

The proximity-dependent biotinylation approach BioID is increasingly used to define proximity interactomes and subcellular organization, but is most often limited to a few easily transfectable cell lines. Here, a versatile set of lentiviral delivery vectors that enable BioID across cell types is described that incorporates features such as inducible expression and selectable markers. Benchmarking of these vectors demonstrates their ease and versatility across primary and immortalized cell systems. The lentiviral vectors are available through the Network Biology Collaborative Centre (nbcc.lunenfeld.ca).
Keywords: Affinity proteomics, Affinity tagging, Cell biology, Cellular organelles, Molecular biology, Protein-Protein Interactions, Spectral counting
Graphical Abstract
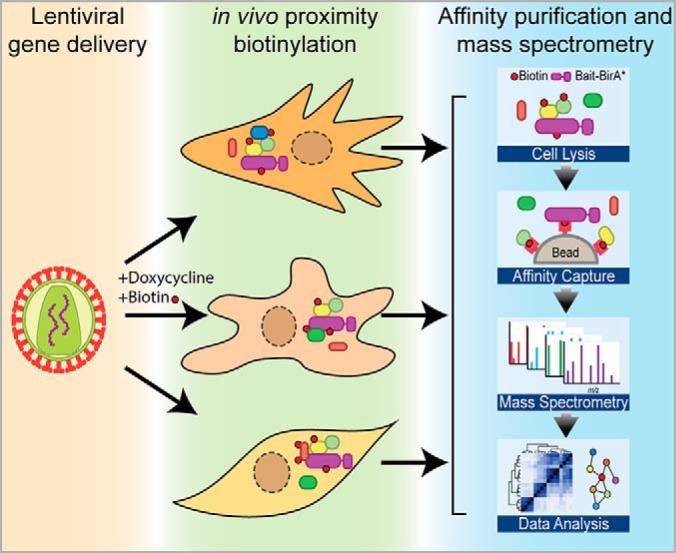
Highlights
Description of a set of lentiviral constructs for doxycycline-inducible BioID.
All vector configurations enable amino- or carboxyl-terminal fusion in a Gateway system.
Vector variants incorporate selectable markers (puromycin, fluorescent protein).
Guidelines for using the vectors and designing BioID experiments are provided.
Abstract
Proximity-dependent biotinylation strategies have emerged as powerful tools to characterize the subcellular context of proteins in living cells. The popular BioID approach employs an abortive E. coli biotin ligase mutant (R118G; denoted as BirA*), which when fused to a bait protein enables the covalent biotinylation of endogenous proximal polypeptides. This approach has been mainly applied to the study of protein proximity in immortalized mammalian cell lines. To expand the application space of BioID, here we describe a set of lentiviral vectors that enable the inducible expression of BirA*-tagged bait fusion proteins for performing proximity-dependent biotinylation in diverse experimental systems. We benchmark this highly adaptable toolkit across immortalized and primary cell systems, demonstrating the ease, versatility and robustness of the system. We also provide guidelines to perform BioID using these reagents.
Understanding the functional relationships between proteins is essential for gaining mechanistic insight into their biological roles. Proteins can engage in stable or dynamic direct interactions, or can participate in indirect interactions mediated through molecules such as other proteins or nucleic acids. Mass spectrometry (MS)-based proteomics approaches have played an integral role in assessing such interactions (1). For example, biochemical fractionation followed by MS can be employed to detect protein complexes that co-fractionate (2, 3). More frequently, MS is coupled with affinity purification (AP) of a selected protein of interest (bait) in a technique commonly referred to as AP-MS1. In that set-up, an affinity reagent specific to the bait protein (e.g. an antibody specific to the bait or an epitope tag fused to the bait) is used to enrich it from a cellular lysate alongside its interaction partners, which are subsequently identified by MS (4, 5). However, with such techniques that involve cellular lysis followed by fractionation or affinity-based enrichment, weak or transient interactions, or protein complexes that are recalcitrant to solubilization under mild lysis conditions, are often not captured (6–8).
To overcome these challenges and to limit the detection of spurious post-lysis interactions, in vivo proximity-dependent labeling approaches have been introduced in the past 5 years (e.g. (9, 10)). Using these approaches, a bait protein of interest is fused to an enzyme and expressed in a physiologically-relevant system where the addition of an enzymatic substrate leads to covalent biotinylation of proteins located near the bait (11, 12). In the case of the BioID approach described here, a mutant form of biotin ligase catalyzes the activation of exogenously-supplied biotin to the reactive intermediate, biotinoyl-5′-AMP (13). The abortive BirA* enzyme, which harbors a R118G mutation, displays a reduced affinity for the activated biotin molecule. Biotin-AMP thus diffuses away from the bait and can covalently modify epsilon amine groups of lysine residues on nearby proteins (14, 15). Because these proximity partners are covalently marked, maintaining protein-protein interactions during lysis and purification is not necessary, and harsh lysis conditions can be employed to maximize solubilization of all cellular structures. Subsequent recovery of the biotinylated proteins via streptavidin affinity purification followed by MS allows identification of the labeled proteins (9, 12). Importantly, the inclusion of proper negative controls in the experimental design (to model both endogenously biotinylated proteins, such as the mitochondrial carboxylases, as well as promiscuous biotinylation resulting from expression of an abortive BirA* enzyme) enables the use of computational tools initially developed for AP-MS (e.g. (16, 17)) to score proximity partners. First introduced to identify new components of the nuclear lamina (9), BioID has since been employed to uncover new components of signaling pathways (18) and their enzyme targets (19), to describe the protein composition of structures such as the centrosome, primary cilia (20, 21), focal adhesions (22), stress granules and P-bodies (23) and has been used to examine contacts between organelles (24), to highlight a few examples.
Importantly, however, most of the BioID studies have so far been performed in easily-transfectable cell lines, including HEK293, U2OS and HeLa cells. Although these cell systems continue to reveal important biological insight, it is also critical to perform some of these studies in different contexts and model systems that are less amenable to transfection, including primary cells. Although there have now been several reports that have used viral delivery from adenovirus (25), lentivirus (26–28), retrovirus (29) or AAV (30) systems, these reagents have so far been limited in their range of application to different workflows. Further, there is a lack of demonstration of optimization and benchmarking to facilitate implementation of BioID across various cell types.
Our motivation was to expand the ease and breadth of applicability of BioID to include diverse experimental systems by developing and optimizing lentiviral delivery reagents and workflows. To achieve this, we cloned a cassette consisting of BirA* (enabling BioID) and a single FLAG epitope (that can be used both to detect bait expression and to perform AP-MS) (31) downstream of a tetracycline-regulatable promoter, enabling inducible gene expression. We inserted this cassette into five lentiviral vector backbones, each harboring different features, including: integrated rtTA (tetracycline-controled transactivator), puromycin selection or mCitrine expression. Each vector is made available as a Gateway-compatible destination vector enabling amino- or carboxy-terminal tagging, and a subset is also available for standard ligase-mediated cloning. We describe the benchmarking of these vectors for performing BioID through lentiviral gene delivery in HeLa cells as well as in mouse and human fibroblasts. The ease and robustness of this approach was compared with BioID performed using stable Flp-In T-REx HeLa cell lines. Importantly, we highlight a major advantage of this approach, which is the overall decrease in experimental timeline (compared with the common practice of stable cells), while maintaining data quality and reproducibility. We also outline strategies for lentiviral backbone selection and considerations for experimental design that will assist the community in the use of these reagents.
EXPERIMENTAL PROCEDURES
Generation of Lentiviral Vector Sequences and Expression Constructs
The region internal to the long terminal repeats (LTRs) of the second generation pLVX-Tight-Puro lentiviral backbone (Takara Bio, Clontech, Mountain View, CA) was modified to generate the five different lentiviral transfer vector backbones (Fig. 1A). The orientation of the inducible P-tight promoter, which consists of tetracycline response element (TRE) along with a minimum Cytomegalovirus (CMV) promoter, was reversed to allow for robust transgene expression (32, 33). The phosphoglycerate kinase 1 (PGK) promoter was also replaced with a shorter fragment (227bp) of the stronger elongation factor-1 alpha (EF1a) promoter (denoted as sEF1a). Downstream of these constitutive promoters, various cassettes were inserted to generate the transfer vectors. For the pSTVH5 transfer vector, mCitrine was amplified by PCR from pLM-mCitrine-2A-Sox2 (a gift from Michel Sadelain, Addgene #23242) (34), fused to rtTA from PB-CA-rtTA Advanced (a gift from Andras Nagy, Addgene #20910) (35) using fusion-PCR, and inserted downstream of the sEF1a promoter. For the pSTV6 transfer vector, the PGK-puromycin-2A-rtTA cassette was transferred by restriction cloning from the pLIX-402 vector (a gift from David Root, Addgene #41394) using the MluI/KpnI sites and substituted for the sEF1a-mCitrine-2A-rtTA of the pSTVH5 vector. For design of the pSTVH7 transfer vector, the puromycin resistance cassette from pLVX-Tight-Puro was placed downstream of the sEF1a promoter. For the pSTVH8 transfer vector, the rtTA from the PB-CA-rtTA Advanced vector was amplified and cloned downstream of the sEF1a promoter.
Fig. 1.

A, Schematic of the five lentivirus transfer vectors pSTV2, pSTVH5, pSTV6, pSTVH7, pSTVH8. P-tight - Tetracycline response element (TRE) along with a minimum Cytomegalovirus (CMV) promoter; BirA*-FLAG-ORF tag; PGK - phosphoglycerate kinase 1 promoter; sEF1a - short variant of the elongation factor-1 alpha; rtTA - reverse tetracycline-controlled transactivator; 2A - cleavage peptide; WPRE - Woodchuck Hepatitis Virus (WHP) Posttranscriptional Regulatory Element. The approximate size of Open Reading Frame (ORF) that can be cloned into the vector and still allow packaging of the RNA into the lentivirus, based on lentiviral packaging limit, is indicated on the right. B, Schematic depicting expression of selectable markers (mCitrine or the puromycin-resistance gene in pSTV6 and pSTVH7) and rtTA from the constitutive promoter which enables doxycycline-dependent expression of the BirA*-FLAG tagged transgene. 1. A constitutive promoter drives expression of the selection marker and the rtTA as a single mRNA. 2. The 2A cleavage peptide separates the selection marker from the rtTA protein. 3. rtTA and doxycycline control the activity of the regulatable promoter. 4, 5. The BirA*-FLAG mRNA, and protein fusion is expressed and can be used for BioID. C, The full human proteome (20,192 reviewed entries) was downloaded from Uniprot (version 2017_07) and binned by expected molecular weights. Based on the 10 kb theoretical packaging limit of a lentiviral particle, the extent of the human proteome that can be analyzed in a viral BioID experiment using the vectors described in this manuscript is highlighted on the graph.
Subsequently, the BirA*-FLAG-Gateway fragments from pcDNA5-FRT/TO-BirA*-FLAG vectors were excised using HindIII/SphI restriction sites and ligated into the pSTVH5-pSTVH8 lentiviral backbones which were also digested with the same enzymes. Additionally, we constructed a minimal packaging virus (pSTV2) in which the PGK-puromycin cassette from the pLVX-Tight-Puro was removed and blunt-end repaired. The BirA*-FLAG Gateway fragment was digested using HindIII/XhoI cut sites and blunt-end repaired and ligated into pSTV2. We also constructed a non-Gateway version of the pSTV2 vector by using the same cloning strategy as above but using the pcDNA5-FRT/TO-BirA*-FLAG-MCS (Multiple cloning sites) vectors (the MCS vectors were a kind gift from Brian Raught) (19, 36).
All vectors were validated through restriction digestion and DNA sequencing of subcloned fragments. The plasmid sequences are available through the Network Biology Collaborative Centre, NBCC, website (maps and sequences available at http://nbcc.lunenfeld.ca/resources). All the Gateway constructs were functionally tested through virus production (See the Testing and Optimization of Lentiviral Vectors section below), infection and immunostaining to assess BirA*-FLAG expression and in vivo biotinylation (See the Immunofluorescence section below).
Gateway-compatible entry clones for the genes LMNA (Genbank accession number EU832167, encoding lamin A), HIST1H2BG (EU446968, encoding histone H2B), TBP (EU831783, Tata Binding Protein) and TUBB (DQ891742, tubulin beta) were transferred in to the pcDNA5-FRT/TO-BirA*-FLAG Gateway destination vector (37) or the pSTV2-BirA*-FLAG Gateway lentivirus destination vector using LR-Gateway cloning. The pcDNA5-based plasmids were used to generate stable cell lines in Flp-In T-REx HeLa cells, as previously described (31), whereas the pSTV2 vectors were used to generate lentivirus for infection-mediated gene delivery. Different control vectors were also generated to allow for background subtraction. “No-BirA*” control samples were infected with a lentivirus vector without BirA*-FLAG. EGFP and EGFP fused to SV40 Nuclear Localization Sequence (EGFP-NLS) were transferred from pEntry vectors (pENTR223, Invitrogen) to the pSTV2-N-BirA*-FLAG destination vectors to create the BirA*-EGFP and BirA*-EGFP-NLS controls, respectively. To deliver rtTA into the cell lines not harboring the rtTA transcriptional activator, a lentivirus driving the expression of rtTA from the constitutive UbC promoter, was also used.
Cell Lines and Virus Production and Infection
The cancer cell lines used are HeLa and Flp-In-T-REx HeLa cells (kind gifts from Laurence Pelletier and Arshad Desai, respectively). Normal human foreskin fibroblasts (BJ-5ta) and Mouse Embryonic Fibroblasts (MEF) isolated from E14.5 ROSA26 Neo-Out rtTA mouse strain (38) were both kindly provided by Jeffrey Wrana. HEK293T cells (American Type and Tissue Collection, ATCC, Manassas, VA; Cat# CRL-3216) were used for virus production. Although a diverse range of DNA concentrations and transfection conditions are described in the literature, we have systematically optimized the protocol for our vectors and transfection reagents used in this study, with the final version detailed below.
Briefly, 1.3 μg of psPAX2 (a gift from Didier Trono, AddGene #12260), 0.8 μg of VSV-G packaging vectors (a gift from Bob Weinberg, AddGene #8454) (39) and 1.3 μg of transfer vectors encoding the gene of interest (such as pSTV2) are transfected into HEK293T cells (at 85% confluence in a 6-well plate) using the jetPRIME reagent as per manufacturer's recommendations (Polyplus-transfection SA, Illkirch-Graffenstaden, France, Cat# 114–01). After 10 h, the media is replaced with 3 ml of virus production media. Virus production media consists of DMEM supplemented with 5% heat-inactivated Fetal Bovine Serum (Gibco, ThermoFisher Scientific, Waltham, MA) and 50 U/ml Penicillin-Streptomycin solution (Corning, Manassas, VA). Virus is harvested at 36–40 h post media change, and cleared by centrifugation (500 rcf, 5 min) and filtering through a 0.45 μm filter. The viral titer is estimated by co-infecting HeLa cells (at 40% confluence in a 24-well plate) with a range of 25–100 μl of the pSTV2-BirA* tagged bait as well as the rtTA viral supernatant. The next day, the medium is replaced by media containing doxycycline (1 μg/ml; Sigma-Aldrich, St. Louis, MO) and biotin (40 μm; BioBasic, Amherst, New York, Cat# 58–85-5) and the cells are incubated for 24 h. Cells are subsequently fixed and analyzed by immunofluorescence (staining for both the FLAG epitope, the biotinylated proteins and the nuclei by DAPI; see the Immunofluorescence section below) to determine the optimal amount of supernatant that yields 75–85% infection rate. These results provide the infection parameters for scaling up of experiments for BioID followed by mass spectrometry.
For all BioID experiments, cells in a single 10 cm dish at ∼35–40% density were infected with the determined amount of virus determined to yield 75–85% infection. For the transfer vectors described here, this corresponded to 750–850 μl of viral supernatant. Cells were then grown until ready for splitting, at which point they were scaled up into 15 cm plates for the BioID experiment. One 15 cm dish was used for each biological replicate. Biological duplicates were prepared for all experiments (alongside negative controls defined below). An aliquot of the cell suspension was also plated onto coverslips for immunofluorescence.
Testing and Optimization of Lentiviral Vectors
Throughout the lentiviral transfer vector design and production, the various plasmids were analyzed by restriction digest and were sequence-validated, followed by functional testing. The functionality of the elements downstream of the constitutive promoters were tested before and after inserting the BirA*-FLAG fragment into the pSTVH5 to pSTVH8 transfer vectors (Fig. 1A). To test the functionality of the rtTA element in our vectors, we co-infected HeLa cells with a lentivirus expressing H2B-mCherry downstream of the tetracycline inducible TRE promoter with our various transfer vectors encoding the rtTA (pSTVH5, pSTV6 and pSTVH8). We next assessed rtTA/doxycycline-dependent expression of H2B-mCherry. The functionality of the selection cassettes was also validated: for pSTVH5, we confirmed expression of mCitrine by fluorescence microscopy. For the vectors harboring the puromycin resistance gene (pSTV6 and pSTVH7), we confirmed cell survival in the presence of puromycin (2 μg/ml) for 3–4 days. The plasmids that tested positive in these assays were then used for cloning the BirA*-FLAG fragments downstream of the P-tight promoter (as described above). The resulting vectors were re-tested by assessing for BirA*-FLAG expression and in vivo biotinylation using immunofluorescence (see below).
Immunofluorescence
Cells were plated on coverslips and induced with doxycycline and biotin as described above. After 24 h, cells were washed once with PBS and fixed in 4% paraformaldehyde in PBS for 10 min. Subsequently cells were washed, permeabilized with 0.25% NP-40 in PBS, and blocked in 2.5% bovine serum albumin (BSA) in PBS. Cells were stained for bait proteins using mouse anti-FLAG antibody (Monoclonal anti-FLAG M2 antibody, Sigma-Aldrich, Cat# F3165; used at 1:2000), in blocking buffer. Secondary detection was performed using goat anti-mouse Alexa FluorTM 488 (Molecular Probes, ThermoFisher Scientific, A11001, used at 1:1000) and Alexa FluorTM 594 streptavidin conjugate (Molecular Probes, ThermoFisher Scientific, Cat# S11227, used at 1:2500) to localize the sites of in vivo biotinylation. DAPI (Sigma Aldrich, 20 mg/ml, used at 1:20,000) was used as a nuclear counterstain. Slides were mounted in ProLong Gold AntiFade (Molecular Probes, ThermoFisher Scientific, Cat# P36930) and imaged on a Leica Spinning disk microscope with a 40X oil-immersion microscope. Images were captured and processed using Volocity software V6.2 (PerkinElmer, Waltham, MA).
Immunoblotting
Cells were lysed as described for BioID (see below) and boiled in Laemmli SDS-PAGE sample buffer. Proteins were resolved on a 10% polyacrylamide gel and transferred to nitrocellulose membrane (GE Healthcare Life Science, Uppsala, Sweden, Cat# 10600001) for immunoblotting. Following Ponceau S staining, membranes were blocked in 4% bovine serum albumin in TBS with 0.1% Tween-20 (TBST). Bait proteins were probed using mouse anti-FLAG antibody (Monoclonal anti-FLAG M2 antibody, Sigma-Aldrich, F3165) at 1:2000, or anti-LMNA (Clone-131C3, Abcam, Cambridge, UK, ab8984) at 1:1000, in blocking buffer, washed in TBST and detected with 1:5000 Sheep anti-Mouse IgG-Horseradish peroxidase (HRP; GE Healthcare Life Science, Cat#NA931). Similarly, biotinylated proteins were probed using HRP-conjugated Streptavidin (GE Healthcare Life Science, Cat# RPN1231vs) at 1:2000 in blocking buffer. Mouse anti-Beta-Tubulin antibody (Developmental Studies Hybridoma Bank, Iowa City, IA, Cat# E7) was used at 1:2500. Membranes were developed using LumiGLO chemiluminescent reagent (Cell Signaling Technology, Danvers, MA, Cat# 7003S) and imaged using a BioRad ChemiDoc (BioRad, Mississauga, ON, Canada).
BioID
Cells at 75% confluence in 15 cm plates were induced with 1 μg/ml doxycycline (Dox) and 40 μm biotin for 24 h. At the end of the induction and labeling phase, cells were washed and harvested in cold PBS and flash-frozen until time of sample processing. Affinity purification was carried out essentially as described elsewhere (31). Briefly, cells were lysed in 1.5 ml of modified RIPA (modRIPA) buffer [50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EGTA, 0.5 mm EDTA, 1 mm MgCl2, 1% NP40, 0.1% SDS, 0.4% sodium deoxycholate, 1 mm PMSF and 1x Protease Inhibitor mixture (Sigma-Aldrich, Cat# P8340)]. Cells were sonicated for 15 s (5 s on, 3 s off for three cycles) at 30% amplitude on a Q500 Sonicator with 1/8” Microtip (QSonica, Newtown, Connecticut, Cat# 4422). Subsequently, 250 U of TurboNuclease (BioVision Inc., Milpitas, CA, Cat# 9207) and 10 μg of RNase A (Bio Basic, Markham, ON, Canada, Cat# RB0473) were added, and samples were rotated at 4 °C for 15 min. Next, the SDS concentration was increased to 0.4% (by the addition of 10% SDS) and the samples were rotated at 4 °C for 5 min. Samples were centrifuged at 15,000 × g for 15 min and the supernatant was used for biotinylated protein capture using 30 μl of pre-washed Streptavidin agarose beads (GE Healthcare Life Science, Cat# 17511301). After 4 h, the beads were washed once with SDS-Wash buffer (25 mm Tris-HCl, pH 7.4, 2% SDS), twice with RIPA (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% NP40, 0.1% SDS, 0.4% sodium deoxycholate), once with TNNE buffer (25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 0.1% NP40), and three times with 50 mm ammonium bicarbonate (ABC buffer), pH 8.0. On-bead digestion was performed with 1 μg of trypsin (Sigma Aldrich, Cat# 6567) in 70 μl of ABC buffer, overnight at 37 °C, followed by further digestion with an additional 0.5 μg of trypsin for 3 h. Supernatants were collected into a new tube. Beads were washed twice with water and this supernatant was pooled with the peptide supernatant, and subsequently dried using vacuum centrifugation. Peptides were re-suspended in 30 μl of 5% formic acid in mass spectrometry grade water and stored at −80 °C until ready for mass spectrometry analysis.
Mass Spectrometry
The LC-MS/MS setup consisted of a TripleTOF 6600 (SCIEX, Framingham, MA, Canada) equipped with a nanoelectrospray ion source connected in-line to a 425 Nano-HPLC system (Eksigent Technologies, Dublin, CA). The fused silica column (15 cm × ID 100 μm, OD 360 μm) had an integrated emitter tip prepared in-house using a laser puller (Sutter Instrument Co., Novato, CA). The column was packed with ∼15 cm of C18 resin (Reprosil-Pur, 3.5 μm, Dr. Maisch HPLC GmbH, Germany). 5 μl of sample was loaded onto the column using the autosampler, and the LC delivered the organic phase gradient at 400 nl/min over 90 min (2–35% acetonitrile with 0.1% formic acid). The MS instrument was operated in data-dependent acquisition mode with 1 MS scan (250 ms; mass range 400–1250 m/z) followed by up to 20 MS/MS scans (50 ms each). Only candidate ions between two and five charge states were considered, and ions were dynamically excluded for 10 s with a 50 mDa window. The isolation width was 0.7 m/z, and minimum threshold was set to 200. Between sample injections, 2 blank samples were injected (5% formic acid), each with 3 rapid gradient cycles at 1500 nl/min over 30 min. Before another sample was injected, system performance was verified with a 30 min BSA quality control run and a 30 min BSA mass calibration run.
Peptide and Protein Identification
Raw files (.WIFF and .WIFF.SCAN) were converted to an MGF format and to an mzML format using ProteoWizard (v3.0.4468) (40) and the AB SCIEX MS Data Converter (V1.3 beta), as implemented within ProHits (41). For human samples, the database used for searches consisted of the human and adenovirus sequences in the RefSeq protein database (version 57), and for mouse samples, the mouse complement of sequences in RefSeq (version 53). Both databases were supplemented with “common contaminants” from the Max Planck Institute (http://141.61.102.106:8080/share.cgi?ssid=0f2gfuB) and the Global Proteome Machine (GPM; http://www.thegpm.org/crap/index.html), and with commonly used epitope tags. The search databases consisted of forward and reverse sequences (labeled “gi 9999” or “DECOY”); in total, 72,226 entries were searched for the human database and 58,206 entries for the mouse database. Spectra were analyzed separately using Mascot (2.3.02; Matrix Science) (42) and Comet [2012.01 rev.3 (43)] with trypsin specificity and up to two missed cleavages; deamidation (Asn or Gln) and oxidation (Met) were selected as variable modifications. The fragment mass tolerance was 0.15 Da, and the mass window for the precursor was ±40 ppm with charges of 2+ to 4+ (both monoisotopic mass). The resulting Comet and Mascot results were individually processed by PeptideProphet (44) and combined into a final iProphet output using the Trans-Proteomic Pipeline (TPP; Linux version, v0.0 Development trunk rev 0, Build 201303061711) (45). TPP options were as follows: general options were -p0.05 -x20 -PPM -dDECOY, iProphet options were -pPRIME, and PeptideProphet options were -pPAEd. All proteins with a minimal iProphet probability of 0.95 were used for analysis.
The entire data set was deposited in ProteomeXchange via partner MassIVE (massive.ucsd.edu), and assigned accession numbers PXD010008, PXD010009 and MSV000082429, MSV000082430 (ftp://massive.ucsd.edu/MSV000082429, ftp://massive.ucsd.edu/MSV000082430) for the mouse and human data sets, respectively.
Identification of High-confidence Proximity Partners
SAINTexpress (version 3.6.1) (16) was used to calculate the probability that identified proteins were enriched above background contaminants. SAINTexpress uses a semi-supervised spectral counting model that compares the detection of putative proximal interactors in a BioID profile of a given bait against a series of negative control runs. For analysis with SAINT, only proteins with an iProphet protein probability of >0.95 were considered, and a minimum of two unique peptides was required. For each cell type, bait proteins were profiled using independent biological duplicates and analyzed alongside six independent negative control runs from the same cell type. Negative control runs consisted of streptavidin purifications from cells expressing BirA*-EGFP only (two replicates; this models promiscuous biotinylation) or BirA*-EGFP-NLS only (two replicates; this models promiscuous biotinylation in the nucleus) or without BirA* transgene expression (two replicates; this models endogenous biotinylation). For running SAINTexpress, the six independent negative controls were compressed to two, meaning that for each prey, its two highest counts across the six negative controls were selected for stringent evaluation with SAINTexpress (as detailed previously; (7)). SAINTexpress scores were averaged across both biological replicate purifications of the baits, and these averaged values were used to calculate a Bayesian False Discovery Rate (FDR); proximity interactions detected with a calculated FDR of 1% or less were deemed of high confidence.
Data Visualization
Dot plots and heat maps were generated using ProHits-viz (prohits-viz.lunenfeld.ca) (46). Prey proteins identified as high-confidence proximal interactors (FDR ≤1%) are provided in supplemental Table S1–S4; In ProHits-viz, once a prey passes the ≤1% FDR threshold with one bait, all its quantitative values across all baits are recovered, and the respective SAINT FDR is represented as the edge color intensity. Quantitation is encoded schematically, with color gradient representing raw spectral counts (capped at 50), with relative bait spectral counts represented by node size. To compare human and mouse data, the mouse protein names were converted to their human orthologs using g:Convert (47) (supplemental Table S4). Gene Ontology (GO) enrichment analysis was performed using g:Profiler within the ProHits-viz analysis module, using default options.
Experimental Design and Statistical Rationale
For each BioID experiment, biological duplicates were employed (each replicate generated through independent infections and harvests). Statistical scoring was performed against six negative controls compressed to two virtual controls using Significance Analysis of INTeractome (SAINT; SAINTexpress 3.6.1 was employed) as described above (“Identification of High-Confidence Proximity Partners”). Control samples (“No-BirA*”, “BirA*-EGFP”, “BirA*-EGFP-NLS”) are described above. The average SAINTexpress score was used to determine the Bayesian FDR, which therefore requires a high-confidence interaction to be detected across both biological replicates. This analysis was performed independently for each cell type.
RESULTS
Lentivirus Transfer Vector Design
To expand the ease and applicability of BioID to various cell types, including those for which clonal selection is not feasible (such as primary cells with limited replicative potential), we sought to create and validate a set of BioID lentiviral vectors. We began by constructing five different vector backbones (pSTV-2, -H5, -6, -H7, -H8), each harboring distinct features that may benefit varying experimental requirements (detailed in the next section) (Fig. 1A). In each of these backbone vectors, the BirA*-FLAG coding sequence was inserted downstream of a tetracycline-inducible promoter. All vectors were engineered for Gateway-compatible cloning (note that a standard multiple cloning site (MCS) version is also available for pSTV2). In addition, because the position of the tag may influence the behavior of the protein or the detection of proximity interactions, each of the vector backbones is provided as either an N-terminal or C-terminal fusion of the BirA*-FLAG cassette to the bait of interest (maps and sequences available at http://nbcc.lunenfeld.ca/resources). Each of the vectors was optimized and tested for basic functionality (see Experimental Procedures), and pSTV2 was further selected for benchmarking purposes (see below). Here we briefly review the key elements of these vectors.
Transgene Promoter Selection
Because high constitutive overexpression levels can result in spurious protein-protein interactions, altered cell behavior or toxicity, we employed the widely used tetracycline-inducible “Tet-ON” promoter system, that allows tight regulation and dose-responsive induction (48–50). In this system, the transgene is driven by a P-tight promoter, which consists of tandem copies of the tetracycline operator (TetO) and a minimum Cytomegalovirus (CMV) promoter. On treatment of cells with doxycycline, a constitutively expressed rtTA (tetracycline-controlled transactivator; a fusion of VP16 and rTetR) binds to the TetO elements and drives transcription of the transgene (Fig. 1B) (51, 52).
Several cell lines (and mouse strains) have been engineered to constitutively express rtTA (38, 53), or alternatively, nontransgenic cells can be co-infected with any of the commonly used rtTA lentivirus vectors (for example rtTA-N144, AddGene #66810, (54)). However, to avoid these requirements, we constructed three “all-in-one” vectors (pSTVH5, pSTV6, and pSTVH8) to enable the delivery of the BirA*-FLAG tagged bait along with the rtTA. In these vectors, the constitutive phosphoglycerate kinase 1 (PGK) promoter or short variant of the elongation factor-1 alpha (sEF1a) promoter drives the expression of rtTA alone or in conjunction with a selectable marker gene (described below), separated by a porcine teschovirus-1 2A cleavage peptide (55). Irrespective of the system selected for rtTA expression, addition of doxycycline leads to recruitment of rtTA to the TRE to induce transcription of the BirA*-FLAG fused transgene (Fig. 1B).
Selection Markers
It is often useful to select the populations of cells that have been infected (e.g. for establishing stable cell lines or enriching for infected cells when infection rate is suboptimal). To provide versatility in selection marker options, we provide two alternative strategies, namely puromycin resistance for drug selection, or mCitrine (yellow fluorescent protein) expression, which can be used to either monitor infection efficiency by microscopy or to sort infected cells by flow cytometry. The selection markers are expressed independently from the BirA*-FLAG-fused transgene via the constitutive PGK or sEF1a promoters.
The vector backbones contain different combinations of selection markers and rtTA elements in the standard lentiviral backbone (Fig. 1A). A key consideration in vector selection is the inherent RNA packaging limitation of lentivirus particles (∼10 kb) (56, 57). For the vectors that do not harbor an rtTA cassette (pSTV2, pSTVH7), rtTA must be provided separately (see above). However, their key advantage is that they enable expression of larger proteins, up to ∼180 kDa (open reading frames of up to 4900 bp) in pSTV2. This upper size limit is compatible with a majority of the human proteome (∼95% of human proteome is under 175 kDa; Fig. 1C). In cases where the transgene of interest is small enough to not interfere with viral packaging, the “all-in-one” vectors pSTVH5, which harbors both the yellow fluorescence protein and the rtTA element (sEF1a-mCitrine-2A-rtTA), pSTV6, which harbors the puromycin resistance gene and the rtTA element, (PGK-PuromycinR-2A-rtTA), or pSTVH8, which harbors just the rtTA element (sEF1a-rtTA) can be used. pSTVH5 and pSTV6 can in theory accommodate proteins as large as ∼101 kDa and ∼107 kDa (Fig. 1C), respectively. Therefore, selection of vector to use should be made with consideration for lentiviral packaging limit and the inverse relationship between viral titer and the size of the RNA being packaged, alongside the features desired in the vector.
Lentiviral Vectors Enable Proximity Biotinylation Across Different Cell Types
To benchmark our lentiviral vector system, we selected four proteins: (1) the cytoskeletal protein beta-tubulin (TUBB); (2) histone H2B (gene name HISTH2BG, here referred to as H2B); (3) TATA-binding Protein (TBP), and; (4) the nuclear envelope protein Lamin A (LMNA). Of the three nuclear proteins used as baits, we anticipate some degree of overlap in proximal protein labeling, though we expect distinct specificity as they are either ubiquitously distributed on the genome (H2B), enriched at transcription start sites (TBP) or predominantly localized to the nuclear envelope (LMNA).
To generate stable cells lines expressing the BirA*-tagged baits, the open reading frame for each of these proteins was cloned into the previously described pcDNA5-FRT/TO-BirA*-FLAG vector, and stable Flp-In T-REx HeLa cell pools were generated, as previously reported (31). This process requires on average 14 to 21 days post-transfection to generate stable cell pools ready for transgene induction and protein labeling. For the purposes of lentiviral mediated delivery of the BirA*-tagged baits, each of the four baits was also cloned into the pSTV2 lentiviral transfer vector, and lentivirus supernatants were produced and used for infecting target cells. Using an experimentally determined volume of the viral supernatant (described in Experimental Procedures), infection was performed overnight prior to using the cells for subsequent induction of protein expression and in vivo biotinylation. It is noteworthy that the lentiviral system saves ∼ 2 weeks compared with the generation of stable lines (Fig. 2A), offering significant economies of time and resources.
Fig. 2.

A, Graphical depiction of a BioID workflow, highlighting the differences in sample preparation protocol: stable cell line generation (such as using the Flp-In system) or lentivirus-mediated transgene delivery. B, BirA*-FLAG-tagged LMNA transgene expression was compared with endogenous Lamin A in the stable cell pools (Flp-In T-REx HeLa) or lentiviral infected cells (MEF, HeLa or BJ). Non-transgene expressing cell lines were used as a negative control. Following 24 h of doxycycline and biotin treatment, cells were lysed in modRIPA buffer and protein was resolved on 10% SDS-PAGE. Rabbit anti-Lamin A/C was used to detect the endogenous protein (white arrows) as well as the transgene BirA*-FLAG-LMNA. Mouse anti-beta-tubulin was used to assess loading. Blots were also probed for biotinylated protein using Streptavidin-conjugated HRP. C, Following 24 h of doxycycline and biotin treatment, cells were fixed and stained to visualize expression of BirA*-FLAG-LMNA transgene (mouse anti-FLAG; green) and site of proximal biotinylation (Streptavidin; red). DAPI was used to stain the DNA.
For benchmarking, BioID was performed in HeLa cells, using both the Flp-In T-REx HeLa stable cells, and virally-delivered transgenes. Additionally, to demonstrate the application of BioID in primary cells, Mouse Embryonic Fibroblasts (MEFs) expressing rtTA (ROSA26-rtTA-NeoOut (38)) and human foreskin fibroblasts (BJ cells; co-infected with an rtTA-expressing vector) were also tested. In all cases, once cells had reached 75% confluence, transgene expression was induced with 1 μg/ml doxycycline (Dox) in the presence of 40 μm biotin for 24 h, followed by processing for immunofluorescence or cell lysis.
Expression levels of BirA*-FLAG-LMNA were monitored using an anti-LMNA antibody (Fig. 2B), allowing for comparison of transgene expression levels to the endogenous protein. BirA*-FLAG-LMNA levels were consistent across cell types, and in the same range as the endogenous protein. Probing of western blots with HRP-conjugated Streptavidin to detect biotinylated proteins revealed extensive banding patterns with LMNA whereas the negative control lanes (No-BirA*) predominantly showed the major endogenously biotinylated proteins (Fig. 2B) (7). Like what we observed for LMNA, all baits used in this study exhibit consistent relative expression levels across cell types (supplemental Fig. S1). As detailed in Experimental Procedures, a stringent background correction strategy utilizing multiple controls per cell line was employed. Controls were: (1) a “No-BirA*” control in which we capture the endogenous biotinylation signature from cells without expression of a BirA* transgene; (2) BirA*-tagged EGFP (BirA*-GFP), which should account for spurious biotinylation in the cytoplasm and to a certain extent the nuclear compartment; and (3) BirA*-tagged EGFP harboring an SV40 Nuclear Localization Sequence (BirA*-EGFP-NLS), which should account for proteins that are spuriously biotinylated in the nucleus (7).
To visualize the distribution of biotinylation in cells expressing BirA*-tagged baits, we performed immunofluorescence on both Flp-In T-REx HeLa stable cells and in the three virally transduced cell types. As previously described (9), irrespective of its expression system and cell type, the LMNA bait protein (visualized using an anti-FLAG antibody) and biotinylated proximity partners (visualized using fluorophore-conjugated streptavidin) were predominantly nuclear, with a pronounced staining at the nuclear envelope (Fig. 2C; supplemental Figs. S2 and S3). Similarly, both the bait and biotinylated proximity partner signals for TBP, H2B, and EGFP-NLS exhibited a clear nuclear localization (supplemental Figs. S2 and S3). Consistent with their expected cellular localization, BirA*-EGFP and BirA*-TUBB displayed a pronounced cytosolic distribution, paralleling the biotinylation signal. We also note that bait protein expression levels in transduced cells was relatively uniform. Taken together, our analysis indicates that the lentiviral toolset enables performing in vivo proximal biotinylation in a bait dependent subcellular compartment. Using these vectors, we and others have also successfully performed viral BioID in other cell types, including cancer cell lines such as MDA-MB-231 (58), DLD-1, MCF10A and 501MEL, and primary cells including HUVEC (A.L. Couzens, J.D.R. Knight, J.P. Lambert, personal communication), confirming that the system is widely applicable.
Lentiviral BioID Yields Highly Reproducible Results
To more directly assess the lentiviral BioID strategy for the identification of proximity partners, cells infected as above (or Flp-In T-REx HeLa cells stably integrated with the inducible transgene) were grown to 75% confluence in a 15-cm dish, and induced with 1 μg/ml doxycycline and 40 μm biotin for 24 h prior to harvesting of the cells for BioID experiments (see Experimental Procedures and (31)). For each cell type, two biological replicates (consisting of independent viral infection, doxycycline/biotin induction and harvesting) of each bait or control protein (empty vector without BirA*, BirA*-EGFP, BirA*-EGFP-NLS) were profiled. The results were analyzed using SAINTexpress (16), with compression of the six controls to two virtual controls to increase stringency in scoring, as in (17); preys detected with a calculated Bayesian FDR ≤ 1% were qualified as high-confidence preys.
To test the reproducibility of the viral BioID pipeline in capturing proximity interactions, the spectral counts for each protein detected across the two biological replicates, or only for the high-confidence preys, were compared within each cell line (Fig. 3). Consistent with previous studies (20, 59), the biological replicates in Flp-In T-REx HeLa lines were highly similar, with R2 = 0.99 across all prey proteins identified and R2 = 0.96 when only the high-confidence preys were considered (Fig. 3A). Importantly, the reproducibility metrics associated with the lentivirally transduced HeLa were also high (R2 = 0.93 across all preys identified and R2 = 0.93 across only high-confidence preys, respectively; Fig. 3B). Primary cells also yielded reproducible data, with R2 of 0.97 and 0.96 in BJs and 0.99 and 0.99 in MEF cells (Fig. 3C and 3D). Taken together, this demonstrates that the lentiviral BioID approach can generate reproducible proximal protein identification.
Fig. 3.

Spectral counts for biological replicate 1 and biological replicate 2 were plotted against each other for each cell type. A, Flp-In T-REx HeLa, B, HeLa, C, BJ human fibroblasts, D, MEF. Graphs on the left show the full set of proteins identified whereas the graphs on the right only show proteins identified at FDR ≤ 1% following SAINTexpress analysis as described in Identification of High-confidence Proximity Partners. supplemental Tables S1–S4 report the SAINTexpress outputs whereas supplemental Table S9 lists the underlying protein evidence.
Lentiviral BioID Detects Biologically Relevant Proximal Interactions In Immortalized and Primary Cells
As detailed below and in supplemental Fig. S4 and supplemental Table S1–S4, the high-confidence proximal partners identified across all cell lines are consistent with the known localization and function of each bait.
LMNA
Lamins are intermediate filament proteins that form a scaffold on the luminal side of the inner nuclear membrane. They provide structural integrity to the nuclear envelope and play important regulatory roles in genomic organization and transcriptional regulation (reviewed in (60, 61)). However, because of their limited solubility, the biochemical purification of these proteins and their associated proteins poses technical challenges, which were overcome using BioID (9). Here, application of BioID to profile LMNA in Flp-In T-REx HeLa stable cell lines or virally transduced cells identified proteins enriched for the expected Gene Ontology Cellular Compartment (GO CC) term “Nuclear Envelope” (supplemental Table S5; GO:0005635, p values ≤ 1.3 × 10−13 across all cell lines). LMNA BioID captured multiple nuclear pore complex (NPC) components (supplemental Fig. S4A), including NUP107, NUP133, NUP160, NUP98, and NUP155 and the associated membrane proteins POM121 and NDC1 (62), but not the cytoplasmically-oriented proteins such as NUP214 and NUP88 (60). We also identified nuclear lamina scaffold proteins (LMNB1, LMNB2) and integral nuclear envelope and inner nuclear membrane proteins (LBR, SUN1, SUN2, EMD, LEMD3, TOR1AIP1, TMPO/LAP2) as well as AHCTF1 (also known as ELYS) (63), which is involved in the assembly of the nuclear pore complex into the nuclear envelope (supplemental Fig. S4A). In agreement with previous BioID work, we also recovered FAM169A (SLAP75) and VRK2 as high-confidence proximity partners for LMNA in most cell types (9, 29).
H2B
Genomic DNA wraps around the nucleosome core particle consisting of two pairs of H2A, H2B, H3, and H4 histones. Through its multiple post-translational modifications (64), the nucleosome is central to the regulation of chromatin compaction and DNA accessibility, the recruitment of regulatory proteins, and ultimately gene regulation (reviewed in (65, 66)). Consistent with these functions, BioID of H2B led to the identification of a set of proteins enriched for the GO CC term “Chromosome” (GO: 0005694; p value ≤ 6.9 × 10−8 across all cell lines) and the GO Biological Process (BP) term “Chromosome Organization” (GO:0006325; p value ≤ 2.0 × 10−15 across all cell lines; supplemental Table S6). These proximal interactors are implicated in diverse aspects of chromatin regulation, and include histone chaperones and nucleosome assembly proteins (RSF1, CHAF1A, NPM1) (67) and proteins involved in DNA damage response (MDC1, PARP1, TRIP12; supplemental Fig. S4B). Additionally, epigenetic code writers such as DNA methyltransferases (DNMT1, DNMT3A) and histone methyltransferases (EHMT1, EH2, NSD1–3), erasers (the demethylases KDM2A, KDM5A) as well as epigenetic readers (BRD2, BRD3, BRD7, CBX4, CBX5, CBX8, and MECP2) are recovered in the H2B BioID across cell types (supplemental Fig. S4B, supplemental Table S1–S4).
TBP
TATA box-Binding Protein (TBP) is a key component of the general transcription factor machinery, and is essential for RNA Polymerase II activity. Binding of TBP to proximal promoters leads to the recruitment of numerous TBP-associated factors (TAFs), followed by the sequential, regulated association of additional general transcription factor complexes (TFIIA-TFIIH) to form the pre-initiation complex (reviewed in (68)). BioID profiling of TBP led to identification of proteins most enriched for the GO CC term “Transcription Factor TFIID Complex” (GO:0005669; p value ≤ 3.67 × 10−5 across all cell lines; supplemental Table S7). Multiple components of the 1.3 mega-dalton TFIID complex (TAF1–6, TAF9) and members of the general transcription machinery (GTF2A1, GTF2B) were recovered (supplemental Fig. S4C, supplemental Table S1–S4) (68). In addition, in different cell types, we also identified several sequence-specific transcription factors that may function at the transcription start site (supplemental Tables S1–S4), though a further investigation of the cell type specificity of these finding was beyond the scope of this technical report.
TUBB
Lastly, BioID of the cytoskeleton protein tubulin beta (TUBB) recovered cytoskeletal proteins (e.g. HN1, HN1L, DPYSL2, DPYSL2). Many of the proximal proteins identified across cell types were chaperones, primarily the CCT complex (also known as TCP1 ring complex, TRiC) that is well-characterized for its role in the folding and assembly of tubulins (69), HSP70 and HSP90 and their co-chaperones, Tubulin-specific chaperones (TBC) (70), and the prefoldin complex (71). The most significant GO CC term was “Microtubule” (GO:0015630; p value ≤ 9.81 × 10−8 across multiple lines; supplemental Table S8), and the most significant GO BP term across all cell types was “Protein Folding” (GO:0006457; p value ≤ 1.23 × 10−11 across multiple lines; supplemental Table S8). Thus, like the nuclear baits, lentiviral BioID recovered meaningful proximity interactomes for this cytoskeletal bait (supplemental Fig. S4D, supplemental Tables S1–S4).
DISCUSSION
The study of protein-protein interactions has been a linchpin of our understanding of molecular mechanisms and rational pathway targeting for therapeutics. The BioID technique is playing a growing role in this endeavor (reviewed in (12)), as it can identify proximal partners that can be difficult to capture by other methods. Our primary motivation for generating lentiviral delivery plasmids for BioID was to expand this useful approach to other cell types, while maintaining the reproducibility and quality of the systems derived from stable inducible expression. We generated a set of lentiviral vectors enabling N- or C-terminal fusion of baits to BirA*-FLAG and tetracycline-regulatable expression. We also describe several versions of the vectors that enable establishment of stable cells, or fluorescence-based sorting of the infected cells. The viral system described here also allows for significant time savings (outlined in Fig. 2A).
As we previously described (7), the number and type of negative controls used influences the scoring of high-confidence interactions. For example, using cells that do not express the BirA* tag serves as an important control to model endogenous biotinylation, which is proportionally more highly detected if baits of interest are expressed at low levels. The analysis of an EGFP-BirA* fusion or BirA* alone helps to identify nonspecific biotinylation events. Lastly, the BirA*-EGFP-NLS fusions help to maximize background estimations in the nuclear compartment, mimicking proteins that are compartmentally restricted such as histones or transcription factors. To facilitate implementation and benchmarking of BioID in other cell types of interest, we recommend re-profiling both the negative controls as well as the test baits used in our study: these are made available to the scientific community in the pSTV2 lentiviral vector backbone.
Although direct comparison of the proximal proteins across multiple cell lines will be of great value and is enabled by our lentiviral systems (this is outside of the scope of this technical manuscript), this requires well-controlled experimental design to minimize technical variations. Besides the inclusion of controls within each experiment and standard good practices, comparison across cell types may require additional experimental considerations. For example, a careful titration of viral infection efficiency for each cell type is critical to ensure that comparable proportions of cells are infected with the transgene. It may also be necessary to adjust the concentrations of doxycycline to enable relatively comparable expression levels across conditions or cell types. Furthermore, because of variations in cell size and growth characteristics, normalizing the input material (either through matched cell pellet mass or ideally protein concentration), may help minimize variations in proximal partner identification and quantitation. In all cases, however, it is important to realize that a likely cause of the presence or absence of a protein in a BioID experiment in a given cell is simply a function of expression profiles or growth condition. Although ideally this can be assessed directly on the cell system used by deep proteome profiling, available resources that aim at providing protein abundance estimates across cell and tissue types (e.g. ProteomicsDB - (72)) may serve as a good proxy. Together with proper normalization strategies, the lentiviral toolkit should enable comparative studies of proximity proteome across conditions, cell types, and different patient-derived cells.
In summary, we have described the generation, implementation and benchmarking of lentiviral delivery vectors to perform BioID experiments in cell types that are difficult to transfect or for which stable cell line generation is not practical. These vectors are shared with the scientific community through the NBCC website (http://nbcc.lunenfeld.ca/resources), and new versions of these vectors, e.g. that incorporate newer enzymes such as BioID2 (73) and TurboID (74), will be distributed through this venue.
DATA AVAILABILITY
Data was deposited in ProteomeXchange via partner MassIVE (massive.ucsd.edu), and assigned accession numbers PXD010008, PXD010009 and MSV000082429, MSV000082430 (ftp://massive.ucsd.edu/MSV000082429, ftp://massive.ucsd.edu/MSV000082430) for the mouse and human data sets, respectively.
Supplementary Material
Acknowledgments
We thank Jeffrey Wrana for the kind gift of BJ fibroblasts and MEFs, Arshad Desai (UCSD) for the Flp-In T-REx HeLa cells, Laurence Pelletier for parental HeLa cells, Brian Raught for the MCS vectors, and our scientific colleagues who provided their DNA constructs through Addgene. We also thank Brett Larsen for expert advice on mass spectrometry, and James Knight and Guomin Liu for computational assistance. We are also grateful to Daniel Schramek, Brian Raught, and members of the Gingras laboratory, and in particular to Karen Colwill, for critical reading of this manuscript.
Footnotes
* A.-C.G. was supported by a Canadian Institutes of Health Research (CIHR) Foundation Grant (FDN 143301). Proteomics work was performed at the Network Biology Collaborative Centre at the Lunenfeld-Tanenbaum Research Institute, a facility supported by Canada Foundation for Innovation funding, by the Ontarian Government and by Genome Canada and Ontario Genomics (OGI-097, OGI-139). PST was supported through a grant to A.-C.G. from Medicine by Design, a program supported by the Canada First Research Excellence Fund. RS was partially supported through an Ontario Student Opportunity Trust Funds (OSOTF) Award. A.-C.G. is the Canada Research Chair (Tier 1) in Functional Proteomics.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- AP-MS
- affinity-purification coupled to mass spectrometry
- BirA*
- E. coli biotin ligase harboring a R118G mutation
- VP16
- herpes simplex virus (HSV) virion protein 16
- rTetR
- reverse tetracycline repressor
- rtTA
- reverse tetracycline-controlled transactivator, rTetR fused to VP16
- PGK
- phosphoglycerate kinase 1 promoter
- sEF1a
- short form of elongation factor-1 alpha (EF1a), promoter
- MEF
- mouse embryonic fibroblast
- MCS
- multiple cloning site
- NLS
- nuclear localization sequence.
REFERENCES
- 1. Gingras A. C., Gstaiger M., Raught B., and Aebersold R. (2007) Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8, 645–654 [DOI] [PubMed] [Google Scholar]
- 2. Stasyk T., and Huber L. A. (2004) Zooming in: fractionation strategies in proteomics. Proteomics 4, 3704–3716 [DOI] [PubMed] [Google Scholar]
- 3. Havugimana P. C., Hart G. T., Nepusz T., Yang H., Turinsky A. L., Li Z., Wang P. I., Boutz D. R., Fong V., Phanse S., Babu M., Craig S. A., Hu P., Wan C., Vlasblom J., Dar V. U., Bezginov A., Clark G. W., Wu G. C., Wodak S. J., Tillier E. R., Paccanaro A., Marcotte E. M., and Emili A. (2012) A census of human soluble protein complexes. Cell 150, 1068–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dunham W. H., Mullin M., and Gingras A. C. (2012) Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics 12, 1576–1590 [DOI] [PubMed] [Google Scholar]
- 5. Ho Y., Gruhler A., Heilbut A., Bader G. D., Moore L., Adams S. L., Millar A., Taylor P., Bennett K., Boutilier K., Yang L., Wolting C., Donaldson I., Schandorff S., Shewnarane J., Vo M., Taggart J., Goudreault M., Muskat B., Alfarano C., Dewar D., Lin Z., Michalickova K., Willems A. R., Sassi H., Nielsen P. A., Rasmussen K. J., Andersen J. R., Johansen L. E., Hansen L. H., Jespersen H., Podtelejnikov A., Nielsen E., Crawford J., Poulsen V., Sorensen B. D., Matthiesen J., Hendrickson R. C., Gleeson F., Pawson T., Moran M. F., Durocher D., Mann M., Hogue C. W., Figeys D., and Tyers M. (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415, 180–183 [DOI] [PubMed] [Google Scholar]
- 6. Li X., Wang W., Wang J., Malovannaya A., Xi Y., Li W., Guerra R., Hawke D. H., Qin J., and Chen J. (2015) Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol. Syst. Biol. 11, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lambert J. P., Tucholska M., Go C., Knight J. D., and Gingras A. C. (2015) Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteomics 118, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Snider J., Kotlyar M., Saraon P., Yao Z., Jurisica I., and Stagljar I. (2015) Fundamentals of protein interaction network mapping. Mol. Syst. Biol. 11, 848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hung V., Zou P., Rhee H. W., Udeshi N. D., Cracan V., Svinkina T., Carr S. A., Mootha V. K., and Ting A. Y. (2014) Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rees J. S., Li X. W., Perrett S., Lilley K. S., and Jackson A. P. (2015) Protein neighbors and proximity proteomics. Mol. Cell Proteomics 14, 2848–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim D. I., and Roux K. J. (2016) Filling the void: proximity-based labeling of proteins in living cells. Trends Cell Biol. 26, 804–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu Y., and Beckett D. (1994) Kinetics of biotinyl-5′-adenylate synthesis catalyzed by the Escherichia coli repressor of biotin biosynthesis and the stability of the enzyme-product complex. Biochemistry 33, 7354–7360 [DOI] [PubMed] [Google Scholar]
- 14. Choi-Rhee E., Schulman H., and Cronan J. E. (2004) Promiscuous protein biotinylation by Escherichia coli biotin protein ligase. Protein Sci. 13, 3043–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cronan J. E. (2005) Targeted and proximity-dependent promiscuous protein biotinylation by a mutant Escherichia coli biotin protein ligase. J. Nutr. Biochem. 16, 416–418 [DOI] [PubMed] [Google Scholar]
- 16. Teo G., Liu G., Zhang J., Nesvizhskii A. I., Gingras A. C., and Choi H. (2014) SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J. Proteomics 100, 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mellacheruvu D., Wright Z., Couzens A. L., Lambert J. P., St-Denis N. A., Li T., Miteva Y. V., Hauri S., Sardiu M. E., Low T. Y., Halim V. A., Bagshaw R. D., Hubner N. C., Al-Hakim A., Bouchard A., Faubert D., Fermin D., Dunham W. H., Goudreault M., Lin Z. Y., Badillo B. G., Pawson T., Durocher D., Coulombe B., Aebersold R., Superti-Furga G., Colinge J., Heck A. J., Choi H., Gstaiger M., Mohammed S., Cristea I. M., Bennett K. L., Washburn M. P., Raught B., Ewing R. M., Gingras A. C., and Nesvizhskii A. I. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Couzens A. L., Knight J. D., Kean M. J., Teo G., Weiss A., Dunham W. H., Lin Z. Y., Bagshaw R. D., Sicheri F., Pawson T., Wrana J. L., Choi H., and Gingras A. C. (2013) Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal 6, rs15. [DOI] [PubMed] [Google Scholar]
- 19. Coyaud E., Mis M., Laurent E. M., Dunham W. H., Couzens A. L., Robitaille M., Gingras A. C., Angers S., and Raught B. (2015) BioID-based identification of Skp Cullin F-box (SCF)beta-TrCP1/2 E3 ligase substrates. Mol. Cell. Proteomics 14, 1781–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gupta G. D., Coyaud E., Goncalves J., Mojarad B. A., Liu Y., Wu Q., Gheiratmand L., Comartin D., Tkach J. M., Cheung S. W., Bashkurov M., Hasegan M., Knight J. D., Lin Z. Y., Schueler M., Hildebrandt F., Moffat J., Gingras A. C., Raught B., and Pelletier L. (2015) A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell 163, 1484–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Firat-Karalar E. N., Rauniyar N., Yates J. R. 3rd, Stearns T. (2014) Proximity interactions among centrosome components identify regulators of centriole duplication. Curr. Biol. 24, 664–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dong J. M., Tay F. P., Swa H. L., Gunaratne J., Leung T., Burke B., and Manser E. (2016) Proximity biotinylation provides insight into the molecular composition of focal adhesions at the nanometer scale. Sci. Signal 9, rs4. [DOI] [PubMed] [Google Scholar]
- 23. Youn J. Y., Dunham W. H., Hong S. J., Knight J. D. R., Bashkurov M., Chen G. I., Bagci H., Rathod B., MacLeod G., Eng S. W. M., Angers S., Morris Q., Fabian M., Cote J. F., and Gingras A. C. (2018) High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell 69, 517–532.e511 [DOI] [PubMed] [Google Scholar]
- 24. van Vliet A. R., Giordano F., Gerlo S., Segura I., Van Eygen S., Molenberghs G., Rocha S., Houcine A., Derua R., Verfaillie T., Vangindertael J., De Keersmaecker H., Waelkens E., Tavernier J., Hofkens J., Annaert W., Carmeliet P., Samali A., Mizuno H., and Agostinis P. (2017) The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol. Cell 65, 885–899.e886 [DOI] [PubMed] [Google Scholar]
- 25. Wang Y., Liu L., Zhang H., Fan J., Zhang F., Yu M., Shi L., Yang L., Lam S. M., Wang H., Chen X., Wang Y., Gao F., Shui G., and Xu Z. (2016) Mea6 controls VLDL transport through the coordinated regulation of COPII assembly. Cell Res. 26, 787–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gimpel P., Lee Y. L., Sobota R. M., Calvi A., Koullourou V., Patel R., Mamchaoui K., Nedelec F., Shackleton S., Schmoranzer J., Burke B., Cadot B., and Gomes E. R. (2017) Nesprin-1alpha-dependent microtubule nucleation from the nuclear envelope via Akap450 is necessary for nuclear positioning in muscle cells. Curr. Biol. 27, 2999–3009.e2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Loh K. H., Stawski P. S., Draycott A. S., Udeshi N. D., Lehrman E. K., Wilton D. K., Svinkina T., Deerinck T. J., Ellisman M. H., Stevens B., Carr S. A., and Ting A. Y. (2016) Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell 166, 1295–1307.e1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fu Y., Lv P., Yan G., Fan H., Cheng L., Zhang F., Dang Y., Wu H., and Wen B. (2015) MacroH2A1 associates with nuclear lamina and maintains chromatin architecture in mouse liver cells. Sci. Rep. 5, 17186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Birendra K., May D. G., Benson B. V., Kim D. I., Shivega W. G., Ali M. H., Faustino R. S., Campos A. R., and Roux K. J. (2017) VRK2A is an A-type lamin-dependent nuclear envelope kinase that phosphorylates BAF. Mol. Biol. Cell 28, 2241–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Uezu A., Kanak D. J., Bradshaw T. W., Soderblom E. J., Catavero C. M., Burette A. C., Weinberg R. J., and Soderling S. H. (2016) Identification of an elaborate complex mediating postsynaptic inhibition. Science 353, 1123–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hesketh G. G., Youn J. Y., Samavarchi-Tehrani P., Raught B., and Gingras A. C. (2017) Parallel exploration of interaction space by BioID and affinity purification coupled to mass spectrometry. Methods Mol. Biol. 1550, 115–136 [DOI] [PubMed] [Google Scholar]
- 32. Golding M. C., and Mann M. R. (2011) A bidirectional promoter architecture enhances lentiviral transgenesis in embryonic and extraembryonic stem cells. Gene. Ther. 18, 817–826 [DOI] [PubMed] [Google Scholar]
- 33. Amendola M., Venneri M. A., Biffi A., Vigna E., and Naldini L. (2005) Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat. Biotechnol. 23, 108–116 [DOI] [PubMed] [Google Scholar]
- 34. Papapetrou E. P., Tomishima M. J., Chambers S. M., Mica Y., Reed E., Menon J., Tabar V., Mo Q., Studer L., and Sadelain M. (2009) Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proc. Natl. Acad. Sci. U.S.A. 106, 12759–12764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Woltjen K., Michael I. P., Mohseni P., Desai R., Mileikovsky M., Hamalainen R., Cowling R., Wang W., Liu P., Gertsenstein M., Kaji K., Sung H. K., and Nagy A. (2009) piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 458, 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Comartin D., Gupta G. D., Fussner E., Coyaud E., Hasegan M., Archinti M., Cheung S. W., Pinchev D., Lawo S., Raught B., Bazett-Jones D. P., Luders J., and Pelletier L. (2013) CEP120 and SPICE1 cooperate with CPAP in centriole elongation. Curr. Biol. 23, 1360–1366 [DOI] [PubMed] [Google Scholar]
- 37. Katzen F. (2007) Gateway((R)) recombinational cloning: a biological operating system. Expert. Opin. Drug Discov. 2, 571–589 [DOI] [PubMed] [Google Scholar]
- 38. Belteki G., Haigh J., Kabacs N., Haigh K., Sison K., Costantini F., Whitsett J., Quaggin S. E., and Nagy A. (2005) Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 33, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stewart S. A., Dykxhoorn D. M., Palliser D., Mizuno H., Yu E. Y., An D. S., Sabatini D. M., Chen I. S., Hahn W. C., Sharp P. A., Weinberg R. A., and Novina C. D. (2003) Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kessner D., Chambers M., Burke R., Agus D., and Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu G., Zhang J., Larsen B., Stark C., Breitkreutz A., Lin Z. Y., Breitkreutz B. J., Ding Y., Colwill K., Pasculescu A., Pawson T., Wrana J. L., Nesvizhskii A. I., Raught B., Tyers M., and Gingras A. C. (2010) ProHits: integrated software for mass spectrometry-based interaction proteomics. Nat. Biotechnol. 28, 1015–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perkins D. N., Pappin D. J., Creasy D. M., and Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 43. Eng J. K., Jahan T. A., and Hoopmann M. R. (2013) Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 [DOI] [PubMed] [Google Scholar]
- 44. Ma K., Vitek O., and Nesvizhskii A. I. (2012) A statistical model-building perspective to identification of MS/MS spectra with PeptideProphet. BMC Bioinformatics 13 Suppl 16, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A. I., and Aebersold R. (2010) A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Knight J. D. R., Choi H., Gupta G. D., Pelletier L., Raught B., Nesvizhskii A. I., and Gingras A. C. (2017) ProHits-viz: a suite of web tools for visualizing interaction proteomics data. Nat. Methods 14, 645–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reimand J., Arak T., Adler P., Kolberg L., Reisberg S., Peterson H., and Vilo J. (2016) g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 44, W83–W89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Loew R., Heinz N., Hampf M., Bujard H., and Gossen M. (2010) Improved Tet-responsive promoters with minimized background expression. BMC Biotechnol. 10, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhu Z., Zheng T., Lee C. G., Homer R. J., and Elias J. A. (2002) Tetracycline-controlled transcriptional regulation systems: advances and application in transgenic animal modeling. Semin. Cell Dev. Biol. 13, 121–128 [DOI] [PubMed] [Google Scholar]
- 50. Yang T., Burrows C., and Park J. H. (2014) Development of a doxycycline-inducible lentiviral plasmid with an instant regulatory feature. Plasmid 72, 29–35 [DOI] [PubMed] [Google Scholar]
- 51. Kamper M. R., Gohla G., and Schluter G. (2002) A novel positive tetracycline-dependent transactivator (rtTA) variant with reduced background activity and enhanced activation potential. FEBS Lett. 517, 115–120 [DOI] [PubMed] [Google Scholar]
- 52. Gossen M., Freundlieb S., Bender G., Muller G., Hillen W., and Bujard H. (1995) Transcriptional activation by tetracyclines in mammalian cells. Science 268, 1766–1769 [DOI] [PubMed] [Google Scholar]
- 53. Casola S. (2010) Mouse models for miRNA expression: the ROSA26 locus. Methods Mol. Biol. 667, 145–163 [DOI] [PubMed] [Google Scholar]
- 54. Richner M., Victor M. B., Liu Y., Abernathy D., and Yoo A. S. (2015) MicroRNA-based conversion of human fibroblasts into striatal medium spiny neurons. Nat. Protoc. 10, 1543–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim J. H., Lee S. R., Li L. H., Park H. J., Park J. H., Lee K. Y., Kim M. K., Shin B. A., and Choi S. Y. (2011) High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE 6, e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kumar M., Keller B., Makalou N., and Sutton R. E. (2001) Systematic determination of the packaging limit of lentiviral vectors. Hum. Gene. Ther. 12, 1893–1905 [DOI] [PubMed] [Google Scholar]
- 57. al Yacoub N., Romanowska M., Haritonova N., and Foerster J. (2007) Optimized production and concentration of lentiviral vectors containing large inserts. J. Gene. Med. 9, 579–584 [DOI] [PubMed] [Google Scholar]
- 58. Knight J. F., Sung V. Y. C., Kuzmin E., Couzens A. L., de Verteuil D. A., Ratcliffe C. D. H., Coelho P. P., Johnson R. M., Samavarchi-Tehrani P., Gruosso T., Smith H. W., Lee W., Saleh S. M., Zuo D., Zhao H., Guiot M. C., Davis R. R., Gregg J. P., Moraes C., Gingras A. C., and Park M. (2018) KIBRA (WWC1) Is a metastasis suppressor gene affected by chromosome 5q loss in triple-negative breast cancer. Cell Rep. 22, 3191–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. St-Denis N., Gupta G. D., Lin Z. Y., Gonzalez-Badillo B., Veri A. O., Knight J. D. R., Rajendran D., Couzens A. L., Currie K. W., Tkach J. M., Cheung S. W. T., Pelletier L., and Gingras A. C. (2016) Phenotypic and interaction profiling of the human phosphatases identifies diverse mitotic regulators. Cell Rep. 17, 2488–2501 [DOI] [PubMed] [Google Scholar]
- 60. Beck M., and Hurt E. (2017) The nuclear pore complex: understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 18, 73–89 [DOI] [PubMed] [Google Scholar]
- 61. Raices M., and D'Angelo M. A. (2012) Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nat. Rev. Mol. Cell Biol. 13, 687–699 [DOI] [PubMed] [Google Scholar]
- 62. Mansfeld J., Guttinger S., Hawryluk-Gara L. A., Pante N., Mall M., Galy V., Haselmann U., Muhlhausser P., Wozniak R. W., Mattaj I. W., Kutay U., and Antonin W. (2006) The conserved transmembrane nucleoporin NDC1 is required for nuclear pore complex assembly in vertebrate cells. Mol. Cell 22, 93–103 [DOI] [PubMed] [Google Scholar]
- 63. Clever M., Funakoshi T., Mimura Y., Takagi M., and Imamoto N. (2012) The nucleoporin ELYS/Mel28 regulates nuclear envelope subdomain formation in HeLa cells. Nucleus 3, 187–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Suganuma T., and Workman J. L. (2011) Signals and combinatorial functions of histone modifications. Annu. Rev. Biochem. 80, 473–499 [DOI] [PubMed] [Google Scholar]
- 65. Ruthenburg A. J., Li H., Patel D. J., and Allis C. D. (2007) Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 8, 983–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li B., Carey M., and Workman J. L. (2007) The role of chromatin during transcription. Cell 128, 707–719 [DOI] [PubMed] [Google Scholar]
- 67. Venkatesh S., and Workman J. L. (2015) Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 16, 178–189 [DOI] [PubMed] [Google Scholar]
- 68. Sainsbury S., Bernecky C., and Cramer P. (2015) Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 16, 129–143 [DOI] [PubMed] [Google Scholar]
- 69. Llorca O., Martin-Benito J., Ritco-Vonsovici M., Grantham J., Hynes G. M., Willison K. R., Carrascosa J. L., and Valpuesta J. M. (2000) Eukaryotic chaperonin CCT stabilizes actin and tubulin folding intermediates in open quasi-native conformations. EMBO J. 19, 5971–5979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tian G., and Cowan N. J. (2013) Tubulin-specific chaperones: components of a molecular machine that assembles the alpha/beta heterodimer. Methods Cell Biol. 115, 155–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lopez-Fanarraga M., Avila J., Guasch A., Coll M., and Zabala J. C. (2001) Review: postchaperonin tubulin folding cofactors and their role in microtubule dynamics. J. Struct. Biol. 135, 219–229 [DOI] [PubMed] [Google Scholar]
- 72. Wilhelm M., Schlegl J., Hahne H., Gholami A. M., Lieberenz M., Savitski M. M., Ziegler E., Butzmann L., Gessulat S., Marx H., Mathieson T., Lemeer S., Schnatbaum K., Reimer U., Wenschuh H., Mollenhauer M., Slotta-Huspenina J., Boese J. H., Bantscheff M., Gerstmair A., Faerber F., and Kuster B. (2014) Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587 [DOI] [PubMed] [Google Scholar]
- 73. Kim D. I., Jensen S. C., Noble K. A., Kc B., Roux K. H., Motamedchaboki K., and Roux K. J. (2016) An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 27, 1188–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Branon T. C., Bosch J. A., Sanchez A. D., Udeshi N. D., Svinkina T., Carr S. A, Feldman J. L., Perrimon N. and Ting A. Y. (2018) Directed evolution of TurboID for efficient proximity labeling in living cells and organisms. Nature Biotechnology, 2018, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data was deposited in ProteomeXchange via partner MassIVE (massive.ucsd.edu), and assigned accession numbers PXD010008, PXD010009 and MSV000082429, MSV000082430 (ftp://massive.ucsd.edu/MSV000082429, ftp://massive.ucsd.edu/MSV000082430) for the mouse and human data sets, respectively.