

Laulimalide (1), also known as figianolide B, a 20-membered macrolide isolated from the Indonesian sponge Hyattella sp., has shown remarkable antitumor activities.1 Recently, Laulimalide was also isolated from an Okinawan sponge Fasciospongia rimosa.2 It displayed potent cytotoxicity against the KB cell line with an IC50 value of 15 ng/mL.1 Furthermore, it has shown cytotoxicity against P388, A549, HT29, and MEL28 cell lines in the range of 10–50 ng/mL (IC50 values).2b The structure of 1 was initially established by NMR studies. Subsequently, its absolute configuration was established by X-ray analysis by Higa and coworkers.2 The significant clinical potential of laulimalide has stimulated considerable interest in its synthesis and structure-function studies.3 Herein, we report the first synthesis of (−)-laulimalide 1.
As outlined in Figure 1, our synthetic strategy of laulimalide is convergent and involves the assembly of C3−C16 segment 2 and C17−C28 segment 3 by Julia olefination, followed by an intramolecular Horner-Emmons reaction between the C19 phosphonoacetate and C3 aldehyde. The sensitive epoxide at C16−C17 was selectively introduced at the final stage of the synthesis by Sharpless epoxidation.4 The construction of both dihydropyran rings of laulimalide was achieved by ring-closing olefin metathesis utilizing Grubbs’ catalyst as the key step.5
Figure 1.
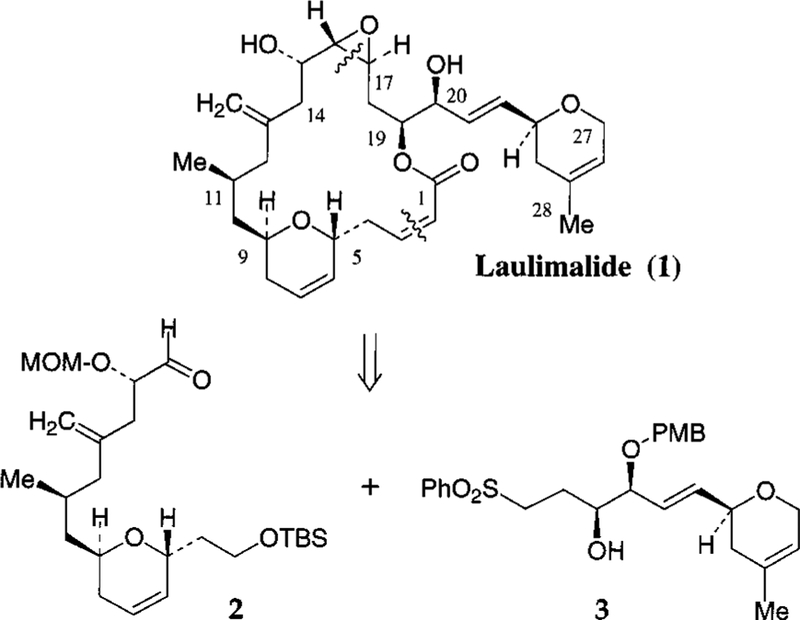
The synthesis of C3−C16 segment is accomplished by modifications of the previously published sequence.3a As outlined in Scheme 1, diastereomerically pure δ-lactone 5 was prepared efficiently by exposure of acryloyl ester 4 to Grubbs’ catalyst (10 mol %) in CH2Cl2.3a DIBAL reduction of 5 at −78 °C in CH2Cl2 followed by reaction with ethanol and CSA afforded the ethyl glycoside.6 Reaction of this ethyl acetal with tert-butyldimethylsilyl vinyl ether and Montmorillonite K-10 as the Lewis acid in CH2Cl2 at 23 °C followed by NaBH4 reduction of the resulting aldehyde afforded the corresponding dihydropyran as a single isomer (by 1H and 13C NMR). Protection of the alcohol with TBSCl and imidazole furnished the TBS ether 6. Removal of the benzyl group by lithium in liquid ammonia provided the alcohol which was subsequently converted to iodide 7. To install the C13 methylene unit and the C15 hydroxyl group, alkylation of lactone 8 was carried out by treatment with NaH in DMF at 0 °C for 15 min followed by reaction with iodide 7 at 23 °C for 15 min and then 60 °C for 12 h, which furnished lactone 9 as a mixture (4.2:1 by 1H NMR) of isomers. Reduction of the mixture of lactone 9 by Red-Al provided the diol 10. Benzoylation of 10 afforded the corresponding dibenzoate which was exposed to Na−Hg in MeOH at −20 °C to provide the olefin 11.7 Protection of the C15 hydroxyl group as a MOM ether, removal of the PMB group and Swern oxidation of the resulting alcohol furnished the C3−C16 segment, aldehyde 2.
Scheme 1a.
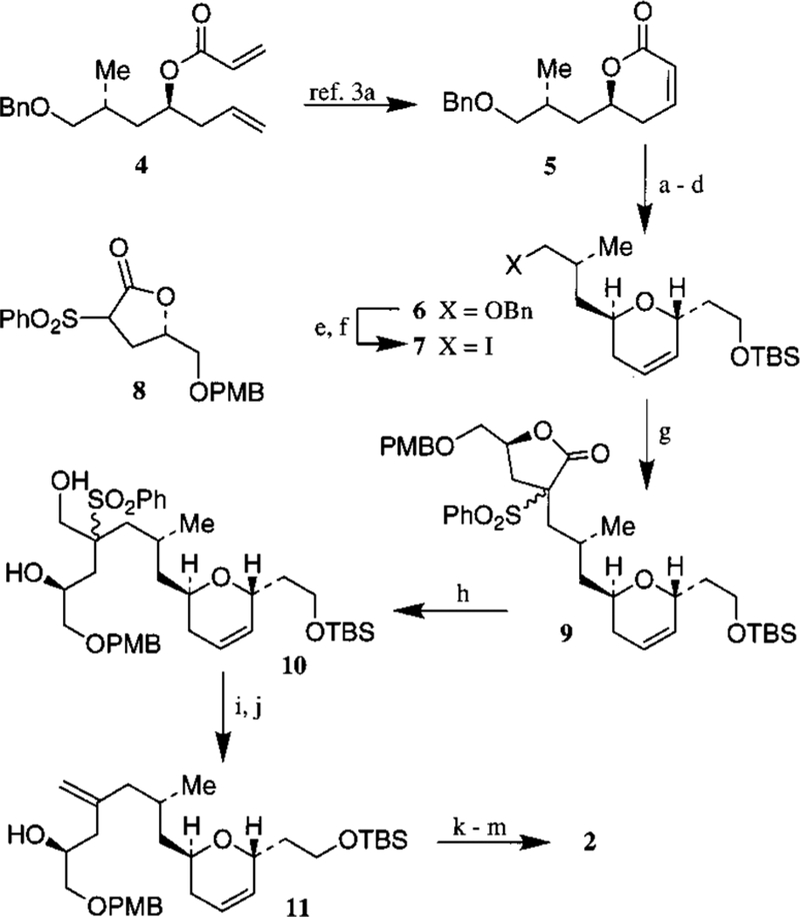
a (a) Dibal-H, −78 °C then CSA, EtOH, 23 °C; (b) K-10, CH2 = CHOTBS, 23 °C; (c) NaBH4, MeOH, 0 °C (54%); (d) TBSCl, imidazole, DMF, 23 °C (75%); (e) Li, NH3 (95%); (f) I2, PPh3, Imidazole (96%); (g) 8, NaH, DMF, 0°C then iodide 7, 60 °C (89%); (h) Red-Al, THF, 0 °C; (i) PhCOCl, Et3N, DMAP (cat.); (j) Na(Hg), Na2HPO4, MeOH, −20° to 23 °C (72%); (k) MOMCl, i-Pr2NEt, 23 °C; (l) DDQ, pH 7 buffer, 23 °C (81%); (m) DMSO, (COCl)2, i-Pr2NEt, −60 °C (85%).
The synthesis of the C17−C28 segment 3 has been achieved by modification of reaction sequences published previously.3b Optically active dihydropyran derivative 12 was prepared in multigram quantity by utilizing ring-closing olefin methathesis5 and Corey-Fuchs’ homologation8 reactions as the key steps. The alkynyl anion of 12 was readily obtained by treatment with n-BuLi at −78 °C for 1 h followed by warming to 23 °C for 1 h (Scheme 2). Reaction of the resulting alkynyl anion with optically active aldehyde 13 at −78 °C furnished a diastereomeric mixture (1.8: 1) of alcohols 14 and 15, which upon oxidation with Dess-Martin periodinane9 gave the corresponding alkynyl ketone. L-Selectride reduction of this ketone at −78 °C furnished the desired synalkynyl alcohol 15 as a single diastereomer (by 1H NMR and 13C NMR). Red-Al reduction of 15 set the C21−C22 trans-olefin geometry. Removal of the C19 PMB group by exposure to TFA10 and subsequent reaction of the resulting diol with p-methoxybenzylidene acetal and CSA provided acetal 16. DIBAL reduction of 16 at −78 °C in CH2Cl2 afforded the C17−C28 segment 3 as a single regio isomer (by 1H and 13C NMR).
Scheme 2a.
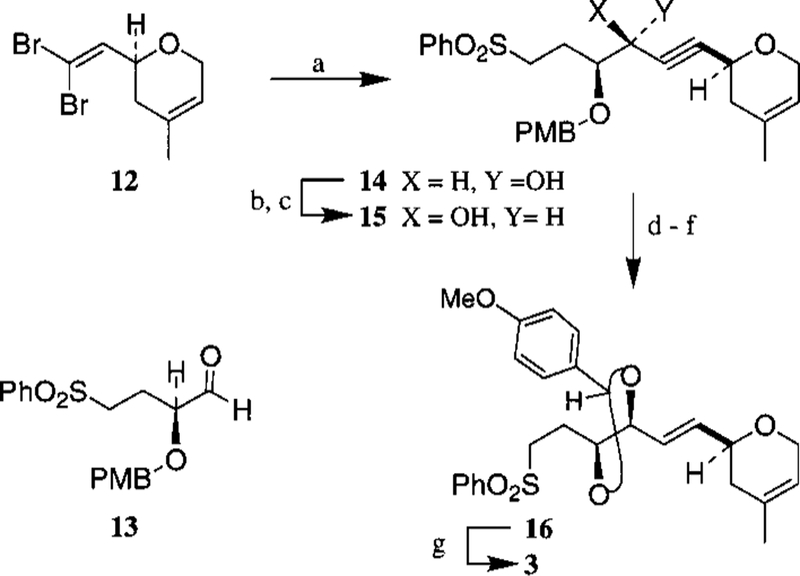
a (a) n-BuLi, −78 °C, 1 h and 23 °C, 1 h then 13, −78 °C (64%); (b) Dess−Martin, CH2Cl2, 23 °C (81%); (c) L-Selectride, THF, −78 °C (87%); (d) Red-Al, THF, −20 °C (81%); (e) CF3CO2H, CH2Cl2, 23 °C; (f) p-MeO-Ph-CH(OMe)2, CSA, CH2Cl2, 23 °C (71%); (g) Dibal-H, CH2Cl2, −78 °C (74%).
Our subsequent synthetic strategy calls for the assembly of fragments 2 and 3 by Julia olefination (Scheme 3). Thus, lithiation of sulfone derivative 3 with 2.1 equiv of n-BuLi in THF at −78 °C for 15 min followed by reaction of the resulting dianion with the aldehyde 2 at −78 °C to −40 °C for 2 h furnished the expected R-hydroxy sulfone derivatives. The hydroxy sulfone was transformed into the corresponding C16−C17 olefin in a two step sequence involving (1) acylation of the hydroxy sulfone derivatives with Ac2O, Et3N and DMAP (cat.) and (2) exposure of the resulting acetates to Na(Hg) in methanol at −20 °C for 2 h followed by warming the reaction to 23 °C for 30 min. The C16−C17 trans olefin 17 was obtained in 34% yield along with 10% cis-olefin, which was readily separated by silica gel chromatography.
Scheme 3a.
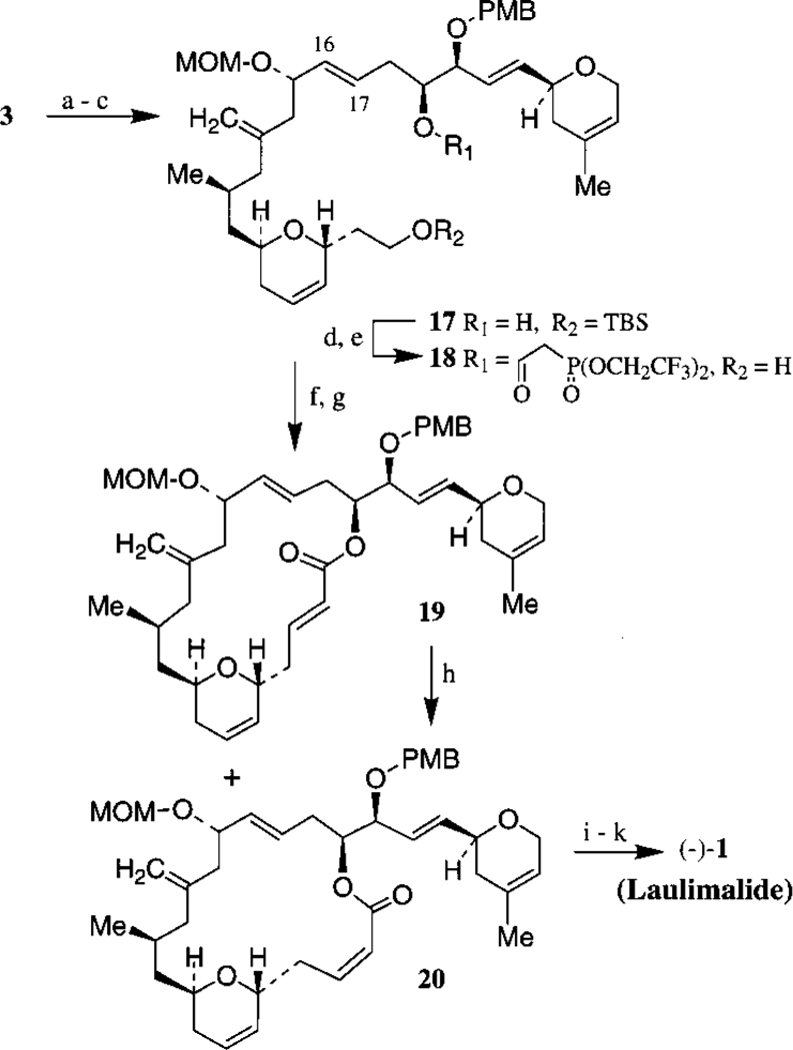
a (a) n-BuLi, −78°C, 15 min, Then 2, −78 to −40 °C, 2 h; (b) Ac2O, Et3N, DMAP (cat.); (c) Na(Hg), Na2HPO4, MeOH, −20 to 23 °C (34%); (d) (CF3CH2O)2P(O)CH2CO2H, Cl3C6H2COCl, i-Pr2NEt, DMAP; (e) AcOH-THF-H2O (3:1:1), 23 °C (99%); (f) Dess−Martin, CH2Cl2, 23 °C (79%); (g) K2CO3, 18-C-6, −20 to 0 °C (84%); (h) hv, Et2O, 50 min (66%); (i) PPTS, t-BuOH, 84 °C (45%) (j) Ti(OiPr)4, (+)-DET, t-BuOOH, −20 °C; (k) DDQ, pH 7, 23 °C (48%).
Subsequent elaboration to the macrolactone possessing C2−C3 cis-olefin geometry proved to be a formidable task. We finally relied upon an intramolecular Horner-Emmons reaction of C19 phosphonoacetate and C3 aldehyde using Still’s protocol.11 Thus, acylation of C19 hydroxyl group with bis-(2,2,2-trifluoroethyl)-phosphonoacetic acid followed by removal of the TBS group by exposure to aqueous acetic acid in THF at 23 °C furnished the acetate derivative 18 in near quantitative yield.12 Oxidation of 18 with Dess−Martin periodinane provided the C3 aldehyde which upon treatment with K2CO3 in the presence of 18-C-6 at −20 °C for 30 min and then at 0 °C for 2.5 h furnished a mixture (2:1) of macrolactones 19 and 20 in 84% yield (Scheme 3). Both cisand trans-lactones were separated by silica gel chromatography. Horner-Emmons reaction of the corresponding (diphenylphosphono)acetate derivative provided slight improvement of the cis-selectivity (cis:trans =1:1.7, 69% isolated yield).13 Further attempts to improve the ratio for 20 by changing reaction conditions or C20 protecting group have been unsuccessful. The overall yield of the desired cis-macrolactone 20 was however improved to 47% after photoisomerization of the trans-macro-lactone 19.14 Thus, irradiation of 19 in ether under UV in a Rayonet photochemical reactor for 50 min afforded a mixture of trans-lactone 19 (33%) and the cis-lactone 20 (33%) which were separated by chromatography. The identity of the cis-olefin geometry of 20 was established by its observed coupling constant (J = 11.6 Hz). Macrolactone 20 was converted to synthetic (−)-laulimalide 1 as follows: removal of the MOM group by refluxing with PPTS in t-BuOH,15 exposure of the resulting alcohol to Sharpless epoxidation4 with (+)-DET and removal of the C20 PMB ether by exposure to DDQ. Spectral data (1H and 13C NMR) of synthetic 1 ([α]23D −196 c 0.23, CHCl3) are identical to that from a sample of natural laulimalide (lit.2a [α]29D −200 c 1.03, CHCl3) kindly provided by Professor Higa.
Thus, a stereocontrolled synthesis of (−)-laulimalide has been achieved. Considering its clinical potential as an antitumor agent, the present synthesis will enable important structure−function studies as well as synthesis of structural variants of laulimalide. Further improvement in synthesis and biological studies are currently in progress.
Supplementary Material
Acknowledgment.
Financial support by the National Institutes of Health (GM 55600) is gratefully acknowledged. We also thank Professor Higa for providing a sample of natural laulimalide and Professor Forsyth for the experimental details of bis-(2,2,2-trifluoroethyl)phosphonoacetic acid.
Footnotes
Supporting Information Available:
Experimental procedures and spectral data for compounds 1−3, 6−11, 15−20; 1H NMR spectra for compounds 1, 3, 11, 15−17, 19, 20; and 13C NMR for compounds 1, 3, 11, 17, and 19 (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) Quinoa E; Kakou Y; Crews P J. Org. Chem 1988, 53, 3642. [Google Scholar]; (b) Corley DG; Herb R; Moore RE; Scheuer PJ; Paul VJ J. Org. Chem 1988, 53, 3644. [Google Scholar]
- (2).(a) Jefford CW; Bernardinelli G; Tanaka J-I; Higa T Tetrahedron Lett 1996, 37, 159. [Google Scholar]; (b) Tanaka J-I; Higa T; Bernardinelli G; Jefford CW Chem. Lett 1996, 255. [Google Scholar]
- (3).(a) Ghosh AK; Wang Y Tetrahedron Lett 2000, 41, 2319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK; Wang Y Tetrahedron Lett 2000, 41, 4705. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mulzer J; Hanbauer M Tetrahedron Lett 2000, 41, 33. [Google Scholar]; (d) Shimizu A; Nishiyama S Synlett 1998, 1209. [Google Scholar]; (e) Ghosh AK; Mathivanan P; Cappiello J Tetrahedron Lett 1997, 38, 2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Johnson RA; Sharpless KB In Catalytic Asymmetric Synthesis; Ojima I; Ed.; VCH Publishers: New York, 1993, p 103–158. [Google Scholar]
- (5).Grubbs RH; Chang S Tetrahedron 1998, 54, 4413 and references therein. [Google Scholar]
- (6).For a related method, see: Dahanukar VH; Rychnovsky SD J. Org. Chem 1996, 61, 8317. [DOI] [PubMed] [Google Scholar]
- (7).Lee GH; Lee HK; Choi EB; Kim BT; Pak CS Tetrahedron Lett 1995, 36, 5607. [Google Scholar]
- (8).Corey EJ; Fuchs PL Tetrahedron Lett 1972, 3769. [Google Scholar]
- (9).Meger SD; Schreiber SL J. Org. Chem 1994, 59, 7549. [Google Scholar]
- (10).Yan L; Kahne D Synlett 1995, 523. [Google Scholar]
- (11).Still WC; Gennari C Tetrahedron Lett 1983, 24, 4405. [Google Scholar]
- (12).This protocol was recently employed in the synthesis of phorboxazole A. See: Forsyth CJ; Ajmed F; Cink RD; Lee CS J. Am. Chem. Soc 1998, 120, 5597. [Google Scholar]
- (13).Ando KJ Org. Chem 1999, 64, 8406 and references therein. [DOI] [PubMed] [Google Scholar]
- (14).Smith AB; Lupo AT; Ohba M; Chen KJ Am. Chem. Soc 1989, 111, 6648 and references therein. [Google Scholar]
- (15).Monti H; Leandri G; Klos-Ringquet M; Corriol C Synth. Commun 1983, 13, 1021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.