

Abstract
Chronic hyperlipidemia causes the dysfunction of pancreatic β-cells, such as apoptosis and impaired insulin secretion, which are aggravated in the presence of hyperglycemia. The underlying mechanisms, such as endoplasmic reticulum (ER) stress, oxidative stress and metabolic disorders, have been reported before; however, the time sequence of these molecular events is not fully understood. Here, using isobaric labeling-based mass spectrometry, we investigated the dynamic proteomes of INS-1 cells exposed to high palmitate in the absence and presence of high glucose. Using bioinformatics analysis of differentially expressed proteins, including the time-course expression pattern, protein-protein interaction, gene set enrichment and KEGG pathway analysis, we analyzed the dynamic features of previously reported and newly identified lipotoxicity- and glucolipotoxicity-related molecular events in more detail. Our temporal data highlight cholesterol metabolism occurring at 4 h, earlier than fatty acid metabolism that started at 8 h and likely acting as an early toxic event highly associated with ER stress induced by palmitate. Interestingly, we found that the proliferation of INS-1 cells was significantly increased at 48 h by combined treatment of palmitate and glucose. Moreover, benefit from the time-course quantitative data, we identified and validated two new molecular targets: Setd8 for cell replication and Rhob for apoptosis, demonstrating that our temporal dataset serves as a valuable resource to identify potential candidates for mechanistic studies of lipotoxicity and glucolipotoxicity in pancreatic β-cells.
Keywords: Diabetes, Quantification, Cell biology, Mass Spectrometry, Molecular biology, Protein Identification, beta Cells, Glucolipotoxicity, Lipotoxicity, Proteome
Pancreatic β-cell failure, one of the core events of type 2 diabetes (T2D)1 mellitus, is the result of a complex interaction between genetic, epigenetic and environmental factors (1–4). Diet excess leading to obesity is considered as the most important environmental risk factor for T2D. It has been proved that chronic higher levels of circulating free fatty acids (FFAs) are commonly associated with the deterioration of β-cell function (5, 6). Furthermore, hyperglycemia can aggravate lipotoxicity mediated by hyperlipidemia (glucolipotoxicity) (7–9).
Great effort has been devoted to understanding the mechanisms of lipotoxicity and glucolipotoxicity, including metabolic disorders, impaired cAMP generation and Ca2+ channel function, ER stress, mitochondrial dysfunction and oxidative stress, and dysregulated autophagy in β-cells (10–22). Metabolic disorders and impaired cAMP generation and Ca2+ channel function can contribute to defective insulin secretion (11, 12). Activation of ER stress and oxidation stress induce β-cell apoptosis (14–16). Moreover, recent studies suggested that the metabolites of acyl-CoAs produced by FFAs and glucose can act as the substrates for post-translational modification (PTM) of proteins, such as palmitoylation and acetylation. These PTMs change protein function, which is highly linked to the dysfunction of β-cells (23, 24). However, the interplay of the above molecular events is not well understood.
In recent decades, to understand the global molecular events of lipotoxicity/glucolipotoxicity, systems biology-oriented strategies, such as transcriptomics, proteomics, and metabolomics, have been successfully adopted to detect abundant changes of genes, proteins and metabolites in pancreatic β-cells or islets exposed to long-term FFA and glucose treatment (10, 25–30). However, these studies primarily focused on the later stages of lipotoxicity/glucolipotoxicity. Because the deterioration of β-cell function is a time-based progress, it is of great interest to look into the temporal sequences of lipotoxicity- and glucolipotoxicity-related biological events.
In this study, which aims to characterize in great detail the time course of lipotoxicity and glucolipotoxicity in pancreatic β-cells, we analyzed the protein changes of INS-1 rat insulinoma cell lines (INS-1 cells) treated with high palmitate without (PA) or with high glucose (PAG) at five consecutive time points (4, 8, 16, 24, and 48 h) by multiplex TMT (Tandem Mass Tag) labeling-based quantitative proteomics approaches. Subsequent bioinformatics analysis of differentially expressed proteins, including time-course pattern clustering, protein-protein interaction networks, gene set enrichment and KEGG pathway analysis, allowed us to recapitulate the lipotoxicity- and glucolipotoxicity-related molecular events in more detail, including previously reported and newly identified features. Moreover, further mining our quantitative data set allowed us to identify two new molecular targets, Setd8 and Rhob. By performing functional experiments, we validated that Setd8 promotes cell proliferation of INS-1 cells induced by PAG and that Rhob accelerates PA-induced β-cell apoptosis. Therefore, our temporal quantitative data set represents a valuable resource for identifying potential candidates for the mechanistic study of lipotoxicity and glucolipotoxicity in pancreatic β-cells.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatment
INS-1 cells were cultured with RPMI 1640 medium containing 11 Mm glucose, 10% fetal bovine serum (FBS), 1 mm sodium pyruvate, 50 μm 2-mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin in a humidified atmosphere of 5% CO2 at 37 °C. PA was dissolved in 0.1 m sodium hydroxide to prepare a stock solution (100 mm). The PA stock solution was diluted in culture medium to which fatty-acid-free BSA had been added in a 1:19 molar ratio to prepare BSA-conjugated PA. Cells were incubated with the BSA-conjugated-PA at 0.5 mm without or with additional 16.7 mm glucose (PA or PAG, respectively) for different times (4, 8, 16, 24, and 48 h).
Proteomics Sample Preparation
INS-1 cells were harvested at different time points under three culture conditions and then were lysed in a buffer containing 8 m urea/100 mm NH4HCO3 (Sigma, cat. no. A6141, St. Louis, MO), pH 8.3, and a protease inhibitor mixture (Sigma, cat. no. P8340). The protein concentration was determined by a bicinchoninic acid assay (BCA). A total of 200 μg of protein was first reduced by adding a final concentration of 10.0 mm dithiothreitol for 60.0 min at 37 °C and was then immediately alkylated by incubating with a final concentration of 20 mm iodoacetamide for 45 min at room temperature in the dark. The resulting protein mixtures were diluted to less than 2 m urea with 100 mm NH4HCO3 prior to enzymatic digestion overnight with sequencing-grade trypsin (Promega, cat. no. V5111, Madison, WI) at a substrate/enzyme ratio of 100:1 (w:w). Protein digestion was quenched via acidification with formic acid (FA). The resulting peptide mixtures were subsequently desalted via solid phase extraction (Sep-pack Vac C18 cartridges, Waters, cat. no. Wat020515, Manchester, UK) followed by vacuum drying.
The dried peptide samples were re-suspended in 100 mm triethylammonium bicarbonate (TEAB) buffer. A 50% aliquot of each sample was chemically labeled with 6-plex TMT reagents (Thermo Fisher, cat. no. 90066, Rockford, IL) according to the manufacturer's instructions. To accommodate all 15 samples in the analysis of a single biological replicate, we created a reference sample by mixing 1/5 of the amount of each sample (15 in total) and then labeling with TMT reagent 131. The remainder of the TMT reagents was used to label the other samples: 126 for BSA_, PA_, PAG_4h, 127 for BSA_, PA_, PAG_8h, 128 for BSA_, PA_, PAG_16h, 129 for BSA_, PA_, PAG_24h, and 130 for BSA_, PA_, PAG_48h. The reference sample was used for data normalization and dataset combination. To ensure that each of the samples contained the same amount of protein, a small 1:1:1:1:1:1 aliquot from the 6-plex reagents was prepared and analyzed by MS. Summed reporter ion ratios informed mixing ratios of the remaining labeled digests. In this way, three sets of 6-plex TMT-labeling peptides were equally pooled and were treated in parallel throughout the following steps.
Before MS analysis, mixed TMT-labeling peptides were fractionated using high-pH reversed-phase chromatography. Briefly, the samples were first desalted using Sep-Pak Vac C18 SPE cartridges (Waters) and dried in a vacuum concentrator. Desalted peptides were dissolved in solution A (2% acetonitrile, pH 10, pH adjusted with ammonium hydroxide) and were then loaded automatically onto a YMC-Triart C18 basic reversed-phase liquid chromatography column (250 × 4.6 mm, 5 μm particles) (cat. no. TA12S05–2546WT, YMC, Kyoto, Japan). Peptides were separated in a binary buffer system of solution A and solution B (98% acetonitrile, without pH adjustment, solution B) in an Ultimate-3000 LC system (Thermo Scientific). The gradient of buffer B was set as follows: 0–5% for 1 min, 5–15% for 5 min, 15–26% for 32 min, 26–40% for 22 min, 40–95% for 2 min, and 95% for 3 min. The first fraction was collected beginning at 5 min, and the remaining eluates were collected at intervals of 100 s. A total of 36 fractions was obtained, and these were concatenated into 12 fractions by merging fractions 1, 13, 25; fractions 2, 14, 26, etc. Then, all 12 peptide fractions were dried in a vacuum concentrator and stored at −20 °C until subsequent nanoLC-MS/MS analysis was performed.
Mass Spectrometry Analysis
NanoLC-MS/MS experiments were performed on a Q-Exactive mass spectrometer (Thermo Scientific) coupled to an Easy-nLC 1000 HPLC system (Thermo Scientific). The peptides were loaded onto a 100-μm id × 2-cm fused silica trap column packed in-house with reversed-phase silica (Reprosil-Pur C18 AQ, 5 μm, Dr. Maisch, GmbH, Ammerbuch, Germany) and then separated on a 75-μm id × 20-cm C18 column packed with reversed-phase silica (Reprosil-Pur C18 AQ, 3 μm, Dr. Maisch, GmbH). The loaded peptides were eluted with a 120-min gradient. Solvent A consisted of 0.1% FA in water solution, and solvent B consisted of 0.1% FA in acetonitrile solution. The following segmented gradient was used at a flow rate of 280 nl/min: 4–12% B, 5 min; 12–22% B, 80 min; 22–32% B, 25 min; 32–90% B, 1 min; and 90% B, 9 min. The mass spectrometer was operated in data-dependent acquisition mode, and full-scan MS data were acquired in the Orbitrap with a resolution of 70,000 (m/z 200) across a mass range of 300–1600 m/z. The target value was 3.0E+06 with a maximum injection time of 60 ms. After the survey scans, the top 20 most intense precursor ions were selected for MS/MS fragmentation with an isolation width of 2 m/z in the HCD collision cell and an optimized normalized collision energy of 32%. Subsequently, MS/MS spectra were acquired in the Orbitrap with a resolution of 17,500 (m/z 200) and a low-mass cut-off setting of 100 m/z. The target value was set as 1.0E+05 with a maximum injection time of 80 ms. The dynamic exclusion time was 50 s. The following nanoelectrospray ion source settings were used: spray voltage of 2.1 kV, no sheath gas flow, and a heated capillary temperature of 320 °C.
For the SPS (synchronous precursor selection)-MS3 mode-based method, nanoLC-MS/MS experiments were performed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) coupled to an Easy-nLC 1200 HPLC system (Thermo Scientific). The peptides were loaded and separated onto a 75-μm id × 20-cm C18 column packed with reversed-phase silica (Reprosil-Pur C18 AQ, 3 μm, Dr. Maisch, GmbH). The loaded peptides were eluted with a 90-min gradient. Solvent A consisted of 0.1% FA in water solution, and solvent B consisted of 0.1% FA in acetonitrile solution. The following segmented gradient was used at a flow rate of 280 nl/min: 3–12% B, 5 min; 12–30% B, 73 min; 30–90% B, 7 min; and 95% B, 5 min. The mass spectrometer was operated in data-dependent acquisition mode, and full-scan MS data were acquired in the Orbitrap with a resolution of 60,000 (m/z 200) across a mass range of 350–1500 m/z. The target value was 4.00E+05 with a maximum injection time of 50 ms. After the survey scans, the top N most intense precursor ions were selected for MS/MS fragmentation with an isolation width of 1.6 m/z in the CID collision cell and an optimized normalized collision energy of 35% (AGC target: 1E4; maximum injection time: 50 ms; and scan mode: rapid). MS3 analysis for each MS2 scan was performed by isolating the five most-intense MS2 fragment ions with a multinotch isolation waveform. MS3 spectra were detected in the Orbitrap (resolution 30,000) after high energy collision-induced dissociation (HCD) (NCE: 65%; AGC: 1E5; maximum injection time: 100 ms; and isolation window of 1.6Th).
MS Data Analysis
The raw MS data were processed with Proteome Discoverer (version 1.4, Thermo Scientific). Briefly, peptide identification was performed with the Sequest HT search engine comparing against a UniProt Rattus norvegicus database (2015.05.27, containing 29,362 entries) supplemented with all frequently observed MS contaminants (containing 247 entries). The following parameters were used for database searching: semi-tryptic, 10 ppm precursor mass tolerance, 0.02 Da fragment ion tolerance, up to two missed cleavages, carbamidomethyl cysteine, TMT modification on amino (N)-term and lysine as fixed modifications, and oxidized methionine as a variable modification. The peptide confidence was set to a high level (q-value < 0.01) for peptide filtering by Percolator (31), resulting in 1% FDR (false discovery rate) at peptide-spectrum match (PSM) level. Proteins (or protein groups) were assembled by at least one unique peptide.
To improve the accuracy and confidence of protein quantification by TMT reporters in MS2 scan, optimized data processing was developed using freely accessible tools and in-house written scripts: (1) msconvert (http://proteowizard.sourceforge.net, ProteoWizard release: 3.0.9974) was first used to perform a deconvolution of the high-resolution MS2 spectra in which all fragment ion isotopic distributions were converted to a m/z value corresponding to the monoisotopic single charge. The signals of TMT reporter ions were extracted with the following requirements: maximum mass accuracy of 15 ppm, detection of all 6 TMT reporter ion channels required. (2) The summed reported ion intensity from each channel for all acquired MS2 spectra was used for sample normalization. (3) To minimize ratio distortion because of the presence of more than one peptide species within a precursor ion isolation width, we also rejected the quantification of MS/MS spectra based on the precursor intensity fraction (PIF). PIF reported by Proteome Discoverer was calculated as a proportion of the background intensity within the isolation window that does not belongs to the precursor itself. For our data set, a PIF of 50% was selected as the optimal trade-off value for both identification and quantification (supplemental Fig. S2A). (4) If the peptides were quantified multiple times, the one(s) with the lowest PIF was/were selected to produce the representative quantitative TMT ratio. The median values of the TMT ratios of peptides from the same protein were calculated as the protein ratios.
Bioinformatics Analysis
Hierarchical clustering analysis (by R-package “pheatmap”), principal component analysis (PCA) (by built-in R function “prcomp”), two-way ANOVA analysis (by built-in R function “anova”) and multiple hypothesis testing (by built-in R function “p. adjust” with the “BH” method) were performed in R, version Windows Rx64, 3.1.3. “kmeans” embedded in “pheatmap” was utilized for protein expression clustering analysis, and the resulting patterns were further constructed by using Circos software (32) followed by DAVID (33) functional annotation analysis to assess their biological relevance. STRING (34) was used for protein-protein interaction pathways and a minimum required interaction score above 0.70 was required for high confidence. The networks were visualized with 3.3.1 (35). An R-package, “fgsea” (36), was used for fast pre-ranked gene set enrichment analysis (GSEA). Both Gene ontology (GO) and KEGG pathways were analyzed. The enriched items with q-values of less than 0.05 were considered significantly different hits. GO functional annotations were further connected as functional networks by calculating the Jaccard Coefficient as follows:
Two GO annotations with Jaccard coefficients above 0.3 were accepted as being connected.
Measurement of Cell Apoptosis
Cell apoptosis was detected by Hoechst 33258 (Sigma-Aldrich, St. Louis, MO) (37). INS-1 cells grown for 48 h were treated with BSA, PA, and PAG for different times, then cultured with 5 μg/ml Hoechst 33258 for 30 min. Fluorescent images were acquired using a confocal laser scanning microscope (FV 1200, Olympus, Tokyo, Japan). The nucleus, which is bright, and shrinkage represent the apoptotic cells. The percentage of apoptotic cells was determined in at least 5000 total cells per condition.
Measurement of Cell Proliferation
Cell proliferation was evaluated using ki67 and EdU assays. For ki67, INS-1 cells grown for 48 h were treated with BSA, PA, and PAG for different times, then fixed in 4% paraformaldehyde for 30 min at room temperature and permeabilized in 0.5% Triton X-100 for 10 min. Cells were washed with PBS and then blocked with 5% goat serum for 1 h, cultured with anti-ki67 (Abcam, Cambridge, MA) antibody for 1 h at room temperature followed by secondary antibodies (Invitrogen, Carlsbad, CA) along with DAPI (Invitrogen) for nuclear staining. In the assay the proliferation phenotype of lipotoxicity and glucolipotoxicity, fluorescent images were acquired using a widefield microscope with a 20 x objective lens (IX81, Olympus, Tokyo, Japan). In the assay the proliferation of setd8-overexpressed INS-1 cells, the fluorescent images were acquired using a confocal laser scanning microscope with 60 x objective lens (FV 1200, Olympus).
For EdU assays (GeneCopoeia, Rockville, MD), cells were plated on coverslips and exposed to 1 mm EdU for 4 h at 37 °C. The cells were then fixed in 4% paraformaldehyde for 30 min at room temperature and permeabilized in 0.5% Triton X-100 for 10 min. Cells were washed with PBS and then incubated with a reaction mixture solution for 30 min. Then, the cells were stained with DAPI and imaged under a confocal laser scanning microscope (FV 1200, Olympus).
ELISA Detection of Insulin
For glucose-stimulated insulin secretion (GSIS), INS-1 cells treated with BSA, PA and PAG for different times were pre-incubated for 2 h in 2.8 mm glucose Krebs-Ringer bicarbonate HEPES buffer (KRBB) (114 mm NaCl, 4.7 mm KCl, 1.2 mm KH2PO4, 1.16 mm MgSO4, 0.5 mm MgCl2, 2.5 mm CaCl2, and 20 mm HEPES with 0.2% BSA, pH 7.4). The cells were then stimulated with 2.8 mm and 16.7 mm glucose KRBB for 1 h. The secreted insulin in KRBB was assayed with the insulin ELISA kit. The protein content in the cells was used for normalization.
For detection of insulin content, INS-1 cells treated with BSA, PA, and PAG for different times were pre-incubated for 2 h in 2.8 mm glucose KRBB, then were lysed with RIPA buffer supplemented with proteinase inhibitor (Sigma-Aldrich) and centrifuged at 10,000 × g for 15 min at 4 °C. The supernatant was assayed with an insulin ELISA kit (Shibayagi, Shibukawa, Japan, AKRIN-011S).
Quantitative PCR Analysis of Gene Expression
Total RNA was extracted using Trizol (Invitrogen) according to the manufacturer's instructions. Reverse transcription (RT) using random hexamers and oligo dT mix primers and SuperRT reverse transcriptase (CWBIO, Beijing, China) was carried out according to the manufacturer's instructions. Quantitative PCR was carried out with a 1:20 dilution of cDNA and 2× SYBR Green PCR Mix in combination with 10 mm specific primers (supplemental Table S1). The GAPDH primer was used for normalization and ΔCt was calculated to derive the relative expression. All quantitative RT-PCR data presented are the mean ± S.E. from three independent experiments.
Western Blotting (WB) Analyses and Antibodies
Whole-cell lysates prepared using RIPA buffer with proteinase inhibitor were separated by SDS-PAGE, transferred onto PVDF membranes (Millipore, Billerica, MA), and incubated with primary antibodies against Rhob (Santa Cruz Biotechnology), Setd8 (Proteintech, Wuhan, China), β-actin (Sigma-Aldrich), β-tubulin (Proteintech) and HA (Thermo Fisher Scientific, Waltham, MA) with the appropriate HRP-conjugated secondary antibodies (Sungene Biotech, Tianjin, China) followed by detection with enhanced luminescence (GE Healthcare, Piscataway, NJ).
Gene Knock-out and Overexpression
The targeted Rhob sgRNA expression oligos were introduced into the CRISPR-Cas9 with green fluorescent protein (GFP) fusion protein expression vector pX458 (Addgene). The sequences of the oligos are shown in supplemental Table S1. The pX458 plasmid DNA containing the target sgRNA sequence was transfected into INS-1 cells with lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 48 h, GFP positive cells were sorted with flow cytometry to generate single clones. Each colony was passaged and genotyped. DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen). The genomic region surrounding the CRISPR/Cas9 target site was PCR amplified, and PCR products were purified using the Gel Extraction Kit (CWBIO, Beijing, China) according to the manufacturer's protocol and sequenced.
HA-Rhob overexpression in INS-1 cell lines was constructed by retroviral plasmid pQCXIP. Briefly, the HA-Rhob PCR fragments were amplified from the cDNA of INS-1 cells using a forward primer bearing an HA tag and a reverse primer (supplemental Table S1), then cloned into the retroviral plasmid pQCXIP vector. The retrovirus was packaged via transfecting Plat E cells with the HA-Rhob plasmids and two packaging plasmids (vsvg and phit). At 48 h post transfection, the virus-containing supernatant was collected, filtered and used to infect INS-1 cells. The infected cells were selected and maintained with puromycin to generate stable cell lines.
Setd8 overexpression in INS-1 cell lines was constructed by modified lentiviral plasmid pCDH-CMV-MCS-EF1-Puro (EGFP-P2A). Setd8 PCR fragments were amplified from the cDNA of INS-1 cells using a forward primer and a reverse primer (supplemental Table S1), then cloned into the modified lentiviral plasmid pCDH-CMV-MCS-EF1-Puro (EGFP-P2A) vector. Lentivirus was packaged via transfecting 293T cells with the EGFP-P2A-Setd8 plasmids and two packaging plasmids (vsvg and Δ3.1). Forty-eight hours post-transfection, the virus-containing supernatant was collected, filtered and used to infect INS-1 cells. The infected cells were selected with puromycin for 1 week. EGFP positive cells were sorted with flow cytometry to generate stable cell lines.
Experimental Design and Statistical Rationale
The purpose of this study was to analyze the temporal proteome in INS-1 cells treated with PA in the presence or absence of high glucose. Because aqueous-soluble palmitic acid was prepared by conjugating to bovine serum albumin (BSA), we selected the culture medium containing BSA as the control. The experimental design comprised three biological replicates. The 6-plex TMT reagents were used for quantitative proteomics. The MS data of all three biological samples were acquired with MS2 mode by a Q-Exactive instrument (Experiment 1, Expt 1), and the same TMT-labeling samples of two biological replicates in Expt 1 were re-analyzed with SPS-MS3 mode by a LUMOUS-fusion instrument (Experiment 2, Expt 2). Protein identification was performed by Proteome Discoverer (PD) 1.4 software. Protein quantification was analyzed by in-house written scripts by either MS2 data pre-filtering by the precursor intensity fraction (PIF ≤ 50%) (38) or MS3 data. The change of protein expression was calculated as an average value from Exp1 and Exp2. Two-way ANOVA was utilized to analyze differentially expressed proteins. All p values were corrected for multiple hypothesis testing by the Benjamini-Hochberg (BH) method.
RESULTS AND DISCUSSION
The Time-course Phenotypes of INS-1 Cells Exposure to PA and PAG
To choose reasonable time points for capturing the molecular events of lipotoxicity and glucolipotoxicity at the proteome level, we first evaluated cell apoptosis and insulin secretion of INS-1 cells. Cell apoptosis and insulin secretion are the two main phenotypes of lipotoxicity/glucolipotoxicity in INS-1 cells treated with PA and PAG at different times (27). As depicted in supplemental Fig. S1A, apoptosis in INS-1 cells induced by PA was significant at 16 h and increased gradually with time. Compared with PA, PAG induced apoptosis in INS-1 cells earlier (8 h) with a higher apoptosis rate. Similarly, PAG treatment inhibited GSIS in INS-1 cells earlier (8 h) and impaired GSIS more severely than PA-treatment (supplemental Fig. S1B–S1D). However, both PA and PAG reduced insulin content of INS-1 cells as early as 8 h (supplemental Fig. S1E). This discoordination of time-course features between GSIS and intracellular insulin content suggests that reduction of insulin content alone cannot account for impaired insulin secretion.
Multiplexed Quantitative Analysis of the Proteome in INS-1 cells Exposure to PA and PAG
Based on the above phenotypic results, we subsequently designed a multiplexed TMT peptide labeling-based quantitative proteomics experiment to analyze the protein expression of INS-1 cells under three different culture conditions (BSA, PA, and PAG) at five consecutive time points (4, 8, 16, 24, and 48 h, Fig. 1A). The period at 4 h was added to capture earlier molecular events. To compare the protein expression changes across 45 samples in all three biological replicates, one reference sample was created by pooling from each sample for data normalization and combination. Off-line high-pH reversed-phase chromatography was carried out to pre-fractionate the combined TMT-labeling peptide mixtures, and each resulting fraction was subjected to 1D-LC-MS/MS analysis. The samples from all three biological replicates were analyzed by Q-Exactive mass spectrometry with MS2 mode (Expt 1), whereas the same samples from two biological replicates were reanalyzed by Orbitrap Fusion Lumos Tribrid Mass Spectrometer with SPS-MS3 mode (Expt 2) for more accurate TMT quantification (39). All MS data files were carefully processed in sequential steps as described in the Experimental Procedures section. Finally, by requiring a maximum FDR of 1% at PSM level by Percolator (31) and at least one unique peptide included, 9065 proteins and 8233 proteins were identified by Expt 1 and Expt 2, respectively (supplemental Fig. S2B, supplemental Table S2), representing deep proteome coverage of INS-1 cells. A total of 5214 proteins (at least two biological replicates and peptides of precursor intensity fraction ≤ 50%) and 4753 proteins (both two biological replicates) were quantified by Expt 1 and Expt 2, respectively. The hierarchical clustering analysis showed the quantitative results in Exp1 and Exp2 is well-consistent (supplemental Fig. S2C). Finally, we obtained 5531 proteins by combining two data sets as quantifiable species for further analysis.
Fig. 1.

A, The temporal quantitative proteomic analysis of INS-1 cells treated by high palmitate without or with high glucose at five different time points (4, 8, 16, 24, and 48 h). INS-1 cells cultured with BSA were used as control. The experimental workflow included protein extraction, tryptic digestion, 6-plex TMT peptide labeling, offline peptide separation via high-pH reversed-phase chromatography, and 1D-LC-MS/MS analysis of each fraction. B, PCA analysis of all quantifiable proteins (n = 5331) showed the effect of different treatments (PC1 axis) and times (PC2 axis) on the proteome of INS-1 cells. The quantitative result of protein was calculated as a relative TMT ratio normalized by reference samples. C, Unsupervised hierarchical clustering of quantitative changes of DE proteins (n = 783) in PA-/PAG-treated versus BSA-treated INS-1 cells at five time points. The Z-score was transformed from log2 fold change of DE protein by mean normalization.
To investigate the effect of PA and PAG on the proteome of INS-1 cells, we next performed a principal component analysis (PCA) of all quantitative data (Fig. 1B). The data from BSA-cultured INS-1 cells were also analyzed serving as the basal change of proteome at five time point. Interestingly, the PC2 axis clearly indicated the time-course behaviors of the proteomes in INS-1 cells from 4 h to 48 h, which were similar under both PA- and PAG-treatment. The PC1 axis demonstrated the degree of the similarity between PA- and PAG-treated INS-1 cells at each time point. The distance of proteomes in INS-1 cells treated with BSA, PA and PAG was relatively close at the early time points of 4 h and 8 h, whereas it was separated at late time points, particularly at 48 h. These results verified the dynamic features of lipotoxicity and glucolipotoxicity at different stages.
Because of our time-resolved expression data set, we were able to analyze the temporal significance of protein expression changes using two-way ANOVA by considering time (5 different time points) and treatment (BSA, PA, and PAG) as the two statistical factors. Using the filtering criteria of both log2-fold changes above 0.2 and adjusted p values by the BH (Benjamini-Hochberg) method less than 0.01, we quantified 783 differentially expressed (DE) proteins (supplemental Table S2). Unsupervised hierarchical clustering (Fig. 1C) analysis of these DE proteins showed that the hierarchical matrix was divided into three groups: one cluster of 4 h and 8 h subdivided into their own subgroups of PA and PAG treatment, a second cluster of PA treatment comprising 16, 24, and 48 h, and a third separate cluster of PAG treatment comprising 16, 24, and 48 h. This co-segregation indicated that, at the protein level, the response of INS-1 cells to PA and PAG was similar for a short treatment at 4 or 8 h but was different when the time of treatment was prolonged for more than 16 h, demonstrating that our data unbiasedly unveiled the dynamic behavior of protein expression during the course of lipotoxicity and glucolipotoxicity in INS-1 cells.
Temporal Characterizations of Lipotoxicity/Glucolipotoxicity in INS-1 Cells
Next, we performed deep analysis of our temporal proteome data using several bioinformatics approaches.
Protein Expression Time-course Pattern Analysis
We first performed the time-course pattern analysis of PA-DE and PAG-DE proteins by k-means clustering method and successfully identified 10 protein expression patterns (PA1-PA5, PAG1-PAG5) (Fig. 2A). For better visualization, we created Circos plots (32) to illustrate the similarity and differences that arose from the pattern analysis. As expected, the overlapped PA- and PAG-DE proteins, displayed as links by Circos, were mostly clustered in a similar time-course pattern, such as PAG1 and PA4, PAG2 and PA3, PAG4 and PA1, and PAG3 and PA5, whereas PA2 and PAG5 were identified as PA- and PAG-specific patterns, respectively. Subsequent DAVID analysis of proteins within each pattern was performed for functional enrichment annotations (supplemental Table S3). Accordingly, we were able to classify the enriched annotations into the following categories: (1) Constant-up proteins (PAG1, PA4), that were upregulated at all time points. The most-emphasized annotation was cholesterol metabolism (Ldlr, Hmgcs1 and Idi1), which was upregulated by both PA and PAG at the early time point. In addition, lysosomes and microtubules were also constantly upregulated in PAG-treated INS-1 cells. (2) Medium-up proteins (PA3 and PAG2), that were upregulated later than 8 h. Fatty acid metabolism including fatty acid degradation (Acsl3, Acsl4, Acadl, Acadvl, Capt1a, Eci2, and Acads), biosynthesis of unsaturated fatty acids (Acot2, Fads1, and Fasd2), and steroid biosynthesis (Msmo1, Hsd17b7, and Nsdhl) were highly enriched and upregulated by both PA and PAG. Chronic oxidative stress as a key mechanism for lipotoxicity and glucolipotoxicity was harmful to pancreatic β-cell function in vivo and in vitro (40, 41). Oxidoreductase and NADP were highly enriched in this pattern. For example, Cat, an important antioxidant enzyme, gradually upregulated by both PA and PAG, serves a protective role against oxidative stress. Moreover, we found three apoptosis-associated proteins, Cd40, Txnip, and Bclaf1, were specifically upregulated in PAG2. Two of these three, Cd40 and Txnip, have been validated as apoptotic contributors to glucolipotoxicity (9, 42), but Bclaf1 has not been investigated, thus, likely serving as a new potential anti-apoptosis target of glucolipotoxicity. (3) Medium-down proteins (PA1 and PAG4) that were downregulated by PA and PAG beginning from 16 h. The most noticeable feature is the downregulation of insulin secretion (Ins1, Ins2, Pdx1, and Prkca), indicating that the development of impairment in GSIS by PA and PAG is a staged process. In addition, we found that Ero1b (endoplasmic reticulum oxidoreductin-1-like β), a pancreas-specific disulfide oxidase for insulin biogenesis (43), was also downregulated in this pattern, indicating that disregulated protein folding of insulin also contributes to defects in GSIS. (4) Early down proteins (PA5 and PAG3) that were downregulated at early time points. Interestingly, two intracellular Ca2+-releasing channels, Itpr1 (Inositol 1,4,5-trisphosphate receptor type 1) and Itpr3 that can modulate β-cell apoptosis and ER stress resulting from Ca2+-leaking (44), were downregulated by PA and PAG. This may attenuate ER stress caused by PA-induced ER Ca2+ depletion (45). (5) Later-down (PAG5), representing a PAG-specific pattern that was downregulated at late time points of 24 or 48 h. The chaperonin-containing CCT-complex (CCT7, TCP1, CCT4, CCT8, CCT2, CCT6A, and CCT3), which is mainly involved in the folding of actin and tubulin (46), was identified as the most enriched function. Compared with a previous proteomic study that identified CCT3 only (27), our study quantified almost all components of CCTs, demonstrating in-depth coverage of our quantitative proteomic data. (6) Fluctuated proteins (PA2), many of which were functionally associated with transcription regulation.
Fig. 2.

A, Time-course dynamic expression pattern analysis of DE proteins. Fold changes of DE proteins in PA- and PAG-treated INS-1 cells were first transformed into Z-scores by mean normalization. k-means clustering was then applied to classify the genes into 6 PA- and PAG-protein patterns, illustrated as a Circos figure. The DE proteins that were identified in both PA and PAG patterns were highlighted as linked lines (pink color). Functional enrichment analyses of the proteins within each pattern were analyzed by DAVID. B, PPI analysis of DE proteins. All DE proteins were submitted to the STRING database and only PPIs with high confidence (minimum required interaction score above 0.70) were retrieved to reconstruct the sub-networks (SN) by Cytoscape. The edges between two nodes (DE proteins) were color coded according to the above protein expression patterns (A) indicated by hierarchical clustering of centered values of each pattern.
We further investigated latent associations among these patterns by submitting all DE proteins to PPI analysis employing the STRING database. Only PPIs with high confidence (minimum required interaction score above 0.70) were retrieved for the reconstruction of PPI (Fig. 2B). Based on DAVID functional annotations of DE proteins, several sub-networks (SN) were highlighted: SN1, CCT complex (PAG5); SN2, fatty acid metabolism (PAG2 and PA3); SN3, cholesterol metabolism (PAG2 and PA3, PAG1 and PA4); SN4, insulin secretion (PAG4 and PA1); SN5, glycerophospholipid biosynthesis (PAG2 and PA3); SN6, mRNA processing (PAG2); SN7, ribosome (PAG5); and SN8, cell cycle (PAG2 and PA5). By integrating the results of time-course patterns and PPI networks, we were able to identify several new features of INS-1 cells treated with PA and PAG. First, interestingly, several cell cycle-associated DE proteins (SN8) were highly interacted. Among these, several proteins (Damp1, Smarca1, Smarca2, Kif20a, and Setd8) were specifically upregulated at 48 h in the PAG-treated cells, whereas the others (Kifc1, Kif20b and Cdca8) were specifically medium downregulated in the PA-treated cells. Such distinctions suggested that cell proliferation of INS-1 cells may differ when treated by PA and PAG. To verify this, we measured the cell proliferation rate of INS-1 cells under the culture medium of BSA (as control), PA and PAG. As expected, compared with PA, PAG promoted the growth of INS-1 cells at 24 h and further increased it at 48 h (supplemental Fig. S1F), strongly supporting the above bioinformatics results. This is consistent with a recent in vivo study reporting that glucose and fatty acids promote cell proliferation in part via direct action on the β-cell and independently from secreted insulin (47). In addition, we identified several DE proteins in SN8 serving as an unbiased resource for additional mechanistic studies of PAG-promoted cell proliferation. Second, time-course pattern analysis revealed cholesterol metabolism (Constant-up, PA4, and PAG1) was upregulated at 4 h, earlier than lipid metabolism (Medium up, PA3 and PAG2) at 8 or 16 h. One recent study demonstrated that cholesterol could injure pancreatic β-cells by the endoplasmic reticulum (ER) stress/autophagy pathway (34). Therefore, we speculate that increased cholesterol metabolism probably was the early cause of PA-induced ER stress in INS-1 cells by changing the ER membrane lipid composition (38). Third, several tightly interacting RNA binding proteins (SN6), responsible for mRNA polyadenylation (Cstf1 and Cpsf4), mRNA splicing (Hnrnpm, Hnrnpu, and Snw1) and nuclear mRNA export (Thoc1, Thoc5, and Zc3h11a) were identified in cluster PAG2. Although little is known about the role of these proteins in glucolipotoxicity, most of these proteins, such as Cpsf4, Snw1, and Thoc1, were previously reported to be associated with cell proliferation (48–51). Therefore, one possible role of RNA binding proteins may be to stabilize and modulate the translation of numerous target mRNAs involved in INS-1 cell proliferation.
Gene Set Enrichment Analysis (GSEA)
We performed GSEA (52), a more extensive annotation foundation than gene ontology enrichment analysis, by considering the ranking of all genes in the data set, to identify enriched GO annotations (supplemental Table S4). To facilitate comparisons, GO networks were further constructed by building connections between two GO Terms, which shared more than 30% DE proteins (Fig. 3A). The resulting time-course patterns clearly showed that lipid and cholesterol metabolism (Group 1) were regulated by PA and PAG in a similar way, whereas regulation of cell division and gene regulation (Group 2 and 3) were different, particularly at the latter time point, when they were downregulated by PA but upregulated by PAG. However, protein kinase cascade and cell communication (Group 4) were only downregulated by PA. Furthermore, we performed GSEA using the KEGG PATHWAY Database (53) and identified 29 pathways significantly enriched (q-value < 0.05) at least one time point by either PA- or PAG-treatment (Fig. 3B, supplemental Table S4). As with our phenotypic data of GSIS, PAG significantly inhibited the insulin secretion pathway than PA, demonstrating the accuracy of our quantitative proteome data. Additionally, we found that the biosynthesis of unsaturated fatty acids (UFAs) was constantly upregulated by both PA and PAG. It was reported that exogenous UFAs are not toxic to rat insulin-producing cells and can even protect cells against PA-induced lipotoxicity (54); therefore, increasing the biosynthesis of endogenous UFAs in the INS-1 cells might play a self-protection role by relieving lipotoxicity/glucolipotoxicity. In addition, when inspecting the details of the KEGG pathway analysis more closely, we found that FFA metabolism-related DE proteins, including Acsl3, Acsl4, Acadl, Cpt1a and Fads2, were mapping into the downstream products of the PPAR pathway, suggesting that PA- and PAG-induced fatty acid metabolism is PPAR pathway-dependent. Glutathione metabolism (Ggt7, Gstm1 and Gstm5) was also upregulated by responding to the oxidative stress of lipotoxicity/glucolipotoxicity.
Fig. 3.

Temporal sequences of gene ontology biological process (A) and KEGG pathways by the GSEA method using the Bioconductor package “fgsea.” GO terms and pathways with adjusted p values (Benjamini and Hochberg method) of less than 0.05 are presented. Enrichment scores representing the average direction of fold change were scaled as either circle (A) or heatmap (B).
Taken together, the above bioinformatics analysis of our temporal proteome dataset allows us to characterize in great depth the dynamic features of lipotoxicity and glucolipotoxicity in INS-1 cells. These features include protein and biological functions/pathways that have been reported in the literature as well as features newly identified in our study, demonstrating that our proteomic data were unbiased for unveiling the molecular mechanisms associated with lipotoxicity and glucolipotoxicity in INS-1 cells.
Biological Validation
Compared with fixed-time data, our temporal data covers the dynamic changes of the proteome during lipotoxicity and glucolipotoxicity in INS-1 cells, so it has potential merits in identifying the new candidates functionally related to lipotoxicity/glucolipotoxicity. Thus, through detailed correlation analysis of the phenotypic and proteomic data, we identified two new molecular targets and further carried out follow-up experimental validation.
Setd8 Promotes PAG-stimulated INS-1 Cell Proliferation
The results of both PPI and GSEA indicated that PAG increased the proliferation of INS-1 cells, which was also validated by our experimental evidence (supplemental Fig. S1F). We therefore proceeded to perform further validation at molecular levels. We first compared our proteome evidence with recent published transcriptomes of replicating pancreatic β-cells in vivo (55). A total of 62 overlapped genes was identified as DE proteins in our study, in which the cell replication marker, Mki67, was included. More interestingly, most of these DE proteins were upregulated at 48 h only by PAG (supplemental Fig. S3). Because histone methylation is one of the remarkable molecular events during the cell cycle, we focused on one protein-lysine N-methyltransferase, Setd8 (Kmat5), for further validation. It was reported that Setd8 can specifically monomethylate 'Lys-20′ of histone H4 (H4K20me1), which subsequently functions as a key player in the epigenetic regulation of genomic integrity (56, 57). We next explored the role of Setd8 in the promotion of proliferation of INS-1 cells by PAG. First, using quantitative RT-PCR and Western blotting, we measured the mRNA and protein expression of Setd8 (Fig. 4A, 4B) to verify the quantitative proteome data. Subsequently, to test the effect of exogenous Setd8 on cell replication of INS-1 cells, we constructed a Setd8-overexpressed INS-1 cell line (Fig. 4C) and then measured the cell proliferation rate by Ki67 immunofluorescence and EdU cell proliferation assay. Compared with control cells, INS-1 cells overexpressing Setd8 were detected more as determined by the EdU+ and Ki67+ cell (Fig. 4D). By contrast, when Setd8 was inhibited by its specific inhibitor, UNC0379 (58–60), Ki67+ INS-1 cells were significantly reduced, regardless of whether the cells were cultured with PAG or not (Fig. 4E). These results indicated that Setd8 can promote cell replication of INS-1 cells induced by PAG.
Fig. 4.

A, Quantitative RT-PCR analysis of Setd8 in INS-1 cells treated with BSA, PA and PAG for 24 h. B, Immunoblotting of Setd8 in INS-1 cells treated with BSA, PA and PAG for 24 h. C, Immunoblotting of Setd8 in the control (Ctr) and Setd8-overexpressed INS-1 cells. D, Immunofluorescence images of Ctr and Setd8-overexpressed INS-1 cells. The replicating INS-1 cells were recognized as Ki67+ (up) and EdU+ (down) cells. E, Immunofluorescence images of INS-1 cells treated with PAG and Setd8 specific inhibitor (UNC0379) for 48 h. The replicating INS-1 cells were recognized as Ki67+ cells. All results shown are the mean ± S.E. from three independent experiments. Statistical analysis was performed with two-tailed Student's t test. *p < 0.05; **p < 0.01.
PA Induced Apoptosis Partially Via Upregulation of Rhob in INS-1 Cells
PA-induced apoptosis resulting in the dysfunction of pancreatic β-cells is one of marked pathogenesis in T2D. Our in vitro phenotypic data showed that apoptosis of INS-1 cells was gradually increased by PA and PAG and that the latter can lead to more serious apoptosis. Accordingly, we extracted 23 DE proteins from our dataset, of which the expression patterns were either positively or negatively correlated with the time-course phenotype of apoptosis (supplemental Fig. S4). Among these, Rhob (Rho-related GTP-binding protein) was upregulated by PA and upregulated even higher by PAG, suggesting it is positively correlated with apoptosis. Therefore, we next explored whether Rhob participated in the process of PA-induced apoptosis. First, quantitative RT-PCR and Western blotting confirmed that PA induced Rhob expression at both mRNA and protein levels (Fig. 5A and 5B). By overexpressing Rhob in INS-1 cells (Fig. 5C), we found that cell apoptosis rate was augmented no matter cells were treated with PA or not (Fig. 5D). Using CRISPR-Cas9 techniques, we created Rhob-knockout (Rhob-KO) INS-1 cells (Fig. 5E) to study the effect of Rhob deficiency on cell apoptosis induced by PA. Compared with wild-type cells, Rhob-deficient INS-1 cells can, in part, attenuate apoptosis induced by PA (Fig. 5F). Therefore, our results suggested that PA induced apoptosis partially via upregulation of Rhob in INS-1 cells.
Fig. 5.
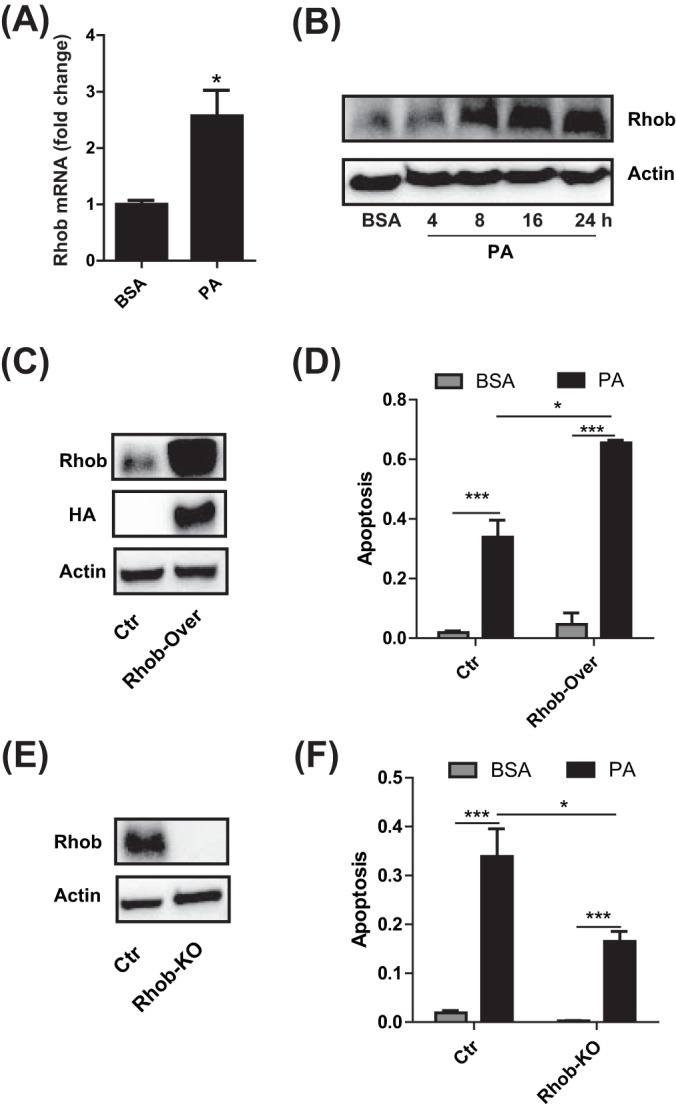
A, Quantitative RT-PCR analysis of RhoB expression in INS-1 cells cultured with BSA and PA for 24 h. B, Immunoblotting of Rhob in INS-1 cells after treatment with BSA and PA at different time points. C, Immunoblotting of Rhob in Ctr and Rhob-overexpressed INS-1 cells. D, The apoptosis rate of Ctr and Rhob-overexpressed INS-1 cell treatment with BSA and PA for 24 h. E, Immunoblotting of Rhob in Ctr and Rhob-KO INS-1 cells. F, The apoptosis rate of Ctr and Rhob-KO INS-1 cell treatment with BSA and PA for 24 h. All results shown are the mean ± S.E. from three independent experiments. Statistical analysis was performed with two-tailed Student's t test. *p < 0.05; **p < 0.01.
CONCLUSION
In this study, using the multiplex-TMT-based quantitative proteomics strategy, we measured the temporal changes of protein expression in INS-1 cells treated by PA and PAG. 783 differentially expressed proteins were identified, representing the high depth of our quantitative proteome data. Following comprehensive bioinformatics analysis of these datasets not only allowed us to recapitulate lipotoxicity/glucolipotoxicity-related proteins and biological pathways as previously reported, validating the accuracy of our proteomics data, but also provided new insights into the molecular events associated with lipotoxicity and glucolipotoxicity time-course profiles. Finally, we were able to identify and validate two new molecular targets from our dataset: Setd8 for cell replication and Rhob for apoptosis, demonstrating that our study offers a valuable data resource for studying the mechanism of lipotoxicity and glucolipotoxicity in pancreatic β-cells.
DATA AVAILABILITY
The MS data were deposited in the ProteomeXchange Consortium through the PRIDE (Proteomics Identifications) (61) partner repository with the data set identifier PXD008154.
Supplementary Material
Acknowledgments
We thank the staff of the Institute of Biophysics Core Facilities, especially, Yan Teng for her technical support with confocal imaging.
Footnotes
* This work was supported by grants from the National Science Foundation of China (31770891, 31400703), the National Basic Research Program of China 973 Program (2014CB910503), the National Key Research and Development Program of China (2016YFC0903301) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12030101).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- T2D
- type 2 diabetes
- PA
- palmitate
- PAG
- palmitate and glucose
- FFA
- free fatty acid
- GSIS
- glucose-stimulated insulin secretion
- TEAB
- triethylammonium bicarbonate
- PIF
- precursor intensity fraction
- TMT
- Tandem Mass Tag
- SPS
- synchronous precursor selection
- HCD
- high energy collision-induced dissociation
- PSM
- peptide-spectrum match
- GSEA
- gene set enrichment analysis
- GO
- gene ontology
- SN
- sub-network.
REFERENCES
- 1. Saisho Y. (2015) beta-cell dysfunction: Its critical role in prevention and management of type 2 diabetes. World J. Diabetes 6, 109–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cnop M., Welsh N., Jonas J. C., Jorns A., Lenzen S., and Eizirik D. L. (2005) Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54, S97–S107 [DOI] [PubMed] [Google Scholar]
- 3. Dedoussis G. V., Kaliora A. C., and Panagiotakos D. B. (2007) Genes, diet and type 2 diabetes mellitus: a review. Rev. Diabet. Stud. 4, 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prasad R. B., and Groop L. (2015) Genetics of type 2 diabetes-pitfalls and possibilities. Genes 6, 87–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shimabukuro M., Zhou Y. T., Levi M., and Unger R. H. (1998) Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc. Natl. Acad. Sci. U.S.A. 95, 2498–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Listenberger L. L., Han X., Lewis S. E., Cases S., Farese R. V. Jr, Ory D. S., and Schaffer J. E. (2003) Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. U.S.A. 100, 3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Poitout V., and Robertson R. P. (2008) Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocrine Rev. 29, 351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guo J., Qian Y., Xi X., Hu X., Zhu J., and Han X. (2010) Blockage of ceramide metabolism exacerbates palmitate inhibition of pro-insulin gene expression in pancreatic beta-cells. Mol. Cell. Biochem. 338, 283–290 [DOI] [PubMed] [Google Scholar]
- 9. Bagnati M., Ogunkolade B. W., Marshall C., Tucci C., Hanna K., Jones T. A., Bugliani M., Nedjai B., Caton P. W., Kieswich J., Yaqoob M. M., Ball G. R., Marchetti P., Hitman G. A., and Turner M. D. (2016) Glucolipotoxicity initiates pancreatic beta-cell death through TNFR5/CD40-mediated STAT1 and NF-kappaB activation. Cell Death Dis. 7, e2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malmgren S., Spegel P., Danielsson A. P., Nagorny C. L., Andersson L. E., Nitert M. D., Ridderstrale M., Mulder H., and Ling C. (2013) Coordinate changes in histone modifications, mRNA levels, and metabolite profiles in clonal INS-1 832/13 beta-cells accompany functional adaptations to lipotoxicity. J. Biol. Chem. 288, 11973–11987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tian G., Sol E. R., Xu Y., Shuai H., and Tengholm A. (2015) Impaired cAMP generation contributes to defective glucose-stimulated insulin secretion after long-term exposure to palmitate. Diabetes 64, 904–915 [DOI] [PubMed] [Google Scholar]
- 12. Hoppa M. B., Collins S., Ramracheya R., Hodson L., Amisten S., Zhang Q., Johnson P., Ashcroft F. M., and Rorsman P. (2009) Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca(2+) channels from secretory granules. Cell Metabolism 10, 455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brun T., and Maechler P. (2016) Beta-cell mitochondrial carriers and the diabetogenic stress response. Biochim. Biophys. Acta 1863, 2540–2549 [DOI] [PubMed] [Google Scholar]
- 14. Biden T. J., Boslem E., Chu K. Y., and Sue N. (2014) Lipotoxic endoplasmic reticulum stress, beta cell failure, and type 2 diabetes mellitus. Trends Endocrinol. Metab. 25, 389–398 [DOI] [PubMed] [Google Scholar]
- 15. Lai E., Bikopoulos G., Wheeler M. B., Rozakis-Adcock M., and Volchuk A. (2008) Differential activation of ER stress and apoptosis in response to chronically elevated free fatty acids in pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 294, E540–E550 [DOI] [PubMed] [Google Scholar]
- 16. Li N., Frigerio F., and Maechler P. (2008) The sensitivity of pancreatic beta-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem. Soc. Trans. 36, 930–934 [DOI] [PubMed] [Google Scholar]
- 17. Las G., and Shirihai O. S. (2010) The role of autophagy in beta-cell lipotoxicity and type 2 diabetes. Diabetes Obes. Metab. 12, 15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mir S. U., George N. M., Zahoor L., Harms R., Guinn Z., and Sarvetnick N. E. (2015) Inhibition of autophagic turnover in beta-cells by fatty acids and glucose leads to apoptotic cell death. J. Biol. Chem. 290, 6071–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giacca A., Xiao C., Oprescu A. I., Carpentier A. C., and Lewis G. F. (2011) Lipid-induced pancreatic beta-cell dysfunction: focus on in vivo studies. Am. J. Physiol. Endocrinol. Metab. 300, E255–E262 [DOI] [PubMed] [Google Scholar]
- 20. Boni-Schnetzler M., Boller S., Debray S., Bouzakri K., Meier D. T., Prazak R., Kerr-Conte J., Pattou F., Ehses J. A., Schuit F. C., and Donath M. Y. (2009) Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 150, 5218–5229 [DOI] [PubMed] [Google Scholar]
- 21. Youn D. Y., Xiaoli A. M., Pessin J. E., and Yang F. (2016) Regulation of metabolism by the Mediator complex. Biophys. Reports 2, 69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen X., Cui Z., Wei S., Hou J., Xie Z., Peng X., Li J., Cai T., Hang H., and Yang F. (2013) Chronic high glucose induced INS-1beta cell mitochondrial dysfunction: a comparative mitochondrial proteome with SILAC. Proteomics 13, 3030–3039 [DOI] [PubMed] [Google Scholar]
- 23. Baldwin A. C., Green C. D., Olson L. K., Moxley M. A., and Corbett J. A. (2012) A role for aberrant protein palmitoylation in FFA-induced ER stress and beta-cell death. Am. J. Physiol. Endocrinol. Metab. 302, E1390–E1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ciregia F., Bugliani M., Ronci M., Giusti L., Boldrini C., Mazzoni M. R., Mossuto S., Grano F., Cnop M., Marselli L., Giannaccini G., Urbani A., Lucacchini A., and Marchetti P. (2017) Palmitate-induced lipotoxicity alters acetylation of multiple proteins in clonal beta cells and human pancreatic islets. Sci. Rep. 7, 13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xiao J., Gregersen S., Kruhoffer M., Pedersen S. B., Orntoft T. F., and Hermansen K. (2001) The effect of chronic exposure to fatty acids on gene expression in clonal insulin-producing cells: studies using high density oligonucleotide microarray. Endocrinology 142, 4777–4784 [DOI] [PubMed] [Google Scholar]
- 26. Cnop M., Abdulkarim B., Bottu G., Cunha D. A., Igoillo-Esteve M., Masini M., Turatsinze J. V., Griebel T., Villate O., Santin I., Bugliani M., Ladriere L., Marselli L., McCarthy M. I., Marchetti P., Sammeth M., and Eizirik D. L. (2014) RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 63, 1978–1993 [DOI] [PubMed] [Google Scholar]
- 27. Maris M., Robert S., Waelkens E., Derua R., Hernangomez M. H., D'Hertog W., Cnop M., Mathieu C., and Overbergh L. (2013) Role of the saturated nonesterified fatty acid palmitate in beta cell dysfunction. J. Proteome Res. 12, 347–362 [DOI] [PubMed] [Google Scholar]
- 28. Hovsepyan M., Sargsyan E., and Bergsten P. (2010) Palmitate-induced changes in protein expression of insulin secreting INS-1E cells. J. Proteomics 73, 1148–1155 [DOI] [PubMed] [Google Scholar]
- 29. Sol E. R., Hovsepyan M., and Bergsten P. (2009) Proteins altered by elevated levels of palmitate or glucose implicated in impaired glucose-stimulated insulin secretion. Proteome Sci. 7, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roomp K., Kristinsson H., Schvartz D., Ubhayasekera K., Sargsyan E., Manukyan L., Chowdhury A., Manell H., Satagopam V., Groebe K., Schneider R., Bergquist J., Sanchez J. C., and Bergsten P. (2017) Combined lipidomic and proteomic analysis of isolated human islets exposed to palmitate reveals time-dependent changes in insulin secretion and lipid metabolism. PloS one 12, e0176391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spivak M., Weston J., Bottou L., Kall L., and Noble W. S. (2009) Improvements to the percolator algorithm for Peptide identification from shotgun proteomics data sets. J. Proteome Res. 8, 3737–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S. J., and Marra M. A. (2009) Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 34. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sergushichev A. (2016) An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv, 10.1101/060012 [DOI] [Google Scholar]
- 37. Kong F. J., Wu J. H., Sun S. Y., and Zhou J. Q. (2017) The endoplasmic reticulum stress/autophagy pathway is involved in cholesterol-induced pancreatic beta-cell injury. Sci. Rep. 7, 44746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hou J., Li Z., Zhong W., Hao Q., Lei L., Wang L., Zhao D., Xu P., Zhou Y., Wang Y., and Xu T. (2017) Temporal transcriptomic and proteomic landscapes of deteriorating pancreatic islets in type 2 diabetic rats. Diabetes 66, 2188–2200 [DOI] [PubMed] [Google Scholar]
- 39. Ting L., Rad R., Gygi S. P., and Haas W. (2011) MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hauck A. K., and Bernlohr D. A. (2016) Oxidative stress and lipotoxicity. J. Lipid Res. 57, 1976–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim J. W., and Yoon K. H. (2011) Glucolipotoxicity in pancreatic beta-cells. Diabetes Metabolism J. 35, 444–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hong K., Xu G., Grayson T. B., and Shalev A. (2016) Cytokines Regulate beta-Cell Thioredoxin-interacting Protein (TXNIP) via Distinct Mechanisms and Pathways. J. Biol. Chem. 291, 8428–8439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zito E., Chin K. T., Blais J., Harding H. P., and Ron D. (2010) ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J. Cell Biol. 188, 821–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luciani D. S., Gwiazda K. S., Yang T. L., Kalynyak T. B., Bychkivska Y., Frey M. H., Jeffrey K. D., Sampaio A. V., Underhill T. M., and Johnson J. D. (2009) Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes 58, 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gwiazda K. S., Yang T. L., Lin Y., and Johnson J. D. (2009) Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am. J. Physiol. Endocrinol. Metab. 296, E690–E701 [DOI] [PubMed] [Google Scholar]
- 46. Bregier C., Kupikowska B., Fabczak H., and Fabczak S. (2008) [CCT chaperonins and their cochaperons]. Postepy biochemii 54, 64–70 [PubMed] [Google Scholar]
- 47. Moulle V. S., Vivot K., Tremblay C., Zarrouki B., Ghislain J., and Poitout V. (2017) Glucose and fatty acids synergistically and reversibly promote beta cell proliferation in rats. Diabetologia 60, 879–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen W., Guo W., Li M., Shi D., Tian Y., Li Z., Wang J., Fu L., Xiao X., Liu Q. Q., Wang S., Huang W., and Deng W. (2013) Upregulation of cleavage and polyadenylation specific factor 4 in lung adenocarcinoma and its critical role for cancer cell survival and proliferation. PloS one 8, e82728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pitzonka L., Wang X., Ullas S., Wolff D. W., Wang Y., and Goodrich D. W. (2013) The THO ribonucleoprotein complex is required for stem cell homeostasis in the adult mouse small intestine. Mol. Cell. Biol. 33, 3505–3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Karamysheva Z., Diaz-Martinez L. A., Warrington R., and Yu H. (2015) Graded requirement for the spliceosome in cell cycle progression. Cell Cycle 14, 1873–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van der Lelij P., Stocsits R. R., Ladurner R., Petzold G., Kreidl E., Koch B., Schmitz J., Neumann B., Ellenberg J., and Peters J. M. (2014) SNW1 enables sister chromatid cohesion by mediating the splicing of sororin and APC2 pre-mRNAs. EMBO J. 33, 2643–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kanehisa M., Furumichi M., Tanabe M., Sato Y., and Morishima K. (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Plotz T., Hartmann M., Lenzen S., and Elsner M. (2016) The role of lipid droplet formation in the protection of unsaturated fatty acids against palmitic acid induced lipotoxicity to rat insulin-producing cells. Nutrition Metabolism 13, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wagner B. K. (2016) The genetic landscape of beta-cell proliferation: toward a road map. Diabetes 65, 1789–1790 [DOI] [PubMed] [Google Scholar]
- 56. Brustel J., Tardat M., Kirsh O., Grimaud C., and Julien E. (2011) Coupling mitosis to DNA replication: the emerging role of the histone H4-lysine 20 methyltransferase PR-Set7. Trends Cell Biol. 21, 452–460 [DOI] [PubMed] [Google Scholar]
- 57. Beck D. B., Oda H., Shen S. S., and Reinberg D. (2012) PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 26, 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ma A., Yu W., Li F., Bleich R. M., Herold J. M., Butler K. V., Norris J. L., Korboukh V., Tripathy A., Janzen W. P., Arrowsmith C. H., Frye S. V., Vedadi M., Brown P. J., and Jin J. (2014) Discovery of a selective, substrate-competitive inhibitor of the lysine methyltransferase SETD8. J. Med. Chem. 57, 6822–6833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Veschi V., Liu Z., Voss T. C., Ozbun L., Gryder B., Yan C., Hu Y., Ma A., Jin J., Mazur S. J., Lam N., Souza B. K., Giannini G., Hager G. L., Arrowsmith C. H., Khan J., Appella E., and Thiele C. J. (2017) Epigenetic siRNA and chemical screens identify SETD8 inhibition as a therapeutic strategy for p53 activation in high-risk neuroblastoma. Cancer cell 31, 50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tanaka H., Takebayashi S. I., Sakamoto A., Igata T., Nakatsu Y., Saitoh N., Hino S., and Nakao M. (2017) The SETD8/PR-Set7 methyltransferase functions as a barrier to prevent senescence-associated metabolic remodeling. Cell Reports 18, 2148–2161 [DOI] [PubMed] [Google Scholar]
- 61. Vizcaino J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS data were deposited in the ProteomeXchange Consortium through the PRIDE (Proteomics Identifications) (61) partner repository with the data set identifier PXD008154.