

Abstract
Angiogenesis, the formation of new blood vessels from pre-existing capillaries, is very tightly regulated and normally does not occur except during developmental and reparative processes. This tight regulation is maintained by a balanced production of positive and negative regulators, and alterations under pathological conditions such as retinopathy of prematurity, diabetic retinopathy, and age-related macular degeneration can lead to growth of new and abnormal blood vessels. Although the role of proangiogenic factors such as vascular endothelial growth factor has been extensively studied, little is known about the roles of negative regulators of angiogenesis in the pathogenesis of these diseases. Here, we will discuss the role of thrombospondin-1 (TSP1), one of the first known endogenous inhibitors of angiogenesis, in ocular vascular homeostasis, and how its alterations may contribute to the pathogenesis of age-related macular degeneration and choroidal neovascularization. We will also discuss its potential utility as a therapeutic target for treatment of ocular diseases with a neovascular component.
Keywords: Choroidal Endothelial Cells, Pigment Epithelium Drived Factor, Retinal Pigment Epithelial Cells, Retinal Vascular Cells, Thrombospondins
INTRODUCTION
Angiogenesis plays a fundamental role in tissue development, regeneration, repair, and pathological responses in the eye. Although numerous studies have focused on the identification of factors that promote angiogenesis, limited studies have examined the physiological and pathological roles of endogenous inhibitors of angiogenesis. We have previously shown that vitreous and aqueous humor samples prepared from the eyes of various species including mouse, rat, bovine, and human contain a significant amount of thrombospondin-1 (TSP1).[1] Furthermore, the level of TSP1 was significantly downregulated in vitreous samples prepared from diabetic animals and humans,[1,2] suggesting an important role for TSP1 in the development and progression of diabetic retinopathy.
TSP1 was one of the first identified endogenous inhibitors of angiogenesis, and it was subsequently demonstrated that its expression is downregulated during the progression and angiogenic switch in a variety of solid tumors.[3] We showed that the TSP1 level is downregulated during the progression of uveal melanoma in the transgenic Tyr-Tag mouse model. This was concomitant with increased tumor vascularity and growth.[4] However, the physiological role that TSP1 plays during vascular development and neovascularization remains unknown. Mice deficient in TSP1 (TSP1-/-) exhibit increased vascular density in many tissues and delayed wound healing.[5] Using these mice, we demonstrated that TSP1 expression is essential for appropriate pruning and remodeling during the late stages of postnatal retinal vasculature development, such that in its absence, an increase in retinal vascular density is observed.[6]
To assess the contribution of TSP1 to retinal neovascularization, we utilized the mouse oxygen-induced ischemic retinopathy (OIR) model. We found that TSP1 expression contributes to blood vessel loss during exposure to hyperoxia (when VEGF level is low), but minimally affects ischemia-mediated retinal neovascularization (when VEGF level is high).[6] These observations are consistent with increased VEGF expression in the retinas during hypoxia phase of OIR, independent of TSP1 status. In addition, exposure to hyperoxia and hypoxia did not affect TSP1 expression in wild-type mice during OIR. Thus, we proposed that threshold levels exist for TSP1 and VEGF during normal retinal vascularization, and the presence or absence of neovascularization is mainly determined by the level of VEGF such that an increase in VEGF (during hypoxia) will drive neovascularization whereas a decrease in VEGF (during hyperoxia) leads to TSP1-mediated elimination of excess blood vessels.[6] This process is important during pruning and remodeling of the developing vasculature, and for maintenance of a mature and quiescence vascular phenotype.
Although a significant increase in retinal neovascularization was not observed in TSP1 null mice during OIR, a significant increase in choroidal neovascularization (CNV) was evident in TSP1 null mice subjected to a mouse model of laser-induced CNV.[7] This was mainly attributed to an increased number of macrophages recruited to the lesions in the absence of TSP1, and was consistent with the anti-inflammatory role proposed for TSP1 in the eye.[8] Thus, the regulatory effect of TSP1 on the retinal vasculature may be significantly different from that in the choroidal vasculature, and needs further investigation.
Mouse retinal vascularization occurs after birth, but the maximum retinal TSP1 expression is not observed until two weeks after birth.[6] To further demonstrate the important role of TSP1 expression in retinal vascular development and neovascularization, we proposed that TSP1 overexpression early during eye development would attenuate retinal vascular development and neovascularization. To test this hypothesis, we generated transgenic mice in which TSP1 expression was driven by the αA-crystallin promoter. This allowed TSP1 expression in the eye around embryonic day 14, in advance of the onset of retinal vascularization. This increase in early TSP1 expression was sufficient to attenuate retinal vascular development and neovascularization during OIR.[9] Furthermore, crossing these transgenic mice with the Tyr-Tag mice dramatically suppressed the development and progression of uveal melanoma.[4]
Our earlier studies indicated that decreased TSP1 production during diabetes may exacerbate the development and progression of diabetic retinopathy.[1] To further demonstrate the importance of TSP1 suppression during diabetes and progression of diabetic retinopathy, we generated a line of diabetic mice that lacked TSP1. We showed that the diabetic mice lacking TSP1 developed more severe retinopathy.[10] Collectively, these studies establish an important role for TSP1 expression in ocular vascular homeostasis, in which its altered production contributes to vascular abnormalities and neovascularization associated with various pathological conditions. Therefore, TSP1 presents as a potential target for the development of new therapeutics for the treatment of eye diseases with a neovascular component. In this review, we will discuss the important roles of TSP1 in ocular vascular homeostasis, its cell autonomous effect on various ocular vascular cell types, and the development of mimetic peptides of TSP1 with antiangiogenic activity for the treatment of eye diseases, wherein changes in TSP1 production play a pivotal role.
THROMBOSPONDINS
Thrombospondins (TSPs) belong to the family of matricellular proteins, which play critical roles in remodeling and re-organization of extracellular matrix components affecting various cellular functions. The TSP family currently consists of 5 members (TSP1-5) encoded by different genes that exhibit a specific temporal and spatial expression pattern during development.[3] They are multi-domain proteins and interact with various cell surface receptors with different affinities and avidities, thereby affecting various cellular functions including proliferation, migration, and apoptosis [Figure 1]. TSP1 and TSP2 form one subfamily, and both contain type 1 repeat domains that confer their antiangiogenic activity. TSP1 and TSP2 normally form homo-trimers. TSP3–5 lack the type 1 repeat domains and are normally pentomeric in their organization. Although this subfamily was initially thought to have limited roles in the modulation of angiogenesis, recent studies using transgenic mice suggest important contributions from this subfamily to vascular integrity and function.[11]
Figure 1.
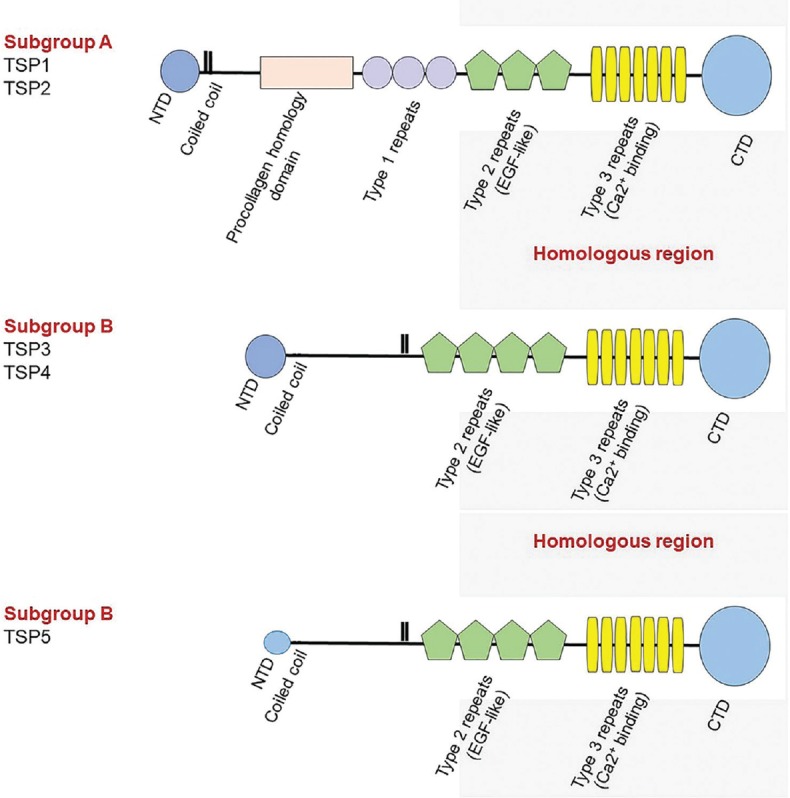
Domain organization of thrombospondins. Schematic diagram of the domain organization of thrombospondin family members. NTD, N-terminal domain; CTD, C-terminal domain.
TSP1 or platelet TSP was the first family member identified in platelets as a major component of the α-granules.[12] Later, it was demonstrated that many cell types including epithelial cells, endothelial cells (ECs), perivascular supporting cells, and fibroblasts produce TSP1.[13,14] However, the cell autonomous function of TSP1 in these cells remains poorly understood. We have shown that various ocular cell types in culture produce TSP1 including retinal ECs, pericytes, astrocytes, choroidal ECs, and retinal pigment epithelial (RPE) cells.[15,16,17,18,19] To assess the cell autonomous impact of TSP1 in various cell types, we compared their phenotypic characteristics with those prepared from TSP1-deficient mice. Here we will briefly discuss the cell autonomous impact of TSP1 expression on the function of ocular vascular cells.
THROMBOSPONDIN-1 EXPRESSION IN RETINAL VASCULAR CELLS
Numerous studies have indicated that vascular cells from vascular bed of various tissues express TSP1. We showed that retinal ECs, pericytes, and astrocytes express TSP1, and compared the characteristics of these cells with those prepared from TSP1-/-mice.[15,16,17] We showed that retinal ECs prepared from TSP1-/-mice maintain a more proliferative and migratory phenotype compared with wild-type cells.[15] These activities were mediated, at least in part, through sustained activation of various cell signaling pathways including MAPK/ERKs and cell cycle regulatory proteins.[20] This proangiogenic phenotype of retinal EC is consistent with increased retinal vascular density and increased number of ECs in retinas from TSP1-deficient mice.[6] Exogenous TSP1 inhibits EC proliferation and migration, and induces EC apoptosis through intrinsic and extrinsic pathways.[21] We also showed that re-expression of TSP1 in a polyomavirus middle-T transformed line of brain ECs (bEND.3) restored the normal phenotype by suppressing the migratory and proliferative phenotype of these cells.[22] Thus, TSP1 expression in EC is essential for keeping their proangiogenic activity in check.
Perivascular-supporting cells including smooth muscle cells (SMCs) express TSP1.[23] In addition, expression of TSP1 by SMC is essential for their proliferative and migratory responses to platelet-derived growth factor (PDGF).[24] Thus, TSP1 expression was proposed to act as an autocrine factor with an important role in proliferation and migration of SMCs. We showed that retinal pericytes also express TSP1, and most importantly, retinal pericytes prepared from TSP1-/-mice were less proliferative and migratory, and failed to respond to PDGF-BB.[17] Thus, TSP1 expression is important for proliferation and migration of retinal pericytes, especially in response to PDGF.[23] However, we did not observe a significant difference in the density and number of pericytes in TSP1-/-mice developing retinal vasculature.[6] Thus, other complimentary pathways may promote the recruitment of pericytes to the developing retina vasculature in the absence of TSP1. However, the identity of these pathways requires further investigations of the regulatory mechanisms that control pericyte migration and proliferation.
We and others have shown that astrocytes also express TSP1.[16,25] However, little was known about the cell autonomous impact of TSP1 on retinal astrocyte function. Recent studies have established a role for TSP1 and TSP2 produced by astrocytes in the synaptogenesis of retinal ganglion cells.[25] To determine the cell autonomous impact of TSP1 expression in retinal astrocytes, we isolated retinal astrocytes from TSP1-/-mice.[16] We showed that TSP1-/-astrocytes are more adherent on fibronectin and vitronectin, but exhibit similar migratory activity and integrin expression. In addition, lack of TSP1 may be compensated by increased production of TSP2, a closely related TSP1 family member, in these cells. This is consistent with lack of any sign of retinal neuronal dysfunction in TSP1-/-mice determined by electroretinography analysis.
Although retinal pericytes also express significant amounts of TSP2, the retinal EC normally exhibit no detectable levels of TSP2. However, we recently showed that increased oxidative stress enhances TSP2 expression in retinal EC and attenuates their ability to undergo capillary morphogenesis.[26] In the retinal vasculature, pericytes are the major source of TSP2 expression, and its expression is further enhanced under oxidative stress.[27,28] However, the cell autonomous role of TSP2 expression in retinal vascular cells and the characteristics of TSP2-null vascular cells compared with wild type cells have yet to be studied, as we have reported for TSP1. However, we recently demonstrated that the germline deletion of TSP2 minimally affects the postnatal retinal vascular development and neovascularization.[28]
The level of TSP1 is dramatically downregulated in ocular samples from diabetic animals and humans.[2] However, the reason for this decrease in vivo remains unknown. We have shown that incubation of retinal EC, under high glucose conditions, results in decreased production of TSP1 in retinal ECs, and it is likely responsible for the enhanced migration of retinal ECs observed under high glucose conditions through activation of MAPK/ERK and PI3kinase pathways.[1,20,29] In contrast, retinal pericytes under high glucose conditions are less migratory and do not respond to enhanced promigratory activity of PDGF, perhaps as a result of increased Bim expression, oxidative stress, and apoptosis.[30] Incubation of retinal astrocytes under high glucose conditions resulted in their increased proliferation, which was concomitant with increased GFAP production.[31] Astrocytes cultured under high glucose conditions were also less migratory and failed to form a network on Matrigel. However, exogenously added PDGF-AA or PDGF-BB restored their migratory activity. High glucose conditions also enhanced the production of both TSP1 and TSP2 in astrocytes concomitant with the activation of NF-κB, ERK, and JNK pathways with a proinflammatory phenotype.[31]
THROMBOSPONDIN-1 EXPRESSION IN RETINAL PIGMENT EPITHELIAL CELLS
Expression of TSP1 by RPE cells has been previously reported, and changes in its expression may be linked to abnormalities associated with RPE cell function and integrity of Bruch's membrane.[18,32,33,34] TSP1 is present in the Bruch's membrane, and its decreased levels in eyes from patients with AMD suggest that the integrity of Bruch's membrane is compromised, leading to CNV under the retinal pigment epithelium.[33,35] Thus, TSP1 expression by RPE cells may play a significant role in proper RPE cell function and the integrity of Bruch's membrane, thereby preventing the growth of choroidal new vessels into the sub-RPE space. Increased expression of TSP1 and activation of TGF-β in response to retinoic acid has been demonstrated to be responsible for attenuation of RPE cell adhesion and migration.[34] In addition, expression of TSP1 is impaired in eyes of patients with AMD, and its reduced levels in Bruch's membrane in the submacular region may contribute to AMD and CNV.[33,35] Furthermore, TSP1-deficient mice exhibit enhanced choroidal neovascularization in a mouse model of laser-induced CNV.[7] Thus, decreased endogenous TSP1 levels produced by RPE cells may contribute to CNV during AMD. However, the cell autonomous impact of TSP1 on RPE and choroidal EC has not been previously addressed.
To better understand the role TSP1 plays in the modulation of RPE cell function, we isolated RPE cells from wild-type and TSP1-/-mice.[18] We showed that TSP1 deficiency had a significant impact on the junctional properties of RPE cells. TSP1-/-RPE cells were more proliferative and adherent, less migratory, and exhibited defects in phagocytosis, perhaps as a result of reduced cathepsin B levels and lysosomal activity. TSP1-/-RPE cells also exhibited increased oxidative stress and an inflammatory phenotype compared with wild-type cells. These results were consistent with the reduced expression of TSP1 and the inflammatory phenotype of eyes from patients with AMD.[35]
THROMBOSPONDIN-1 EXPRESSION IN CHOROIDAL ENDOTHELIAL CELL
Endothelial cells from many vascular beds express TSP1. However, whether choroidal EC express TSP1 and how TSP1 affects their cell autonomous function remains unknown. We have recently cultured choroidal ECs from wild-type and TSP1-/-mice.[19] We showed that TSP1-/-choroidal ECs grow at a slower rate and exhibit increased basal levels of apoptosis as well as when challenged with H2O2. TSP1-/-choroidal ECs were also less adherent and migratory compared to wild-type cells, and failed to undergo capillary morphogenesis in Matrigel. These results are in contrast to those reported in TSP1-/-retinal EC, which were more proliferative and migratory.[20] Wild-type choroidal ECs produced a significant amount of TSP1 that was mainly cell-associated. Although TSP1-/-choroidal EC did not express TSP1, as expected, they expressed significantly higher levels of TSP2 compared with wild-type cells.[19] In contrast, wild type retinal EC do not express detectable levels of TSP2.[26] TSP1-/-choroidal EC also showed increased levels of iNOS and NO but similar levels of VEGF compared to wild-type cells. Thus, TSP1 expression in choroidal ECs modulates their angioinflammatory phenotype, which is distinct from the effects of TSP1 on retinal ECs. These differences may be associated, at least in part, with the fenestrated phenotype of choroidal EC and deserves further investigation.
PATHOGENESIS OF AMD
AMD is characterized by a progressive degeneration of the macula and severe irreversible vision loss. Development of AMD is associated with an abnormal anatomy of the photoreceptor-RPE, Bruch's membrane, and choriocapillaris complex. Two morphologic forms have been defined for AMD including the dry form, which consists of drusen and geography atrophy (GA), and the wet or exudative form, which is associated with CNV. Pathological hallmarks of AMD are mostly presented in the central area of the retina, specifically in the macula. The appearance of drusen bodies between the basement membrane of RPE and Bruch's membrane has been considered an early clinical sign of AMD.[36] Drusen alone do not lead to a significant loss of visual acuity in AMD, but can cause a decline in macular function including decreased sensitivity of the central visual field and color contrast as well as spatiotemporal sensitivity.[37,38,39] A few small drusen may not lead to diminished vision; however, large drusen with an increased number may result in distortion of vision and increase the possibility of progression to late stages of AMD.[40]
Geographic atrophy (GA) occurs when there is loss of RPE cells associated with the overlying photoreceptor layer. On fluorescein angiography, areas of GA are distinguished by the presence of increased fluorescence from diffuse and irregular patches indicating the development of GA. GA gradually contributes to advanced stages of vision loss, most likely through RPE degeneration and loss of photoreceptors in the macula leading to tissue atrophy or death.[40,41] Choroidal vasculature, RPE, and photoreceptors can be involved in different stages of the disease.
Visual impairment initially arises from RPE degeneration, and may be complicated by the secondary effect of CNV.[41,42,43] Growth of new blood vessels originate from the choroid. However, the detailed mechanisms underlying CNV pathogenesis are not well understood. The early phase of CNV is associated with migration and proliferation of choroidal EC into the sub-RPE space, rupturing through the Bruch's membrane. In the active phase, CNV starts to expand either by remaining beneath the RPE or by entering the sub-retinal space. Ultimately, in the end stage of untreated exudative AMD, CNV becomes fibrotic and forms disciform scars.[40,44,45] The diameter and thickness of these disciform scars might be influenced by the degree of RPE and photoreceptor cell degeneration.[45]
The dysregulation of VEGF expression has been identified as an essential factor in the development and progression of AMD and CNV. Donor eyes obtained from AMD patients have revealed over-expression of VEGF in the RPE, consistent with higher VEGF levels in vitreous samples of patients with exudative AMD.[46,47,48] There are also many other angiogenic factors whose alterations could contribute to the development of CNV including insulin-like growth factor (IGF), fibroblast growth factor (FGF), interleukins, and angiopoietins.[49,50,51,52,53] VEGF promotes vascular permeability and functions as a specific EC mitogen.[46,50,51,54] It also acts as a chemotactic factor for recruitment of macrophages that actively enhance the production of VEGF and various cytokines in the early phase of CNV.[55,56] The penetration of CNV through Bruch's membrane is enhanced via production of matrix metalloproteinases (MMPs) of macrophages and vascular endothelium.[57] At the late phase, maturation and stabilization of these new blood vessels is stimulated by transforming growth factor β (TGFβ), tissue inhibitor of metalloproteinases 3 (TIMP3), and other inhibitors of angiogenesis including TSP1, which ultimately result in the formation of disciform scars at the end stage.
RETINAL PIGMENT EPITHELIUM AND BRUCH’S MEMBRANE
Retinal detachment and RPE degeneration are major contributors to the loss of central vision acuity.[58] RPE is a single layer of polarized epithelial cells located at the base of the retina. These hexagonal cells are attached to each other by tight junctions, which form the outer blood retinal barrier blocking nonspecific transportation of various materials from the choroid to the retina. On its basolateral surface, the RPE firmly attaches to Bruch's membrane, which intercepts the RPE from the choroid.[40] The RPE plays crucial roles in retinal hemostasis through several activities including light absorption, phagocytosis, transepithelial transport, and vascular homeostasis.[59]
RPE cells supply nutrients required for maintenance of the visual function through photoreceptor outer segments. Moreover, RPE cells transport metabolic end products, water, and ions from the sub-retinal space to the choroid.[60] RPE cells act as a crucial element of the waste disposal system for the retina. They are involved in the phagocytosis of shed outer segments and photoreceptor debris by either recycling or completely degrading them, and also exocytosis of the remains to the choroid for clearance from the retina.[61] In addition, RPE cells produce many factors including FGFs, IGF-I, transforming growth factor-β (TGF-β), ciliary neurotrophic factor (CNTF), lens epithelium-derived growth factor (LEDGF), platelet-derived growth factor (PDGF), members of the interleukin family, VEGF, TSP1, and PEDF, all of which have important functions in maintaining retinal homeostasis.
Curcio et al suggested that photoreceptor degeneration and loss occurs in the RPE-Bruch's membrane complex in the early stages of AMD.[62] Induction of photoreceptor and RPE cell apoptosis occurs in human AMD.[63,64] Furthermore, these apoptotic cells are mainly found at the edges of atrophy, where extended atrophic regions are associated with vision loss in patients with GA.[40,63] Fas/Fas ligand system has been postulated to be a potential mechanism involved in the photoreceptor apoptosis in AMD.[40,63] RPE cell degeneration has been identified as a critical element involved in the loss of photoreceptor cells. Maeda et al showed that intravitreal ornithine-induced degeneration of RPE had a direct effect on the loss of photoreceptors. RPE degeneration leads to severe abnormal photoreceptor function.[65] The aging RPE monolayer has approximately 0.3% rate of cell loss every year, which may lead to high metabolic demand for each cell.[66,67] Furthermore, aging RPE cells present alterations in pigmentation, decreased melanosomes, reduced cell density, and increased number of lipofuscin granules.[68] These alterations contribute to elevated stress levels in RPE cells, where some of this oxidative stress[60] could be linked to decreased levels of TSP1 production by RPE cells. Declining protective mechanisms of RPE cells associated with induced oxidative stress or active photo-oxidative reaction species were shown to be major contributors to the pathogenesis of AMD.
Lipofuscin granules present in the RPE cells provide a source of reactive oxygen species (ROS).[59,69,70,71,72] Lipofuscin, when excited by the presence of light, generates reactive oxygen intermediates, which can contribute to lysosomal dysfunction, leading to lipid peroxidation and ultimately RPE cell atrophy.[73,74,75] Although not much is known about the potential causes of lipofuscin accumulation, age-related alterations in RPE cells including lysosomal enzyme dysfunction and increased oxidative stress were similar to those observed in TSP1-/-RPE cells,[18] and a reduction of antioxidants and pigmentary changes have been suggested to be major contributors. TSP1 levels have also been reported to be decreased in Bruch's membrane in eyes of patients with AMD.[35,76]
Accumulation of advanced-glycation end products (AGEs) in RPE cells and in the Bruch's membrane can also result in oxidative stress.[77,78,79] AGEs are found in drusen bodies, which are considered early clinical signs of AMD.[80] They can also contribute to the formation of CNV. Incomplete degradation of metabolic end products from RPE and/or photoreceptor cells including lipoproteins, phospholipids, triglycerides, fatty acids, and other hydrophobic materials may result in deposit formation in the Bruch's membrane or basal lamina, which may indicate failure of aged Bruch's membrane in transporting the materials.[40,81,82,83,84] Some of the age-related changes in the Bruch's membrane contributing to AMD pathologies include increased thickness caused by basal laminar deposits, drusen and basal linear deposits, decreased macular elastic layer thickness, declined solubility of Bruch's membrane collagens, followed by reduced activity of collagenolytic enzymes and oxidative modification of proteins, lipid peroxidation, and a reduction of water transport activity (hydraulic conductivity) leading to decreased permeability of the Bruch's membrane.[59,85,86,87,88,89,90,91,92,93] Collectively, changes in Bruch's membrane most likely have a key role in RPE cell dysfunction related to aging and/or AMD.
CHOROIDAL VASCULATURE
The choroid plays a crucial role in retinal homeostasis, dissipating heat and nourishing the outer retina including the RPE and photoreceptor cells.[94] The choroid is composed of posterior ciliary arteries branching from the ophthalmic artery outside of the globe, transpiring the sclera at multiple sites. These arteries convey terminal arterioles generating exclusive, non-overlapping choriocapillaris lobules. The choriocapillaris (CC), the capillary component of the choroid, is located on the posterior portion of the Bruch's membrane and runs in a single layer beneath the RPE cells.[40] The feeding arterioles of CC are only present at the inner portion of the choroid. The CC and medium-sized vessels supply the inner choroidal vessels located between the apical RPE and outer choroidal melanocytes.[40,94] The inner surface of the CC (i. e., the surface facing the RPE cells) and its basement membrane (posterior layer of Bruch's membrane) are composed of fenestrated EC, which are involved in secretion and/or filtration.[40,94,95,96] The other layers of the choroid include Sattler's layer, which contains medium-sized vessels and interposing connective tissue close to the CC, as well as the Haller's layer composed of larger vessels that feed and drain arterioles and venules of Sattler's layer.[94]
Extensive choroidal perfusion might be crucial due to the massive oxygen consumption by the photoreceptors and neural retina. Studies suggest that CC, as a very dense vessel bed, supports the enormous utilization of oxygen by the retina. Although photoreceptors and neural retina utilize a large amount of oxygen, the oxygen depletion from the CC is limited.[94] This unique characteristic of the choroid is extremely crucial for proper photoreceptor function and survival.[40]
Various studies suggest that choroidal vascular changes, such as decreasing density and diameter of the CC and intermediate-sized vessels, are associated with aging and early stages of AMD.[94,97] Several factors have been identified as critical elements for choroidal microvascular dysfunction in AMD. The localization of complement complex within the CC in aging eyes suggests a potential mechanism for development of vascular injury in AMD.[98,99] Secondly, vascular alteration in both the choroid and peripheral vasculature insinuate endothelial dysfunction as a contributing factor to the development of AMD.[100,101] Other important factors include morphometric changes in the choroidal vascular density and choroidal neovascularization that originate from capillaries in the choroid with aging and AMD.[40,102]
Abnormal choroidal blood flow has been shown in patients with AMD.[103] These patients demonstrated reduced filling speed for capillary, arterial, and venous vessels associated with decreased capillary density, suggesting declining choroidal blood flow.[104] It has been postulated that the reduced choroidal blood flow and abnormal venous drainage is due to the formation of atherosclerotic plaques associated with increased scleral rigidity.[105] Subsequently, this resulted in changes in the RPE function with accumulation of lipoproteins that ultimately lead to RPE cell degeneration and altered permeability of the Bruch's membrane as observed in early AMD.[106]
Although changes in TSP1 levels were observed in AMD patient samples and Bruch's membrane, the underlying cellular changes and the mechanisms involved remain largely unknown. Our studies indicated that TSP1-/-mice develop significantly larger lesions of neovascularization when subjected to laser-induced choroidal neovascularization.[7] The enhanced CNV was associated with increased accumulation of macrophages at the lesion site. Therefore, the anti-inflammatory action of TSP1 may play an important role in maintaining choroidal/RPE homeostasis. However, decreased levels of TSP1 in response to oxidative stress may exacerbate the inflammatory processes driving the pathogenesis of AMD and CNV. Thus, restoration of TSP1 levels using mimetic peptides with anti-inflammatory and anti-angiogenic activity may provide beneficial therapeutics for treatment of exudative AMD.[7]
TREATMENTS FOR DRY AMD
Current strategies for management of dry AMD mostly rely on the prophylactic supplementation and inhibition of the disease progression. Studies published from multicenter controlled trials by Age Related Disease Study (AREDS) and AREDS2, reported the protective effect of oral supplementation of a combination of vitamin E and C, lutein, cupric oxide, and zinc oxide in patients with intermediate or advanced AMD, lowering the chance of disease progression in the other eye within the next five years.[107] The Antioxydants, Lipides Essentiels, Nutrition et maladies OculaiRes (ALIENOR) Study suggested omega-3 poly unsaturated fatty acid (PUFA), which are long chain omega-3 fatty acids, may delay late stages of AMD by inhibition of the oxidative and inflammatory processes that damage the retina;[108,109,110,111,112] however, the omega-3 PUFA supplements did not have a protective effect in the AREDS2 trial. Other protective and preventative supplements for AMD are macular carotenoid pigments including zeaxanthin and lutein, which can prevent lipofuscin photooxidation.[113,114,115] Although these nutritional supplementations can delay the development of end-stage AMD, there is not sufficient scientific data to support the administration of routine nutritional supplements as sufficient for the primary prevention of AMD. However, it is recommended that patients with intermediate or advanced AMD in one eye take AREDS2-type supplements for slowing the progression of AMD in the other eye.
Vitamin D is also another protective supplement and its deficiency is linked to progression of AMD in the elderly population. Vitamin D is a potent inhibitor of retinal neovascularization and acts through its receptor, thereby attenuating the promigratory and proliferative capacity of perivascular-supporting cells and promotes the quiescent and mature vascular phenotype.[116] How vitamin D affects choroidal vascular cells and its potential role in the inhibition of CNV needs further investigation. Vitamin D could also prevent the development of AMD through its reanimated effect, which can decrease inflammation and clear amyloid β.[117,118,119,120] Accumulation of amyloid β in various tissues occurs during the aging process, including in drusen in patients with AMD.[121,122,123] Macrophage and microglia recruitment to the site of damaged RPE is another important risk factor for AMD development, whose clearance is affected by interaction of TSP1 and its receptor CD47 and modulated by complement factor H, a risk factor for AMD.[124] Thus, a better understanding of these processes and their underlying molecular mechanisms will provide potential targets for prevention and treatment of AMD.
Increased risk of AMD has also been associated with polymorphisms in the CX3CR1 chemokine receptor on microglia and macrophages.[125,126] Recent studies have also suggested that neuroprotective therapy with promoted cell survival effects and inhibition of cell death signaling could provide suitable candidates for treatment of dry AMD. Some of the drugs that are currently being tested in patients with dry AMD include: 1) 4-hydroxy (phenyl) retinamide fenretinide; 2) brimonidine Brimo PS DDS; 3) compstatin derivate PDT-4 (reversibly targets C3, which is a point of convergence for all three pathways of complement activation); 4) ciliary neurotrophic factor CNTF; 5) Eculizumab, which is a humanized monoclonal antibody specifically binding C5; and 6) fluocinolone acetonide Iluvien®.[127]
Unfortunately, targeting of various components of the complement pathway has had limited success, but targeting of NLRP3 inflammasome signaling may provide a more suitable alternative.[128] Another available option to halt AMD progression is improvement of the choroidal blood flow to facilitate the disposal of the metabolic waste from RPE, Bruch's membrane, and photoreceptor cells. MC-1101, a FDA-approved antihypertensive drug, results in accelerated recovery of retinal function in ischemic eyes of the rats, and increased choroidal blood flow in an ocular hypertensive rabbit model. A phase Ib clinical trial showed that topical instillation of MC-1101 had no ocular toxicity or significant impact on the cardiovascular system. In addition, no significant effect on the blood–eye barrier was observed.[129,130] This drug has been under investigation in a phase II trial.
RPE cell transplantation has also been suggested as a potential therapeutic option for AMD treatment. RPE can be differentiated from human induced pluripotent stem cells (iPSCs) or human embryonic stem cells (hESCs).[131,132,133] The therapeutic potential of RPE transplantation was demonstrated in the Royal College of Surgeons (RCS) rat model of retinal degeneration.[134] The retinal atrophy caused by inherited phagocytosis defects in the RCS rat model was prevented after labeled RPE suspension grafts were delivered.[135] Transplantation of RPE appears to be a convincing approach that promises great benefits for restoring vision in patients with AMD. However, a successful transplantation involves a complex and challenging process that demands the following criteria: 1) effective techniques providing safe delivery of transplants, 2) survival of transplanted tissue within the host, 3) avoiding graft rejection, 4) absence of trans-differentiation from the normal phenotype in the grafted cells, 5) ability to restore the normal photoreceptor and retinal function, and 6) preventing the disease progression and improving vision.
The safety and tolerability of hESC-derived RPE cell grafts have been evaluated in patients with dry AMD in phase I trials. These studies reported functional recovery in the patients with no signs of rejection and trans-differentiation such as hyper-proliferation or tumorigenesis.[136,137,138] These results were consistent with two more phase I/II trials that showed improvement of the best corrected visual acuity (BCVA) and vison-related quality of life in patients with atrophic AMD. Another option being tested in stem cell therapy for dry AMD involves clonogenic human central nervous system stem cells (HuCNS-SC).[139] Based on preclinical data, transplantation of the HuCNS-SC protected photoreceptors and visual function in rats with retinal degeneration.[140,141]
Collectively, medical management of dry AMD can be divided into two strategies. The primary strategy is preventative and is focused on AREDS formulations, which monitor inflammatory pathways, oxidative stress, and RPE degeneration, and byproducts of the visual cycle that ultimately decrease the risk of AMD progression by 25–30%. Secondary strategies explore different pathophysiological approaches including restoration of choroidal perfusion, and replenishing RPE cells with stem cell-derived RPE cells that provide medium-and long-term safety, graft survival, and possible biological activity demonstrated by improvement of vision acuity in patients. Although several preclinical studies and clinical trials are attempting to identify an effective therapeutic factor to prevent or treat dry AMD, GA is still a devastating blinding disease without an effective cure.
TREATMENTS FOR EXUDATIVE AMD
Current treatments for exudative AMD are mainly focused on the attenuation of CNV and prevention of blood vessel leakiness using anti-VEGF agents. Laser treatment, including photodynamic therapy (PDT) for subfoveal CNV or laser photocoagulation for extrafoveal CNV, may also be used to delay further CNV progression in patients with exudative AMD.[142,143,144] However, their effectiveness in visual improvement has been limited.[142,145] The limitations with PDT include a low rate of vision improvement in patients (only 14%) and high incidence of CNV recurrence after laser treatment.[142,145]
Extensive research on the molecular and cellular mechanisms involved in AMD pathogenesis and CNV has provided a better understanding of disease development, which has the benefit of preventing CNV progression without the potential disadvantages associated with laser photocoagulation or PDT treatment. Clinical and laboratory studies have indicated that VEGF acts as a key proangiogenic factor in the pathogenesis of different ocular vascular diseases including AMD.[146] For this reason, the majority of the available treatments for neovascular eye diseases are focused on blocking VEGF activity directly or indirectly. Four different isoforms of VEGF-A have been identified in humans, including (VEGF-165,-206,-189, and-121).[147,148] Although all the VEGF isoforms have the ability to induce EC proliferation, only VEGF-165 and-189 have the potency to induce neovascularization.[149] The intravitreal injection of VEGF inhibitors has greatly halted CNV progression and improved management of exudative AMD in many patients. However, the need for frequent injections of VEGF blockers can be a major treatment burden for patients with exudative AMD, and not all patients respond to the anti-VEGF treatment due to unknown reasons.
The first anti-VEGF drug introduced to the market was pegaptanib, a 28-base ribonucleic acid aptamer that binds and blocks the activity of extracellular VEGF (VEGF-165).[150] Following that, bevacizumab was used to treat CNV in patients with AMD, after its original approval for colorectal cancer.[151] Bevacizumab (Avastin) is a full-length humanized antibody that binds to all isoforms of VEGF. Different prospective and retrospective studies have reported bevacizumab efficacy for exudative AMD with a low rate of complications.[152,153,154,155,156,157,158,159,160,161] Ranibizumab (Lucentis) is another anti-VEGF drug available in the market, which was specifically designed to easily penetrate the retina after intravitreal injection.[146,162] Ranibizumab is a humanized fab fragment of 48 kDa that binds to all VEGF-A isoforms. The efficacy of Ranibizumab has been evaluated in various trials, either combined with or without PDT treatment. The phase III trial of Ranibizumab in the treatment of exudative AMD (MARINA) indicated 30–40% improvement in maintaining vision acuity in the patients receiving 0.3 and 0.5 mg of ranibizumab compared with the control patients. In addition, 25–34% of the treated eyes showed vision improvement by 15 ETDRS letters.[163] In Antibody for the Treatment of Predominantly Classic Choroidal Neovascularization in Age-Related Macular Degeneration (ANCHOR) trial, ranibizumab was compared with PDT, and similar results were reported. Majority of patients receiving ranibizumab (90–96%) maintained vision compared with patients receiving PTD treatment (64–66%).[164,165] However, the Comparisons of Age-Related Macular Degeneration Treatment Trials (CATT) reported no difference in the visual acuity outcome between bevacizumab and ranibizumab, and therefore supported the use of bevacizumab over ranibizumab for treatment of exudative AMD given the economic benefits of bevacizumab compared to ranibizumab. Another multicenter randomized clinical trial, the Avastin (Bevacizumab) for choroidal neovascular age-related macular degeneration (ABC) trial also suggested bevacizumab for the treatment of exudative AMD; however, this treatment is not FDA-approved for ophthalmic use.[159,166] Retrospective studies suggest that combination of photodynamic and anti-VEGF therapy may be beneficial for patients with the potential for less frequent intravitreal injections.[167] Retrospective analysis of bevacizumab alone or along with PDT treatment results showed no difference in the visual acuity outcome, but decreased number of retreatments was observed with the combined regimen.[168,169,170]
Aflibercept (Eylea) is a newer anti-VEGF drug with a broader anti-VEGF activity that inhibits VEGF-A and placental growth factor (PlGF).[162] PIGF is a member of the VEGF family, which is involved in the pathogenesis of exudative AMD. A phase I trial of aflibercept reported no serious adverse events with the improvement of visual acuity by 4.3 ETDRS letters and reduced foveal thickness on OCT in patients with exudative AMD.[171] In the phase II trial, patients with subfoveal CNV receiving 0.5, 2, or 4 mg aflibercept showed a significant decrease in the foveal thickness on OCT along with improved vision acuity of 5.3 letters.[172] In addition, in a phase III trial, aflibercept showed a comparable safety profile with ranibizumab, both favorable, along with a similar mean change in visual acuity.[173]
Endogenous inhibitors of angiogenesis including angiostatin, endostatin, TSP1, and PEDF have been suggested as potential candidates for anti-angiogenesis therapy. PEDF has only been currently tested in the phase I trial. Adenoviral vector transfer of the human PEDF gene showed no adverse events in the patients with advanced AMD.[174] We have shown that TSP1 mimetic peptide ABT-898 is a potent inhibitor of CNV in a preclinical model.[7] Therefore, TSP1 as well as PEDF peptides may have utility as a single standing or in combination for treatment of neovascular AMD, and deserve further consideration.
Collectively, preferred therapies for exudative AMD include intravitreal injection of anti-VEGF agents, which are currently the standard for care. The efficacy of combined anti-VEGF regimens with PDT for treatment of CNV is still under investigation by current clinical trials. Lastly, new emerging therapies including, Ang-2 inhibition, are starting phase 3 clinical trials. Endogenous inhibitors of angiogenesis may provide additional alternatives for attenuation of CNV and need further exploration.
SUMMARY AND FUTURE PERSPECTIVE
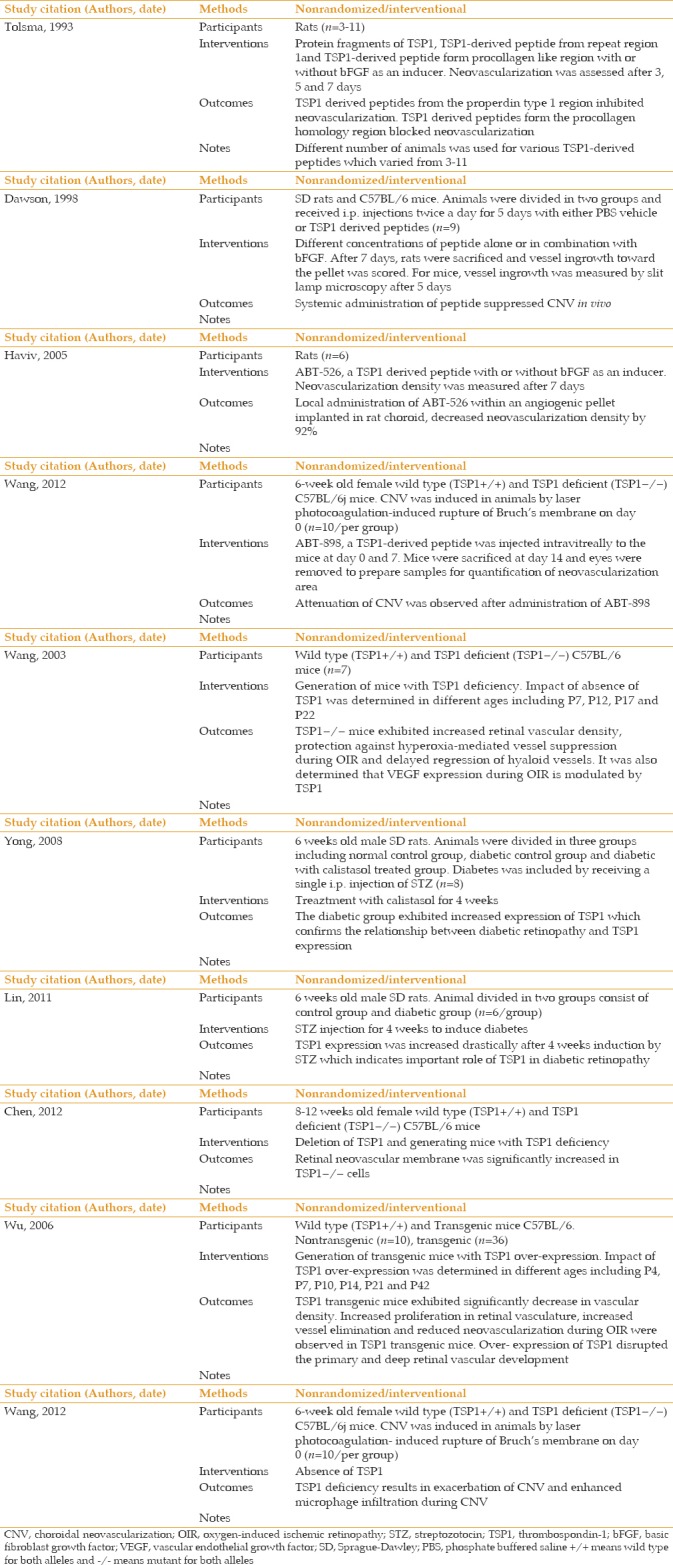
The availability of various preclinical models, advances in molecular genetics, and availability of transgenic animals and appropriate cell culture systems have helped to advance our understanding of the underlying mechanisms that contribute to the pathogenesis of AMD and CNV. Our studies have established an important role for TSP1 expression in ocular vascular homeostasis, whose altered production contribute to vascular abnormalities and neovascularization associated with various ocular pathological conditions with a neovascular component. The fact that TSP1 is an endogenous inhibitor of angiogenesis, along with its significant clinical impact aroused from studies to explore its therapeutic applications, have provided a better understanding of its detailed mechanisms of action. Recent discoveries derived from these studies will facilitate the design of novel therapeutic strategies to optimize its function and efficacy. Either upregulation of endogenous TSP1, use of its synthetic mimetic peptides, and/or recombinant proteins derived from the antiangiogenic fragment of TSP1 can be used to evaluate its therapeutic effects.[4,175,176,177] The synthetic peptides from TSP1 have been extensively used in various preclinical tumor models in vitro and in vivo [Table 1].[4,139,178,179,180] These investigations have resulted in the development of anti-angiogenic agents as a therapeutic target for various malignancies and vascular disorders [Table 2]. Introducing a new therapeutic target for AMD may produce promising results in terms of suppression of clinical complications of this devastating disease, which considerably impacts both patients and society. Thus, angiogenic inhibitors, particularly TSP1 alone or combined with other existing therapeutic agents, can be a promising treatment with established approaches that offer a more efficacious therapy for ocular neovascularization as a major complication of exudative AMD in the near future.
Table 1.
Thrombospondin-1 peptides in human clinical trials
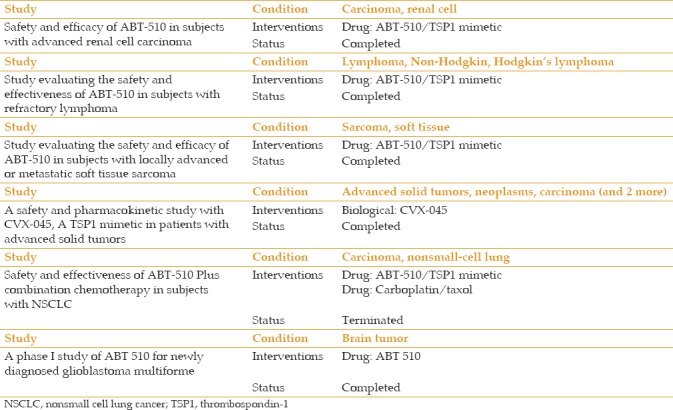
Table 2.
Evaluation of Thrombospondin-1-derived peptides effect on choroidal neovascularization and diabetic retinopathy
Financial Support and Sponsorship
Nil.
Conflicts of Interest
There are no conflicts of interest.
Acknowledgements
This work was supported by an unrestricted award from Research to Prevent Blindness to the Department of Ophthalmology and Visual Sciences, Retina Research Foundation, P30 EY016665, P30 CA014520, EPA 83573701, R24 EY022883, and R01 EY026078. CMS is supported by the RRF/Daniel M. Albert Chair. NS is a recipient of RPB Stein Innovation Award.
REFERENCES
- 1.Sheibani N, Sorenson CM, Cornelius LA, Frazier WA. Thrombospondin-1, a natural inhibitor of angiogenesis, is present in vitreous and aqueous humor and is modulated by hyperglycemia. Biochem Biophys Res Commun. 2000;267:257–261. doi: 10.1006/bbrc.1999.1903. [DOI] [PubMed] [Google Scholar]
- 2.Wang S, Gottlieb JL, Sorenson CM, Sheibani N. Modulation of thrombospondin 1 and pigment epithelium-derived factor levels in vitreous fluid of patients with diabetes. Arch Ophthalmol. 2009;127:507–513. doi: 10.1001/archophthalmol.2009.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawler PR, Lawler J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med. 2012;2:a006627. doi: 10.1101/cshperspect.a006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang S, Neekhra A, Albert DM, Sorenson CM, Sheibani N. Suppression of thrombospondin-1 expression during uveal melanoma progression and its potential therapeutic utility. Arch Ophthalmol. 2012;130:336–341. doi: 10.1001/archopthalmol.2011.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am J Pathol. 2002;161:831–839. doi: 10.1016/S0002-9440(10)64243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S, Wu Z, Sorenson CM, Lawler J, Sheibani N. Thrombospondin-1-deficient mice exhibit increased vascular density during retinal vascular development and are less sensitive to hyperoxia-mediated vessel obliteration. Dev Dyn. 2003;228:630–642. doi: 10.1002/dvdy.10412. [DOI] [PubMed] [Google Scholar]
- 7.Wang S, Sorenson CM, Sheibani N. Lack of thrombospondin 1 and exacerbation of choroidal neovascularization. Arch Ophthalmol. 2012;130:615–620. doi: 10.1001/archopthalmol.2011.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masli S, Sheibani N, Cursiefen C, Zieske J. Matricellular protein thrombospondins: Influence on ocular angiogenesis, wound healing and immuneregulation. Curr Eye Res. 2014;39:759–774. doi: 10.3109/02713683.2013.877936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu Z, Wang S, Sorenson CM, Sheibani N. Attenuation of retinal vascular development and neovascularization in transgenic mice over-expressing thrombospondin-1 in the lens. Dev Dyn. 2006;235:1908–1920. doi: 10.1002/dvdy.20837. [DOI] [PubMed] [Google Scholar]
- 10.Sorenson CM, Wang S, Gendron R, Paradis H, Sheibani N. Thrombospondin-1 deficiency exacerbates the pathogenesis of diabetic retinopathy. J Diabetes Metab. 2013;10(Suppl 12):104172/2155–6156S12-005. doi: 10.4172/2155-6156.S12-005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frolova EG, Pluskota E, Krukovets I, Burke T, Drumm C, Smith JD, et al. Thrombospondin-4 regulates vascular inflammation and atherogenesis. Circ Res. 2010;107:1313–1325. doi: 10.1161/CIRCRESAHA.110.232371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lawler JW, Slayter HS, Coligan JE. Isolation and characterization of a high molecular weight glycoprotein from human blood platelets. J Biol Chem. 1978;253:8609–8616. [PubMed] [Google Scholar]
- 13.Sheibani N, Frazier WA. Thrombospondin-1, PECAM-1, and regulation of angiogenesis. Histol Histopathol. 1999;14:285–294. doi: 10.14670/HH-14.285. [DOI] [PubMed] [Google Scholar]
- 14.Lawler J. The functions of thrombospondin-1 and-2. Curr Opin Cell Biol. 2000;12:634–640. doi: 10.1016/s0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- 15.Su X, Sorenson CM, Sheibani N. Isolation and characterization of murine retinal endothelial cells. Mol Vis. 2003;9:171–178. [PubMed] [Google Scholar]
- 16.Scheef E, Wang S, Sorenson CM, Sheibani N. Isolation and characterization of murine retinal astrocytes. Mol Vis. 2005;11:613–624. [PubMed] [Google Scholar]
- 17.Scheef EA, Sorenson CM, Sheibani N. Attenuation of proliferation and migration of retinal pericytes in the absence of thrombospondin-1. Am J Physiol Cell Physiol. 2009;296:C724–C734. doi: 10.1152/ajpcell.00409.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farnoodian M, Kinter JB, Yadranji Aghdam S, Zaitoun I, Sorenson CM, Sheibani N, et al. Expression of pigment epithelium-derived factor and thrombospondin-1 regulate proliferation and migration of retinal pigment epithelial cells. Physiol Rep. 2015;3 doi: 10.14814/phy2.12266. pii: e12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fei P, Zaitoun I, Farnoodian M, Fisk DL, Wang S, Sorenson CM, et al. Expression of thrombospondin-1 modulates the angioinflammatory phenotype of choroidal endothelial cells. PLoS One. 2014;9:e116423. doi: 10.1371/journal.pone.0116423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Wang S, Sheibani N. Enhanced proangiogenic signaling in thrombospondin-1-deficient retinal endothelial cells. Microvasc Res. 2006;71:143–151. doi: 10.1016/j.mvr.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 21.Volpert OV, Zaichuk T, Zhou W, Reiher F, Ferguson TA, Stuart PM, et al. Inducer-stimulated fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat Med. 2002;8:349–357. doi: 10.1038/nm0402-349. [DOI] [PubMed] [Google Scholar]
- 22.Sheibani N, Frazier WA. Thrombospondin 1 expression in transformed endothelial cells restores a normal phenotype and suppresses their tumorigenesis. Proc Natl Acad Sci U S A. 1995;92:6788–6792. doi: 10.1073/pnas.92.15.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majack RA, Cook SC, Bornstein P. Control of smooth muscle cell growth by components of the extracellular matrix: Autocrine role for thrombospondin. Proc Natl Acad Sci U S A. 1986;83:9050–9054. doi: 10.1073/pnas.83.23.9050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Majack RA, Goodman LV, Dixit VM. Cell surface thrombospondin is functionally essential for vascular smooth muscle cell proliferation. J Cell Biol. 1988;106:415–422. doi: 10.1083/jcb.106.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 26.Tang Y, Scheef EA, Wang S, Sorenson CM, Marcus CB, Jefcoate CR, et al. CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood. 2009;113:744–754. doi: 10.1182/blood-2008-03-145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palenski TL, Sorenson CM, Jefcoate CR, Sheibani N. Lack of cyp1b1 promotes the proliferative and migratory phenotype of perivascular supporting cells. Lab Invest. 2013;93:646–662. doi: 10.1038/labinvest.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fei P, Palenski TL, Wang S, Gurel Z, Hankenson KD, Sorenson CM, et al. Thrombospondin-2 expression during retinal vascular development and neovascularization. J Ocul Pharmacol Ther. 2015;31:429–444. doi: 10.1089/jop.2014.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang Q, Sheibani N. High glucose promotes retinal endothelial cell migration through activation of src, PI3K/Akt1/eNOS, and ERKs. Am J Physiol Cell Physiol. 2008;295:C1647–C1657. doi: 10.1152/ajpcell.00322.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin ES, Huang Q, Gurel Z, Palenski TL, Zaitoun I, Sorenson CM, et al. STAT1-mediated bim expression promotes the apoptosis of retinal pericytes under high glucose conditions. Cell Death Dis. 2014;5:e986. doi: 10.1038/cddis.2013.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin ES, Huang Q, Gurel Z, Sorenson CM, Sheibani N. High glucose alters retinal astrocytes phenotype through increased production of inflammatory cytokines and oxidative stress. PLoS One. 2014;9:e103148. doi: 10.1371/journal.pone.0103148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carron JA, Hiscott P, Hagan S, Sheridan CM, Magee R, Gallagher JA, et al. Cultured human retinal pigment epithelial cells differentially express thrombospondin-1, -2, -3, and -4. Int J Biochem Cell Biol. 2000;32:1137–1142. doi: 10.1016/s1357-2725(00)00065-0. [DOI] [PubMed] [Google Scholar]
- 33.He S, Incardona F, Jin M, Ryan SJ, Hinton DR. Thrombospondin-1 expression in RPE and choroidal neovascular membranes. Yan Ke Xue Bao. 2006;22:265–274. [PubMed] [Google Scholar]
- 34.Uchida H, Hayashi H, Kuroki M, Uno K, Yamada H, Yamashita Y, et al. Vitamin A up-regulates the expression of thrombospondin-1 and pigment epithelium-derived factor in retinal pigment epithelial cells. Exp Eye Res. 2005;80:23–30. doi: 10.1016/j.exer.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 35.Uno K, Bhutto IA, McLeod DS, Merges C, Lutty GA. Impaired expression of thrombospondin-1 in eyes with age related macular degeneration. Br J Ophthalmol. 2006;90:48–54. doi: 10.1136/bjo.2005.074005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gass JD. Drusen and disciform macular detachment and degeneration 1972. Retina. 2003;23:409–436. [PubMed] [Google Scholar]
- 37.Stangos N, Voutas S, Topouzis F, Karampatakis V. Contrast sensitivity evaluation in eyes predisposed to age-related macular degeneration and presenting normal visual acuity. Ophthalmologica. 1995;209:194–198. doi: 10.1159/000310612. [DOI] [PubMed] [Google Scholar]
- 38.Sunness JS, Johnson MA, Massof RW, Marcus S. Retinal sensitivity over drusen and nondrusen areas. A study using fundus perimetry. Arch Ophthalmol. 1988;106:1081–1084. doi: 10.1001/archopht.1988.01060140237032. [DOI] [PubMed] [Google Scholar]
- 39.Tolentino MJ, Miller S, Gaudio AR, Sandberg MA. Visual field deficits in early age-related macular degeneration. Vision Res. 1994;34:409–413. doi: 10.1016/0042-6989(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 40.Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol Aspects Med. 2012;33:295–317. doi: 10.1016/j.mam.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: Etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257–293. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- 42.Chiang A, Regillo CD. Preferred therapies for neovascular age-related macular degeneration. Curr Opin Ophthalmol. 2011;22:199–204. doi: 10.1097/ICU.0b013e32834597d9. [DOI] [PubMed] [Google Scholar]
- 43.Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358:2606–2617. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- 44.Green WR. Histopathology of age-related macular degeneration. Mol Vis. 1999;5:27. [PubMed] [Google Scholar]
- 45.Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman lecture. Ophthalmology. 1993;100:1519–1535. doi: 10.1016/s0161-6420(93)31466-1. [DOI] [PubMed] [Google Scholar]
- 46.Frank RN, Amin RH, Eliott D, Puklin JE, Abrams GW. Basic fibroblast growth factor and vascular endothelial growth factor are present in epiretinal and choroidal neovascular membranes. Am J Ophthalmol. 1996;122:393–403. doi: 10.1016/s0002-9394(14)72066-5. [DOI] [PubMed] [Google Scholar]
- 47.Freund KB, Yannuzzi LA, Sorenson JA. Age-related macular degeneration and choroidal neovascularization. Am J Ophthalmol. 1993;115:786–791. doi: 10.1016/s0002-9394(14)73649-9. [DOI] [PubMed] [Google Scholar]
- 48.Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 49.Ishibashi T, Hata Y, Yoshikawa H, Nakagawa K, Sueishi K, Inomata H, et al. Expression of vascular endothelial growth factor in experimental choroidal neovascularization. Graefes Arch Clin Exp Ophthalmol. 1997;235:159–167. doi: 10.1007/BF00941723. [DOI] [PubMed] [Google Scholar]
- 50.Kvanta A, Algvere PV, Berglin L, Seregard S. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Invest Ophthalmol Vis Sci. 1996;37:1929–1934. [PubMed] [Google Scholar]
- 51.Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1996;37:855–868. [PubMed] [Google Scholar]
- 52.Otani A, Takagi H, Oh H, Koyama S, Ogura Y, Matumura M, et al. Vascular endothelial growth factor family and receptor expression in human choroidal neovascular membranes. Microvasc Res. 2002;64:162–169. doi: 10.1006/mvre.2002.2407. [DOI] [PubMed] [Google Scholar]
- 53.Wada M, Gelfman CM, Matsunaga H, Alizadeh M, Morse L, Handa JT, et al. Density-dependent expression of FGF-2 in response to oxidative stress in RPE cells in vitro . Curr Eye Res. 2001;23:226–231. doi: 10.1076/ceyr.23.3.226.5467. [DOI] [PubMed] [Google Scholar]
- 54.Grossniklaus HE, Martinez JA, Brown VB, Lambert HM, Sternberg P, Jr, Capone A, Jr, et al. Immunohistochemical and histochemical properties of surgically excised subretinal neovascular membranes in age-related macular degeneration. Am J Ophthalmol. 1992;114:464–472. doi: 10.1016/s0002-9394(14)71859-8. [DOI] [PubMed] [Google Scholar]
- 55.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marmé D, et al. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- 56.Clauss M, Gerlach M, Gerlach H, Brett J, Wang F, Familletti PC, et al. Vascular permeability factor: A tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J Exp Med. 1990;172:1535–1545. doi: 10.1084/jem.172.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grossniklaus HE, Green WR. Choroidal neovascularization. Am J Ophthalmol. 2004;137:496–503. doi: 10.1016/j.ajo.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 58.Sarraf D, Gin T, Yu F, Brannon A, Owens SL, Bird AC, et al. Long-term drusen study. Retina. 1999;19:513–519. doi: 10.1097/00006982-199911000-00006. [DOI] [PubMed] [Google Scholar]
- 59.Binder S, Stanzel BV, Krebs I, Glittenberg C. Transplantation of the RPE in AMD. Prog Retin Eye Res. 2007;26:516–554. doi: 10.1016/j.preteyeres.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 60.Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 61.Marshall J. The ageing retina: Physiology or pathology. Eye (Lond) 1987;1(Pt 2):282–295. doi: 10.1038/eye.1987.47. [DOI] [PubMed] [Google Scholar]
- 62.Curcio CA. Photoreceptor topography in ageing and age-related maculopathy. Eye (Lond) 2001;15:376–383. doi: 10.1038/eye.2001.140. [DOI] [PubMed] [Google Scholar]
- 63.Dunaief JL, Dentchev T, Ying GS, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- 64.Xu GZ, Li WW, Tso MO. Apoptosis in human retinal degenerations. Trans Am Ophthalmol Soc. 1996;94:411–430. [PMC free article] [PubMed] [Google Scholar]
- 65.Maeda H, Ogata N, Yi X, Takeuchi M, Ohkuma H, Uyama M, et al. Apoptosis of photoreceptor cells in ornithine-induced retinopathy. Graefes Arch Clin Exp Ophthalmol. 1998;236:207–212. doi: 10.1007/s004170050066. [DOI] [PubMed] [Google Scholar]
- 66.Panda-Jonas S, Jonas JB, Jakobczyk-Zmija M. Retinal pigment epithelial cell count, distribution, and correlations in normal human eyes. Am J Ophthalmol. 1996;121:181–189. doi: 10.1016/s0002-9394(14)70583-5. [DOI] [PubMed] [Google Scholar]
- 67.Dorey CK, Wu G, Ebenstein D, Garsd A, Weiter JJ. Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci. 1989;30:1691–9. [PubMed] [Google Scholar]
- 68.Delori FC, Goger DG, Hammond BR, Snodderly DM, Burns SA. Macular pigment density measured by autofluorescence spectrometry: Comparison with reflectometry and heterochromatic flicker photometry. J Opt Soc Am A Opt Image Sci Vis. 2001;18:1212–1230. doi: 10.1364/josaa.18.001212. [DOI] [PubMed] [Google Scholar]
- 69.Brizzee KR. Cellular features, regional accumulation, and prospectsof modification of age pigments in mammals. In: Sohal SR, editor. Age Pigments. Amsterdam: Elsevier/North- Holland Biomedical Press; 1981. pp. 101–54. [Google Scholar]
- 70.Elleder MS. Chemical Characterization of Age Pigments. Age Pigments. Amsterdam: Elsevier, North Holland Biomedical Press; 1981. pp. 203–41. [Google Scholar]
- 71.Siakotos AN, Koppang N. Procedures for the isolation of lipopigments from brain, heart and liver, and their properties: A review. Mech Ageing Dev. 1973;2:177–200. doi: 10.1016/0047-6374(73)90016-x. [DOI] [PubMed] [Google Scholar]
- 72.Yin D. Biochemical basis of lipofuscin, ceroid, and age pigment-like fluorophores. Free Radic Biol Med. 1996;21:871–888. doi: 10.1016/0891-5849(96)00175-x. [DOI] [PubMed] [Google Scholar]
- 73.Wassell J, Davies S, Bardsley W, Boulton M. The photoreactivity of the retinal age pigment lipofuscin. J Biol Chem. 1999;274:23828–23832. doi: 10.1074/jbc.274.34.23828. [DOI] [PubMed] [Google Scholar]
- 74.Boulton M, Rozanowska M, Rozanowski B, Wess T. The photoreactivity of ocular lipofuscin. Photochem Photobiol Sci. 2004;3:759–764. doi: 10.1039/b400108g. [DOI] [PubMed] [Google Scholar]
- 75.Sparrow JR, Boulton M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res. 2005;80:595–606. doi: 10.1016/j.exer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 76.Bhutto IA, Uno K, Merges C, Zhang L, McLeod DS, Lutty GA, et al. Reduction of endogenous angiogenesis inhibitors in Bruch's membrane of the submacular region in eyes with age-related macular degeneration. Arch Ophthalmol. 2008;126:670–678. doi: 10.1001/archopht.126.5.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Handa JT, Verzijl N, Matsunaga H, Aotaki-Keen A, Lutty GA, te Koppele JM, et al. Increase in the advanced glycation end product pentosidine in Bruch's membrane with age. Invest Ophthalmol Vis Sci. 1999;40:775–779. [PubMed] [Google Scholar]
- 78.Uchiki T, Weikel KA, Jiao W, Shang F, Caceres A, Pawlak D, et al. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics) Aging Cell. 2012;11:1–13. doi: 10.1111/j.1474-9726.2011.00752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weikel KA, Fitzgerald P, Shang F, Caceres MA, Bian Q, Handa JT, et al. Natural history of age-related retinal lesions that precede AMD in mice fed high or low glycemic index diets. Invest Ophthalmol Vis Sci. 2012;53:622–632. doi: 10.1167/iovs.11-8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–1467. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sarks JP, Sarks SH, Killingsworth MC. Evolution of geographic atrophy of the retinal pigment epithelium. Eye (Lond) 1988;2(Pt 5):552–577. doi: 10.1038/eye.1988.106. [DOI] [PubMed] [Google Scholar]
- 82.Sunness JS. The natural history of geographic atrophy, the advanced atrophic form of age-related macular degeneration. Mol Vis. 1999;5:25. [PubMed] [Google Scholar]
- 83.Holz FG, Sheraidah G, Pauleikhoff D, Bird AC. Analysis of lipid deposits extracted from human macular and peripheral bruch's membrane. Arch Ophthalmol. 1994;112:402–406. doi: 10.1001/archopht.1994.01090150132035. [DOI] [PubMed] [Google Scholar]
- 84.Ruberti JW, Curcio CA, Millican CL, Menco BP, Huang JD, Johnson M, et al. Quick-freeze/deep-etch visualization of age-related lipid accumulation in bruch's membrane. Invest Ophthalmol Vis Sci. 2003;44:1753–1759. doi: 10.1167/iovs.02-0496. [DOI] [PubMed] [Google Scholar]
- 85.Hogan MJ. Histology of the Human Eye. Philadelphia: WB Saunders; 1971. [Google Scholar]
- 86.Hogan MJ. Bruch's membrane and disease of the macula. Role of elastic tissue and collagen. Trans Ophthalmol Soc U K. 1967;87:113–161. [PubMed] [Google Scholar]
- 87.Okubo A, Rosa RH, Jr, Bunce CV, Alexander RA, Fan JT, Bird AC, et al. The relationships of age changes in retinal pigment epithelium and Bruch's membrane. Invest Ophthalmol Vis Sci. 1999;40:443–449. [PubMed] [Google Scholar]
- 88.Ramrattan RS, van der Schaft TL, Mooy CM, de Bruijn WC, Mulder PG, de Jong PT, et al. Morphometric analysis of Bruch's membrane, the choriocapillaris, and the choroid in aging. Invest Ophthalmol Vis Sci. 1994;35:2857–22864. [PubMed] [Google Scholar]
- 89.Sarks SH, Arnold JJ, Killingsworth MC, Sarks JP. Early drusen formation in the normal and aging eye and their relation to age related maculopathy: A clinicopathological study. Br J Ophthalmol. 1999;83:358–368. doi: 10.1136/bjo.83.3.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grindle CF, Marshall J. Ageing changes in Bruch's membrane and their functional implications. Trans Ophthalmol Soc U K. 1978;98:172–175. [PubMed] [Google Scholar]
- 91.Löffler KU, Lee WR. Basal linear deposit in the human macula. Graefes Arch Clin Exp Ophthalmol. 1986;224:493–501. doi: 10.1007/BF02154735. [DOI] [PubMed] [Google Scholar]
- 92.Pauleikhoff D, Zuels S, Sheraidah GS, Marshall J, Wessing A, Bird AC, et al. Correlation between biochemical composition and fluorescein binding of deposits in Bruch's membrane. Ophthalmology. 1992;99:1548–15453. doi: 10.1016/s0161-6420(92)31768-3. [DOI] [PubMed] [Google Scholar]
- 93.Moore DJ, Hussain AA, Marshall J. Age-related variation in the hydraulic conductivity of Bruch's membrane. Invest Ophthalmol Vis Sci. 1995;36:1290–1297. [PubMed] [Google Scholar]
- 94.Whitmore SS, Sohn EH, Chirco KR, Drack AV, Stone EM, Tucker BA, et al. Complement activation and choriocapillaris loss in early AMD: Implications for pathophysiology and therapy. Prog Retin Eye Res. 2015;45:1–29. doi: 10.1016/j.preteyeres.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burns MS, Hartz MJ. The retinal pigment epithelium induces fenestration of endothelial cells in vivo . Curr Eye Res. 1992;11:863–873. doi: 10.3109/02713689209033484. [DOI] [PubMed] [Google Scholar]
- 96.Mancini MA, Frank RN, Keirn RJ, Kennedy A, Khoury JK. Does the retinal pigment epithelium polarize the choriocapillaris? Invest Ophthalmol Vis Sci. 1986;27:336–345. [PubMed] [Google Scholar]
- 97.Moreira-Neto CA, Moult EM, Fujimoto JG, Waheed NK, Ferrara D. Choriocapillaris loss in advanced age-related macular degeneration. J Ophthalmol 2018. 2018:8125267. doi: 10.1155/2018/8125267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103:2328–2333. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aredo B, Li T, Chen X, Zhang K, Wang CX, Gou D, et al. Achimeric cfh transgene leads to increased retinal oxidative stress, inflammation, and accumulation of activated subretinal microglia in mice. Invest Ophthalmol Vis Sci. 2015;56:3427–3440. doi: 10.1167/iovs.14-16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hoffmann S, Friedrichs U, Eichler W, Rosenthal A, Wiedemann P. Advanced glycation end products induce choroidal endothelial cell proliferation, matrix metalloproteinase-2 and VEGF upregulation in vitro . Graefes Arch Clin Exp Ophthalmol. 2002;240:996–1002. doi: 10.1007/s00417-002-0568-6. [DOI] [PubMed] [Google Scholar]
- 101.Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO. Vascular endothelial growth factors and angiogenesis in eye disease. Prog Retin Eye Res. 2003;22:1–29. doi: 10.1016/s1350-9462(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 102.Zamora DO, Riviere M, Choi D, Pan Y, Planck SR, Rosenbaum JT, et al. Proteomic profiling of human retinal and choroidal endothelial cells reveals molecular heterogeneity related to tissue of origin. Mol Vis. 2007;13:2058–2065. [PubMed] [Google Scholar]
- 103.Berenberg TL, Metelitsina TI, Madow B, Dai Y, Ying GS, Dupont JC, et al. The association between drusen extent and foveolar choroidal blood flow in age-related macular degeneration. Retina. 2012;32:25–31. doi: 10.1097/IAE.0b013e3182150483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ciulla TA, Harris A, Kagemann L, Danis RP, Pratt LM, Chung HS, et al. Choroidal perfusion perturbations in non-neovascular age related macular degeneration. Br J Ophthalmol. 2002;86:209–213. doi: 10.1136/bjo.86.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Friedman E. Choroidal blood flow.Pressure-flow relationships. Arch Ophthalmol. 1970;83:95–99. doi: 10.1001/archopht.1970.00990030097018. [DOI] [PubMed] [Google Scholar]
- 106.Friedman E. The pathogenesis of age-related macular degeneration. Am J Ophthalmol. 2008;146:348–349. doi: 10.1016/j.ajo.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 107.Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with Vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no.8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Merle B, Delyfer MN, Korobelnik JF, Rougier MB, Colin J, Malet F, et al. Dietary omega-3 fatty acids and the risk for age-related maculopathy: The alienor study. Invest Ophthalmol Vis Sci. 2011;52:6004–6011. doi: 10.1167/iovs.11-7254. [DOI] [PubMed] [Google Scholar]
- 109.Merle B, Delyfer MN, Korobelnik JF, Rougier MB, Malet F, Goff ML, et al. Plasma omega3 fatty acids and the risk for age-related macular degeneration: The Alienor Study. Invest Ophthalmol Vis Sci. 2012;53:3813. [Google Scholar]
- 110.Delyfer MN, Buaud B, Korobelnik JF, Rougier MB, Schalch W, Etheve S, et al. Association of macular pigment density with plasma ω-3 fatty acids: The PIMAVOSA study. Invest Ophthalmol Vis Sci. 2012;53:1204–1210. doi: 10.1167/iovs.11-8721. [DOI] [PubMed] [Google Scholar]
- 111.Stahl A, Krohne TU, Sapieha P, Chen J, Hellstrom A, Chew E, et al. Lipid metabolites in the pathogenesis and treatment of neovascular eye disease. Br J Ophthalmol. 2011;95:1496–1501. doi: 10.1136/bjo.2010.194241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shalev V, Sror M, Goldshtein I, Kokia E, Chodick G. Statin use and the risk of age related macular degeneration in a large health organization in Israel. Ophthalmic Epidemiol. 2011;18:83–90. doi: 10.3109/09286586.2011.560746. [DOI] [PubMed] [Google Scholar]
- 113.Seddon JM, Ajani UA, Sperduto RD, Hiller R, Blair N, Burton TC, et al. Dietary carotenoids, Vitamins A, C, and E, and advanced age-related macular degeneration. Eye disease case-control study group. JAMA. 1994;272:1413–1420. [PubMed] [Google Scholar]
- 114.Moeller SM, Parekh N, Tinker L, Ritenbaugh C, Blodi B, Wallace RB, et al. Associations between intermediate age-related macular degeneration and lutein and zeaxanthin in the carotenoids in age-related eye disease study (CAREDS): Ancillary study of the women's health initiative. Arch Ophthalmol. 2006;124:1151–1162. doi: 10.1001/archopht.124.8.1151. [DOI] [PubMed] [Google Scholar]
- 115.Tan JS, Wang JJ, Flood V, Rochtchina E, Smith W, Mitchell P, et al. Dietary antioxidants and the long-term incidence of age-related macular degeneration: The blue mountains eye study. Ophthalmology. 2008;115:334–341. doi: 10.1016/j.ophtha.2007.03.083. [DOI] [PubMed] [Google Scholar]
- 116.Jamali N, Wang S, Darjatmoko SR, Sorenson CM, Sheibani N. Vitamin D receptor expression is essential during retinal vascular development and attenuation of neovascularization by 1, 25(OH) 2D3. PLoS One. 2017;12:e0190131. doi: 10.1371/journal.pone.0190131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Parekh N, Chappell RJ, Millen AE, Albert DM, Mares JA. Association between Vitamin D and age-related macular degeneration in the Third National Health and Nutrition Examination Survey, 1988 through 1994. Arch Ophthalmol. 2007;125:661–669. doi: 10.1001/archopht.125.5.661. [DOI] [PubMed] [Google Scholar]
- 118.Millen AE, Voland R, Sondel SA, Parekh N, Horst RL, Wallace RB, et al. Vitamin D status and early age-related macular degeneration in postmenopausal women. Arch Ophthalmol. 2011;129:481–489. doi: 10.1001/archophthalmol.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Morrison MA, Silveira AC, Huynh N, Jun G, Smith SE, Zacharaki F, et al. Systems biology-based analysis implicates a novel role for Vitamin D metabolism in the pathogenesis of age-related macular degeneration. Hum Genomics. 2011;5:538–568. doi: 10.1186/1479-7364-5-6-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee V, Rekhi E, Hoh Kam J, Jeffery G. Vitamin D rejuvenates aging eyes by reducing inflammation, clearing amyloid beta and improving visual function. Neurobiol Aging. 2012;33:2382–2389. doi: 10.1016/j.neurobiolaging.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 121.Ohno-Matsui K. Parallel findings in age-related macular degeneration and Alzheimer's disease. Prog Retin Eye Res. 2011;30:217–238. doi: 10.1016/j.preteyeres.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 122.Armstrong RA. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer's disease. Folia Neuropathol. 2009;47:289–299. [PubMed] [Google Scholar]
- 123.Isas JM, Luibl V, Johnson LV, Kayed R, Wetzel R, Glabe CG, et al. Soluble and mature amyloid fibrils in drusen deposits. Invest Ophthalmol Vis Sci. 2010;51:1304–1310. doi: 10.1167/iovs.09-4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Calippe B, Augustin S, Beguier F, Charles-Messance H, Poupel L, Conart JB, et al. Complement factor H inhibits CD47-mediated resolution of inflammation. Immunity. 2017;46:261–272. doi: 10.1016/j.immuni.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 125.Tuo J, Smith BC, Bojanowski CM, Meleth AD, Gery I, Csaky KG, et al. The involvement of sequence variation and expression of C×3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:1297–1299. doi: 10.1096/fj.04-1862fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Combadière C, Feumi C, Raoul W, Keller N, Rodéro M, Pézard A, et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zając-Pytrus HM, Pilecka A, Turno-Kręcicka A, Adamiec-Mroczek J, Misiuk-Hojło M. The dry form of age-related macular degeneration (AMD): The current concepts of pathogenesis and prospects for treatment. Adv Clin Exp Med. 2015;24:1099–1104. doi: 10.17219/acem/27093. [DOI] [PubMed] [Google Scholar]
- 128.Dolgin E. Age-related macular degeneration foils drugmakers. Nat Biotechnol. 2017;35:1000–1001. doi: 10.1038/nbt1117-1000. [DOI] [PubMed] [Google Scholar]
- 129.Hanus J, Zhao F, Wang S. Current therapeutic developments in atrophic age-related macular degeneration. Br J Ophthalmol. 2016;100:122–127. doi: 10.1136/bjophthalmol-2015-306972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jiang W, Chiou GC. Effects of hydralazine on ocular blood flow andlaser-induced choroidal neovascularization. Int J Ophthalmol. 2009;2:324–327. [Google Scholar]
- 131.Carr AJ, Vugler AA, Hikita ST, Lawrence JM, Gias C, Chen LL, et al. Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS One. 2009;4:e8152. doi: 10.1371/journal.pone.0008152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Buchholz DE, Hikita ST, Rowland TJ, Friedrich AM, Hinman CR, Johnson LV, et al. Derivation of functional retinal pigmented epithelium from induced pluripotent stem cells. Stem Cells. 2009;27:2427–2434. doi: 10.1002/stem.189. [DOI] [PubMed] [Google Scholar]
- 133.Kokkinaki M, Sahibzada N, Golestaneh N. Human induced pluripotent stem-derived retinal pigment epithelium (RPE) cells exhibit ion transport, membrane potential, polarized vascular endothelial growth factor secretion, and gene expression pattern similar to native RPE. Stem Cells. 2011;29:825–835. doi: 10.1002/stem.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li LX, Turner JE. Inherited retinal dystrophy in the RCS rat: Prevention of photoreceptor degeneration by pigment epithelial cell transplantation. Exp Eye Res. 1988;47:911–917. doi: 10.1016/0014-4835(88)90073-5. [DOI] [PubMed] [Google Scholar]
- 135.Lopez R, Gouras P, Kjeldbye H, Sullivan B, Reppucci V, Brittis M, et al. Transplanted retinal pigment epithelium modifies the retinal degeneration in the RCS rat. Invest Ophthalmol Vis Sci. 1989;30:586–588. [PubMed] [Google Scholar]
- 136.Becker S, Jayaram H, Limb GA. Recent advances towards the clinical application of stem cells for retinal regeneration. Cells. 2012;1:851–873. doi: 10.3390/cells1040851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Aharoni R. Immunomodulation neuroprotection and remyelination – The fundamental therapeutic effects of glatiramer acetate: A critical review. J Autoimmun. 2014;54:81–92. doi: 10.1016/j.jaut.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 138.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 139.Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, et al. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cuenca N, Fernández-Sánchez L, McGill TJ, Lu B, Wang S, Lund R, et al. Phagocytosis of photoreceptor outer segments by transplanted human neural stem cells as a neuroprotective mechanism in retinal degeneration. Invest Ophthalmol Vis Sci. 2013;54:6745–6756. doi: 10.1167/iovs.13-12860. [DOI] [PubMed] [Google Scholar]
- 141.McGill TJ, Cottam B, Lu B, Wang S, Girman S, Tian C, et al. Transplantation of human central nervous system stem cells – Neuroprotection in retinal degeneration. Eur J Neurosci. 2012;35:468–477. doi: 10.1111/j.1460-9568.2011.07970.x. [DOI] [PubMed] [Google Scholar]
- 142.Bressler NM. Treatment of Age-Related Macular Degeneration with Photodynamic Therapy (TAP) Study Group. Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin: Two-year results of 2 randomized clinical trials-tap report 2. Arch Ophthalmol. 2001;119:198–207. [PubMed] [Google Scholar]
- 143.Subfoveal neovascular lesions in age-related macular degeneration. Guidelines for evaluation and treatment in the macular photocoagulation study. Macular photocoagulation study group. Arch Ophthalmol. 1991;109:1242–1257. [PubMed] [Google Scholar]
- 144.Persistent and recurrent neovascularization after laser photocoagulation for subfoveal choroidal neovascularization of age-related macular degeneration. Macular photocoagulation study group. Arch Ophthalmol. 1994;112:489–499. doi: 10.1001/archopht.1994.01090160065024. [DOI] [PubMed] [Google Scholar]
- 145.Bressler NM, Silva JC, Bressler SB, Fine SL, Green WR. Clinicopathologic correlation of drusen and retinal pigment epithelial abnormalities in age-related macular degeneration. Retina. 1994;14:130–142. [PubMed] [Google Scholar]
- 146.Michels S, Schmidt-Erfurth U, Rosenfeld PJ. Promising new treatments for neovascular age-related macular degeneration. Expert Opin Investig Drugs. 2006;15:779–793. doi: 10.1517/13543784.15.7.779. [DOI] [PubMed] [Google Scholar]
- 147.Plate KH, Warnke PC. Vascular endothelial growth factor. J Neurooncol. 1997;35:365–372. doi: 10.1023/a:1005845307160. [DOI] [PubMed] [Google Scholar]
- 148.Lange T, Guttmann-Raviv N, Baruch L, Machluf M, Neufeld G. VEGF162, a new heparin-binding vascular endothelial growth factor splice form that is expressed in transformed human cells. J Biol Chem. 2003;278:17164–17169. doi: 10.1074/jbc.M212224200. [DOI] [PubMed] [Google Scholar]
- 149.Küsters B, de Waal RM, Wesseling P, Verrijp K, Maass C, Heerschap A, et al. Differential effects of vascular endothelial growth factor A isoforms in a mouse brain metastasis model of human melanoma. Cancer Res. 2003;63:5408–5413. [PubMed] [Google Scholar]
- 150.Gragoudas ES, Adamis AP, Cunningham ET, Jr, Feinsod M, Guyer DR VEGF Inhibition Study in Ocular Neovascularization Clinical Trial Group. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351:2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- 151.Michels S, Rosenfeld PJ, Puliafito CA, Marcus EN, Venkatraman AS. Systemic bevacizumab (Avastin) therapy for neovascular age-related macular degeneration twelve-week results of an uncontrolled open-label clinical study. Ophthalmology. 2005;112:1035–1047. doi: 10.1016/j.ophtha.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 152.Spaide RF, Laud K, Fine HF, Klancnik JM, Jr, Meyerle CB, Yannuzzi LA, et al. Intravitreal bevacizumab treatment of choroidal neovascularization secondary to age-related macular degeneration. Retina. 2006;26:383–390. doi: 10.1097/01.iae.0000238561.99283.0e. [DOI] [PubMed] [Google Scholar]
- 153.Yoganathan P, Deramo VA, Lai JC, Tibrewala RK, Fastenberg DM. Visual improvement following intravitreal bevacizumab (Avastin) in exudative age-related macular degeneration. Retina. 2006;26:994–998. doi: 10.1097/01.iae.0000244380.34082.67. [DOI] [PubMed] [Google Scholar]
- 154.Avery RL, Pieramici DJ, Rabena MD, Castellarin AA, Nasir MA, Giust MJ, et al. Intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration. Ophthalmology. 2006;113:363–372.e5. doi: 10.1016/j.ophtha.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 155.Bashshur ZF, Haddad ZA, Schakal A, Jaafar RF, Saab M, Noureddin BN, et al. Intravitreal bevacizumab for treatment of neovascular age-related macular degeneration: A one-year prospective study. Am J Ophthalmol. 2008;145:249–256. doi: 10.1016/j.ajo.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 156.Weigert G, Michels S, Sacu S, Varga A, Prager F, Geitzenauer W, et al. Intravitreal bevacizumab (Avastin) therapy versus photodynamic therapy plus intravitreal triamcinolone for neovascular age-related macular degeneration: 6-month results of a prospective, randomised, controlled clinical study. Br J Ophthalmol. 2008;92:356–360. doi: 10.1136/bjo.2007.125823. [DOI] [PubMed] [Google Scholar]
- 157.Lazic R, Gabric N. Intravitreally administered bevacizumab (Avastin) in minimally classic and occult choroidal neovascularization secondary to age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2007;245:68–73. doi: 10.1007/s00417-006-0466-4. [DOI] [PubMed] [Google Scholar]
- 158.Algvere PV, Steén B, Seregard S, Kvanta A. A prospective study on intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration of different durations. Acta Ophthalmol. 2008;86:482–489. doi: 10.1111/j.1600-0420.2007.01113.x. [DOI] [PubMed] [Google Scholar]
- 159.Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, et al. CATT Research Group. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011;364:1897–1908. doi: 10.1056/NEJMoa1102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wu L, Martínez-Castellanos MA, Quiroz-Mercado H, Arevalo JF, Berrocal MH, Farah ME, et al. Twelve-month safety of intravitreal injections of bevacizumab (Avastin): Results of the pan-american collaborative retina study group (PACORES) Graefes Arch Clin Exp Ophthalmol. 2008;246:81–87. doi: 10.1007/s00417-007-0660-z. [DOI] [PubMed] [Google Scholar]
- 161.Jyothi S, Chowdhury H, Elagouz M, Sivaprasad S. Intravitreal bevacizumab (Avastin) for age-related macular degeneration: A critical analysis of literature. Eye (Lond) 2010;24:816–824. doi: 10.1038/eye.2009.219. [DOI] [PubMed] [Google Scholar]
- 162.Lally DR, Gerstenblith AT, Regillo CD. Preferred therapies for neovascular age-related macular degeneration. Curr Opin Ophthalmol. 2012;23:182–188. doi: 10.1097/ICU.0b013e328352411c. [DOI] [PubMed] [Google Scholar]
- 163.Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- 164.Kaiser PK, Blodi BA, Shapiro H, Acharya NR MARINA Study Group. Angiographic and optical coherence tomographic results of the MARINA study of ranibizumab in neovascular age-related macular degeneration. Ophthalmology. 2007;114:1868–1875. doi: 10.1016/j.ophtha.2007.04.030. [DOI] [PubMed] [Google Scholar]
- 165.Sadda SR, Stoller G, Boyer DS, Blodi BA, Shapiro H, Ianchulev T, et al. Anatomical benefit from ranibizumab treatment of predominantly classic neovascular age-related macular degeneration in the 2-year anchor study. Retina. 2010;30:1390–1399. doi: 10.1097/IAE.0b013e3181e44599. [DOI] [PubMed] [Google Scholar]
- 166.Tufail A, Patel PJ, Egan C, Hykin P, da Cruz L, Gregor Z, et al. Bevacizumab for neovascular age related macular degeneration (ABC trial): Multicentre randomised double masked study. BMJ. 2010;340:c2459. doi: 10.1136/bmj.c2459. [DOI] [PubMed] [Google Scholar]
- 167.Cruess AF, Zlateva G, Pleil AM, Wirostko B. Photodynamic therapy with verteporfin in age-related macular degeneration: A systematic review of efficacy, safety, treatment modifications and pharmacoeconomic properties. Acta Ophthalmol. 2009;87:118–132. doi: 10.1111/j.1755-3768.2008.01218.x. [DOI] [PubMed] [Google Scholar]
- 168.Wan MJ, Hooper PL, Sheidow TG. Combination therapy in exudative age-related macular degeneration: Visual outcomes following combined treatment with photodynamic therapy and intravitreal bevacizumab. Can J Ophthalmol. 2010;45:375–380. doi: 10.3129/i10-011. [DOI] [PubMed] [Google Scholar]
- 169.Potter MJ, Claudio CC, Szabo SM. A randomised trial of bevacizumab and reduced light dose photodynamic therapy in age-related macular degeneration: The VIA study. Br J Ophthalmol. 2010;94:174–179. doi: 10.1136/bjo.2008.155531. [DOI] [PubMed] [Google Scholar]
- 170.Lim JY, Lee SY, Kim JG, Lee JY, Chung H, Yoon YH, et al. Intravitreal bevacizumab alone versus in combination with photodynamic therapy for the treatment of neovascular maculopathy in patients aged 50 years or older: 1-year results of a prospective clinical study. Acta Ophthalmol. 2012;90:61–67. doi: 10.1111/j.1755-3768.2009.01841.x. [DOI] [PubMed] [Google Scholar]
- 171.Nguyen QD, Shah SM, Browning DJ, Hudson H, Sonkin P, Hariprasad SM, et al. A phase I study of intravitreal vascular endothelial growth factor trap-eye in patients with neovascular age-related macular degeneration. Ophthalmology. 2009;116:2141–2180. doi: 10.1016/j.ophtha.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 172.Ba R. Bayer and Regeneron Report Positive Top-Line Results of Two Phase 3 Studies with VEGF Trap-Eye in Wet Age-related Macular Degeneration. [Last accessed on 2010 Nov 22]. Available from: http://www.newsroomregeneroncom/releasedetailcfm?ReleaseID=532099 .
- 173.Mitchell P, Korobelnik JF, Lanzetta P, Holz FG, Prünte C, Schmidt-Erfurth U, et al. Ranibizumab (Lucentis) in neovascular age-related macular degeneration: Evidence from clinical trials. Br J Ophthalmol. 2010;94:2–13. doi: 10.1136/bjo.2009.159160. [DOI] [PubMed] [Google Scholar]
- 174.Campochiaro PA, Nguyen QD, Shah SM, Klein ML, Holz E, Frank RN, et al. Adenoviral vector-delivered pigment epithelium-derived factor for neovascular age-related macular degeneration: Results of a phase I clinical trial. Hum Gene Ther. 2006;17:167–176. doi: 10.1089/hum.2006.17.167. [DOI] [PubMed] [Google Scholar]
- 175.Zhang X, Lawler J. Thrombospondin-based antiangiogenic therapy. Microvasc Res. 2007;74:90–99. doi: 10.1016/j.mvr.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rüegg C, Hasmim M, Lejeune FJ, Alghisi GC. Antiangiogenic peptides and proteins: From experimental tools to clinical drugs. Biochim Biophys Acta. 2006;1765:155–177. doi: 10.1016/j.bbcan.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 177.Armstrong LC, Bornstein P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003;22:63–71. doi: 10.1016/s0945-053x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 178.Haviv F, Bradley MF, Kalvin DM, Schneider AJ, Davidson DJ, Majest SM, et al. Thrombospondin-1 mimetic peptide inhibitors of angiogenesis and tumor growth: Design, synthesis, and optimization of pharmacokinetics and biological activities. J Med Chem. 2005;48:2838–2846. doi: 10.1021/jm0401560. [DOI] [PubMed] [Google Scholar]
- 179.Gologorsky D, Thanos A, Vavvas D. Therapeutic interventions against inflammatory and angiogenic mediators in proliferative diabetic retinopathy. Mediators Inflamm 2012. 2012:629452. doi: 10.1155/2012/629452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hoekstra R, de Vos FY, Eskens FA, de Vries EG, Uges DR, Knight R, et al. Phase I study of the thrombospondin-1-mimetic angiogenesis inhibitor ABT-510 with 5-fluorouracil and leucovorin: A safe combination. Eur J Cancer. 2006;42:467–472. doi: 10.1016/j.ejca.2005.08.040. [DOI] [PubMed] [Google Scholar]