

Abstract
The dynamic modification of intracellular proteins by O-linked β-N-acetylglucosamine (O-GlcNAcylation) plays critical roles in many cellular processes. Although various methods have been developed for O-GlcNAc detection, there are few techniques for monitoring glycosylation stoichiometry and state (i.e., mono-, di-, etc., OGlcNAcylated). Measuring the levels of O-GlcNAcylation on a given substrate protein is important for understanding the biology of this critical modification and for prioritizingsubstrates for functional studies. One powerful solution to this limitation involves the chemoenzymatic installation of polyethylene glycol polymers of defined molecular mass onto O-GlcNAcylated proteins. These “mass tags” produce shifts in protein migration during sodium dodecyl sulfate−polyacrylamide gel electrophoresis (SDS-PAGE) that can be detected by Western blotting. Broad adoption of this method by the scientific community has been limited, however, by a lack of commercially available reagents and well-defined protein standards. Here, we develop a “click chemistry” approach to this method using entirely commercial reagents and confirm the accuracy of the approach using a semisynthetic O-GlcNAcylated protein. Our studies establish a new, expedited experimental workflow and standardized methods that can be readily utilized by non-experts to quantify the O-GlcNAc stoichiometry and state on endogenous proteins in any cell or tissue lysate.
Graphical Abstract
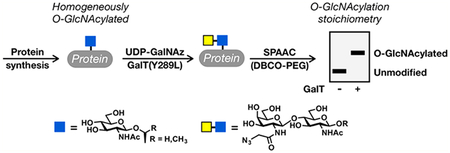
O-GlcNAc glycosylation (or O-GlcNAcylation) is the covalent addition of the monosaccharide N-acetylglucosamine to serine and threonine residues of intracellular proteins.1–3 This dynamic, inducible posttranslational modification is critical for vertebrate development and mammalian cell survival.4–6 Moreover, the levels of O-GlcNAcylation are altered in many human diseases, including cancer, diabetes, and neuro-degenerative disorders.7,8 Several established and commercially available reagents have been developed for the identification and visualization of O-GlcNAcylated proteins.9–11 Despite the power of these methods, however, measuring the O-GlcNAc stoichiometry and state (mono-, di-, etc., O-GlcNAcylated) of a given protein remains challenging. Such information is often critical for identifying the key glycosylation events within a particular protein or pathway, such as those that are inducible, highly occupied, or turned over rapidly, and for elucidating the molecular functions of O-GlcNAc. To address this challenge, we developed a chemoenzymatic method for the direct detection and quantification of endogenous O-GlcNAc modification levels.12,13 Cell or tissue lysates are first reacted with a recombinant, mutant version of a bovine galactosyltransferase, GalT(Y289L), which transfers nonnatural analogues of N-acetylgalactosamine from the corresponding uridine diphosphate (UDP) sugar donors to the C-4 hydroxyl group of O-GlcNAc. These analogues contain bioorthogonal reactive groups that can be used to selectively install polyethylene glycol (PEG) polymer tags of defined molecular mass. The “mass tags” will cause the O-GlcNAcylated fraction of any protein to migrate slower during sodium dodecyl sulfate−polyacrylamide gel electrophoresis (SDS−PAGE), thus enabling the determination of O-GlcNAc stoichiometry and state upon Western blotting. Previously, we extensively characterized a version of this chemoenzymatic strategy that involves the transfer of 2-keto-galactose, which is then appended using aminooxy-functionalized PEG via oxime formation (Figure 1A).14 However, the UDP-2-keto-galactose sugar is not commercially available, which limits broader adoption of this method by the scientific community. As such, we and others have focused on using GalT(Y289L) to transfer N-azidoacetylgalactosamine (GalNAz) because the corresponding UDP-GalNAz donor sugar is available from various commercial sources. In this case, the PEG tag can be installed using copper-catalyzed azide−alkyne cycloaddition (CuAAC)15–17 or strain-promoted azide−alkyne cycloaddition (SPAAC) chemistry18–20 using commercially available re-agents (Figure 1B). The enzymatic modification step of this method has been well established, and commercial kits based on those procedures for tagging O-GlcNAcylated proteins with small molecules such as biotin or fluorophores are now available.13 However, no commercial vendor has established a protocol for labeling proteins with PEG polymers to determine O-GlcNAc stoichiometries, and the reaction conditions for CuAAC and SPAAC used in academic laboratories have varied considerably. Furthermore, the lack of pure O-GlcNAcylated protein standards of defined stoichiometry has made it difficult to systematically evaluate and optimize this method. Here, we first used protein semisynthesis to prepare a homogeneous OGlcNAcylated protein. We then investigated and optimized the SPAAC chemoenzymatic mass tagging protocol for the quantitation of the modification stoichiometry and state on both this protein standard and endogenous proteins in mammalian cell lysates. We found that previously published conditions using SPAAC can underrepresent O-GlcNAc stoichiometry and demonstrate complete labeling at the protein level using our significantly improved workflow. Our studies establish standardized methods using entirely commercially available reagents that can be readily utilized by non-experts to quantify O-GlcNAc stoichiometry on endogenous proteins in any cell or tissue lysate.
Figure 1.

Quantification of O-GlcNAc stoichiometry by chemoenzymatic mass tagging. (A) Endogenous O-GlcNAc modifications are first enzymatically modified by 2-keto-galactose before installation of a PEG mass tag via oxime chemistry. The O-GlcNAcylated fraction of any given protein can then be measured by visualizing the slower-migrating “mass-tagged” species by Western blotting. (B) Here, we optimize the conditions for a similar procedure that utilizes strain-promoted cycloaddition (SPAAC) chemistry.
RESULTS
Previous reports using SPAAC to install PEG tags on modified proteins have used different concentrations of the cyclooctynefunctionalized PEG, ranging from 100 to 2000 μM.18–20 Furthermore, they have differed in their inclusion of a cysteine alkylation step (i.e., iodoacetamide treatment), which can be used to reduce the background reactivity of common SPAAC tags with thiols.21 However, these reaction conditions, to the best of our knowledge, have not been directly compared or systematically evaluated for the chemoenzymatic quantification of O-GlcNAcylation stoichiometry. To accomplish this goal, we first used an expressed protein ligation (EPL) strategy to prepare a homogeneously modified protein (Figure 2A).22 Briefly, HA-tagged ubiquitin was heterologously expressed in Escherichia coli as a genetic fusion to a DnaE intein from Anabaena variabilis,23 resulting in the production of a recombinant ubiquitin thioester. In parallel, we used solid-phase peptide synthesis to prepare an O-GlcNAcylated peptide (NH2-CGKSIAKgSIA-NH2, where gS = O-GlcNAcylated serine), whose sequence was chosen for ease of ligation. An EPL reaction was then used to generate O-GlcNAcylated ubiquitin, which was purified by reversed-phase high-performance liquid chromatography (RP-HPLC) and characterized by electrospray ionization mass spectrometry (ESI-MS) (Figure S1), yielding highly pure protein of the correct mass. This OGlcNAcylated ubiquitin protein standard was added to cell lysates, and the O-GlcNAcylated proteins were enzymatically modified with GalNAz by incubating with GalT(Y289L) and UDP-GalNAz.24 After precipitation to remove excess UDPGalNAz, the lysates were resuspended and incubated with different concentrations (250−1000 μM) of a 5-kDa PEG chain linked to dibenzocylcootyne (DBCO-PEG, Click Chemistry Tools). The reactions were terminated after 16 h by the addition of SDS loading buffer, and the samples subjected to SDS−PAGE. Unfortunately, without a cysteine alkylation step, these samples could not be transferred to a PVDF membrane using a semidry transfer system, presumably because of the high background levels of protein PEGylation. We therefore repeated this experiment with the addition of an iodoacetamide treatment step directly after enzymatic modification. Precipitation was again used to remove the excess UDP-GalNAz and iodoacetamide before reaction with DBCO-PEG, SDS−PAGE, and Western blotting (Figure 2B). At all concentrations of DBCO-PEG tested, we observed a notable amount of PEGylation of O-GlcNAcylated ubiquitin. However, the mass tagging was not complete at concentrations of <1000 μM, with the measured stoichiometry steadily increasing from 88% at 250 μM DBCO-PEG to 99% at 1000 μM (Figure 2B). This suggests that previous reports using only 100 μM DBCO-PEG likely underestimated the O-GlcNAc stoichiometry of proteins. Additionally, the quantitative labeling of O-GlcNAcylated ubiquitin using 1000 μM DBCO-PEG demonstrates that the enzymatic transfer of UDP-GalNAz and subsequent SPAAC reaction indeed go to completion at the protein level.
Figure 2.

Optimization of the mass tagging protocol. (A) Synthesis of a site-specifically O-GlcNAcylated control protein. A recombinant, HA-tagged ubiquitin thioester was prepared using intein chemistry and was subsequently ligated to an O-GlcNAcylated peptide prepared by solid-phase peptide synthesis. (B) High concentrations of DBCOPEG are required for quantitative mass shifting of the OGlcNAcylated control protein. O-GlcNAcylated ubiquitin was added to cell lysates before enzymatic modification with GalNAz and reaction with the indicated concentrations of DBCO-PEG and analysis by Western blotting. (C) High concentrations of DBCO-PEG are also required for complete mass shifting of endogenous proteins. Cell lysates were chemoenzymatically modified, and the known OGlcNAcylated proteins Nup62 and CREB were visualized by Western blotting.
We next repeated the chemoenzymatic labeling protocol on cell lysates without the addition of O-GlcNAcylated ubiquitin. Again, the proteins were subjected to SPAAC using a range of DBCO-PEG concentrations and resolved by SDS−PAGE. The mass tagging of two known O-GlcNAcylated proteins, nucleoporin 62 (Nup62) and cyclic AMP-response element binding protein (CREB), was visualized by Western blotting (Figure 2C). Essentially all Nup62 in the cell has been shown to be O-GlcNAcylated at ≥10 different sites, while approximately 30% of CREB is known to be O-GlcNAcylated in various brain tissues.14,25 Again, we found that treatment with DBCO-PEG concentrations of <1000 μM resulted in incomplete labeling and underestimation of the O-GlcNAc stoichiometry. More specifically, the amounts of CREB modification increased from only 3% at 100 μM DBCO-PEG to 12% at 1000 μM in H1299 cells. Interestingly, as the mass tagging became more quantitative on Nup62, the overall amount of shifted Nup62 was reduced. We interpreted this result as being due to interference from excess PEG with SDS− PAGE or Western blot. Therefore, we repeated the analysis of Nup62 and CREB with or without an additional precipitation step before SDS−PAGE to remove the unreacted PEG (Figure S2). Consistent with our hypothesis, we found that this final precipitation improved the overall signal and therefore incorporated this step into all of our subsequent analyses. However, a fraction of the total Nup62 signal appears to be lost as a result of the mass shifting protocol. Notably, we do not see this with CREB or the semisynthetic protein, indicating that this issue is most likely confined to high-molecular weight proteins or ones with a large number of O-GlcNAc modifications and could result from inefficient transfer or antibody recognition during the Western blot.
With these optimized conditions in hand, we then set out to compare the SPAAC method to the CuAAC and oxime formation strategies.14,15,17 In the case of CuAAC, we subjected the GalNAz-labeled proteins to CuAAC conditions with alkyne-PEG (Click Chemistry Tools).17 For the oxime chemistry, we followed our previously published protocol15 for the enzymatic transfer of 2-keto-galactose, followed by reaction with 5 kDa aminooxy-PEG (Nanocs Inc.). First, we used lysates that had been doped with O-GlcNAcylated ubiquitin and found that even aggressive CuAAC reaction conditions (24 h at 37°C) resulted in substoichiometric labeling (Figure 3A). Consistent with our previous results, the oxime chemistry resulted in essentially quantitative mass shifting that compares very well to that seen with SPAAC (Figure 3A). These results were also true for our analysis of the endogenous OGlcNAcylated protein CREB, where the SPAAC and oxime chemistries again resulted in very similar measurements of OGlcNAc stoichiometry (23 and 20%, respectively), and less mass tagging (11%) could be detected using CuAAC (Figure 3B). The increase in this stoichiometry compared to that in Figure 2C could be due to inclusion of a final precipitation step to remove the excess PEG (Figure 2S) or changes in CREB glycosylation as a part of normal H1299 cell function.
Figure 3.

Comparison of mass tagging with SPAAC with CuAAC or oxime formation. (A) Cell lysates were doped with O-GlcNAcylated ubiquitin and subjected to enzymatic modification with either GalNAz (CuAAC and SPAAC) or 2-keto-galactose (Oxime) before bioorthogonal reaction with the appropriate PEG tag and visualization by Western blotting. (B) Cell lysates were subjected to the procedure described for panel A, and the endogenously O-GlcNAcylated proteins Nup62 and CREB were visualized by Western blotting. The quantification is consistent in three different biological replicates.
To ensure that our protocols were standardized using entirely commercially available reagents, we also compared our mass tagging results, which used GalT(Y289L) expressed in our laboratory, to those found with the commercially available chemoenzymatic labeling kit from ThermoFisher Scientific (Click-iT O-GlcNAc Enzymatic Labeling System). We found that both sources of enzyme resulted in similar levels of mass tagging on both Nup62 and CREB (Figure 4A). The fact that GalT(Y289L) is expressed in E. coli raises the possibility that contaminating bacterial proteins might cross-react with the antibodies employed to detect mammalian proteins of interest. To test this possibility, we immunoblotted four independent recombinant preparations of GalT(Y298L) using a handful of antibodies and found that, indeed, several antibodies recognize bacterial proteins of different molecular weights (Figure S3). We believe that these proteins are low levels of impurities due to incomplete washing of the inclusion bodies during GalT(Y298L) expression and purification. Therefore, we strongly recommend that researchers test their antibodies of choice with appropriate control experiments to rule out any misidentification of mass-tagged bands.
Figure 4.

Further characterization of chemoenzymatic mass tagging by SPAAC. (A) Commercial reagents are sufficient for O-GlcNAc quantitation. Cell lysates were subjected to the SpAAC mass tagging protocol using purified GalT or a commercially available enzyme (ThermoFisher Scientific) before Western blotting. (B) SPAAC proceeds selectively and quantitatively in 5 min at 98°C. OGlcNAcylated ubiquitin was added to H1299 lysates and enzymatically labeled. SPAAC was then performed at either room temperature for 16 h or 98°C for 5 min. Notably, we found that 98°C for 5 min results in quantitative labeling and does not increase the nonspecific background.
To improve the overall efficiency of the method, we next asked whether the SPAAC reaction could be accelerated by heating, without negatively affecting its selectivity. Accordingly, we performed chemoenzymatic labeling of O-GlcNAcylated ubiquitin or cell lysates as previously described, followed by SPAAC at 25°C for 16 h as described above or at 98°C for 5 min. Remarkably, we found that these “boiling” conditions result in selective SPAAC and indistinguishable mass shifts (Figure 4B). No mass shifts were observed with an O GLcNAc-deficient mutant of CREB25 using either room-temperature or boiling conditions (Figure S4), further confirming the selectivity of the approach. The ability to achieve quantitative chemoenzymatic labeling within minutes reduces the overall time required for the mass tagging procedure by about one half.
Finally, DBCO-PEG has limited solubility in DMSO. Therefore, increasing its concentration resulted in higher percentages of DMSO in the SPAAC reactions described above (≤10% DMSO at 1000 μM). This raised the possibility that increased amounts of DMSO could result in improved mass shifting at lower DBCO-PEG concentrations. To examine this further, we repeated the chemoenzymatic mass shifting of O-GlcNAcylated ubiquitin with DBCO-PEG (100−1000 μM) in the presence of 10% DMSO at room temperature for 16 h (Figure S5A). We observed a small improvement in the SPAAC reaction at lower concentrations of DBCO-PEG (compare to Figure 2B) but found that complete mass shifting still required 1000 μM DBCO-PEG. Notably, this was also true when we performed the SPAAC reaction at 98°C for 5 min (Figure S5B).
In summary, we have developed an SPAAC-based approach for the determination of O-GlcNAc stoichiometry and state using entirely commercially available reagents. This method was significantly improved, systematically optimized, and validated for the first time on the protein level using a pure, semisynthetic O-GlcNAcylated protein standard. Our results suggest that several previously published SPAAC protocols may result in an underestimation of O-GlcNAcylation stoichiometry. Importantly, the protocol developed here does not require mass spectrometry or other advanced instrumentation and can be performed using commercially available reagents from ThermoFisher Scientific and Click Chemistry Tools, which will empower non-experts to explore their own proteins of interest. We believe that these expedited, highly accessible procedures will both standardize and expand the application of this powerful mass tagging strategy for the analysis of O-GlcNAcylation stoichiometry and state.
DETAILED PROTOCOL FOR SPAAC
Buffers and reagents needed:
0.05% SDS buffer [0.05% SDS, 150 mM NaCl, and 10 mM TEA (pH 7.4)]
4% SDS buffer [4% SDS, 150 mM NaCl, and 10 mM TEA (pH 7.4)]
1% SDS buffer [1% SDS, 150 mM NaCl, and 10 mM TEA (pH 7.4)]
1% SDS GalT buffer [1% SDS with 20 mM HEPES (pH7.9)]
GalT as supplied by Invitrogen Click-IT Kit or produced in house Labeling buffer [2.5×; 5% NP-40 (or IGEPAL CA-630), 125 mM NaCl, and 50 mM HEPES (pH 7.9)], supplied by Invitrogen Click-IT Kit or made in house (see the Supporting Information)
MnCl2 (100 mM in H2O), supplied by Invitrogen Click-IT Kit or made in house
UDP-GalNAz [0.5 mM in 10 mM HEPES (pH 7.9)], supplied by Invitrogen Click-It Kit or made in house (see the Supporting Information)
Iodoacetamide (made fresh, 600 mM stock in H2O)
DBCO-PEG-5K (10 mM stock in DMSO) from Click Chemistry Tools
2× loading buffer [20% glycerol, 0.2% bromophenol blue, and 1.4% β-mercaptoethanol (pH 6.8)]
cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail from Roche Biosciences
Benzonase from EMD Millipore
Control antibodies for Western blotting: anti-CREB1 (9104S, Cell Signaling Technology) and anti-Nup62 (610497, BD Biosciences)
Step-by-step protocol:
Collect cells and wash twice with PBS (1 cm × 10 cm dish at ≥80% confluency)
Add 26 μL of H2O containing 10× cOmplete Mini Protease Inhibitor Cocktail (Sigma-Aldrich)
Add 50 μL of 0.05% SDS buffer and resuspend cells
Add 1 μL of benzonase and incubate on ice for 30 min Add 200 μL of 4% SDS buffer
Vortex and pellet any debris by centrifugation at 10000g for 10 min at 15°C
Perform the BCA assay and normalize the protein concentration to 1 μg/μL using 1% SDS buffer
To 1 mg of protein (1000 μL)
I. Add 3000 μL of methanol and vortex
II. Add 750 μL of chloroform and vortex
III. Add 2000 μL of H2O and vortex
IV. Centrifuge at 5000g for 5 min at 15°C
V. Discard the upper aqueous phase, while leaving the interface layer interacting
VI. Add 2250 μL of methanol and vortex
VII. Centrifuge at 5000g for 10 min at 15°C and discard the supernatant
VIII. Allow the pellet to air-dry for 5 min. Note: Do not allow the protein pellet to “over-dry”, as this will make the proteins difficult to resuspend. This is true for all subsequent drying steps.
Resuspend protein in 100 μL of 1% SDS GalT buffer
Bath sonicate until proteins are dissolved
Perform the BCA assay and normalize the protein concentration to 2.5 μg/μL using 1% SDS GalT buffer
Add enzymatic reagents in the following order for the “+GalT + UDP sample”
I. 40 μL of lysate
II. 49 μL of H2O
III. 80 μL of labeling buffer
IV. 11 μL of MnCl2
V. 10 μL of UDP-GalNAz
VI. 10 μL of GalT enzyme
Add enzymatic reagents in the following order for the “− GalT + UDP sample”
I. 40 μL of lysate
II. 59 μL of H2O
III. 80 μL of labeling buffer
IV. 11 μL of MnCl2
V. 10 μL of UDP-GalNAz
Optional: Add enzymatic reagents in the following order for the “+GalT − UDP sample”
I. 40 μL of lysate
II. 59 μL of H2O
III. 80 μL of labeling buffer
IV. 11 μL of MnCl2
V. 10 μL of the GalT enzyme
Incubate samples at 4°C for 20 h
Add 7.5 μL of freshly made iodoacetamide (600 mM) to each sample
Incubate for 30 min in the dark
To each sample
I. Add 622 μL of methanol and vortex
II. Add 156 μL of chloroform and vortex
III. Add 415 μL of H2O and vortex
IV. Centrifuge at 10000g for 5 min at 15°C
V. Discard the upper aqueous phase, while leaving the interface layer intact
VI. Add 467 μL of methanol to tube and vortex
VII. Centrifuge at 10000g for 10 min at 15°C and discard the supernatant
VIII. Allow the pellet to air-dry for 5 minAdd 90 μL of 1% SDS and bath sonicate until proteins are dissolved
Add 10 μL of DBCO-PEG-5K and vortex
Incubate for 16 h at room temperature or boil for 5 min at 98°C
To each sample
I. Add 300 μL of methanol and vortex
II. Add 75 μL of chloroform and vortex
III. Add 200 μL of H2O and vortex
IV. Centrifuge at 10000g for 5 min at 15°C
V. Discard upper aqueous phase while leaving the interface layer intact
VI. Add 225 μL of methanol to tube and vortex
VII. Centrifuge at 10000g for 10 min at 15°C and discard the supernatant
VIII. Allow the pellet to air-dry for 5 min
Add 25 μL of 4% SDS and bath sonicate until proteins are dissolved
Add 25 μL of 2× SDS and boil for 5 min at 98°C
Load ~40 μg per lane on SDS−PAGE for Western blotting
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Science Foundation (Grant CHE-1506503 to M.R.P.), the National Institutes of Health (Grants R01GM114537 and R01GM125939 to M.R.P., R01GM084724 and R01AG060540–13 to L.C.H.-W., and T32GM008042, T32GM007616, and F30AG055314 to J.W.T.), and the UCLA-Caltech Medical Scientist Training Program (J.W.T.). The authors also thank Dr. P. Qasba (National Cancer Institute at Frederick, Frederick, MD) for generously providing the GalT(Y289L) construct.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00648.
Experimental methods, supporting figures, and data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Bond MR, and Hanover JA (2015) A little sugar goes a long way: the cell biology of O-GlcNAc. J. Cell Biol 208, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Levine ZG, and Walker S (2016) The Biochemistry of OGlcNAc Transferase: Which Functions Make It Essential in Mammalian Cells? Annu. Rev. Biochem 85, 631–657. [DOI] [PubMed] [Google Scholar]
- (3).Yang X, and Qian K (2017) Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol 18, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Shafi R, Iyer SP, Ellies LG, O’Donnell N, Marek KW, Chui D, Hart GW, and Marth JD (2000) The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. U. S. A 97, 5735–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).O’Donnell N, Zachara NE, Hart GW, and Marth JD (2004) Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol. Cell. Biol 24, 1680–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sinclair DAR, Syrzycka M, Macauley MS, Rastgardani T, Komljenovic I, Vocadlo DJ, Brock HW, and Honda BM (2009) Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc). Proc. Natl. Acad. Sci. U. S. A 106, 13427–13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ma Z, and Vosseller K (2013) O-GlcNAc in cancer biology. Amino Acids 45, 719–733. [DOI] [PubMed] [Google Scholar]
- (8).Yuzwa SA, and Vocadlo DJ (2014) O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer’s disease and beyond. Chem. Soc. Rev 43, 6839–6858. [DOI] [PubMed] [Google Scholar]
- (9).Banerjee PS, Hart GW, and Cho JW (2013) Chemical approaches to study O-GlcNAcylation. Chem. Soc. Rev 42, 4345–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chuh KN, and Pratt MR (2015) Chemical methods for the proteome-wide identification of posttranslationally modified proteins. Curr. Opin. Chem. Biol 24, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chuh KN, Batt AR, and Pratt MR (2016) Chemical Methods for Encoding and Decoding of Posttranslational Modifications. Cell Chemical Biology 23, 86–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Khidekel N, Arndt S, Lamarre-Vincent N, Lippert A, Poulin-Kerstien KG, Ramakrishnan B, Qasba PK, and Hsieh-Wilson LC (2003) A Chemoenzymatic Approach toward the Rapid and Sensitive Detection of O-GlcNAc Posttranslational Modifications.J. Am. Chem. Soc 125, 16162–16163. [DOI] [PubMed] [Google Scholar]
- (13).Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, and Hsieh-Wilson LC (2008) Direct In-Gel Fluorescence Detection and Cellular Imaging of O-GlcNAc-Modified Proteins. J. Am. Chem. Soc 130, 11576–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rexach JE, Rogers CJ, Yu S-H, Tao J, Sun YE, and Hsieh-Wilson LC (2010) Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags. Nat. Chem. Biol 6, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Clark PM, Rexach JE, and Hsieh-Wilson LC (2013) Visualization of O-GlcNAc glycosylation stoichiometry and dynamics using resolvable poly(ethylene glycol) mass tags. Curr. Protoc Chem. Biol 5, 281–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Nagel AK, and Ball LE (2014) O-GlcNAc modification of the runt-related transcription factor 2 (Runx2) links osteogenesis and nutrient metabolism in bone marrow mesenchymal stem cells. Mol. Cell. Proteomics 13, 3381–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Thompson JW, Griffin ME, and Hsieh-Wilson LC (2018) Methods for the Detection, Study, and Dynamic Profiling of O-GlcNAc Glycosylation. Methods Enzymol 598, 101–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ortiz-Meoz RF, Merbl Y, Kirschner MW, and Walker S (2014) Microarray discovery of new OGT substrates: the medulloblastoma oncogene OTX2 is O-GlcNAcylated. J. Am. Chem. Soc 136, 4845–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lund PJ, Elias JE, and Davis MM (2016) Global Analysis of O-GlcNAc Glycoproteins in Activated Human T Cells. J. Immunol 197, 3086–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang S, Yang F, Petyuk VA, Shukla AK, Monroe ME, Gritsenko MA, Rodland KD, Smith RD, Qian W-J, Gong C-X, and Liu T (2017) Quantitative proteomics identifies altered OGlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J. Pathol 243, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kim EJ, Kang DW, Leucke HF, Bond MR, Ghosh S, Love DC, Ahn J-S, Kang D-O, and Hanover JA (2013) Optimizing the selectivity of DIFO-based reagents for intracellular bioorthogonal applicaitons. Carbohydr. Res 377, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Muir TW, Sondhi D, and Cole PA (1998) Expressed protein ligation: a general method for protein engineering. Proc. Natl. Acad. Sci. U. S. A 95, 6705–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Shah NH, Dann GP, Vila-Perello M, Liu Z, and Muir TW (2012) Ultrafast Protein Splicing is Common among Cyano-bacterial Split Inteins: Implications for Protein Engineering. J. Am. Chem. Soc 134, 11338–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ramakrishnan B, and Qasba PK (2002) Structure-based Design of beta 1,4-Galactosyltransferase I (beta 4Gal-T1) with Equally Efficient N-Acetylgalactosaminyltransferase Activity. J. Biol. Chem 277, 20833–20839. [DOI] [PubMed] [Google Scholar]
- (25).Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC, and Hsieh-Wilson LC (2012) Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat. Chem. Biol 8, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.