (2, 3).
Current therapeutic agents for CHB are interferon (IFN)-α and nucleos(t)ide analogs (NAs) (4–6). IFN-α is a cytokine that possesses antiviral and immunomodulatory effects, which promotes control and eradication of viral infection. The advantages of using IFN-α for CHB treatment are the decreased incidence of viral resistance following this treatment and the finite duration of therapy with a higher rate of seroconversion of viral antigens, i.e. HBeAg and HBsAg, compared with NAs. However, the drawbacks of IFN-α treatment are its inconvenient route of administration and its adverse effects such as influenza-like symptoms, nausea, vomiting, cytopenia, and psychiatric disorders. Regarding the latter agent, NAs against CHB, the lack of a 3′-hydroxyl group in these compounds allows competitive incorporation into the viral genome resulting in termination of viral replication (7). Although NAs can directly suppress HBV replication and have fewer unfavorable side effects compared with IFN-α, CHB patients usually require a long-term treatment of these agents, thus increasing the chance of the emergence of viral resistance. In addition, the low rate of HBeAg and HBsAg seroconversion is another limitation of NAs. Therefore, new drugs that overcome restrictions of current anti-HBV treatments are still needed.
Several lines of reasoning suggest that IFN-λ could have a superior combination of efficacy and reduced side-effects. IFN-λ or type III IFN is a cytokine in the class II cytokine family that has recently been used in clinical trials of CHB treatment (8, 9). IFN-λ has 4 subtypes, namely IFN-λ1, -λ2, -λ3, and –λ4. IFN-λ3 was shown to have superior antiviral potency compared with other IFN-λ subtypes (10). IFN-λ receptor is composed of an IFNLR1 and IL10R2 dimer; hence upon ligand binding, the receptor activates both IFN and IL-10-like signaling pathways. The type I IFN-receptors also form a heterodimer (IFNAR1 and IFNAR2), however, the sequences of all these receptors do diverge, particularly at the C terminus (11, 12). IFNLR1 is expressed only in epithelial cells including hepatocytes in contrast to IFN-α receptor, which is widely expressed in many cell types throughout the body; thus IFN-λ treatment results in fewer side-effects when compared with IFN-α treatment (9, 13, 14). IFN-λ has been shown to activate the JAK-STAT pathway, inducing formation of the ISGF3 transcription complex, leading to expression of interferon-stimulated genes (ISGs) in similar fashion to type I IFN, but with a different temporal profile compared with those induced by type I IFN (15–17). A limited number of reports regarding IL-10-like signaling pathways have been published, with evidence of STAT3/5 biological activities (18, 19). In fact, no reports have comprehensively investigated the downstream molecular signaling effects of IFN-λ.
To better understand molecular mechanisms underlying direct antiviral and immunomodulatory effects of IFN-λ3, a comprehensive catalogue of protein effectors regulated by IFN-λ3 was compiled using quantitative proteomics analysis in the well-established HBV-transfected hepatoblastoma cell line model viz. HepG2.2.15 cells (20). HepG2.2.15 cells have been widely used as a model of chronic hepatitis B because they support HBV replication and virion secretion (21–27). These new findings could improve the treatment strategy of HBV infection and expand potential applications of IFN-λ3.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
Mixing of dimethyl-labeled samples corresponding to IFN-λ3, IFN-α2a, and control (PBS) treatments were performed at n = 5 versus the typical n = 3 to emphasize depth. Only biological replicates were performed. The normality of all proteomics, western-blotting and qPCR data was evaluated with the Shapiro-Wilk test. In all cases, the unpaired Student's t test or one-way ANOVA was selected when the distribution of the data was normal; otherwise the Mann-Whitney U test was applied. For proteomic analysis, peak intensity log2 ratios (L/M, L/H, M/H) were compared against a value of 0 (no change, log2(1)). Significance was based on the following criterion: p value < 0.05 or cases where proteins were detected in only one condition making standard statistical analysis inapplicable, with the requirement that these proteins must be identified in at least 4 of 5 experiments. Proteins not fulfilling the above criteria as well as those appearing in fewer than 3 experiments, were excluded from downstream analysis. Regarding western-blotting and qPCR experiments, n = 3 was set. One-way ANOVA was performed for these experiments with p value < 0.05 considered significant. In the case of DAVID enrichment analysis, all proteins identified by mass spectrometry were input as background. When Fisher-based enrichment analysis was performed against an in-house database of proteomic/transcriptomic studies, resulting log (p values) were conservatively adjusted by subtracting the log value corresponding to the total of all possible study/study combinations in the database.
Cell Culture and Cell Stimulation
HepG2.2.15 cells are stable HBV-transfected cells derived from a hepatoblastoma HepG2 cell line (20). These cells contain a plasmid which expresses the complete genome of HBV, which integrates into host cellular DNA (20, 28). Because HBV within HepG2.2.15 replicates and secretes HBsAg, HBeAg and HBV DNA into the culture media, HepG2.2.15 has been widely used as a model of chronic hepatitis B (22, 28). The HepG2.2.15 cell line was kindly provided from Professor Antonio Bertoletti (Singapore Institute for Clinical Sciences, A*Star). These cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM; Gibco, MA) supplemented with 10% Fetal Bovine Serum (FBS; Gibco), 1% MEM Non-Essential Amino Acids (MEM-NEAA; Gibco), 1% Penicillin/Streptomycin (Gibco) and Geneticin (G418; Gibco) at a final concentration of 150 μg/ml. The cultured cells were grown in a humidified incubator at 37 °C with 5% CO2. One million HepG2.2.15 cells were seeded into 6-well plates with 1 ml media and maintained in complete DMEM for 24 h. For determining the effects of IFN-λ3 on HBV replication, these cells were left untreated or treated with 1, 10, 100 or 1000 ng/ml of IFN-λ3 (5259-IL-025, R&D Systems, MN) and incubated for another 24 h. For proteomics analysis, HepG2.2.15 cells were plated at 5 × 106 cells in T-75 flasks and grown in complete DMEM for 24 h at 37 °C. These cells were subsequently cultured in media with 100 ng/ml of IFN-λ3 or 100 ng/ml of IFN-α2a (11100–1, pbl assay science, NJ) or PBS (control) for another 24 h. For further qPCR experiments with an optimized concentration of IFN-λ3, HepG2.2.15 cells were stimulated with or without 100 ng/ml of IFN-λ3 for 0, 8, 16, and 24 h.
RNA Isolation, Reverse Transcription, and qPCR for Gene Expression
TRIzol Reagent (Thermo, MA) was used to extract total RNA as specified in the accompanying manual. Complementary DNA (cDNA) synthesis was carried out using the Taqman Reverse transcription kit (Applied Biosystems, MA). Conditions for reverse transcription were as specified in the manual. Relative gene expression was measured with the ABI Prism 7500 sequence detection system (Applied Biosystems). All primers and probes were designed with the “primer express 3” program (Applied Biosystems), and are shown in supplemental Table S1. To monitor gene amplification, the intensity of fluorescence from the 18S housekeeping gene was monitored via Taqman probe whereas all target gene levels were monitored via SYBR green dye (Applied Biosystems). To test the specificity of SYBR green dye, melting curve analysis was conducted for all target genes. The condition of amplification for both target and housekeeping genes was 1 cycle at 95 °C for 5 min followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. Relative gene expression was calculated with the 2−ddCt method. The Student's t test and one-way ANOVA were used to compare the relative expressions of target genes in cells treated with various doses of IFN-λ3. A p value less than 0.05 was considered significant.
DNA Extraction and Absolute qPCR
After trypsinization and PBS-washing, cellular DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, MD) according to manufacturer's instructions. Quantification of HBV viral load was performed by absolute quantitative real-time PCR using the ABI Prism 7500 sequence detection system (Applied Biosystems). For ease of plasmid amplification, we used plasmids containing only the HBV gene preS1 as a surrogate marker for HBV DNA (preS1 was chosen because it is highly conserved across all HBV genotypes). First, preS1 plasmids were extracted from E. coli transformants with the GeneJET Plasmid Miniprep Kit (Fermentas, MA). The concentration of extracted plasmids was measured by spectrophotometer (Nanodrop, Thermo) and copy/μl was determined. The plasmid concentration was adjusted and diluted in a range of 107, 106, 105, 104, 103, and 102 copy/μl. These concentrations were used to construct a standard curve. Both standard and sample preS1 were amplified at the same time using conditions described above. The fluorescent intensities were specified as Ct values. For standards, Ct values at the above concentrations were used to plot a standard curve. This curve and sample Ct values were used to calculate the amount of HBV DNA in the samples. The Student's t test and one-way ANOVA were used to compare viral loads in cells treated with various levels of IFN-λ3, as well as untreated cells. A p value less than 0.05 was considered significant.
MTT Assay
HepG2.2.15 cells were seeded in 96-well plates at a density of 1 × 104 cells per well and incubated for 24 h. The culture media was removed and replaced with fresh complete media in the absence or presence of IFN-λ3 (1, 10, 100 and 1000 ng/ml). The cells were further incubated for 24 h followed by addition of 10 μl 5 mg/ml MTT (Sigma, MO) solution in each well with gentle shaking. After 4h incubation, the resulting purple formazan crystals were dissolved with dimethyl sulfoxide (DMSO, Riedel-deHaën, NJ) and subsequently measured with ELISA plate reader (Thermo) at wavelength 570 nm. The absorbance values of cells treated with each concentration of drug were compared with that of control (untreated) cells and % cell viability was calculated. This experiment was performed in triplicate.
Protein Extraction and In-solution Digestion
After trypsinization, the cells were lysed with 5% sodium deoxycholate (SDC) and 1X protease inhibitor (Thermo) mixture, followed by sonication. All cell debris was removed by centrifugation and supernatant protein concentrations of each sample were measured via BCA Protein Assay (Thermo). Equal amounts of protein from treated and untreated HepG2.2.15 were reduced and alkylated by dithiothreitol (DTT) treatment for 30 min at 37 °C and iodoacetamide (IA) treatment for 30 min at room temperature in the dark, respectively. These samples were further quenched with DTT at least 15 min at room temperature before incubating with trypsin at a ratio of 1:50 at 37 °C overnight. These mixtures were incubated with 0.5% trifluoroacetic acid (TFA) for 30 min and then centrifuged to remove SDC precipitate. The amount of tryptic peptide of each sample was determined with the Pierce Quantitative Fluorometric Peptide Assay (Thermo).
Dimethyl Labeling and Fractionation
Peptides from untreated, IFN-α2a treated, and IFN-λ3 treated HepG2.2.15 were labeled with light reagents (formaldehyde and cyanoborohydride), medium reagents (formaldehyde-d2 and cyanoborohydride), and heavy reagents (deuterated and 13C-labeled formaldehyde and cyanoborodeuteride), respectively, for an hour at room temperature. Ammonia solution and formic acid (FA) were sequentially used to stop the reaction. Labeling efficiency was tested, and we found that greater than 99% of peptides were labeled (data not shown). After combining these three samples, the mixed labeled-peptides were dried in a SpeedVac centrifuge at room temperature. Next, the pooled peptides were separated into 10 fractions to reduce complexity using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo). Eluates of each fraction were dried in a SpeedVac centrifuge before LC-MS/MS analysis.
LC-MS/MS and Analysis
The fractionated samples were resuspended in 0.1% FA (Sigma) to a final volume of 15 μl prior to MS injection. The peptides were then analyzed via an EASY-nLC1000 system (Thermo) coupled to a Q-Exactive Orbitrap Plus mass spectrometer (Thermo) equipped with a nano-electrospray ion source (Thermo). The peptides were eluted in 5–40% acetonitrile in 0.1% FA for 70 min followed by 40–95% acetonitrile in 0.1% FA for 20 min at a flow rate of 300 nl/min. The MS methods included a full MS scan at a resolution of 70,000 followed by 10 data-dependent MS2 scans at a resolution of 17,500. The normalized collision energy of HCD fragmentation was set at 32%. An MS scan range of 350 to 1400 m/z was selected and precursor ions with unassigned charge states, a charge state of +1, or a charge state of greater than +8 were excluded. A dynamic exclusion of 30 s was used. The peaklist-generating software used in this study was Proteome Discoverer™ Software 2.1 (Thermo). The SEQUEST-HT search engine was employed in data processing. MS raw data files were searched against the Human Swiss-Prot Database (20,219 proteins, June 2017) and the Hepatitis B Virus Swiss-Prot Database (225 proteins, June 2017), as well as a list of common protein contaminants (www.thegpm.org/crap/). The following parameters were set for the search: (1) digestion enzyme: trypsin; (2) maximum allowance for missed cleavages: 2; (3) maximum of modifications: 4; (4) fixed modifications: carbamidomethylation of cysteine (+57.02146 Da), as well as light, medium, and heavy dimethylation of N termini and lysine (+28.031300, +32.056407, and +36.075670 Da); (5) variable modifications: oxidation of methionine (+15.99491 Da). The mass tolerances for precursor and fragment ions were set to 10 ppm and 0.02 Da, respectively. Known contaminant ions were excluded. The Proteome Discoverer decoy database together with the Percolator algorithm were used to calculate the false positive discovery rate of the identified peptides based on Q-values which were set to 1%. The Precursor Ions Quantifier node in Proteome Discoverer™ Software was employed to quantify the relative MS signal intensities of dimethyl labeled-peptides. The control channels were used as denominators to generate abundance ratios of IFN-λ3/control and IFN-α2a/control. Log2 of the normalized ratio was used to calculate the mean and standard deviation of fold change across all five biological replicates. When these ratios were found in less than three experiments, the relevant proteins were excluded. Significantly differentially regulated proteins were determined by Mann-Whitney U test and unpaired t-tests with p value < 0.05 considered significant.
Bioinformatics
We compiled a list of defense response to virus using a variety of resources as follows. The online resource Database for Annotation, Visualization and Integrated Discovery (DAVID, v6.8, https://david.ncifcrf.gov/) and Reactome (https://reactome.org/) were employed to classify the proteins regulated by IFN-λ3 into functional categories using all proteins identified by MS as background (for DAVID). We used terms such as “antiviral,” “antigen processing/presentation” to help extract a custom list of broad antiviral proteins. Additionally, the list contains proteins involved in the HBV life-cycle that were derived from manual literature curation. Further analysis of up- and down-regulated proteins was performed against an in-house database currently under assembly. The goal is simply to expand on DAVID's method by emphasizing data sets from individual MS and RNA-seq studies. Probabilities presented here are generated using Fisher's exact test with a background proteome size of 10,000 and are unadjusted. For consistency, lists of proteins with altered expression in our own HBV work are generated by filtering out all cases where differential expression is not accompanied by a p value < 0.05 (i.e. fold-change is not a factor); the lists are then subjected to Fisher analysis.
Western Blotting for MS Confirmation
Fifteen milligrams of protein from IFN-λ3-treated and untreated HepG2.2.15 was subjected to SDS-PAGE (10%) electrophoresis. Proteins were transferred onto nitrocellulose membranes (Bio-Rad, CA) using the Trans-Blot Turbo Transfer System (Bio-Rad). The membranes were blocked with Odyssey Blocking Buffer (LICOR-Biosciences, NE) for an hour at room temperature, washed three times with TBST and probed with anti-OAS3 (ab64163, abcam, MA), anti-SAMHD1 (#12361, CST, MA), anti-STAT1 (#9175, CST), or anti-GAPDH (#5174, CST, MA) antibodies at 4 °C overnight. After washing three times with TBST, the probed membranes were incubated with IRDye 680RD secondary antibody (LICOR-Biosciences) at 1:10,000 dilution for 1 h in the dark followed by three washes with TBST. The membranes were visualized using Odyssey CLx (LICOR-Biosciences).
RESULTS
Validation of Responses to IFN-λ3 Treatment in HBV-transfected Hepatoblastoma Cell Line Model
HepG2.2.15 is a hepatoblastoma cell line that contains a stable HBV expression plasmid that has been validated as a chronic HBV infection model in previous reports (20, 28). The response to type III IFN treatment in HepG2.2.15 cells has not been reported. To determine whether HepG2.2.15 cells respond to IFN-λ3, we performed qPCR to investigate the expression of the classical ISGs, namely OAS1, MX1 and ISG15 in HepG2.2.15 cells treated with various amounts of IFN-λ3 for 24h. Fig. 1 shows that IFN-λ3 could significantly increase the expression of these 3 ISGs in a dose-dependent manner. These results indicated that HepG2.2.15 cells responded to IFN-λ3 stimulation.
Fig. 1.

Relative quantification of ISG transcripts in HepG2. 2.15 treated with IFN-λ3 for 24 h. The relative expression of OAS1 (A), MX1 (B) and ISG15 (C) genes in HepG2.2.15 cells 24h post-stimulation with different doses of IFN-λ3 is shown using Mean ± S.E. As with type I IFNs, all 3 ISGs were significantly elevated by IFN-λ3 treatment (p < 0.001) at all doses compared with a control (PBS). These experiments were performed in triplicate.
For thoroughness, we investigated the anti-HBV effects of IFN-λ3 by determining the differential changes at 3 points in the viral life-cycle including the levels of preS1 (typically used as a representative gene for HBV transcripts, given its high conservation across all genotypes), replicative intermediate pre-genomic RNA (pgRNA), and intracellular HBV DNA (both rcDNA and cccDNA). qPCR was performed on HepG2.2.15 RNA following treatment with various amounts of IFN-λ3 for 24 h. As shown in Fig. 2A and 2B, IFN-λ3 reduced both preS1 and pgRNA expression compared with control in a dose-dependent manner. Measurement of intracellular HBV DNA showed that copy numbers of virus in IFNλ3-treated HepG2.2.15 cells were diminished in a dose-dependent manner relative to control (Fig. 2C). The reduction reached significant levels when the doses of IFN-λ3 were 100 ng/ml (p value = 0.04) and 1,000 ng/ml (p value = 0.0134). Collectively, these results indicated that IFN-λ3 inhibits HBV replication in HepG2.2.15 at the given time point.
Fig. 2.

Effects of IFN-λ3 on HBV replication. HepG2.2.15 cells were incubated with IFN-λ3 (1, 10, 100 and 1000 ng/ml) or treated with PBS for 24 h. The relative transcript expression and the amount of intracellular HBV DNA are shown as Mean ± S.E. IFN-λ3 significantly inhibited preS1 and pgRNA expression at doses equal to or greater than 10 ng/ml (A and B). IFN-λ3 significantly suppressed viral propagation at doses equal to or greater than 100 ng/ml (C). These experiments were performed in triplicate. (* represents a p value less than 0.05 and ** represents a p value less than 0.01).
Before we investigated the cellular response to IFN-λ3, the toxicity of this drug was considered. The MTT cytotoxicity assay was performed to determine HepG2.2.15 viability under distinct concentrations of IFN-λ3. The percentage of viable cells is illustrated in Fig. 3. The increasing doses of IFN-λ3 significantly promoted cell death only at the highest two doses in this experiment, where the viable HepG2.2.15 cells reduced to 94% and 84% in 100 ng/ml and 1000 ng/ml conditions, respectively. Thus, we settled on 100 ng/ml of IFN-λ3 for further experiments because this dose showed the ability to significantly inhibit HBV replication with minimal cytotoxicity on HepG2.2.15 cells.
Fig. 3.
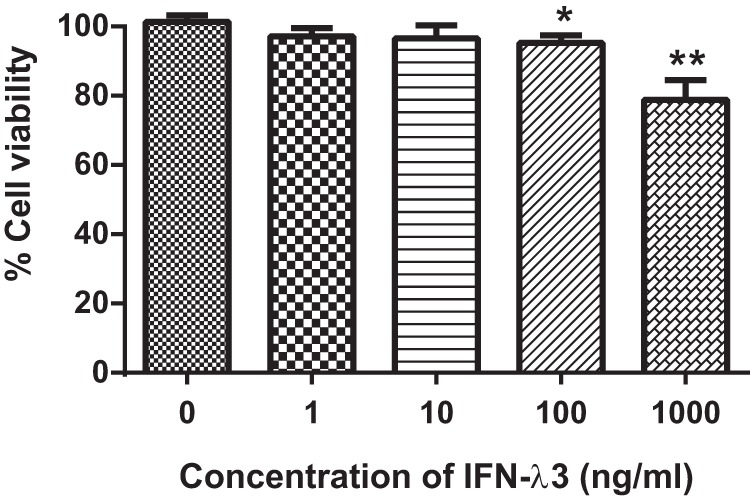
IFN-λ3 cytotoxicity assay. The viability of HepG2.2.15 after IFN-λ3 treatment was determined by MTT assay. The percentage of cell viability is shown as Mean ± S.E. IFN-λ3 showed minimal effect on cell viability when the doses were less than 1,000 ng/ml. These experiments were performed in triplicate. (* represents a p value less than 0.05 and ** represents a p value less than 0.01).
Quantitative Proteomics Analysis of IFN-λ3 Responses in HepG2.2.15
Fig. 4 shows the schematic workflow of this study. Briefly, untreated, IFN-α2a-treated, and IFN-λ3-treated HepG2.2.15 cells were lysed and digested with trypsin. The tryptic peptides of these groups were labeled with light, medium, and heavy dimethyl reagents and then fractionated for subsequent LC-MS/MS analysis. In total, 4670 proteins were identified at a false discovery rate (FDR) of less than 1%, with 1471 proteins identified based on a single peptide (all information regarding peptide sequences assigned and protein identifications are supplied in supplemental Table S2 and S3). For the IFN-λ3 treatment condition, 2904 proteins were identified in at least 3 of 5 replicates allowing evaluation of significance, shown in the corresponding volcano plot (Fig. 5). Seven hundred thirty-seven proteins showed significant changes in abundance, with a slight bias toward down-regulated proteins in response to IFN-λ3 stimulation (see Fig. 5). Table I displays a list of significantly regulated proteins with |log2(IFN-λ3/Ctrl)| ratios of greater than 1. We should point out that our primary intention in this work is to explore IFN-λ3 effects on HepG2.2.15 cells; the effects of IFN-α2a would be secondary, for the sake of comparison with IFN-λ3 effects, and will be presented at the last part of the Results section. All data can be accessed from supplemental Table S4.
Fig. 4.

Quantitative proteomic workflow. HepG2.2.15 cells were treated with 100 ng/ml of IFN-λ3 and IFN-α2a and PBS for 24 h. Cell lysates of each group were digested and then labeled with different dimethyl reagents. After combining the 3 samples, these peptides were fractionated and then analyzed by LC-MS/MS.
Fig. 5.

Volcano plot. Volcano plot shows the distribution of identified proteins according to p value and fold change, indicating significance with a dashed line (p value < 0.05).
Table I. A list of significantly upregulated (A) and downregulated (B) proteins with |log2(IFN-λ3/Control)| ratios of greater than 1.
A.
| Accession number | Description | Gene ID | Average Log2 ratios | Pathway |
|---|---|---|---|---|
| O14879 | Interferon-induced protein with tetratricopeptide repeats 3 | IFIT3 | 4.74 | Antiviral defense (Inhibit viral protein synthesis) |
| Q96AZ6 | Interferon-stimulated gene 20 kDa protein | ISG20 | 3.28 | Antiviral defense (Degrade viral RNA) |
| P05161 | Ubiquitin-like protein ISG15 | ISG15 | 3.27 | Antiviral defense |
| Q15646 | 2′-5′-oligoadenylate synthase-like protein | OASL | 2.58 | RLR signaling pathway |
| O95786 | Probable ATP-dependent RNA helicase DDX58 | DDX58 | 2.47 | RLR signaling pathway |
| Q9Y6K5 | 2′-5′-oligoadenylate synthase 3 | OAS3 | 2.42 | Antiviral defense (Degrade viral RNA) |
| Q29960 | HLA class I histocompatibility antigen, Cw-16 alpha chain | HLA-C | 2.24 | Antigen processing and presentation |
| P09914 | Interferon-induced protein with tetratricopeptide repeats 1 | IFIT1 | 2.19 | Antiviral defense (Inhibit viral protein synthesis) |
| P42224 | Signal transducer and activator of transcription 1-alpha/beta | STAT1 | 2.13 | Type I and III IFN signaling |
| Q63HN8 | Isoform 2 of E3 ubiquitin-protein ligase RNF213 | RNF213 | 1.95 | Ubiquitin proteasome pathway |
| Q01628 | Interferon-induced transmembrane protein 3 | IFITM3 | 1.93 | Antiviral defense (Inhibit viral entry) |
| P52630 | Signal transducer and activator of transcription 2 | STAT2 | 1.65 | Type I and III IFN signaling |
| P29590 | Protein PML | PML | 1.63 | Antiviral defense (Inhibit viral transcription) |
| P28838 | Cytosol aminopeptidase | LAP3 | 1.61 | Antigen processing and presentation |
| Q10589 | Bone marrow stromal antigen 2 | BST2 | 1.55 | Antiviral defense (Inhibit viral egress) |
| Q6IA86 | Isoform 6 of Elongator complex protein 2 | ELP2 | 1.46 | Regulation of transcription |
| Q08380 | Galectin-3-binding protein | LGALS3BP | 1.39 | Cell adhesion |
| Q9H0P0 | Cytosolic 5′-nucleotidase 3A | NT5C3A | 1.38 | Antiviral defense (Inhibit reverse transcription) |
| P35527 | Keratin, type I cytoskeletal 9 | KRT9 | 1.34 | Intermediate filament organization |
| P41226 | Ubiquitin-like modifier-activating enzyme 7 | UBA7 | 1.32 | Ubiquitin proteasome pathway |
| Q9Y3Z3 | Deoxynucleoside triphosphate triphosphohydrolase SAMHD1 | SAMHD1 | 1.19 | Antiviral defense (Inhibit reverse transcription) |
| Q9BQE5 | Apolipoprotein L2 | APOL2 | 1.22 | Movement of lipids in the cytoplasm |
| P01892 | HLA class I histocompatibility antigen, A-2 alpha chain | HLA-A | 1.16 | Antigen processing and presentation |
| Q9Y6A9 | Signal peptidase complex subunit 1 | SPCS1 | 1.13 | Proteolysis |
| B. | ||||
| Q9BX93 | Group XIIB secretory phospholipase A2-like protein | PLA2G12B | -1.05 | Lipid catabolic process |
| Q15427 | Splicing factor 3b subunit 4 | SF3B4 | −1.05 | mRNA processing |
| Q9NPA8 | Transcription and mRNA export factor ENY2 | ENY2 | −1.18 | Regulation of transcription |
| P62158 | Calmodulin | CALM3 | −1.22 | Regulation of synaptic vesicle exocytosis |
| O75410 | Isoform 2 of Transforming acidic coiled-coil-containing protein 1 | TACC1 | −1.25 | Cell proliferation |
| O75438 | Isoform 2 of NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 1 | NDUFB1 | −1.47 | Mitochondrial electron transport, NADH to ubiquinone |
| Q12929 | Epidermal growth factor receptor kinase substrate 8 | EPS8 | −1.57 | Actin polymerization-dependent cell motility |
| Q9NZD2 | Glycolipid transfer protein | GLTP | -1.62 | Intermembrane lipid transfer |
| Q13126 | Isoform 2 of S-methyl-5′-thioadenosine phosphorylase | MTAP | −1.76 | Nucleobase-containing compound metabolic process |
| Q58FF7 | Putative heat shock protein HSP 90-beta-3 | HSP90AB3P | −3.58 | Protein folding and response to stress |
All HBV proteins were identified in this study. However, calculation of significance could not be performed because of absence in multiple MS runs, except for putative X-Core fused protein (which showed no significant change), see supplemental Table S4. Based on spectral counts, all the HBV peptides were apparently expressed at very low levels relative to the entire HepG2.2.15 proteome (below 1 ppm, see in PRIDE partner repository as mentioned in Data Availabilty). This finding is in agreement with a previous study that specifically examined intracellular HBV proteins in HepG2.2.15 cells but found them to be undetectable (29). Note that all HBV proteins, except HBV pol and HBx, are secreted (30–32), likely causing intracellular levels of these proteins to be scarce.
To confirm the results from MS analysis, three proteins known to be differentially regulated in response to HBV infection, namely 2′-5′-oligoadenylate synthase 3 (OAS3) (33), Sterile α motif (SAM) and histidine/aspartate (HD)-containing protein 1 (SAMHD1) (34–36), and signal transducer and activator of transcription 1 (STAT1) (37, 38) were selected for validation by Western blot analysis. Consistent with MS results, OAS3, SAMHD1 and STAT1 were up-regulated as a result of IFN-λ3 treatment (Fig. 6).
Fig. 6.

Validation of altered proteins by immunoblotting assay. SDS-PAGE was performed on treated and untreated lysates, followed by membrane transfer and incubation with anti-OAS3, anti-SAMHD1, anti-STAT1, and anti-GAPDH overnight. The proteins of interest were visualized using the LI-COR Odyssey system. Consistent with proteomic results, the expression of OAS3, SAMHD1 and STAT1 increased after IFN-λ3 treatment. These experiments were performed in triplicate.
To further explore the possible roles of transcriptional regulation for significantly altered proteins, we selected several proteins involved in antiviral processes for qPCR analysis. Fig. 7 show that up-regulation was seen at both protein and RNA expression levels without exception. However, no clear pattern emerged when down-regulated proteins were examined for transcript levels.
Fig. 7.

Correlation of mRNA and protein levels of up- and down-regulated proteins. Various up- and down-regulated proteins were selected to investigate their transcript expressions by qPCR. Clear correlations between protein and mRNA expression were seen in up-regulated proteins (A) but not in down-regulated proteins (B).
Bioinformatics Analysis and Antiviral/Immunomodulatory Process Mapping
The DAVID bioinformatics tool was used to classify and cluster significant functions of all up- and down-regulated proteins. Upon IFN-λ3 treatment, we found several biological processes expected to be canonically regulated on general IFN stimulation, such as immune response and viral infection defense. Additionally, however, several biological processes not previously emphasized upon interferon stimulation emerged from this work, including metabolism of RNA, major pathway of rRNA processing in the nucleolus and cytosol, selenoamino acid metabolism, peptide chain elongation, translation, prefoldin mediated transfer of substrate to CCT/TriC, transcription-coupled nucleotide excision repair (TC-NER), unfolded protein response (UPR), interleukin-23 signaling, and hypusine synthesis from eIF5A-lysine.
Following enrichment analysis, we conducted a protein-by-protein search for relevance to the viral processes. We manually searched the viral literature, as well as using DAVID's antiviral defense groups and antigen processing/presentation groups, to construct a map of points at which these proteins may partake in viral processes (Fig. 8) (as described under Experimental Procedures). We also built a map displaying steps at which IFN-λ3 treatment could promote antigen processing and presentation of viral proteins. Finally, we constructed a map that highlights points at which IFN-λ3 stimulation may heighten expression of proteins in the RIG-I pathway that have been shown to be inhibited via HBV infection. Unless otherwise stated, we found no proteins that contradict the patterns that we report below.
Fig. 8.

Illustration of HBV life-cycle mapped to antiviral proteins that were identified in this study. Antiviral proteins and host cellular factors of interest identified in this study were mapped into the HBV-life cycle. These proteins were identified at most steps of HBV replication. Proteins in red and green text represent up- and down-regulated proteins, respectively.
Regarding the HBV life cycle, we identified many proteins which may be involved at numerous points (Fig. 8). At the early steps of viral attachment, entry, and cytosolic release from endosomes (Fig. 8, step 1 and 2), we detected an increased expression of IFITM3, known to alter intracellular cholesterol homeostasis, preventing viral fusion with endosomes in at least 4 out of 7 of the Baltimore viral groups (39). RAB5C and RAB7A, proteins previously implicated in viral trafficking (Fig. 8, step 2), were down-regulated upon IFN-λ3 treatment (40), though RAB7A did not reach statistical significance. Two other trafficking proteins not mentioned in the viral literature, AP2B1 and EEA1, were significantly down-regulated. Regarding cytosolic-to-nuclear transport of the HBV capsid along microtubules (Fig. 8, step 4), we found that several molecules involved in microtubule assembly and function evinced decreased expression after IFN-λ3 treatment, consistent with the literature. These proteins were DCTN1, DCTN2, KIF5B, MAP4, and MACF1 (41, 42). For nuclear import (Fig. 8, step 5), all identified proteins known to play roles herein (42, 43) were found to decrease because of IFN-λ3 stimulation (IPO5, IPO7, CALR, CALM, RANBP1, and TPR). Within the nucleus (Fig. 8, step 7), all identified ISG products shown previously to limit or degrade viral mRNA (44–46) were observed to be up-regulated: ADAR1, OAS3, ISG20, PML, XRN2, ZC3HAV1, and TRIM25. At the level of RNA export (Fig. 8, step 8), ENY2 and CALR were down-regulated. Regarding translation (Fig. 8, step 9), antiviral proteins (EIF2AK2, IFIT1, and IFIT2) involved in protein synthesis increased in treated cells, as shown in previous work with type I IFN treatment (44, 46, 47). In addition, almost all identified translation initiation factors (EIF1AY, EIF2S3, EIF2B1, EIF3B, EIF4G1, EIF4B, EIF4G2, and EIF5A) decreased in this study. Encapsidation follows translation of viral proteins (Fig. 8, step 10). Interestingly, RIG-I was up-regulated and has been shown to interfere with the interaction between pgRNA and HBV polymerase, required for encapsidation (48). Other effects of RIG-I up-regulation are discussed below. Reverse transcription, which occurs within the capsid (Fig. 8, step 11), has been demonstrated to be inhibited by SAMHD1 (HBV and HIV) (35, 49) and NT5C3A (IAV and CSFV) (50, 51); these proteins were up-regulated in our study upon treatment. Proteins that might interfere with viral assembly (Fig. 8, step 12.1) were not identified in this study. Finally, at the egress step (Fig. 8, step 13), two up-regulated proteins thought to play a role in inhibiting HBV export, BST2 and C4A, were detected after IFN-λ3 stimulation.
Regarding antigen processing and presentation, we identified 22 proteins that are involved in this process (Fig. 9). Among proteins in the degradation-related ubiquitination cascade, we found only up-regulation, specifically UBA7 (E1), UBE2L6 (E2), and SYVN1 (E3) after IFN-λ3 stimulation. Two ubiquitination enzymes were down-regulated but have not been reported to instigate degradation. All deubiquitinating enzymes (DUBs) identified in this study, i.e. USP14, UCHL3, UCHL5, and OTUB1, decreased because of IFN-λ3 treatment. Downstream of these processes, core proteasome components, PSMA7, PSMB7, and PSMG2, were up-regulated with no exceptions. Importantly, both cap components (PSME1 and PSME2) associated with the immunoproteasome were up-regulated, whereas all significantly altered cap components of the constitutive proteasome were down-regulated. Evidence of possible post-proteasome processing was seen in the up-regulation of a cytosol aminopeptidase, LAP3. Next, we saw up-regulation of proteins involved in transport of peptides from the cytosol to the ER as well as assembly of peptide-HLA class I complexes; these proteins included TAP2, TAPBP, HLA-A, HLA-B, and HLA-C. Other proteins involved in the above step did not reach statistical significance but tended to increase expression following IFN-λ3 treatment; TAP1, PDIA3 (ERP57), and B2M. CALR, a player in this process, was down-regulated, the reasons for which will be discussed later.
Fig. 9.

IFN-λ3 enhanced antigen processing/presentation. IFN-λ3 not only up-regulated HLA class I expression but it also increased the expression of other effector molecules involved in antigen processing and antigen presentation. Ubiquitinating enzymes that promote protein degradation were found to be up-regulated whereas deubiquitinating enzymes known to remove ubiquitin were found to decrease after IFN-λ3 treatment. Although all identified subunits of the constitutive proteasome cap were down-regulated, the 2 subunits of the immunoproteasome cap as well as some subunits of the proteasome core were elevated in expression. Several effector molecules involved in peptide loading on class I HLA as shown in red text were up-regulated with the exception of CALR, which was down-regulated as shown in green text. The italic red text represents proteins with increased expression levels that failed to reach statistical significance.
One of the important functions of the innate immune response against HBV is the sensing of viral RNA, leading to IFN activation, which includes components such as toll-like receptors, RIG-I, and MDA5. Here, we saw significant up-regulation of RIG-I upon treatment, which could counteract the known suppression of RIG-I by HBV. Enhancers of RIG-I activity, such as OASL, TRIM25, and IFIT3 (RIG-G) were also significantly up-regulated. Increased levels of type I and III IFNs, which are activated by RIG-I, would be expected, but were not observed via MS. However, qPCR work confirmed significant up-regulation of these IFNs at the transcript level (Fig. 10). It is likely that the increase in IFN I and III gene expression because of RIG-I up-regulation could lead to more production of IFNs at the protein level and in turn activate IFN receptors as positive feedback. All three canonical ISGF3 components (STAT1, STAT2, and IRF9) downstream from IFN receptor activation were up-regulated, probably as a result of both initial IFN-λ3 treatment and further generation of type I and III IFNs (Fig. 11).
Fig. 10.

qPCR analysis of IFN genes. Total RNA of HepG2.2.15 cells treated with IFN-λ3 for 8, 16, and 24 h or left untreated was extracted and then converted to cDNA. The expression levels of IFNB, IFNL1, and IFNL3 (A, B, and C) were found to be up-regulated in a time-dependent manner after IFN-λ3 treatment.
Fig. 11.

IFN-λ3 rescued the RIG-I signaling pathway. IFN-λ3 up-regulated the expression of RIG-I and IFIT3, which have been reported to be suppressed by HBV. IFN-λ3 also elevated the expression of OASL and TRIM25. These proteins promote type I and type III IFN production. These IFNs, in turn, activate the JAK-STAT pathway and induce the expression of ISGs. It is likely, then, that IFN-λ3 provides positive feedback to amplify ISG expression to control HBV replication. Proteins in red text represent up-regulated proteins, whereas the blue text represents the up-regulated transcripts.
We found several likely antiviral proteins that do not conveniently fit into the above viral processes. Firstly, three 14-3-3 proteins (YWHAZ, YWHAH, and SFN) were found to be down-regulated following IFN-λ3 treatment. Another down-regulated protein of interest was cyclophilin D (PPIF). Finally, CHID1 was the single most up-regulated protein in our study on IFN-λ3 treatment. Though not prominent in the viral literature, CHID1 is known to bind LPS (52) and is seen to be down-regulated in several viral studies (53–56).
After removal of all antiviral proteins, enrichment analysis of the remainder produced an interesting result. RNA-binding proteins, particularly spliceosome components, were both up- and down-regulated on IFN-λ3 treatment versus control. For example, the splicing factor U2AF1 was significantly up-regulated on treatment (p = 0.00165). Searching through individual data sets from an in-house database containing a variety of studies and applying Fisher's exact test (see Experimental Procedures) showed a strong tendency toward both up-regulation and down-regulation of proteins that associate with the non-coding RNA NORAD (log p = −5 and −13, respectively) (57). The same pattern applies to proteins shown to bind to the splicing factor U2AF2 (log p = −6 and −9, respectively) (58).
Though our primary intent is to elaborate on IFN-λ3's potential, comparative studies against the best characterized interferon, IFN-α, are essential. Given the known parallelism in broad modes of action between the two IFNs, significant overlaps should be seen in sets of up- and down-regulated proteins upon treatment, offering a simple validation of IFN-λ3's activity. At the same time, and most intriguingly, the comparison would allow us to suggest possible differences in effect. To take advantage of the depth of our data, we constructed a tree depicting all 27 possible differential expression outcomes resulting from 3 pairs of treatment comparisons (i.e. IFN-λ3 versus Ctrl, IFN-α2a versus Ctrl, IFN-λ3 versus IFN-α2a, see Fig. 12). A simple comparison between IFN-λ3 versus IFN-α2a could lead to errors in interpretation. For example, the observation that IFN-λ3 treatment results in up-regulation or down-regulation of a protein versus IFN-α2a renders an uninteresting result if IFN-λ3 does not have any significant effect versus Ctrl. This scenario can be seen in outcomes #12 and #16 in Fig. 12. Another problem in interpretation arises when both IFNs have the same directional effect on cells but one effect is significantly more potent than the other; this would not be noticed upon mere IFN-λ3 versus IFN-α2 comparison. This scenario can be seen in outcomes #3 and #25 in Fig. 12. The tree shows the number of MS-derived proteins at their appropriate outcome branch points and shows proteins associated with various biological processes (as described under “Experimental procedures”) for each outcome. As one simple example of the power of this approach, note that IFITM3 was significantly up-regulated upon treatment with both IFNs, however this effect was significantly stronger upon IFN-λ3 treatment (outcome #1). Another example was in outcome #4 where IFN-λ3 treatment caused up-regulation of proteins involved in antigen processing and presentation (SYVN1, LAP3, PSMA7, and PSMB7), whereas IFN-α2a did not produce any significant effects. Interestingly, the mirror image of this branch, outcome #24 where IFN-λ3 caused down-regulation whereas IFN-α2a did not, was also populated with a subset of proteins involved in antigen processing and presentation, specifically those associated with the constitutive proteasome cap (PSMC4, PSMD4, PSMD7, PSMD13, and PSMD14). A priori, one would expect the most interesting outcomes to be found in cases in which IFN-α2a and IFN-λ3 have clearly opposite effects (outcomes #7 and #21). We did not identify proteins clearly related to antiviral processes in these two outcome groups. This reason pointed to broader unbiased analysis. Hence, we performed “Reactome” enrichment analysis against all outcome groups for complete enrichment analysis beyond the aforementioned antiviral processes. The resulting Reactome enrichment groups were subjected to 2-D cluster analysis (Fig. 13 and underlying data in supplemental Table S4). This analysis offers new insights. Firstly, we note the aforementioned cases in which IFN-λ3 and IFN-α2a showed opposing effects. In outcome #7, where IFN-λ3 exclusively caused up-regulation, the “peptide chain elongation,” “metabolism of RNA,” “major pathway of rRNA processing in the nucleolus and cytosol,” “selenoamino acid metabolism,” and “translation” terms are highlighted, whereas in outcome #21, where IFN-λ3 exclusively caused down-regulation, the “interleukin-23 signaling” term is emphasized. Next, we observe that in most cases where proteins were up-regulated upon IFN-α2a treatment (outcomes#1, #2, #3, #11, and #21), “interferon signaling” and/or “class I MHC mediated antigen processing & presentation” terms were apparent, reinforcing expectations regarding IFN-α's potency on these pathways. Interestingly, specific terms related to “translation” and “metabolism of RNA” were significantly associated with 13 out of 17 different outcome groups. To our best knowledge, these biological processes have been not mentioned as consequences of general IFN activation. Finally, enrichment terms that are largely unexplored with regard to IFN treatment include “transcription-coupled nucleotide excision repair (TC-NER)” (1 significant occurrence), “interleukin-23 signaling” (1), “unfolded protein response (UPR) (2),” “prefoldin mediated transfer of substrate to CCT/TriC (4),” and “hypusine synthesis from eIF5A-lysine (1).” Interestingly, the final enrichment term was only associated with a single case, that of proteins down-regulated in all treatment pairs (outcome #27). These results could help point toward differential effects of IFN-λ3 versus IFN-α2a.
Fig. 12.

Outcome tree. To compare the effects of IFN-λ3, IFN-α2a, and PBS control, we generated a tree representing the 27 possible outcomes. The boxes in red and green represent proteins that are significantly up- and down-regulated, respectively. Groups of proteins with insignificantly changed expression are represented by yellow boxes. As an example, the topmost outcome represents proteins in which IFN-λ3 treatment causes significant up-regulation versus control, IFN-α2a treatment causes significant up-regulation versus control, and IFN-λ3 treatment causes significant up-regulation versus IFN-α2a. The number in parentheses refers to the number of identified proteins in the group. The proteins in each group that might be involved in suppressing HBV replication are shown in colored boxes specifying various processes, particularly antiviral.
Fig. 13.
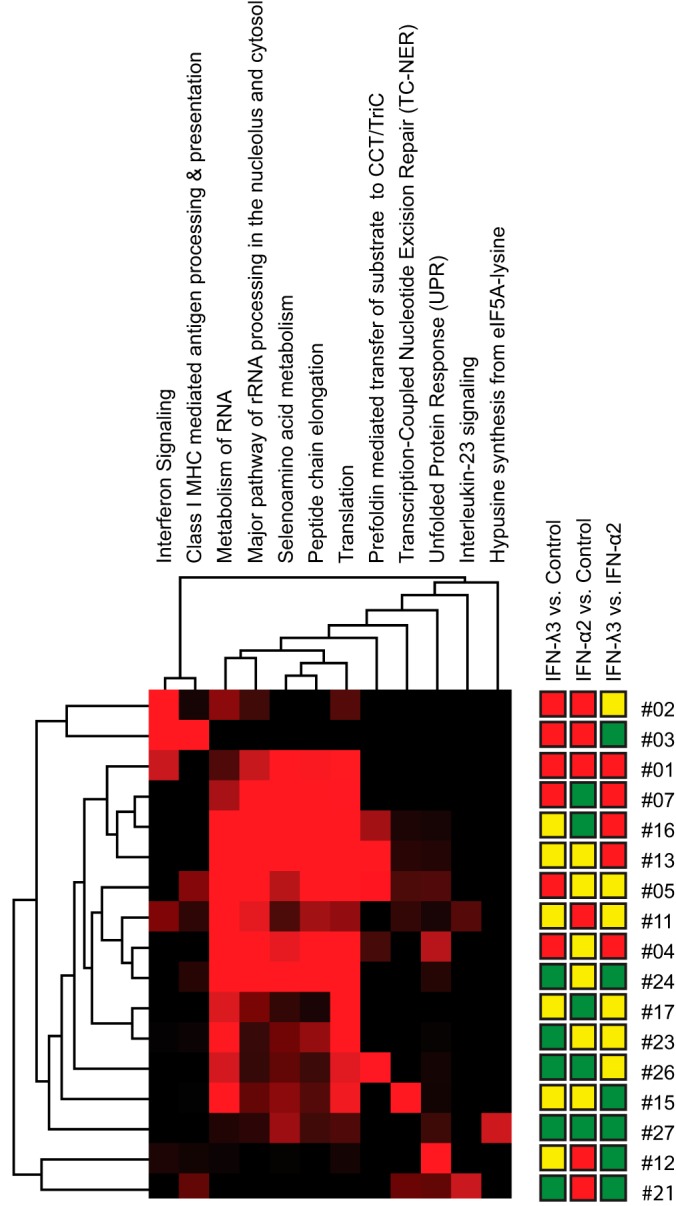
Enrichment heatmap. Reactome enrichment analysis was performed on the tree outcomes that contained at least 9 members, excluding outcome #14 in which no significant alterations were seen on any treatment. Probabilities (Reactome's hypergeometric test, -log(p value)) were generated for every intersection of these tree outcomes with the specified Reactome groups. This matrix of probabilities was then submitted for two-dimensional cluster analysis using Cluster 3.0 and Java Treeview. The result is displayed as a heatmap. Red color intensity indicates the level of significance.
DISCUSSION
Type III IFN has been shown to exert antiviral effects on several viruses including EMCV, IAV, HSV, VSV, HIV, HCV, and HBV (17, 46, 59–64). In clinical trials, peg IFN-λ treatment showed reduced HBV DNA, HBsAg and HBeAg in CHB patients at levels greater than or comparable to those treated with peg IFN-α during on-treatment and at end-of-treatment (9). Although the virological, serological, and biochemical responses of peg IFN-λ at week 24 after treatment were not superior to peg-IFN-α, adverse events associated with drug treatment were mostly seen in patients treated with peg-IFN-α. Numerous HCV studies also illustrate this tendency toward diminished side-effects (13, 14). Another clinical trial demonstrated that patients treated with peg IFN-λ had improved anti-HBV immunity through an increase in poly-functional NK cells and maintenance of both HBV-specific CD4+ and CD8+ T cells, important arms of immunity for viral elimination (8). The mechanisms involved in these immunomodulatory effects remain to be elucidated. At the level of cell culture, only two studies (59, 65) have focused on the effects of IFN-λ treatment of HBV-transfected cells, one of which offers a comparison against a type I IFN, IFN-β. Here, a comparison against IFN-β showed that IFN-β exhibited greater potency in viral elimination at the 6 h time point, but potency was equivalent at 24 h.
Surprisingly, the effects of IFN-λ treatment have not been studied with high-resolution mass spectrometry to date, possibly because it is assumed that type I and type III IFNs share the same signaling pathways. In fact, modern, deep proteomic studies of the effects of IFN treatment in general are lacking, perhaps because of an underlying assumption that RNA-seq results should parallel those generated via MS. Here, we used a modern high-resolution LC-MS/MS system with upstream high-pH reversed phase fractionation to yield a total of 127,989 peptides (25,181 unique peptides) representing 4670 proteins. The peptides were labeled with dimethyl isotopes, allowing accurate quantification under different treatments. This study provides the most comprehensive examination of changes in protein abundance under IFN treatments to date.
To provide objective measures for study-study comparisons, in the discussion below we perform numerous comparisons between our studies and others with Fisher's exact test (see “Experimental procedures”). As expected, canonical IFN-stimulated biological processes were prominent in IFN-λ3 treatment. Proteins significantly up-regulated upon IFN-λ3 treatment versus Ctrl (hereafter λ3/c) overlapped with the GO “antiviral defense” term at p value = 10−14 (with the α/c condition giving p value = 10−12). Similarly, the λ3/c list significantly overlapped (p value = 10−13) transcripts whose up/down-regulation patterns (i.e. “co-expression”) mirror those of RIG-I transcripts reported in the ARCHS4 resource (66); the α/c list gave p value = 10−18, perhaps illustrating the expected difference in potency. Comparing proteins significantly up-regulated in λ3/c against those up-regulated in α/c, we derive p = 10−138, a clear demonstration of similarities in effect versus control. Stepping outside the bounds of our own work, Bolen's transcriptomic comparison of the effects of 5 interferons, including IFN-λ3, on Huh7 cells can be examined against our proteomic results (16). Here, a list of highly up-regulated transcripts derived from Bolen's 6, 12, and 24 h λ3/c time-points significantly intersected with our own λ3/c results (p value = 10−25). Of interest, the degree of overlap (p value = 10−5) was less significant when performing the same exercise with down-regulated transcripts versus proteins, paralleling the tendency of lower correlation seen in our own qPCR results for several down-regulated proteins (Fig. 7). Other comparisons illustrating the expected canonical effects would be an α/c result in Huh7 (67) (a λ3/c experiment was not performed) where up-regulated transcripts align with our own λ3/c results (p value = 10−30), and a list of genes likely to be activated by STAT2 based on curated STAT2 chip-seq results that significantly overlaps with our λ3/c results (p value = 10−25) (68).
In addition to acting on viral replication directly, we propose that the proteolytic pathway may be induced by IFN-λ3 to inhibit HBV replication. Robek et al. demonstrated that small molecule-based inhibition of proteasome activity could restrain the anti-HBV activity of type I IFN in vitro, suggesting that antiviral effects of IFN might relate to the proteasome (69). Yao et al. demonstrated that suppression of HBV replication was accompanied by an increase of 5 proteasome subunits in HepG2.2.15 cells in response to interleukin-4 (IL-4) treatment (70). In our study, we also note up-regulation of immunoproteasome members (cap components PSME1 and PSME2) on IFN-λ3 and IFN-α2a treatment. The immunoproteasome is distinct from the constitutive proteasome in the composition of its catalytic subunits, its cap, and its peptide processing capacity (71). Interestingly, all significantly altered cap components of the constitutive proteasome were down-regulated only upon IFN-λ3 treatment, suggesting coordinated modulation toward the antigen processing form of the proteasome on IFN-λ3 treatment. To our knowledge, this is the first report suggesting a shift from proteasome constitutive degradation mode to antigen processing/presentation mode in response to type III IFN treatment. HBV has been termed a “stealth” virus because innate immune responses in chimpanzees are not observed shortly after infection (72). The synthesis of type I IFNs is induced by the activation of RIG-I-like receptor (RLR) signaling pathways in response to viral infection. Previous studies have shown that HBV polymerase and HBx proteins interfere with the interaction of RIG-I and downstream signaling molecules, resulting in diminished type I IFN production (73–75).
HBV has also been shown to alter RIG-I expression by inducing up-regulation of miR146a, which may directly downregulate RIG-I and RIG-G transcripts (76). IFN treatment has not previously been shown to up-regulate RIG-I pathway components in HBV infected cells. We found, for the first time, that treating HepG2.2.15 with IFN-λ3 increased the expression of RIG-I. In addition, upon IFN-λ3 treatment, we observed elevated expression of RIG-G, known to enhance the activity of RIG-I, as well as TRIM25 and OASL, which promote the interaction between RIG-I and IPS1 on mitochondria or peroxisomes, leading to increased IFN production (77, 78). We propose that IFN-λ3 restores RIG-I and associated downstream signaling to produce type I IFN in HBV infected cells and likely other viruses. Although we did not find any IFNs in our proteomic work, possibly because of low abundances, qPCR verified increases in type I IFN (IFN-β) and type III IFN (IFN-λ1 and IFN-λ3) upon IFN-λ3 stimulation.
Several biological processes not mentioned above emerged from this work, including the involvement of 14-3-3 proteins, cyclophilin D, calreticulin, and cell motility proteins. These subjects are explored in detail below. In our study, the 14-3-3 proteins YWHAZ, YWHAH, and SFN were found to be down-regulated following IFN-λ3 treatment. Several studies have shown that 14-3-3 proteins may facilitate the replication of viruses (79, 80). The 14-3-3 proteins are highly conserved regulatory molecules having multiple functions, including involvement in signal transduction via kinases and phosphatases, the cell cycle, and apoptosis. These proteins have been shown to play important roles in viral infection. For example, complex formation of cdc25 with the HIV accessory protein Vpr, an event that alters the host cell life cycle, is facilitated by 14-3-3 proteins (79). Also, HCV core protein interacts with 14-3-3 proteins resulting in enhancement of Raf-1 kinase activity together with control of hepatocyte growth (80). Interestingly, sequence analysis revealed a 14-3-3 binding domain in HBx protein (81), suggesting that 14-3-3 proteins could directly interact with HBV. Notably, we could not observe significant alterations in 14-3-3 proteins upon IFN-α2a treatment; the differential effects of IFN-λ versus IFN-α on 14-3-3 levels would be worthy of further investigation.
The down-regulation of cyclophilin D upon IFN-λ3 treatment observed in our work suggests that this molecule may play a role in inhibition of HBV replication. One of the pathways affected by HBx is calcium signaling, as Ca2+ is necessary for viral replication and core assembly (82, 83). In addition to interference with RIG-I signaling, the HBx protein can interact with and modulate the mitochondrial permeability transition pore (MPTP) leading to the release of mitochondrial calcium into the cytoplasm (82). The opening of the MPTP anion channel is regulated by cyclophilin D (84), thus down-regulation of cyclophilin D would serve to counteract Ca2+-assisted viral replication and core assembly. Increasing levels of cytosolic calcium causes the stimulation of PYK2 kinase, in turn activating Src kinase signal transduction to promote HBV reverse transcription, replication, and core assembly (83, 85). In several studies, cyclosporine A (CsA) was used as an MPTP specific blocker, thus reducing core assembly and inhibiting HBV replication (30, 84, 85). Consistent with CsA treatment, the calcium ion chelating agent BAPTA-AM also suppressed HBV propagation (85).
Calreticulin (CALR), down-regulated upon IFN-λ3 treatment in our study, is an ER calcium-binding chaperone involved in the regulation of calcium homoeostasis, the folding of newly synthesized proteins, nuclear import, and peptide loading on MHC class I (86–88). In HBV-transfected cells, Yue et al. demonstrated that the inhibition of IRF-7 translocation into the nucleus was induced by CALR, resulting in suppression of type I IFN production (89). Moreover, this group showed that CALR inhibited JAK-STAT pathway induction by IFN-α by inhibiting STAT1 phosphorylation, diminishing expression of PKR and OAS. Thus, the observation of down-regulation of CALR would be expected to have an antiviral effect. However, the down-regulation of CALR would also seem to interfere with antigen processing/presentation. We note that most antigen processing/presentation molecules were up-regulated upon treatment in our work, possibly compensating for this effect. Overall, the observed IFN-λ3-induced down-regulation of CALR in our study may have an inhibitory effect on HBV replication.
Enrichment analysis (DAVID) showed that the most significantly down-regulated proteins following IFN-λ3 treatment were involved in cell motility and cell adhesion. We believe these processes could be relevant to the effects of IFN-λ3 treatment on liver cancer. Tan et al. demonstrated that HBV-transfected cells showed morphological changes because of formation of filopodia and lamellipodia, leading to cell migration. Of interest, CHB patients have 5–15-fold increased risk of HCC, a characteristic of which is increased cell motility (90, 91). CDC42, down-regulated on IFN-λ3 treatment, has been reported to control cell proliferation, adhesion, and metastases. High expression of this protein was observed in several types of cancers including HBV-related HCC. Inhibition of CDC42 by CRISPR/Cas9 knockout and treatment with a specific CDC42 inhibitor in HBx-Huh7 cells (92) reduced cell proliferation and promoted apoptosis in these cells. In addition to CDC42 down-regulation, we also found that its downstream effectors, actin and IQGAP1, showed decreased expression following IFN-λ3 treatment. Also, proteins involved in cell movement and focal adhesion such as ACTN1, ACTN4, RDX, CFL1, PIP4K2C, and ROCK2 were down-regulated.
We have shown that IFN-λ3's effects were largely in accord with those expected of canonical IFN activity. Nevertheless, our tree and cluster analyses (Fig. 12 and 13) highlight several differential effects between IFN-λ3 and IFN-α2a worthy of further investigation. Most obviously, despite the division of treatment outcomes into 27 non-intersecting protein groups, unbiased Reactome enrichment analysis repeatedly elicited an RNA-metabolism/translation theme. This broad theme can be divided into Reactome sub-groups (e.g. metabolism of RNA, major pathway of rRNA processing in the nucleolus and cytosol, translation, transcription-coupled nucleotide excision repair (TC-NER), and peptide chain elongation). However, we could not specifically find IFN-λ3- or IFN-α2a dominant patterns in these sub-groups, suggesting that investigation of this broad theme might best be undertaken at the level of individual proteins. Perhaps the strongest general observation would be a tendency for involvement of ER-associated proteins (GO “establishment of protein localization to endoplasmic reticulum,” which is primarily composed of ribosomal proteins) in cases where IFN-λ3 treatment results in significantly greater protein abundance versus IFN-α2a; in the 5 outcomes where such an effect would be discernable, -log(p) = 4.7, 3.2, 4.8, 8.2, and 5.3. On the other hand, such significance (-log(p) = 4.1) is seen only once out of six outcomes where IFN-α2a treatment results in significantly more abundant protein expression. This result could imply differential regulation of these ER targeting proteins on IFN-λ3 versus IFN-α2a treatment. Altered regulation of these ER proteins could interfere with viral replication (93, 94). It is interesting to note that some viruses utilize the ER as a replication compartment, whereas others do not. Thus, our work suggests that in addition to IFN′s role in regulating ER-targeting, one IFN treatment might be more appropriate than another for infections. Of other Reactome groups that emerged during clustering, we find the “hypusine synthesis from eIF5A-lysine” case especially interesting, as it is only associated with a single outcome group, that in which λ3<c, α2a <c, λ3< α2a (i.e. the two IFNs cause down-regulation, but IFN-λ3's effect is most potent). Hypusine, a rare amino acid, is apparently found only once in the human proteome, as a modification of eukaryotic initiation factor 5 (eIF5A), with proviral implications for HIV and Ebola (95). Thus, IFN-λ3's antiviral effect may be especially potent in down-regulating hypusination. The case of hypusination also illustrates how IFN-λ3-induced alterations to the RNA-metabolism/translation machinery could target viruses.
At the level of individual studies, we note that, in addition to the aforementioned λ3/α2a comparison, Bolen's data (16) also allows a λ3/λ2 comparison; here, up-regulated transcripts aligned strongly with our own λ3/c up-regulated proteins (p value = 10−27), suggesting possible differential effects even within the IFN-λ subgroup. As a side note, IFITM3, strongly up-regulated in our own λ3/α2a comparison (outcome #1), was the single most significantly up-regulated transcript in the λ3/λ2 comparison (p value = 10−10). Having noted possible IFN-λ3 versus IFN-α2a differences, we should reiterate the points that: 1) our experimental design did not allow for discrimination of kinetic effects (e.g. those caused over time by differing receptor/ligand affinities and half-lives) versus substantial alterations at the level of pathways and 2) the effects of IFN-λ3 and IFN-α2a were indeed substantially similar, especially with regard to canonical IFN effects such as regulation of antiviral proteins, antigen processing/presentation, and the RIG-I signaling pathway. A recent work does make a strong case for a differential λ3/α2a effect on murine intestinal epithelial cells (96), but the effect is pronounced only on polarization of cells, and the underlying microarray data does not correlate with our proteomic data. Thorough studies focused on possible differential effects on PTMs may help resolve these questions; the immediate result of IFN-receptor/ligand interaction is, after all, a phosphorylation event. Comprehensive identification of phosphorylation events and other kinds of PTMs will help us to elucidate possible differential effects in different IFN treatment.
In conclusion, our study found that IFN-λ3 exhibited anti-HBV activities by significant inhibition of HBV replication and expression of HBV RNA. We used high-throughput quantitative proteomics to obtain a comprehensive understanding of molecular events upon treatment of HepG2.2.15 with IFN-λ3. To our knowledge, in fact, this study is the most comprehensive proteomics-based analysis of IFN treatment to date. For the first time, we reported significant up-regulation of immunoproteasome components, restoration of HBV-inhibited RIG-I pathway proteins, as well as several proteins not previously associated with IFN-λ3 treatment. Further study of these proteins is required. Clearly, IFN-λ3 exhibited both antiviral and immunomodulatory effects to inhibit HBV replication; therefore, IFN-λ3 is an attractive novel candidate for CHB treatment and the altered proteins might be new therapeutic targets in CHB infection.
DATA AVAILABILITY
The mass spectrometry proteomics data, including annotated spectra for all modified peptides and proteins identified on the basis of a single peptide, have been deposited to: (1) the ProteomeXchange Consortium via the PRoteomics IDEntifications (PRIDE) partner repository with the data set identifier PXD007896 and (2) the MS-Viewer (http://msviewer.ucsf.edu/prospector/cgi-bin/msform.cgi?formmsviewer) with the following keys: dhjzinh2g0, tjnx2fzkzu, 3xzzxfanwm, ac4wmxx0tv, and ao9nga6qqm.
Supplementary Material
Footnotes
* This work was supported by Chulalongkorn Academic Advancement into Its 2nd Century (CUAASC) Project, Thailand Research Fund (TRF) for Research Career Development Grant (RSA 5880014), Government Budget to Chulalongkorn University (Fiscal year 2015), The 100th Anniversary Chulalongkorn University Fund for Doctoral Scholarship, The 90th Anniversary Chulalongkorn University Fund (Ratchadaphiseksomphot Endowment Fund), Center of Excellence in Systems Biology, and Center of Excellence in Immunology and Immune-mediated Diseases. KH's research is supported by Rachadapisek Sompot Fund for Postdoctoral Fellowship, Chulalongkorn University.
 This article contains supplemental Tables.
This article contains supplemental Tables.
1 The abbreviations used are:
- HBV
- hepatitis B virus
- CHB
- chronic hepatitis B
- HCC
- hepatocellular carcinoma
- pgRNA
- pregenomic RNA
- IFN-α2a
- interferon-alpha2a
- IFN-λ3
- interferon-lambda3
- ISG
- interferon-stimulated gene
- SDC
- sodium deoxycholate
- qPCR
- quantitative polymerase chain reaction
- GO
- gene ontology
- FDR
- false discovery rate.
REFERENCES
- 1. Schweitzer A., Horn J., Mikolajczyk R. T., Krause G., and Ott J. J. (2015) Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 386, 1546–1555 [DOI] [PubMed] [Google Scholar]
- 2. Grimm D., Thimme R., and Blum H. E. (2011) HBV life cycle and novel drug targets. Hepatol. Int. 5, 644–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wright T. L. (2006) Introduction to chronic hepatitis B infection. Am. J. Gastroenterol. 101, S1–6 [DOI] [PubMed] [Google Scholar]
- 4. (2017) EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 67, 370–398 [DOI] [PubMed] [Google Scholar]
- 5. Yuen M. F., and Lai C. L. (2011) Treatment of chronic hepatitis B: Evolution over two decades. J. Gastroenterol. Hepatol. 26, 138–143 [DOI] [PubMed] [Google Scholar]
- 6. Lau D. T. Y., and Bleibel W. (2008) Current status of antiviral therapy for Hepatitis B. Therap. Adv. Gastroenterol. 1, 61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fung J., Lai C. L., Seto W. K., and Yuen M. F. (2011) Nucleoside/nucleotide analogues in the treatment of chronic hepatitis B. J. Antimicrobial Chemother. 66, 2715–2725 [DOI] [PubMed] [Google Scholar]
- 8. Phillips S., Mistry S., Riva A., Cooksley H., Hadzhiolova-Lebeau T., Plavova S., Katzarov K., Simonova M., Zeuzem S., Woffendin C., Chen P.-J., Peng C.-Y., Chang T.-T., Lueth S., De Knegt R., Choi M.-S., Wedemeyer H., Dao M., Kim C.-W., Chu H.-C., Wind-Rotolo M., Williams R., Cooney E., and Chokshi S. (2017) Peg-interferon lambda treatment induces robust innate and adaptive immunity in chronic Hepatitis B patients. Front. Immunol. 8, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan H. L. Y., Ahn S. H., Chang T. T., Peng C. Y., Wong D., Coffin C. S., Lim S. G., Chen P. J., Janssen H. L. A., Marcellin P., Serfaty L., Zeuzem S., Cohen D., Critelli L., Xu D., Wind-Rotolo M., and Cooney E. (2016) Peginterferon lambda for the treatment of HBeAg-positive chronic hepatitis B: A randomized phase 2b study (LIRA-B). J. Hepatol. 64, 1011–1019 [DOI] [PubMed] [Google Scholar]
- 10. Dellgren C., Gad H. H., Hamming O. J., Melchjorsen J., and Hartmann R. (2009) Human interferon-lambda3 is a potent member of the type III interferon family. Genes Immunity 10, 125–131 [DOI] [PubMed] [Google Scholar]
- 11. Pestka S. (1997) The interferon receptors. Sem. Oncol. 24, S9-18-S19-40 [PubMed] [Google Scholar]
- 12. de Weerd N. A., Samarajiwa S. A., and Hertzog P. J. (2007) Type I Interferon Receptors: Biochemistry and Biological Functions. J. Biol. Chem. 282, 20053–20057 [DOI] [PubMed] [Google Scholar]
- 13. Miller D. M., Klucher K. M., Freeman J. A., Hausman D. F., Fontana D., and Williams D. E. (2009) Interferon lambda as a potential new therapeutic for hepatitis C. Ann. N.Y. Acad. Sci. 1182, 80–87 [DOI] [PubMed] [Google Scholar]
- 14. Ramos E. L. (2010) Preclinical and clinical development of pegylated interferon-lambda 1 in chronic hepatitis C. J. Interferon Cytokine Res. 30, 591–595 [DOI] [PubMed] [Google Scholar]
- 15. Jilg N., Lin W., Hong J., Schaefer E. A., Wolski D., Meixong J., Goto K., Brisac C., Chusri P., Fusco D. N., Chevaliez S., Luther J., Kumthip K., Urban T. J., Peng L. F., Lauer G. M., and Chung R. T. (2014) Kinetic Differences in the Induction of Interferon Stimulated Genes by Interferon-α and IL28B are altered by Infection with Hepatitis C Virus. Hepatology 59, 1250–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bolen C. R., Ding S., Robek M. D., and Kleinstein S. H. (2014) Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 59, 1262–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marcello T., Grakoui A., Barba Spaeth –G, Machlin E. S., Kotenko S. V., Macdonald M. R., and Rice C. M. (2006) Interferons α and λ inhibit Hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 131, 1887–1898 [DOI] [PubMed] [Google Scholar]
- 18. Dumoutier L., Tounsi A., Michiels T., Sommereyns C., Kotenko S. V., and Renauld J. C. (2004) Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: similarities with type I interferon signaling. J. Biol. Chem. 279, 32269–32274 [DOI] [PubMed] [Google Scholar]
- 19. Donnelly R. P., Sheikh F., Kotenko S. V., and Dickensheets H. (2004) The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J. Leukocyte Biol. 76, 314–321 [DOI] [PubMed] [Google Scholar]
- 20. Sells M. A., Chen M. L., and Acs G. (1987) Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. U.S.A. 84, 1005–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Witt-Kehati D., Bitton Alaluf M., and Shlomai A. (2016) Advances and challenges in studying Hepatitis B virus in vitro. Viruses 8, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verrier E. R., Colpitts C. C., Schuster C., Zeisel M. B., and Baumert T. F. (2016) Cell culture models for the investigation of Hepatitis B and D virus infection. Viruses 8, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J., Jiang D., Zhang H., Lv S., Rao H., Fei R., and Wei L. (2009) Proteome responses to stable hepatitis B virus transfection and following interferon alpha treatment in human liver cell line HepG2. Proteomics 9, 1672–1682 [DOI] [PubMed] [Google Scholar]
- 24. Otsuka M., Aizaki H., Kato N., Suzuki T., Miyamura T., Omata M., and Seki N. (2003) Differential cellular gene expression induced by hepatitis B and C viruses. Biochem. Biophys. Res. Commun. 300, 443–447 [DOI] [PubMed] [Google Scholar]
- 25. Xin X. M., Li G. Q., Guan X. R., Li D., Xu W. Z., Jin Y. Y., and Gu H. X. (2008) Combination therapy of siRNAs mediates greater suppression on hepatitis B virus cccDNA in HepG2.2.15 cell. Hepato-gastroenterology 55, 2178–2183 [PubMed] [Google Scholar]
- 26. Li G. Q., Xu W. Z., Wang J. X., Deng W. W., Li D., and Gu H. X. (2007) Combination of small interfering RNA and lamivudine on inhibition of human B virus replication in HepG2.2.15 cells. World J. Gastroenterol. 13, 2324–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ding X. R., Yang J., Sun D. C., Lou S. K., and Wang S. Q. (2008) Whole genome expression profiling of hepatitis B virus-transfected cell line reveals the potential targets of anti-HBV drugs. Pharmacogenomics J. 8, 61–70 [DOI] [PubMed] [Google Scholar]
- 28. Wang L. Y., Li Y. G., Chen K., Li K., Qu J. L., Qin D. D., and Tang H. (2012) Stable expression and integrated hepatitis B virus genome in a human hepatoma cell line. Gen. Mol. Res. 11, 1442–1448 [DOI] [PubMed] [Google Scholar]
- 29. Fang C., Zhao C., Liu X., Yang P., and Lu H. (2012) Protein alteration of HepG2.2.15 cells induced by iron overload. Proteomics 12, 1378–1390 [DOI] [PubMed] [Google Scholar]
- 30. Xie H. Y., Xia W. L., Zhang C. C., Wu L. M., Ji H. F., Cheng Y., and Zheng S. S. (2007) Evaluation of hepatitis B virus replication and proteomic analysis of HepG2.2.15 cell line after cyclosporine A treatment. Acta Pharmacol. Sinica 28, 975–984 [DOI] [PubMed] [Google Scholar]
- 31. Venkatakrishnan B., and Zlotnick A. (2016) The Structural Biology of Hepatitis B Virus: Form and Function. Ann. Rev. Virol. 3, 429–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu J., and Liu K. (2017) Complete and Incomplete Hepatitis B Virus Particles: Formation, Function, and Application. Viruses 9, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park I. H., Kwon Y. C., Ryu W. S., and Ahn B. Y. (2014) Inhibition of hepatitis B virus replication by ligand-mediated activation of RNase L. Antiviral Res. 104, 118–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jeong G. U., Park I. H., Ahn K., and Ahn B. Y. (2016) Inhibition of hepatitis B virus replication by a dNTPase-dependent function of the host restriction factor SAMHD1. Virology 495, 71–78 [DOI] [PubMed] [Google Scholar]
- 35. Chen Z., Zhu M., Pan X., Zhu Y., Yan H., Jiang T., Shen Y., Dong X., Zheng N., Lu J., Ying S., and Shen Y. (2014) Inhibition of Hepatitis B virus replication by SAMHD1. Biochem. Biophys. Res. Commun. 450, 1462–1468 [DOI] [PubMed] [Google Scholar]
- 36. Sommer A. F., Riviere L., Qu B., Schott K., Riess M., Ni Y., Shepard C., Schnellbacher E., Finkernagel M., Himmelsbach K., Welzel K., Kettern N., Donnerhak C., Munk C., Flory E., Liese J., Kim B., Urban S., and Konig R. (2016) Restrictive influence of SAMHD1 on Hepatitis B Virus life cycle. Sci. Rep. 6, 26616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu X., Wang J., Jin X., Huang Y., Zeng W., and Zhu J. (2015) IFN-CSP Inhibiting Hepatitis B Virus in HepG2.2.15 Cells Involves JAK-STAT Signal Pathway. BioMed Res. Int. 2015, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robek M. D., Boyd B. S., Wieland S. F., and Chisari F. V. (2004) Signal transduction pathways that inhibit hepatitis B virus replication. Proc. Natl. Acad. Sci. U.S.A. 101, 1743–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Amini-Bavil-Olyaee S., Choi Y. J., Lee J. H., Shi M., Huang I. C., Farzan M., and Jung J. U. (2013) The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 13, 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Macovei A., Petrareanu C., Lazar C., Florian P., and Branza-Nichita N. (2013) Regulation of Hepatitis B Virus Infection by Rab5, Rab7, and the Endolysosomal Compartment. J. Virol. 87, 6415–6427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kann M., Schmitz A., and Rabe B. (2007) Intracellular transport of hepatitis B virus. World J. Gastroenterol. 13, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Osseman Q., and Kann M. (2017) Intracytoplasmic transport of Hepatitis B virus capsids. Methods Mol. Biol. 1540, 37–51 [DOI] [PubMed] [Google Scholar]
- 43. Gallucci L., and Kann M. (2017) Nuclear import of Hepatitis B virus capsids and genome. Viruses 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Samuel C. E. (2001) Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sadler A. J., and Williams B. R. G. (2008) Interferon-inducible antiviral effectors. Nature Reviews. Immunology 8, 559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schoggins J. W., and Rice C. M. (2011) Interferon-stimulated genes and their antiviral effector functions. Curr. Opinion Virol. 1, 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pei R., Qin B., Zhang X., Zhu W., Kemper T., Ma Z., Trippler M., Schlaak J., Chen X., and Lu M. (2014) Interferon-induced proteins with tetratricopeptide repeats 1 and 2 are cellular factors that limit hepatitis B virus replication. J. Innate Immunity 6, 182–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sato S., Li K., Kameyama T., Hayashi T., Ishida Y., Murakami S., Watanabe T., Iijima S., Sakurai Y., Watashi K., Tsutsumi S., Sato Y., Akita H., Wakita T., Rice C. M., Harashima H., Kohara M., Tanaka Y., and Takaoka A. (2015) The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132 [DOI] [PubMed] [Google Scholar]
- 49. Ryoo J., Choi J., Oh C., Kim S., Seo M., Kim S. Y., Seo D., Kim J., White T. E., Brandariz-Nunez A., Diaz-Griffero F., Yun C. H., Hollenbaugh J. A., Kim B., Baek D., and Ahn K. (2014) The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 20, 936–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fu Y., Gaelings L., Soderholm S., Belanov S., Nandania J., Nyman T. A., Matikainen S., Anders S., Velagapudi V., and Kainov D. E. (2016) JNJ872 inhibits influenza A virus replication without altering cellular antiviral responses. Antiviral Res. 133, 23–31 [DOI] [PubMed] [Google Scholar]
- 51. Wang X., Li Y., Li L. F., Shen L., Zhang L., Yu J., Luo Y., Sun Y., Li S., and Qiu H. J. (2016) RNA interference screening of interferon-stimulated genes with antiviral activities against classical swine fever virus using a reporter virus. Antiviral Res. 128, 49–56 [DOI] [PubMed] [Google Scholar]
- 52. Meng G., Zhao Y., Bai X., Liu Y., Green T. J., Luo M., and Zheng X. (2010) Structure of human stabilin-1 interacting chitinase-like protein (SI-CLP) reveals a saccharide-binding cleft with lower sugar-binding selectivity. J. Biol. Chem. 285, 39898–39904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rubins K. H., Hensley L. E., Wahl-Jensen V., Daddario DiCaprio K. M., Young H. A., Reed D. S., Jahrling P. B., Brown P. O., Relman D. A., and Geisbert T. W. (2007) The temporal program of peripheral blood gene expression in the response of nonhuman primates to Ebola hemorrhagic fever. Gen. Biol. 8, R174–R174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zapata J. C., Carrion R. Jr, Patterson J. L., Crasta O., Zhang Y., Mani S., Jett M., Poonia B., Djavani M., White D. M., Lukashevich I. S., and Salvato M. S. (2013) Transcriptome analysis of human peripheral blood mononuclear cells exposed to Lassa virus and to the attenuated Mopeia/Lassa reassortant 29 (ML29), a vaccine candidate. PLoS Neglected Tropical Dis. 7, e2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ioannidis I., McNally B., Willette M., Peeples M. E., Chaussabel D., Durbin J. E., Ramilo O., Mejias A., and Flano E. (2012) Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J. Virol. 86, 5422–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mitchell H. D., Eisfeld A. J., Sims A. C., McDermott J. E., Matzke M. M., Webb-Robertson B. J., Tilton S. C., Tchitchek N., Josset L., Li C., Ellis A. L., Chang J. H., Heegel R. A., Luna M. L., Schepmoes A. A., Shukla A. K., Metz T. O., Neumann G., Benecke A. G., Smith R. D., Baric R. S., Kawaoka Y., Katze M. G., and Waters K. M. (2013) A network integration approach to predict conserved regulators related to pathogenicity of influenza and SARS-CoV respiratory viruses. PloS ONE 8, e69374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee S., Kopp F., Chang T. C., Sataluri A., Chen B., Sivakumar S., Yu H., Xie Y., and Mendell J. T. (2016) Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell 164, 69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Whisenant T. C., Peralta E. R., Aarreberg L. D., Gao N. J., Head S. R., Ordoukhanian P., Williamson J. R., and Salomon D. R. (2015) The Activation-Induced Assembly of an RNA/Protein Interactome Centered on the Splicing Factor U2AF2 Regulates Gene Expression in Human CD4 T Cells. PloS ONE 10, e0144409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hong S. H., Cho O., Kim K., Shin H. J., Kotenko S. V., and Park S. (2007) Effect of interferon-lambda on replication of hepatitis B virus in human hepatoma cells. Virus Res. 126, 245–249 [DOI] [PubMed] [Google Scholar]
- 60. Liu M. Q., Zhou D. J., Wang X., Zhou W., Ye L., Li J. L., Wang Y. Z., and Ho W. Z. (2012) IFN-lambda3 inhibits HIV infection of macrophages through the JAK-STAT pathway. PloS ONE 7, e35902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ank N., West H., and Paludan S. R. (2006) IFN-lambda: novel antiviral cytokines. J. Interferon Cytokine Res 26, 373–379 [DOI] [PubMed] [Google Scholar]
- 62. Pagliaccetti N. E., and Robek M. D. (2010) Interferon-lambda in the immune response to hepatitis B virus and hepatitis C virus. J. Interferon Cytokine Res 30, 585–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Donnelly R. P., and Kotenko S. V. (2010) Interferon-Lambda: A New Addition to an Old Family. J. Interferon Cytokine Res. 30, 555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Marcello T., Grakoui A., Barba-Spaeth G., Machlin E. S., Kotenko S. V., MacDonald M. R., and Rice C. M. (2006) Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 131, 1887–1898 [DOI] [PubMed] [Google Scholar]
- 65. Robek M. D., Boyd B. S., and Chisari F. V. (2005) Lambda interferon inhibits hepatitis B and C virus replication. J. Virol. 79, 3851–3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lachmann A., Torre D., Keenan A. B., Jagodnik K. M., Lee H. J., Silverstein M. C., Wang L., Ma and ayan A. (2018) Massive mining of publicly available RNA-seq data from human and mouse. Nature Communications 9, 1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Maiwald T., Schneider A., Busch H., Sahle S., Gretz N., Weiss T. S., Kummer U., and Klingmuller U. (2010) Combining theoretical analysis and experimental data generation reveals IRF9 as a crucial factor for accelerating interferon alpha-induced early antiviral signalling. FEBS J. 277, 4741–4754 [DOI] [PubMed] [Google Scholar]
- 68. Rouillard A. D., Gundersen G. W., Fernandez N. F., Wang Z., Monteiro C. D., McDermott M. G., and Ma'ayan A. (2016) The harmonizome: a collection of processed data sets gathered to serve and mine knowledge about genes and proteins. Database 2016, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Robek M. D., Wieland S. F., and Chisari F. V. (2002) Inhibition of hepatitis B virus replication by interferon requires proteasome activity. J. Virol. 76, 3570–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yao Y., Li J., Lu Z., Tong A., Wang W., Su X., Zhou Y., Mu B., Zhou S., Li X., Chen L., Gou L., Song H., Yang J., and Wei Y. (2011) Proteomic analysis of the interleukin-4 (IL-4) response in hepatitis B virus-positive human hepatocelluar carcinoma cell line HepG2.2.15. Electrophoresis 32, 2004–2012 [DOI] [PubMed] [Google Scholar]
- 71. Ferrington D. A., and Gregerson D. S. (2012) Immunoproteasomes: structure, function, and antigen presentation. Progress Mol. Biol. Translational Sci. 109, 75–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wieland S., Thimme R., Purcell R. H., and Chisari F. V. (2004) Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. U.S.A. 101, 6669–6674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yu S., Chen J., Wu M., Chen H., Kato N., and Yuan Z. (2010) Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J. General Virol. 91, 2080–2090 [DOI] [PubMed] [Google Scholar]
- 74. Jiang J., and Tang H. (2010) Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 1, 1106–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kumar M., Jung S. Y., Hodgson A. J., Madden C. R., Qin J., and Slagle B. L. (2011) Hepatitis B Virus Regulatory HBx Protein Binds to Adaptor Protein IPS-1 and Inhibits the Activation of Beta Interferon. J. Virol. 85, 987–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hou Z., Zhang J., Han Q., Su C., Qu J., Xu D., Zhang C., and Tian Z. (2016) Hepatitis B virus inhibits intrinsic RIG-I and RIG-G immune signaling via inducing miR146a. Sci. Rep. 6, 26150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhu J., Zhang Y., Ghosh A., Cuevas R. A., Forero A., Dhar J., Ibsen M. S., Schmid-Burgk J. L., Schmidt T., Ganapathiraju M. K., Fujita T., Hartmann R., Barik S., Hornung V., Coyne C. B., and Sarkar S. N. (2014) Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 40, 936–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gack M. U., Shin Y. C., Joo C. H., Urano T., Liang C., Sun L., Takeuchi O., Akira S., Chen Z., Inoue S., and Jung J. U. (2007) TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920 [DOI] [PubMed] [Google Scholar]
- 79. Kino T., Gragerov A., Valentin A., Tsopanomihalou M., Ilyina-Gragerova G., Erwin-Cohen R., Chrousos G. P., and Pavlakis G. N. (2005) Vpr protein of human immunodeficiency virus type 1 binds to 14-3-3 proteins and facilitates complex formation with Cdc25C: implications for cell cycle arrest. J. Virol. 79, 2780–2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Aoki H., Hayashi J., Moriyama M., Arakawa Y., and Hino O. (2000) Hepatitis C virus core protein interacts with 14-3-3 protein and activates the kinase Raf-1. J. Virol. 74, 1736–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fu H., Subramanian R. R., and Masters S. C. (2000) 14-3-3 proteins: structure, function, and regulation. Ann. Rev. Pharmacol. Toxicol. 40, 617–647 [DOI] [PubMed] [Google Scholar]
- 82. Yang B., and Bouchard M. J. (2012) The hepatitis B virus X protein elevates cytosolic calcium signals by modulating mitochondrial calcium uptake. J. Virol. 86, 313–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bouchard M. J., Wang L. H., and Schneider R. J. (2001) Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 294, 2376–2378 [DOI] [PubMed] [Google Scholar]
- 84. Xia W. L., Shen Y., and Zheng S. S. (2005) Inhibitory effect of cyclosporine A on hepatitis B virus replication in vitro and its possible mechanisms. Hepatobiliary Pancreatic Dis. Int. 4, 18–22 [PubMed] [Google Scholar]
- 85. Choi Y., Gyoo Park S., Yoo J. H., and Jung G. (2005) Calcium ions affect the hepatitis B virus core assembly. Virology 332, 454–463 [DOI] [PubMed] [Google Scholar]
- 86. Coppolino M. G., and Dedhar S. (1998) Calreticulin. Int. J. Biochem. Cell Biol. 30, 553–558 [DOI] [PubMed] [Google Scholar]
- 87. Raghavan M., Wijeyesakere S. J., Peters L. R., and Del Cid N. (2013) Calreticulin in the immune system: ins and outs. Trends Immunol. 34, 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Michalak M., Corbett E. F., Mesaeli N., Nakamura K., and Opas M. (1999) Calreticulin: one protein, one gene, many functions. Biochem. J. 344 Pt 2, 281–292 [PMC free article] [PubMed] [Google Scholar]
- 89. Yue X., Wang H., Zhao F., Liu S., Wu J., Ren W., and Zhu Y. (2012) Hepatitis B virus-induced calreticulin protein is involved in IFN resistance. J. Immunol. 189, 279–286 [DOI] [PubMed] [Google Scholar]
- 90. Arjonen A., Kaukonen R., and Ivaska J. (2011) Filopodia and adhesion in cancer cell motility. Cell Adhesion Migration 5, 421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhao R., Wang T. Z., Kong D., Zhang L., Meng H. X., Jiang Y., Wu Y. Q., Yu Z. X., and Jin X. M. (2011) Hepatoma cell line HepG2.2.15 demonstrates distinct biological features compared with parental HepG2. World J. Gastroenterol.: WJG 17, 1152–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Xu Y., Qi Y., Luo J., Yang J., Xie Q., Deng C., Su N., Wei W., Shi D., Xu F., Li X., and Xu P. (2017) Hepatitis B Virus X Protein Stimulates Proliferation, Wound Closure and Inhibits Apoptosis of HuH-7 Cells via CDC42. Int. J. Mol. Sci. 18, 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Romero-Brey I., and Bartenschlager R. (2016) Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 8, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Inoue T., and Tsai B. (2013) How Viruses Use the Endoplasmic Reticulum for Entry, Replication, and Assembly. Cold Spring Harbor Perspectives Biol. 5, a013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Olsen M. E., and Connor J. H. (2017) Hypusination of eIF5A as a target for antiviral therapy. DNA Cell Biol. 36, 198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Selvakumar T. A., Bhushal S., Kalinke U., Wirth D., Hauser H., Koster M., and Hornef M. W. (2017) Identification of a predominantly interferon-lambda-induced transcriptional profile in murine intestinal epithelial cells. Front. Immunol. 8, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data, including annotated spectra for all modified peptides and proteins identified on the basis of a single peptide, have been deposited to: (1) the ProteomeXchange Consortium via the PRoteomics IDEntifications (PRIDE) partner repository with the data set identifier PXD007896 and (2) the MS-Viewer (http://msviewer.ucsf.edu/prospector/cgi-bin/msform.cgi?formmsviewer) with the following keys: dhjzinh2g0, tjnx2fzkzu, 3xzzxfanwm, ac4wmxx0tv, and ao9nga6qqm.