

Abstract
The broad-based legalization of cannabis use has created a strong need to understand its impact on human health and behavior. The risks that may be associated with cannabis use, particularly for sensitive subgroups such as pregnant women, are difficult to define because of a paucity of dose-response data and the recent increase in cannabis potency. Although there is a large body of evidence detailing the mode of action of Δ9‐tetrahydrocannabinol (THC) in adults, little work has focused on understanding how cannabis use during pregnancy may impact the development of the fetal nervous system and whether additional plant-derived cannabinoids might participate. This manuscript presents an overview of the historical and contemporary literature focused on the mode of action of THC in the developing brain, comparative pharmacokinetics in both pregnant and nonpregnant model systems and neurodevelopmental outcomes in exposed offspring. Despite growing public health significance, pharmacokinetic studies of THC have focused on nonpregnant adult subjects and there are few published reports on disposition parameters during pregnancy. Data from preclinical species show that THC readily crosses the placenta although fetal exposures appear lower than maternal exposures. The neurodevelopmental data in human and preclinical species suggest that prenatal exposure to THC may lead to subtle, persistent changes in targeted aspects of higher-level cognition and psychological well-being. There is an urgent need for well-controlled studies in humans and preclinical models on THC as a developmental neurotoxicant. Until more information is available, pregnant women should not assume that using cannabis during pregnancy is safe.
Keywords: cannabis, exposure, pregnancy, pharmacokinetics, child development
1. Introduction
The history of cannabis use and its impact on human health and society is complicated and rapidly changing (National Academies Press, 2017). Throughout the world, this flowering plant remains the most commonly used illicit drug and there is a strong shift towards the legalization of its medical and recreational use (Azofeifa et al., 2016). According to the 2015 National Survey on Drug Use and Health, 22.2 million Americans currently use cannabis and in the last twelve months, 2.6 million individuals aged 12 or older tried cannabis for the first time (Center for Behavioral Health Statistics and Quality, 2016; Lipari et al., 2015). This sobering statistic translates into approximately 7,100 new users each day. Attitudes about cannabis use are changing and this is particularly apparent in adolescents and young adults. With significant profits at stake, the legal cannabis market has implemented selective growing methods to boost psychoactive potency. Over the last two decades, the average THC content of cannabis (potency) has increased from approximately 4% to 12% (ElSohly et al., 2016), but levels as high as 30% have recently been documented in legal cannabis grown for recreational use (American Chemical Society, 2015). The cultivation of cannabis is evolving and dramatic increases in potency make it difficult to understand the health risks that may be associated with contemporary use, particularly for sensitive subgroups within the general population.
The increased availability and quality of cannabis, paired with relaxed attitudes about use in younger populations, will likely result in a rise of cannabis use in women of childbearing age. In fact, data collected from 2002–2014 in the U.S. indicate that 7.5% of pregnant women between 18 and 25 years of age use cannabis, while the rate of use in all pregnant women is approximately 4% (Brown et al., 2016). This statistic places cannabis firmly in the bull’s-eye of public health concerns and suggests that thousands, if not millions, of infants will be prenatally exposed to this chemically-complex compound over the coming decades. Women who become pregnant may continue to use cannabis for a variety of reasons. For example, a survey of women in Vancouver, Canada found that up to 77% of medicinal cannabis use during pregnancy was related to nausea; over 50% of respondents also reported cannabis use to treat a lack of appetite, general pain, insomnia, anxiety, depression and fatigue (Westfall et al., 2009). Despite knowledge of potential fetal health risks, cannabis use in pregnant women is becoming more commonplace and the need for clear messaging on the safety of use during pregnancy is urgently needed (Mark et al., 2017).
Given the possibility of increased use in pregnant women and the fact that cannabis is being widely investigated as a novel treatment for a variety of diseases, including epilepsy, multiple sclerosis and cancer, it is imperative that significant efforts be immediately dedicated to evaluating the potential consequences of exposure for the fetus. For example, the oromucosal spray (Sativex) has been approved in Europe to treat spasticity due to multiple sclerosis. While cannabis use during pregnancy has been studied in humans and several animal model systems, defining the risk of fetal cannabis exposure has been complicated by independent factors such as concurrent maternal use of drugs with their own toxicity profile and an absence of quantitative markers of cannabis exposure. Studies in human and preclinical model systems are needed to generate mechanistic data on the maternal-fetal kinetics and toxicity of cannabis that can be interpreted within the context of fetal pharmacology and developmental psychobiology. The consequences of prenatal cannabis exposure will not be elucidated without the methodological control of confounding factors such as tobacco/alcohol use and the quantitative measurements of exposure to generate critical dose-response information. This review was written to bridge our current understanding of 1) THC pharmacokinetics in adults with a focus on pregnancy 2) the consequences of fetal exposure at the molecular and cellular levels and 3) the effects of prenatal exposure on child neurodevelopmental outcomes from birth through adolescence. The marriage of pharmacokinetics with neurodevelopmental data provides an interdisciplinary framework to generate data-driven messages about fetal risk and highlight directions for future research objectives.
2. What is Cannabis?
Cannabis is a dioecious plant that grows wild in many tropical parts of the world. It is one of the world’s oldest crops and the history of cannabis dates back about 12,000 years (Warf, 2014). Cannabis was widely used in ancient China and records of medical applications appeared about 4,000 years ago, originally in relation to its use as a surgical anesthetic. Plant-derived cannabinoids (CB) are referred to as phyto-cannabinoids (phyto-CB) and THC is the most famous, representing the primary psychoactive ingredient produced by the cannabis plant (see Figure 1). Specific strains of cannabis may also produce high levels of a second phyto-CB, cannabidiol (CBD), often referred to as non-psychoactive. Although not associated with cannabis-induced euphoria or intoxication (Grotenhermen et al., 2017), CBD exposure is psychoactive and affects several brain functions and behaviors, including neuronal activity, seizure incidence and social interactions (Renard et al., 2017a; Todd and Arnold, 2016). Accordingly, CBD has been linked to influencing a wide range of clinical outcomes such as epilepsy and neuropsychiatric disorders. Additional phyto-CBs that exhibit a certain level of bioactivity include cannabinol, cannabigerol, and cannabichromine (Rosenthaler et al., 2014; Turner et al., 2017). These compounds are synthesized by a family of enzymes expressed by the cannabis plant and their biological activity and mechanism of action have not yet been studied in great detail. Thus, the cannabis plant produces a family of structurally related compounds, the phyto-CB, that produce a wide array of biological effects, many of which remain to be studied.
Figure 1:
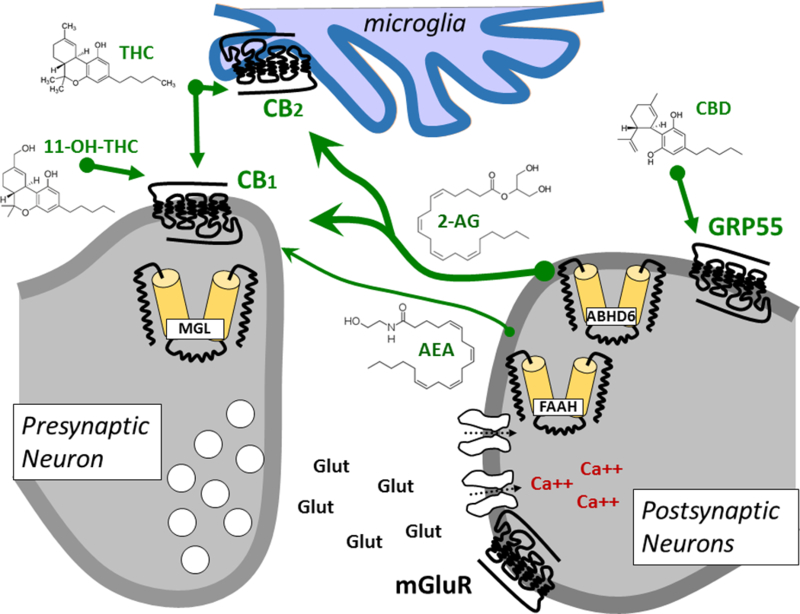
Endogenous signaling system is used by multiple cell types in brain and periphery. It encompasses cannabinoid receptors CB1 and CB2 (primarily expressed by neurons and immune cells microglia, respectively), the two endocannabinoids anandamide (arachidonolyl ethanolamine, AEA) and 2-AG (2-arachidonoyl glycerol) and the enzymes that produce them (not shown) and inactivate them, namely fatty acid amide hydrolase (FAAH), monoacylglycerol lipase (MGL), α/β-hydrolase domain 6 (ABHD6) and cyclooxygenase 2 (COX2, not shown). Additional molecular components are GPR55 that is modulated by cannabidiol (CBD).
3. Phyto-cannabinoids, Cannabinoid Receptors and Endocannabinoid Signaling
The two main phyto-CBs, THC and CBD, and several of their metabolites (e.g. 11-hydroxy- Δ9-THC (11-OH-THC, Figure 1)) bind and differentially activate cannabinoid receptors (Devane et al., 1988). Cannabinoid receptors 1 (CB1R) are G protein-coupled receptors (GPCR) abundantly expressed in the central nervous system (CNS) by many neuronal and glial cell types (Stella, 2010). A key function of CB1R in mature neurons is to modulate the release of neurotransmitters such as glutamate and GABA (Bloomfield et al., 2016; Katona and Freund, 2008, 2012). Central to this review, activation of CB1R during neuronal development influences cell proliferation, migration, and differentiation through control of the expression of key factors, including brain-derived nerve factor (Calvigioni et al., 2014; de Salas-Quiroga et al., 2015; Marsicano et al., 2003). CB1R are also expressed by cells in the periphery, including in reproductive tissues (Pertwee et al., 2010). Thus, most of the psychoactive responses produced by THC and 11-OH-THC exposure are mediated through CB1R expressed by a complex network of neuronal and glial cells in the CNS but circulating phyto-CBs and their metabolites also exhibit significant activity on both the CNS and peripheral tissues.
Cannabinoid receptors 2 (CB2R) are closely-related GPCRs of CB1R and are expressed by hematopoietic cells, as well as by select neurons and cells in the periphery, including cells in reproductive tissues (Pertwee et al., 2010). CB2R also couple to Gi/o proteins and their activation modulates similar signaling pathways and cell functions as CB1R (Pertwee, 2008). Accordingly, both CB1R and CB2R mediate the biological activity of different phyto-CBs, resulting in pronounced changes in several neural and immune functions.
Cannabinoid receptor activity is regulated by endocannabinoids (eCB), anandamide and 2-arachidonoyl glycerol (2-AG), two signaling lipids that are produced and inactivated by distinct lipases and hydrolases, respectively (Castillo et al., 2012; Piomelli, 2003). Increased cannabinoid receptor activation allows for rapid modulation of neuronal functions by modifying synaptic circuits (Katona and Freund, 2008). eCBs are produced on-demand by many different cell types in response to increased activity (typically associated with increases in intracellular calcium) via lipase activation that releases the eCBs from their membrane precursors (Di Marzo et al., 1994; Stella et al., 1997; Stella and Piomelli, 2001). Biological inactivation of eCBs occurs in two steps: a rapid uptake into cells via active transport, followed by FAAH-mediated hydrolysis for anandamide (Cravatt et al., 2001; Fu et al., 2011; McKinney and Cravatt, 2005) and hydrolysis or chemical modification of 2-AG by mono acyl glycerol lipase (MGL), α/β-hydrolase domain 6 (ABHD6) and cyclooxygenase 2 (COX2) (see Figure 1) (Dinh et al., 2002; 2004; Karlsson et al., 1997; Nomura et al., 2011; Tornqvist and Belfrage, 1976). Thus, members of the eCB signaling system include CB1R and CB2R, anandamide and 2-AG, and enzymes that produce and inactivate these two main eCBs. Note that both anandamide and 2-AG modulate the activity of additional targets that play fundamental roles in cell function involved in development and regulation of homeostasis and metabolic pathways. For example, anandamide is a low-efficacy agonist of TRPV1 and GPR55, and high concentrations of 2-AG activate peroxisome proliferator-activated receptor-α (PPARα) and PPARγ (Abood et al., 2012; Pertwee et al., 2010). Thus, eCB hydrolyzing enzymes represent cardinal molecular hubs within lipid signaling networks that control the levels and action of eCBs on their various targets (Marrs and Stella, 2007; Stella, 2012).
While THC remains the most famous phyto-CB thus far, the pharmacological activity of CBD is being studied in greater detail and was recently shown to modulate several non-CB1/CB2 receptors. For example, emerging in vitro evidence suggests that CBD might modulate CB1R through an allosteric site (Laprairie et al., 2015). A large body of work based on mouse genetics studies show that CBD modulates the activity of GPR55, a GPCR expressed by cells in both CNS and peripheral tissue (Ross, 2009; Sharir and Abood, 2010). As mentioned above, much less is known about the mechanism of action of cannabinol, cannabigerol, and cannabichromine.
Over 50 years of medicinal chemistry efforts have led to the development of several classes of synthetic cannabinoids that exhibit remarkable potency and selectivity profiles at the various targets mentioned above. For example, the indole-based compound, JWH-018, is a high potency selective agonist at CB1R (Atwood et al., 2010). In recent years, the human use of recreational cannabis-based products that contain indole-based cannabinoids (such as JWH-018 in K2 and Spice) has greatly increased and its impact on human health is only starting to be reported. Concerning reports indicate that JWH-018 is associated with a toxicity profile that is radically different from phyto-CBs and characterized by renal failure and seizures (Buser et al., 2014; Lapoint et al., 2011). Together, this evidence depicts a complex bioactivity profile linked to the use of cannabinoids that is due to their polypharmacological activity at multiple targets that regulate cardinal physiological functions.
4. eCB Signaling in Development
The eCB signaling system plays an overarching regulatory role during the initial stages of embryo development, implantation and ensuing prenatal development and differentiation. It undergoes a drastic switch in function from the prenatal determination of cell fate to the homeostatic regulation of metabolic pathways and transmission in the mature CNS. The functional role of CB1R and CB2R during this early stage of embryogenesis is not well understood but is likely linked to their ability to control cell proliferation and differentiation (Galve-Roperh et al., 2013).
During brain development, the proliferation and asymmetric division of neural progenitor cells, as well as their positioning and molecular diversification into neuronal and glial progenies, are all tightly regulated by both morphogenetic signaling molecules that contribute to building complex tissues. CB1R and CB2R are expressed by divergent pluripotent cells, including neuronal stem and progenitor cells where they differentially regulate cell proliferation and differentiation (Galve-Roperh et al., 2009; Maccarrone et al., 2014). For example, activation of CB1R expressed by neural stem cells isolated from embryos enhances differentiation into neurons without affecting astrocytes and oligodendrocytes, as evidenced by increased neurite outgrowth and expression in neuronal markers (de Salas-Quiroga et al., 2015). By contrast, activation of CB1R expressed by post-natal radial and neuronal stem cells controls differentiation in the adult brain by promoting astroglial differentiation of newly born cells (Aguado et al., 2005, 2006). Given that CB1R expressed by neural progenitor cells in the developing forebrain regulates the ratio of neurons to astroglial cells in areas such as the hippocampus and cerebral cortex, changes in CB1R during this critical period are likely to influence the connectivity of these brain regions (Berghuis et al., 2007; Maccarrone et al., 2014). Evidence suggests that focal eCB gradients are probably generated to control the direction of cell migration (Miller and Stella, 2008; Tortoriello et al., 2014). Accordingly, the downregulation of DAGLα and DAGLβ expression (enzymes involved in 2-AG release from membranes) following neuronal specification is likely to represent an essential step to increase the reliance of post-mitotic neurons on extracellular 2-AG produced by pyramidal cells in the cortical plate and thus function as positional cues (Tortoriello et al., 2014). Additional molecular mechanisms involving eCB signaling that regulate directed migration include the tropic action of growth factors. Indeed, specific receptor tyrosine kinases are transactivated following stimulation of CB1R, and conversely CB1R are sensitized by growth factors (e.g. BDNF) (Marsicano et al., 2003). In line with these studies, 2-AG signaling through CB1Rs represents an essential effector molecular step of neurotrophic factor signaling (De Chiara et al., 2010; De March et al., 2008; López-Gallardo et al., 2012). This body of work identified a novel physiological role of eCB signaling system in providing signaling cues involved in the regulation of neural stem and progenitor cell differentiation and ensuing function. These studies also highlight the importance of determining how prolonged activation of CB1R in neural progenitors by phyto-CBs intake impact newly born cells.
5. Impact of Cannabinoids on the Developing Brain
The developing brain undergoes substantial structural remodeling that makes it particularly vulnerable to the harmful effects of bioactive ingredients (Chambers et al., 2003; Crews et al., 2007). Such remodeling occurs in many brain areas involved in vital neuronal function, including sensory inputs and the control of body temperature, as well as higher-order cognitive processes such as learning, memory and decision making (Wise, 2004). CB1R signaling modulates long-range neuronal connectivity, including corticofugal connectivity (Katona and Freund, 2008; Tortoriello et al., 2014). Accordingly, THC administration to pregnant mice during a restricted time window interferes with subcerebral projection neuron generation, thereby altering corticospinal connectivity and producing long-lasting alterations in the fine motor performance of the adult offspring (de Salas-Quiroga et al., 2015).
Mechanistically, such impairments are reminiscent of those elicited by the genetic ablation of CB1R and accordingly regimented THC administration to pregnant mice leads to down-regulation of CB1R signaling through desensitization (Berghuis et al., 2007; Castelli et al., 2007; Keimpema et al., 2011; Vitalis et al., 2008). This impairment of long-range neuronal connectivity occurs for dorsal telencephalic glutamatergic neurons but not for forebrain GABAergic neurons (Mulder et al., 2008). Importantly, repeated CB1R activation during such sensitive developmental period of CNS development affects the expression and functionality of multiple additional receptors, including dopaminergic receptors that are critically involved in higher cognitive functions (Renard et al., 2017b). In mice, in utero exposure to THC leads to CB1R activation and neuronal rewiring through the degradation of the molecular effector, SCG10/statmin 2, known to regulate microtubule dynamics in axons (Tortoriello et al., 2014). A telling example is provided by results showing that CB1Rs activation on the axonal surface induces repulsive growth cone turning and eventual collapse in in vitro model systems (Harkany et al., 2008a; 2008b; Maccarrone et al., 2014). The molecular mechanism of CB1R-mediated cytoskeletal instability in growth cones involves select signaling pathways, including RHO-family GTPases, RAS, and PI3K–AKT–β-catenin signaling (Alpár et al., 2014; Díaz-Alonso et al., 2012; Maccarrone et al., 2014). Accordingly, THC exposure leads to ectopic formation of filipodia and alterations in axon morphology, together limiting the computational power of neuronal circuits involved in high cognitive function in affected offspring (Cristino and Di Marzo, 2014; Tortoriello et al., 2014).
An interesting cellular component of the impact of THC on brain development that has not been studied in detail is whether repeated activation of cannabinoid receptors expressed by glial cells will also lead to their down-regulation or desensitization and whether this response might affect normal brain maturation and result in persisting impairments. In accordance with the tripartite synapse hypothesis, which states an involvement of astrocytes in synaptic transmission, peri-synaptic astrocytes that express MAGL should form a barrier limiting 2-AG spread and action on its targets to 20–100 μm and not beyond its immediate site of action (Maccarrone et al., 2014; Metna-Laurent and Marsicano, 2015; Navarrete and Araque, 2010; Oliveira da Cruz et al., 2016; Stella, 2010). This MAGL expression pattern is likely to both limit axonal spread in the prospective internal capsule and help delineate migratory routes for CB1R-expressing neurons, such as exemplified by cortical interneurons (Alpár et al., 2014; Keimpema et al., 2010; Wu et al., 2010). Thus, pharmacological manipulation of eCB signaling and its highjack by phyto-CB during crucial periods of synaptogenesis and/ or postnatal pruning might precipitate or predispose an individual to neuropsychiatric disease-like phenotypes.
6. Pharmacokinetics of THC in Humans
There is strong evidence that THC pharmacology has considerable impacts on neuronal signaling and development and as such, it is plausible to hypothesize that THC exposures during pregnancy could lead to long-term changes in neuronal development. However, a critical component in understanding the potential consequences of cannabis consumption during pregnancy is the duration of exposure, the overall magnitude of exposure and the extent to which the fetus and fetal brain are exposed to THC after maternal cannabis consumption. While the pharmacokinetics (PK) of THC and its metabolites have been studied in adult humans following intravenous (iv), oral and inhalation administration, little is known about the changes in cannabis PK during pregnancy and the maternal-fetal transfer and fetal PK of THC. In addition, it is possible that the route of cannabis consumption (oral, inhalation, and different ways of smoking) will have an impact on the overall fetal toxicity. The absorption pathways between smoked and edible cannabis products are distinctly different. Following oral administration, THC absorption is typically greater than 90% and not affected by formulation (Parikh et al., 2016), but the bioavailability is limited to <20% (e.g. 10–20% in gelatin capsules) (Wall et al., 1983) and 6 ± 3% when ingested in a chocolate cookie (Ohlsson et al., 1980) due to significant liver first pass metabolism. In contrast, smoked cannabis is not subject to liver first pass metabolism. Loss of THC in side stream smoke and in the butt of the cigarette, as well as loss via pyrolysis, result in low absorption of THC from smoking (Grotenhermen, 2003) and overall highly variable bioavailability of 2–56% (mean 18 ± 6%) (Huestis, 2007; Ohlsson et al., 1980).
An important difference between oral and smoked cannabis is also the rate of THC absorption. Following smoking, THC is rapidly absorbed and its peak concentrations (Cmax) are reached within minutes (Kauert et al., 2007; Ohlsson et al., 1980). In comparison, absorption from oral capsules is considerably slower and maximum THC concentrations are reached 1–3 hrs after dosing (Ahmed et al., 2015; Ohlsson et al., 1980; Schwilke et al., 2009). As expected from the faster rate of absorption from smoked cannabis, the average Cmax values for THC following smoking are somewhat higher than those observed after oral consumption if similar THC content is consumed. The average peak concentrations of THC in serum reached after smoking cannabis cigarettes containing 18.2 mg (0.25 mg/kg body weight) and 36.5 mg (0.5 mg/kg body weight) of THC were 48 μg/L and 79 μg/L (Kauert et al., 2007). In a study in occasional users who smoked a cannabis cigarette with 4% THC (20 mg dose) with tobacco, the average Cmax was 25.8 ± 42.9 μg/L with an average time to maximum concentration of 0.2 hrs. (Marsot et al., 2016). The range of individual peak concentrations (1.6–160 μg/L) emphasizes the large inter-individual variability in THC exposures. After oral dosing in daily cannabis users, the Cmax of THC was 16.5 μg/L after 20 mg THC orally, (Schwilke et al., 2009). In elderly patients the Cmax was , 0.41 μg/L after 0.75 mg oral dose and 1 μg/L after a 1.5 mg oral dose (Ahmed et al., 2015). These differences in absorption kinetics and first pass metabolism may ultimately contribute to different toxicity profiles for oral and smoked cannabis, especially if toxicity is related to peak concentrations and/or if metabolites formed in the liver contribute to fetal pharmacology.
The site of action of THC’s psychoactive effects is in the CNS and hence distribution to the site of action is critical for effects. Indeed, THC distributes extensively into tissues with a steady state volume of distribution of 523–626 L (7.5–8.9 L/kg) (Hunt and Jones, 1980; Wall et al., 1983). The distribution of THC into the brain is, however, delayed and the initial distribution volume after iv bolus is estimated between 2.6 L (0.04 L/kg) (Hunt and Jones, 1980) and 22.8 L (0.33 L/kg) (Ohlsson et al., 1980). Following iv administration, the maximum rated psychological “high” is reached at 15 minutes after the dose (Ohlsson et al., 1980). This corresponds to the time required to reach distribution equilibrium (minutes to an hour) (Hunt and Jones, 1980; Ohlsson et al., 1980) at the site of action and results in a hysteresis loop that describes the relationship between THC concentrations in plasma and the observed pharmacological effect with time (see also Grotenhermen, 2003). After peak effects are reached, the effects decline slowly due to the long terminal half-life between 20 and 57 hours (Hunt and Jones, 1980; Lemberger et al., 1971) suggesting prolonged exposures and pharmacological effects even after single use.
In humans, THC is extensively metabolized (Fig. 2) with a systemic clearance of 12–36 L/h (Hunt and Jones, 1980; Wall et al., 1983). The clearance is somewhat restricted by plasma protein binding (THC unbound fraction of 3%). The majority of THC clearance in humans is thought to be hepatic, although metabolism of THC exists in the gut mucosa, lung and heart, at least in preclinical species (Grotenhermen, 2003). After iv dosing in humans, <0.05% of the THC dose is recovered as unchanged Δ9-THC in urine or feces as the vast majority of THC is eliminated as metabolites either in urine (20%) or via biliary secretion of the metabolites in feces (25–40%) (Hunt and Jones, 1980; Lemberger et al., 1971; Wall et al., 1983). Over 80 metabolites of THC have been identified to date, but only some of these metabolites are quantitatively important and pharmacologically active, including 11-OH-THC, 11-nor- Δ9-THC-9-carboxylic acid (11-nor-THC-COOH) and 8-OH- Δ9-THC (see Figure 2) (Dinis-Oliveira, 2016; Grotenhermen, 2003).11-OH-Δ9-THC is even more pharmacologically active than THC (Christensen et al., 1971) but the activity of 8-OH-THC is not known. In vitro and in vivo data suggest that 11-OH-THC is formed predominantly by CYP2C9 while 8-OH-THC is mainly formed by CYP3A4 (Bland et al., 2005; Bornheim et al., 1992; Stott et al., 2013; Watanabe et al., 2007). The 11-nor-THC-COOH is formed from 11-OH-THC by microsomal alcohol dehydrogenase enzymes (Narimatsu et al., 1988). Both 11-OH-THC and 11-nor-THC-COOH undergo glucuronidation by UGT1A9 and UGT1A10 (11-OH-THC) and UGT1A1 and UGT1A3 (11-nor-THC-COOH) (Mazur et al., 2009). As the 11-nor-THC-COOH, together with its acyl glucuronide conjugate, account for the majority (30–65%) of THC elimination (Glaz-Sandberg et al., 2007), altered CYP and UGT activity, as occurs during pregnancy, may significantly alter THC and metabolite exposures and pharmacology. Of note, the route of THC consumption will also alter the exposures to the metabolites. Following iv administration of THC or smoking of cannabis, 11-OH-THC plasma concentrations are much lower than those of THC (Grotenhermen, 2003; Wall et al., 1983) declining with the same half-life as THC (25–33 hr). By contrast following oral THC administration, 11-OH-THC plasma concentrations can exceed those of THC but decline with a shorter half-life (12 hrs) than THC suggesting that most of 11-OH-THC is formed during first pass in the liver (Grotenhermen, 2003; Wall et al., 1983). Because of the low clearance of 11-nor-THC-COOH (5.5 L/h), it is the main circulating compound following any route of THC administration (Glaz-Sandberg et al., 2007). Overall, these data show that the route of consumption of THC may result in distinctly different exposures and pharmacology. Further research is needed to determine the role of peak concentrations, duration of exposures and role of metabolites in THC pharmacology and toxicity.
Figure 2:

Simplified metabolic pathway of ∆9-tetrahydrocannabinol (THC), or ∆1-tetrahydrocannabinol, under the monoterpenoid naming system, in humans. Both 11-hydroxy- ∆9-tetrahydrocannabinol (11-OH-THC), or 7-hydroxy- ∆1-tetrahydrocannabinol, and 8-hydroxy- ∆9-tetrahydrocannabinol (8-OH-THC), or 6-hydroxy- ∆1-tetrahydrocannabinol, are active metabolites.
6.1. THC Pharmacokinetics in Preclinical Species
The disposition of THC has been studied in several animal model systems, including mice, rats, rabbits, dogs and nonhuman primates (Freudenthal et al., 1972; Garrett and Hunt, 1977; Ginsburg et al., 2014; Leuschner et al., 1986). These species generally reproduce the distribution kinetics observed in humans characterized by a relatively rapid distribution phase and long terminal half-life predominantly driven by the extensive distribution of THC into adipose tissue. Unfortunately, due to low analytical sensitivity in the early studies, and oftentimes insufficient duration of sample collection, the terminal half-lives of THC have not yet been thoroughly characterized in most species. A terminal half-life of 8 days was reported in dogs based on radiolabeled THC (Garrett and Hunt, 1977) while terminal half-lives ranging from 41 hrs to 76 hrs were documented in rabbits (Leuschner et al., 1986). In rodents, the terminal half-life of petroleum ether extractable THC related radioactivity was 21 hrs (Klausner and Dingell, 1971). It is likely that these terminal half-lives include both THC and its non-polar metabolites and hence should not be compared to humans.
In contrast to human studies, information on THC metabolite disposition in preclinical species is sparse. The formation of 11-OH-THC is largely conserved across species as a major metabolite and consistent with data in humans, rat CYP2C enzymes (CYP2C11 in males and CYP2C6 in females) predominantly form the 11-OH-THC (Narimatsu et al., 1990, 1992). THC is almost exclusively eliminated via metabolism in rats, and similar to what is observed in humans, only 10% of the dose is excreted in urine. The majority of the iv dose of THC is found in feces as metabolites of THC (Klausner and Dingell, 1971). The role of biliary secretion in rats was confirmed using the isolated perfused rat liver model in which 90% of the perfused THC radioactivity was secreted into the bile. Studies in rats also suggested that enterohepatic circulation of THC metabolites may occur (Klausner and Dingell, 1971). Whether the metabolite exposures in rats reflect those observed in humans is not known. In nonhuman primates (rhesus macaque monkeys) exposed to 0.1 mg/kg iv dose, the plasma disposition of THC is similar to humans and the human metabolites 11-OH-THC and 11-nor-THC-COOH are also detected. 11-nor-THC-COOH is present in the monkey plasma at similar concentrations as 11-OH-THC, but unlike in humans, the concentrations of 11-nor-THC-COOH are lower than THC in monkey plasma. The difference could be due to either lower efficiency of 11-nor-THC-COOH formation in the monkey compared to humans or higher clearance of the 11-nor-THC-COOH in the monkey. These minor species differences are, however, unlikely to affect the validity of the model species as 11-nor-THC-COOH is an inactive metabolite. Together, these studies show that we still have only a basic understanding of THC disposition and metabolism in both human and relevant preclinical model systems and further studies are urgently needed in this area.
6.2. THC Pharmacokinetics during Pregnancy and Maternal-Fetal Distribution
While THC and its metabolites can be reliably detected in meconium and infant urine as an indicator of maternal cannabis use in humans, very few studies have been conducted to evaluate maternal-fetal disposition of THC and its metabolites following maternal smoking or oral consumption of THC products. In addition, infant exposure to THC via passive smoking or via breastmilk has not been systematically assessed. THC is highly lipophilic and as such readily crosses the placenta but fetal exposure may be limited to some degree by active transport in the placenta. Observational studies in a small number of humans have demonstrated that THC distributes into the fetal compartment and readily crosses the placenta, although the overall extent of fetal distribution in humans has not been evaluated (Blackard and Tennes, 1984; Boskovic et al., 2001). Interestingly, based on meconium and hair samples of monozygotic and dizygotic twins, it was shown that THC levels in dizygotic twins can be discrepant (i.e. observed in one fetus but not in the other). This result suggests that placental and possibly fetal factors are the predominant factors controlling fetal THC exposures at constant maternal exposure.
While some epidemiological studies have incorporated maternal measures of exposure in the design of their study, none have provided data on the relationship between maternal and fetal measurements of cannabis exposure. A study of Spanish women undergoing voluntary pregnancy terminations has provided important, initial information on this subject (Falcon et al., 2012). In this study, samples of maternal hair, placenta and fetal tissue were collected and THC and THC-COOH were detected in maternal hair. In the placenta, THC tissue concentrations averaged 196.8±110.1 ng/g (range 101.1–432.8), while the mean THC level in fetal remains was 118.5±97.9 ng/g (range from 3.9–281.7). These results document the transplacental passage of THC during the embryonic and fetal period but are not sufficient to establish a quantitative relationship between maternal cannabis exposure and fetal cannabis levels.
To date, no studies on the disposition of THC in humans during pregnancy have been conducted. Based on the significant contribution of CYP2C9 and CYP3A4 to THC metabolism and their known increase in expression during pregnancy (Isoherranen and Thummel, 2013), one could suggest up to 2-fold increase in THC clearance during pregnancy and potentially a decrease in THC exposure for a given consumption level. However, one cannot easily predict that maternal or fetal exposures to the metabolites of THC are also increased during pregnancy as a result of enhanced CYP activity, as we still do not know how pregnancy may change the metabolite glucuronidation and clearance. However, considering the high intra-individual variability in THC disposition and the ease of altering consumption (dose), we can postulate that pregnancy-mediated changes in maternal THC disposition are unlikely to be of clinical significance.
In preclinical model systems of human developmental exposure, fetal concentrations are generally lower than those observed in the mother and may be dependent on route of administration. In pregnant mice, fetal liver concentrations of THC and its metabolites were similar to maternal liver based on autoradiograms that were taken 2 hours after iv dosing of THC (Freudenthal et al., 1972). When THC was administered orally, a similar analysis yielded higher concentrations in the fetus than maternal liver. However, presence of THC metabolites may confound these measurements. In contrast, in pregnant female dogs administered 0.5 mg/kg THC iv, maternal brain concentrations of THC related radioactivity were 3-fold higher than those observed in the fetal brains 30 min after dosing, although overall distribution of THC to maternal and fetal brain regions was similar (Martin et al., 1977). In pregnant rats given chronic oral doses of THC at 15 mg/kg or 50 mg/kg throughout gestation, fetal THC concentrations reached only 9–13% of the maternal concentrations and fetal exposure to THC increased proportionately to the dose (Hutchings et al., 1989). However, due to the distribution kinetics of THC to the fetus and the potential slow elimination from the fetus, these single point concentrations may not reflect the overall fetal exposure. In pregnant ewes (~130 days gestation) exposed via inhalation to cannabis smoke containing 3.2% THC, maternal THC concentrations peaked rapidly at 10 min after inhalation (Abrams et al., 1984) while the concentrations in the fetal blood peaked at 90 min. Consistent with the rodent data, the fetal concentrations remained lower than those in the mothers during the 24 hr study indicating potential placental efflux transport of THC. In 3 pregnant rhesus macaque monkeys administered iv doses of 0.3 mg/kg THC, distribution to the fetal compartment was faster than that observed in the ewes (Bailey et al., 1987) and the fetal plasma THC concentrations peaked at 15 min after iv administration. Consistent with all other species, the fetal concentrations reached only 28% of the concentrations observed in maternal plasma at this sampling time point. Alarmingly, the half-life of THC in the fetal compartment appears longer than that in the dam with fetal THC concentrations reaching those of maternal plasma at 180 min after dosing (Bailey et al., 1987). Unfortunately, this study does not provide data on longer periods of sampling which prevents full pharmacokinetic analysis of this pattern. However, this study measured fetal tissue THC concentrations and found that the thymus, adrenals and bile had the highest THC concentrations at 3 hrs after THC dosing to the pregnant dam (about 5 times those observed in fetal plasma) (Bailey et al., 1987). In other tissue samples, such as fetal brain, liver and kidney, THC concentrations were either similar or slightly higher than those observed in fetal plasma. Together, these results indicate a broad distribution of THC to fetal tissues. The studies in rhesus macaques also show that 11-nor-THC-COOH is not detectable in the fetal plasma and that the maternal concentrations of 11-nor-THC-COOH were lower than those of THC up to 180 min post-exposure.
These preclinical animal studies show that THC concentrations in the fetus appear lower than in the mother and thus suggest that active transport in the placenta limits fetal exposure to THC. However, most of the studies are limited to single time points and further detailed studies are needed to carefully characterize the rate and extent of THC distribution into the fetus. For example, it is critical to understand whether peak concentrations in the fetal brain are similar to those observed in the mother and whether the exposure in the fetus is prolonged in comparison to the mother even after single doses of cannabis. In addition, mechanistic studies are needed to identify placental transporters responsible for THC transport to support our understanding of potential interindividual variability in THC distribution into the fetus and to identify individuals at high-risk for fetal THC toxicities. Variability in maternal-fetal THC disposition and transport may be partially responsible for inconsistent reports of neurodevelopmental effects from studies of THC exposure during pregnancy, outlined in detail below.
7. Impact of Prenatal Cannabis Exposure on Neurodevelopmental Outcomes in Humans
There are numerous publications focused on the reproductive and developmental effects of cannabis. Excellent review articles describing a range of reproductive effects in both males and females have been published recently and will not be reviewed here (Plessis et al., 2015; Brents, 2016).
Early chemical or drug exposure can result in subtle injuries to the developing CNS that are expressed as changes in postnatal development (Bellinger et al., 2016; Grant and Rice, 2008). Over the past four decades, a number of prospective studies have found changes in the developmental trajectory of children prenatally exposed to cannabis. The demographic characteristics of subjects in these studies as well as exposure and outcome measures are summarized in Table 1. Most studies were conducted in urban environments with economically-disadvantaged families. The most common metric to estimate use of cannabis during pregnancy is maternal self-report on frequency of use (e.g. # joints/day), while fewer studies have collected biological samples to more accurately estimate real-world levels of exposure. The wide variation in cannabis potency and individual smoking habits make the interpretation of the developmental literature challenging but a careful review reveals certain common themes surrounding the fetal risks associated with prenatal exposure. Much of what is known about maternal cannabis use and child development is based on data collected from 3 longitudinal birth cohort studies; the Ottawa Prenatal Prospective Study, the Maternal Health Practices and Child Development Project and the Generation R Study (McLemore and Richardson, 2016), but other longitudinal and cross-sectional studies focused on the developmental neurotoxicity of this compound have also made important contributions. In this section of the review, we opted to separate the developmental outcomes of cannabis exposure into four domains: physical growth/maturation, neonatal behaviors, cognition and psychological health/adaptive behavior.
Table 1:
↑ indicates significant increase in outcome measure. ↓ indicates significant decrease in outcome measure. ↔ indicates no significant change in outcome measure.
| Study Name | Location | # of Children |
Maternal Demographics. |
Exposure | Growth | Behavior | Cognition | Physical Health /Adaptive Behavior |
|---|---|---|---|---|---|---|---|---|
| Ottawa Prenatal Prospective Study (OPPS) Fried, 1980 Fried et al., 1983, 1987, 1992a, 1992b, 1997, 1998, 1999, 2001, 2003 Fried & O’Connell, 1987 Fried & Watkinson, 1988, 1990, 2001 Fried & Smith, 2001 Smith et al., 2006 Smith et al., 2016 |
Ottawa, CA | 190 | Middle SES Predominantly Caucasian/white |
irregular (≤1) moderate (2–5) heavy (> 5) |
↓ birthweight (neonate) ↔ growth (13–18 y) ↔ pubertal milestones (13–18 y) |
↓stimuli habituation (newborn) ↑ tremors (newborns) ↑ startle (newborn) |
↓verbal/memory processing (3–4, 9–12, 13–18 y) ↔ verbal/memory processing (5–6 y) ↔ reading/language (9–12 y) ↔ IQ (9–12,13–18 y) ↓ attention (9–12 y) ↓visual analysis /hypothesis testing (9–12, 13–18 y) ↓impulse control (9–12 y) ↑ activity in frontal, occipital, parahippocampal gyri and cerebellum (8–22 y)* ↓ activity in right anferior and middle frontal gyri (8–22 y)* |
|
| Maternal Health Practices and Child Development Project (MHPCD) Scher et al., 1988 Richardson et al., 1989, 1995, 2002 Dahl et al., 1995 Day et al., 1994, 2011 Goldschmidt et al., 2000, 2004, 2008, 2016 Gray et al; 2005 Leech et al.; 2006 |
Pittsburgh, PA, USA | 763 | Lower SES Half sample Caucasian/white, half black Most single |
light (0–2.8) moderate (2.8- 7.0) heavy use (>7.0) |
↔ growth (5–6 y) | ↑ nighttime arousal (newborns, 3 y) ↓time sleeping (newborn, 3 y) ↔ reflexes (newborn) |
↓ cognitive performance /IQ(1–2, 3, 5–6 y) ↓ attention (3–4 y) ↓ verbal-memory processing (3–4 y) |
↑ depression symptoms (6–10 y) ↑ anxiety symptoms (6–10 y) ↔ drug use (15–18 y) ↓impulse control (6–10 y) ↑ hyperactivity (6–10 y) ↑ delinquent behavior (6–10, 11–16 y) |
| Generation R Jaddoe et al., 2008, 2010 El Marroun et al., 2009, 2011, 2015 |
Rotterdam, NL | 4,000 | Middle-upper SES Predominantly Dutch/ European; multi-ethnic Most married |
Not defined | ↓ birthweight (newborn) ↓ head circumference (fetal) |
↓ attention in females (18 mths) ↓ thickness of frontal cortex (6 y)* |
↑ aggressive behavior in females (18 mths) |
|
| Astley and Little, 1990 | Seattle, WA, USA | 136 | Middle SES Predominantly Caucasian/white Most married |
# days used during pregnancy and lactation |
↔ early cognition (1–2 y) | |||
| Avon Longitudinal Study of Parents and Children Fergusson et al., 2002 Zammit et al., 2009 Mathews et al., 2014 |
Avon, UK | 12,129 | Middle to slightly above average SES Predominantly Caucasian/white Most married |
<1x /wk ≥1x/wk |
↓ birthweight (newborn) ↓ birth length (newborn) |
↑ Tourette syndrome (12 y) ↔ psychosis-like symptoms (12 y) |
||
| Hurd et al., 2005 | Pittsburgh, PA, USA | 139 | Low-Ave SES Mostly nonwhite |
Light (0–2.8) - heavy use(>7.0) |
↓ birthweight (newborn) ↓ foot length (newborn) |
|||
| Noland et al., 2005 | Cleveland, OH, USA | 330 | Low SES Predominantly African American Mostly single |
Mean = 2.8 | ↓ attention (4 y) | |||
| van Gelder et al., 2010 | United States | 5,871 | Lower SES Predominantly Caucasian/white and nonwhite Hispanic |
Not defined | ↔ birthweight (newborn) ↔ gestational age (newborn) |
|||
| Frank et al., 2014 | Boston, MA, USA | 157 | Lower-Middle SES Predominantly non- Hispanic black |
Light and heavy users |
↔ early cannabis use (18 y) ↔ substance abuse disorders (18 y) |
|||
| Saurel-Cubizolles et al., 2014 | France | 13,454 | Middle SES Predominantly Caucasian/white |
Less than once a month vs once a month or more |
↓ birthweight (newborn) | |||
| Varner et al., 2014 | United States | 3,224 | Middle SES | Positive toxicology assay |
↑ stillbirths (newborn) | |||
| Conner et al., 2015 | St Louis, MO, USA | 8,106 | Lower SES Predominantly non- Hispanic black |
At least once during pregnancy vs did not use |
↔ birthweight (newborn) | ↔ APGAR score (newborn) | ||
| Warshak et al., 2015 | Cincinnati, OH, USA | 6,488 | Predominantly non- Hispanic black |
Not defined | ↓ birthweight (newborn) | ↑ NICU admissions (newborn) |
As mentioned above, a frequently used approach to the measurement of cannabis exposure is maternal self-report of use during pregnancy. This approach, while commonly employed, may not provide an accurate evaluation of in utero exposure due to underreporting of drug use by pregnant women (Garg et al., 2016). Reluctance to report cannabis use is commonly linked to feelings of guilt, the fear of being arrested and concern over repercussions in child custody cases. This makes it difficult, if not impossible, to characterize biologically-based dose-response relationships for cannabis-related developmental effects. Few studies have collected biological samples to augment maternal self-report estimates of use and for those that have, the information has been primarily used to determine incidence of drug exposure, not dose-response relationships. The laboratory analysis of cannabis exposure from biological mediums most often relies on samples of maternal urine and blood (Musshoff and Madea, 2006), but more recently, maternal hair, placenta and fetal meconium have been utilized (Falcon et al., 2012). Due to the longer half-life of THC metabolites when compared to THC, modeling of urinary and blood metabolite to parent drug ratios would allow development of quantitative markers of timing and magnitude of THC exposures. Development of such measures is highly recommended to improve the understanding of cannabis exposures during pregnancy.
7.1. Physical Growth and Maturation
In utero exposure to cannabis does not typically result in congenital birth defects (Warner et al., 2014, Linn et al, 1983, van Gelder et al. 2009) and there is no phenotypic signature of this compound in newborns. Effects on physical growth at birth and during the neonatal period have been reported in some studies (see below) but not others (Bada et al., 2006; Conner et al., 2015; van Gelder et al., 2010). In a study of maternal cannabis use and effects on fetal growth, decrements in birthweight and neonatal head circumference were associated with prenatal exposure but only when data were restricted to women with a positive urine assay for cannabis (Zuckerman et al., 1989a). When maternal self-report data were used for analysis, no significant relationship between cannabis exposure and early growth was detected. In a retrospective records study, maternal use of cannabis, as determined by either self-report or a positive urine assay for THC, was associated with decrements in fetal growth (e.g. small-for-gestational age) and an increase of 54% in neonatal intensive care unit admissions (Warshak et al., 2015). This investigation is particularly noteworthy as women who used tobacco during pregnancy were not included in the study population. Because subjects were classified only as cannabis users or nonusers, it is not possible to glean information about dose-response relationships for these effects. Exposure-related changes in early growth were also detected in a study where fetal meconium, maternal self-report and urine were collected from women undergoing voluntary saline-induced abortions (Hurd et al., 2005). The anthropometric examination of fetuses of varying gestational ages revealed a significant exposure-related decrement in fetal foot length, a standard marker of physical maturation at birth. This effect was observed as early as mid-gestation (weeks 17–22) and statistical trends in the data showed that offspring of women who were heavy users of cannabis during early pregnancy (~1 or more joints/day) were most likely to be affected. In contrast, a study of over 8,000 women that relied on either self-report or a positive THC urine screen to determine fetal exposure found no relationship between maternal cannabis use during pregnancy and a composite score of neonatal morbidity composed of birthweight, APGAR score (health status of newborn immediately after birth ) and umbilical artery pH data (Conner et al., 2015).
Changes in physical growth and development have also been documented in studies relying solely on measures of maternal self-report to estimate cannabis use. Ultrasound images collected from thousands of pregnant women demonstrated that maternal cannabis use was not related to adverse neonatal outcomes, such as perinatal death, but was associated with small but detectable reductions in birthweight and fetal head circumference (El Marroun et al., 2009). Changes in weight and growth trajectories were primarily observed in infants whose mothers reported using cannabis on a weekly or daily basis. In separate studies using maternal self-report, the use of cannabis during pregnancy has been linked to an increased risk of having a small-for-gestational age infant (Saurel-Cubizolles et al., 2014) and reductions in birthweight (Fergusson et al., 2002).
Our reading of the literature on prenatal cannabis exposure and early physical development indicates equivocal results. Despite some research supporting a significant relationship between cannabis use during pregnancy and decrements in fetal growth, there is no strong evidence that cannabis has a long-term negative impact on physical maturation (Fried and O’Connell, 1987). Longitudinal tracking of children with a history of prenatal cannabis exposure revealed normal physical growth trajectories at the time of school entrance (age 5–6) and during adolescence (Day et al., 1994; Fried et al., 1999) and key pubertal milestones such as age at menstruation in females and shaving in males were also not affected (Fried et al., 1999, 2001).
7.2. Neonatal Behaviors
Neurobehavioral effects of in utero cannabis exposure have been detected in some studies during the newborn period. Infants born to moderate and heavy users of cannabis during pregnancy (≥2 joints/week, maternal self-report) showed increased tremors/startles and poorer habituation to visual stimuli (Fried, 1980; Fried et al., 1987). The authors note that these behavioral findings are consistent with a mild narcotic withdrawal syndrome and may portend exposure-related changes in CNS functioning. Some women who reduced or quit using cannabis during pregnancy showed a reduced risk of delivering an infant with clinical symptomology. Gestational cannabis exposure has also been associated with changes in postnatal cortical activity. Specifically, a study of neonatal electroencephalography (EEG) sleep patterns found that in utero exposure to cannabis was associated with increased body movements and decreased time in a quiet sleep state (Sher et al., 1988). This effect was most widely observed in infants born to women who used cannabis on a daily basis. When children in this cohort reached 3 years of age, a similar pattern of EEG sleep disturbances was documented (Dahl et al., 1995). These results suggest that the neurophysiological mechanisms that control infant/toddler arousal and sleep cycling may be disrupted by cannabis use during pregnancy. This behavioral change in affected infants may reflect subtle chemical injury to the brain stem, particularly in neurons that comprise the raphe nuclei. The long-term significance of these effects, if any, is unknown.
7.3. Cognition
Learning and memory are perhaps the most consequential outcome measures in developmental cannabis research but studies are relatively few and findings are inconsistent. With few exceptions (e.g. Noland et al., 2005), the central limitation of studies investigating neurocognitive endpoints is their methodological reliance on maternal self-report of cannabis use during pregnancy to estimate fetal exposure. Several studies focused on early cognitive outcomes have reported that maternal cannabis use during pregnancy was not related to performance on infant tests of mental development (Astley and Little, 1990; Fried and Watkinson, 1988). Other studies however, have reported that heavy maternal use is associated with a significant decline in early cognitive performance (Richardson et al., 1995). The reduction in test scores for 9 month-old infants with the highest levels of maternal cannabis use (>1 joint per day) was a disquieting 10 points, providing some evidence for dose-related effects in early mental test performance. When these infants were re-evaluated at 19 months using the same exam, fetal THC exposure was no longer related to language and cognitive scores.
During the preschool period of development (3–4 years), results from child assessment studies found that prenatal cannabis exposure was related to adverse effects on sustained attention, short-term memory and verbal processing, although it is important to note that decrements in performance were frequently subtle and limited in scope (Day et al., 1994; Fried and Watkinson, 1990; Noland et al., 2005). At school age (5–6 years), one prospective, birth-cohort study found no evidence of an adverse effect of prenatal cannabis exposure on any cognitive outcome, including global intelligence quotient (IQ) scores (Fried et al., 1992a). Additional testing with these children did, however, reveal small deficits in sustained attention and increased levels of impulsivity and hyperactivity (Fried et al., 1992b). The number of lapses in attention (omission errors) during a vigilance task was greatest in children born to heavy users of cannabis during pregnancy (>6 joints/week). In contrast, a separate longitudinal study found that heavy maternal cannabis use during pregnancy (~1 or more joints/day) was associated with diminished scores on a standardized IQ test at age 6, including deficits in short-term memory processing, and the effects varied by trimester of exposure (Goldschmidt et al., 2008).
By middle childhood and adolescence, a pattern of neurocognitive results highlights the resiliency of global IQ and the possible sensitivity of attention and memory to prenatal cannabis exposure. Between 9 and 12 years of age, the data suggest that fetal cannabis exposure is not associated with composite IQ scores or performance on broad-based reading and language exams (Fried et al., 1997,1998). However, the heavy use of cannabis during pregnancy (~1 or more joints/day) has been linked with decreased scores on tests of academic achievement, impulse control, visual analysis/hypothesis testing and learning/memory in exposed children (Fried et al., 1998; Goldschmidt et al., 2004; Richardson et al., 2002). Longitudinal tracking of a birth cohort through adolescence (13–16 years) demonstrated that global IQ scores remain unaffected by fetal cannabis exposure but certain aspects of cognition, particularly those related to sustained attention and visual working memory, may continue to be negatively impacted (Fried et al., 2003).
Two cannabis research programs have paired behavioral protocols with in vivo visualization of the brain using functional magnetic resonance imaging (fMRI) (Smith et al., 2004, 2006, 2016). Prenatal cannabis exposure in subjects ranging in age from 8–22 was not related to decrements in performance on a visuospatial cognitive task but fMRI scans revealed increased neural activity in the frontal gyri, parahippocampal gyrus, occipital gyrus and cerebellum and decreased activity in the right inferior and middle frontal gyri in exposed subjects. Brain imaging techniques were also utilized in a study of 6 year old children to investigate cannabis-related changes in brain morphology (El Marroun et al., 2015). Using MRI technology to compare prenatally exposed and nonexposed children, no differences in brain volume were detected but there were significant differences in cortical thickness. While the mechanism and functional significance of these findings remains unknown, thicker cortices in the frontal regions of both hemispheres suggest exposure-driven changes in the maturation of the frontal cortex.
A collective examination of the body of knowledge on fetal cannabis exposure and childhood neurocognitive development suggests that heavy maternal use of cannabis during pregnancy does not result in a reduction in global IQ but rather may act to diminish performance on tasks that require the harnessing and implementation of executive function skills; a top-down set of cognitive processes that are used to manage attention, exert inhibitory control and plan goal-directed behavior (Fried and Smith, 2001). Functional losses in executive function skills may place children with in utero cannabis exposure at a disadvantage for long-term success in school, in the community and in the workplace (Diamond and Lee, 2011).
7.4. Psychological Health and Adaptive Behavior
On the continuum of cannabis-related developmental neurotoxicity, there is growing evidence that psychological health may be particularly vulnerable to the adverse effects of in utero exposure. A study of infant social behavior demonstrated that maternal cannabis use during pregnancy was related to a significant increase in aggressive behavior and attentional problems in 18 month-old girls (El Marroun et al., 2011). In middle childhood, prenatal exposure was predictive of damaging or maladaptive behaviors such as increases in hyperactivity, impulsivity and delinquent behavior (Goldschmidt et al., 2000). In children born to heavy cannabis users (~1 or more joints/day), the risk of scoring in the borderline clinical range for delinquent behavior was 2.4 times that of children born to nonusers. Increased reporting of depressive symptoms and anxiety has also been documented in children with a history of heavy prenatal cannabis exposure during the first trimester (Gray et al., 2005; Leech et al., 2006).
A similar pattern of results has been observed in adolescence where rates of delinquency varied by prenatal exposure history (41% non-exposed, 50% light to moderate exposure and 61% heavy exposure) (Day et al., 2011). It is useful to note that cannabis-exposed children who expressed depressive symptoms at age 10 were at the highest risk of reporting delinquent behaviors during puberty. In separate studies focused on mental health and adaptive behavior during young adulthood, maternal cannabis use during pregnancy was not predictive of non-clinical psychopathology (Zammit et al., 2009) but was related to an increased risk for diagnosis of Tourette syndrome or chronic tic disorder (Mathews et al., 2014). Recent studies have suggested that prenatal exposure predicts the early onset of cannabis use in young adults (22 years of age), but this effect was primarily observed in subjects born to heavy users (~1 or more joints/day) (Sonon et al., 2015). While intriguing, a positive relationship between prenatal cannabis exposure and the early onset of cannabis use was not found in a study that utilized both maternal self-report and infant meconium to measure levels of gestational exposure (Frank et al., 2014).
8. Insights from Experimental Work with Preclinical Species
Preclinical animal model systems provide an important bridge between brain and behavior, allowing for the identification of neural pathways and processes that underlie postnatal changes in exposed offspring (Thompson et al., 2009). One of the most important aspects of preclinical work is the ability to control many of the confounding environmental and genetic factors that can adversely affect neurodevelopmental outcomes in human populations; a condition that is essential to determining the independent contribution of drug exposure (Fried, 2002). Most investigations have used rodent models to study oral or subcutaneous (sc) routes of exposure but a small number of investigations have employed inhalation or intravenous dosing (see Table 2). THC doses in preclinical studies range from 0.1 to 150 mg/kg. An oral dose of 5 mg/kg THC in rats is thought to correspond to moderate levels of drug exposure in humans (García-Gil et al., 1999). Because comprehensive reviews of the behavioral and neuroendocrine effects of prenatal cannabis exposure in preclinical models are available (e.g. Campolongo et al., 2011), the present discussion of the rodent and primate literature is focused on studies which bring translational value to the human research findings outlined above.
Table 2:
↑ indicates significant increase in outcome measure. ↓ indicates significant decrease in outcome measure. ↔ indicates no significant change in outcome measure.
| Study | Species/Strain | n | Dose | Route | Age at Dose | Physical Growth and Maturation | Cognition | Emotionality and Adaptive Behavior | Physical Activity |
|---|---|---|---|---|---|---|---|---|---|
| Gianutsos & Abbatiello, 1972 | Wistar rats | 75 | 250 mg/kg cannabis extract |
sc | GD 8–11 | ↓ leaning/memory via Lashley III maze (PND 65) |
|||
| Borgen et al., 1973 | Wistar rats | 48 | 10 mg/kg THC | sc | GD 10–12 | ↓ birthweight (PND 0) | ↑ locomotion via open field (PND 9) ↔locomotion via elevated plus maze (PND 21) |
||
| Fried, 1976 | Wistar rats | 32 | ~19 mg/kg | inhalation | GD 1–19 | ↓ birthweight (PND 0) ↓ bodyweight (PND 0–100) ↔ postnatal mortality |
↓ locomotion via open field (PND 7) ↔locomotion via open field (PND 1, 14) |
||
| Abel, 1984 | Long Evans hooded rats |
10–14 /test |
50–150 mg/kg THC |
po | GD 1–23 | ↓ birthweight (PND 0) | ↔ rotarod performance (PND 36) | ||
| Asch and Smith, 1986 | Rhesus macaques |
47 | 2.5 mg/kg | im | GD 156–163 | ↔ birthweight (PND 1) | |||
| Brake et al, 1987 | Wistar rats | 38 | 15 and 50 mg/kg THC |
po | GD 8–22 | ↔ locomotion via open field (PND 2–32) ↔ nipple attachment (PND 2–14) |
|||
|
Hutchings et al., 1987, 1989, 1991a, 1991b |
Wistar rats | 35 | 15 and 30 mg/kg THC |
po | GD 2–22 | ↔ birthweight (PND 1) ↑ postnatal mortality |
↔ startle response | ||
| Navarro et al., 1995 | Wistar rats | 40/test | Hashish w/~20 mg/kg THC |
po | GD 5 – PND 1 | ↔ weight (PND 1–70) ↔ litter size (PND 1) |
↑ anxiety via social interaction and novel environments (PND 70) |
↑ locomotion via open field (PND 1, 20) | |
| Moreno et al, 2003 | Wistar rats | 192 | 0.1–2 mg/kg THC | po | GD 5 – PND 24 | ↑ immobility via open field (PND 70) ↓ locomotion via open field (PND 70) |
|||
| O’Shea and Mallet, 2005 | Wistar rats | 12 | 5 mg/kg THC | sc | PND 4–14 | ↓ bodyweight (PND 4–52) | ↔ motivation and learning via spatial discrimination (PND 59) ↓ working memory via delayed alternation (PND 59) |
||
| Campolongo et al., 2007 | Wistar rats | 23–28 | 5 mg/kg THC | po | GD 15 – PND 9 | ↔ birthweight (PND 1) ↔ litter size (PND 1) ↔ postnatal mortality |
↓ long-term memory via inhibitory avoidance (PND 80) ↓ short term memory via social discrimination (PND 80) |
||
| Newsom and Kelly, 2008 | Long-Evans hooded rats (males only) |
24 | 2 mg/kg THC | sc | 2x/day GD 1–22 and PND 2–10 |
↔ birthweight (PND 1) ↔ postnatal mortality |
↑ anxiety/fear via open field activity (PND 90) ↓ anxiety via social interaction (PND 90) ↔ depression via forced swim test (PND 90) |
||
| Trezza et al., 2008 | Wistar rats | 50 | 5 mg/kg THC | po | GD 15 – PND 9 | ↔ birthweight (PND 1) ↔ litter size (PND 1) ↔ postnatal mortality |
↑ anxiety via ultrasonic vocalizations (PND 12) ↑ anxiety via elevated plus maze (PND 80) ↓ social play (PND 35) |
↔ locomotion via elevated plus maze (PND 80) |
|
| Silva et al., 2012 | Sprague Dawley rats |
40–96 | 0.15 mg/kg THC | iv | Before conception to GD 21 |
↔ birthweight (PND 1) | ↓ attention (PND 55) ↓ learning/memory in males via active avoidance (PND 45) |
↓ locomotion activity when challenged with amphetamines (PND 60) |
|
| de Salas-Quiroga et al., 2015 | WT mice | 10–24 | 3 mg/kg THC | ip | ED 12.5 −16.5 | ↓ motor skills activity (PND 60) ↓ time to seizures when administered PTZ (PND 60) |
|||
| Benevenuto et al., 2016 | Balb/C mice | 20 | 5 mg/kg cannabis | inhalation | Daily from conception |
↓ fetal weight (GD 18.5) ↑ placental weight (GD 18.5) ↑ number male pups (GD 18.5) ↓ male fetal:placental weight ratio (GD 18.5) |
8.1. Physical growth and maturation
The effects of THC on fetal growth and neonatal outcomes were among the first investigational topics to be addressed in preclinical modeling research. In a study of chronic oral THC exposure (2.4 mg/kg/day) that was undertaken in pregnant macaque monkeys, THC readily crossed the placenta but was not related to changes in fetal growth or infant birthweight (Asch and Smith, 1986). In an early and ground-breaking longitudinal research program using rats, gravid animals were exposed to THC via gastric intubation (15 or 50 mg/kg/day) on gestation day (GD) 2–22 and offspring were evaluated on a series of neurodevelopmental metrics (Hutchings et al., 1987). While there were significant increases in offspring mortality at both doses, broad-based behavioral testing (including the rest-activity cycle, latency to attach to a nipple and ontogeny of locomotor activity) did not reveal adverse effects of early THC exposure ( Brake et al., 1987; Hutchings et al., 1987, 1989, 1991a, 1991b). In a recent inhalation mouse study, a dose of ~ 0.5 mg/kg/day THC smoke from GD 5.5–17.5 produced deficits in fetal growth and reduced birthweights in cannabis-exposed offspring (Benevenuto et al., 2017). Pups with a history of gestational THC exposure showed a surprising 9.9% drop in birthweight and significant decrements in the weight of lungs, brain, thymus, and liver. In general, male fetuses appeared more susceptible to cannabis-related disruptions in early physical growth. These results suggest that low-dose exposure to cannabis for periods as little as 5 min a day via inhalation can have a compromising effect on fetal development.
8.2. Cognition
Cognitive endpoints have also been evaluated in animals prenatally exposed to THC, and like results from human studies, the evidence is equivocal. While some results with animal models suggest adverse effects on learning and memory, it is important to remember that there are multiple studies that did not identify prenatal cannabis exposure as a risk factor for short-or long-term neurocognitive effects (e.g. Abel, 1984). The original discovery work in this field was plagued by methodological and translational issues that detracted from the overall significance of the experimental results, including the role of maternal toxicity in producing false-positives on behavioral assessments in exposed offspring (Hutchings and Dow-Edwards, 1991). With that said, impairments in learning abilities were among the earliest reported effects of prenatal cannabis exposure (Fried, 1976; Gianutsos and Abbatiello, 1972). More contemporary research with preclinical species has demonstrated that maternal oral exposure to 5 mg/kg/day THC during pregnancy produces measurable deficits in learning and short-term olfactory memory in exposed offspring (Campolongo et al., 2007). Cognitive impairments in exposed animals were accompanied by changes in cortical gene expression that suggest alterations in glutamatergic neurotransmission. In a recent mouse study targeting early CNS development, gravid mice were exposed to intraperitoneal (ip) injections of 3.0 mg/kg/day THC from GD 12.5–16.5 and offspring showed reductions in skilled motor activity and an increased vulnerability to seizures (de Salas-Quiroga et al., 2015). The authors theorized that fetal THC exposure may impede the normal development of corticospinal connectivity and increase seizure susceptibility by interfering with CB1R-dependent regulation of both glutamatergic and GABAergic neuron development.
Consistent with preclinical findings on physical growth and maturation, the effects of developmental THC exposure on cognition are more pronounced in males. Offspring of rats exposed to 0.15 mg/kg/day iv injections of THC throughout gestation showed reduced performance on a test of learning and long-term memory with the most pronounced deficits occurring in male offspring (Silva et al., 2012). This investigation is commendable for tracking animals from weaning through adulthood and the findings suggest emergent memory processes may be particularly vulnerable to perturbation from in utero exposure. Additional evidence of cognitive-based impairments was obtained in a rat study using 5 mg/kg/day THC sc on GD 4–14, a period of major synaptogenesis and analogous to the 3rd trimester in humans (O’Shea and Mallet, 2005). As adults, exposed animals committed more errors and took longer to reach a level of proficient performance on a test of spatial learning and memory. Neurocognitive testing across preclinical studies with rats and mice have identified cognitive effects in exposed offspring that align with cognitive findings from prenatally exposed children, providing corroboration that memory-in-action, or working memory, may be the seat of cannabis-induced cognitive impairment.
8.3. Emotionality and Adaptive Behavior
Given that prenatal cannabis exposure may be associated with increased anxiety and depressive symptomology in children, emotionality is an important outcome measure in studies with animals. Auditory startle in adulthood, a sensitive measure of CNS functioning, was not impaired in rat pups exposed to either 15 or 50 mg/kg/day THC via gastric intubation on GD 2–22 (Hutchings et al., 1991a). Conversely, results from a longitudinal study of 2.5–5 mg/kg/day oral THC exposure from GD 15- Postnatal day 9 in rats demonstrated that perinatal exposure to THC was associated with an increased number of adverse effects including ultrasonic distress calls from pups, inhibited social behavior during adolescence and anxiety-like symptoms during adulthood (Trezza et al., 2008). These findings led investigators to hypothesize that developmental exposure to cannabinoids may exert long-term effects on select brain regions that control emotional development and cognition (Trezza et al., 2012). Interestingly, changes in adult social behavior expressed as increased interactions with peers have been reported after gestational and early postnatal exposure to 2 mg/kg/day THC sc (Newsom and Kelly, 2008). The theoretical premise that cannabinoid exposure may impact the modulation of emotional states, including sociability, is bolstered by two research findings 1) CB1R are highly expressed in brain regions that regulate anxiety (e.g. cortex, hippocampus, lateral septum, nucleus accumbens, amygdala) and 2) the eCB signaling system controls the release of several neurotransmitters (e.g. serotonin, dopamine) involved with emotionality (Campolongo et al., 2011).
8.4. Physical Activity
Findings from modeling studies suggest that fetal exposure to THC may produce transient effects on postnatal physical activity (i.e. body movement). In rodent studies using oral doses of 5 or 10 mg/kg/day THC during gestation, increases (Borgen et al., 1973; Mereu et al., 2003; Navarro et al., 1995), decreases (Fried, 1976) and no changes (Brake et al., 1987; Trezza et al., 2008) in psychomotor activity levels have been documented. While results are inconsistent, some data suggest that prenatal exposure to cannabinoids may impact the development of brain areas involved in motor behavior, a finding that is relevant to the increased hyperactivity and impulsivity observed in exposed children and adolescents. Oral administration of THC from GD 5 – Postnatal day 24 in gravid rats led to significant changes in offspring physical activity levels at doses as low as 0.1 mg/kg (range 0.1– 2 mg/kg/day) (Moreno et al., 2003). Offspring perinatally exposed to THC spent significantly more time in an immobile state and displayed reduced levels of activity in an open field test apparatus when compared to controls.
9. Conclusions and Directions of Future Research Objectives
Much of the research conducted thus far has focused on understanding the MOA of THC and role of the eCB signaling system in mature brain. Much less is known about the MOA mediating the effects of THC on the developing brain and whether glial cells play a role in the impact of THC and how this is influenced by additional phyto-CB. Almost nothing is known about the impact of phyto-CB on the developing brain when exposure occurs in combination with other drugs. There is a strong need to establish the fetal neurotoxic effects of phyto-CB exposure to properly evaluate the safety profile of cannabis use during pregnancy. In addition, a key gap in translating preclinical findings of cannabis toxicity to humans is the lack of detailed knowledge of the pharmacokinetics and maternal-fetal transfer mechanisms of THC and its metabolites in humans and in animal models. Detailed pharmacokinetic studies and quantitative modeling of cannabis pharmacokinetics are needed to develop methods that allow interspecies scaling and determination of human maternal and fetal exposures from spot urine or blood samples. It is also notable that no studies have been conducted to evaluate the potential fetal toxicity of the metabolites of THC and such studies are critically needed. In terms of neurodevelopmental effects, the current evidence is contradictory but certain patterns can be gleaned from the data. Cannabis does not act as a classical teratogen and is not associated with morphological abnormalities at birth. Fetal exposure has been associated with changes in physical growth and maturation early in life but long-term growth, including pubertal milestones, are unaffected. Global intelligence scores in children with a history of in utero cannabis exposure are typically not affected but aspects of cognition involved with executive functioning (e.g. attention, inhibitory control, planning) can be negatively impacted. Effects of exposure also include higher levels of depression and anxiety during adolescence, suggesting that psychological outcomes may be particularly sensitive to the disrupting influence of gestational cannabis exposure. Results from preclinical modeling studies have confirmed that in other mammalian species, fetal exposure to THC does not result in changes in long-term physical growth but may negatively impact certain aspects of cognition, and heighten the occurrence of behaviors that are consistent with anxiety. While the neurodevelopmental effects of in utero cannabis exposure are subtle, they are persistent and have been observed in more than one species. Our overall conclusion is that there is a public health need for well-controlled scientific studies to elucidate the pattern of neurotoxicity that may be associated with fetal exposure and until such time more information is available, pregnant women should not assume that it is safe to use cannabis during pregnancy.
Acknowledgments:
This work was supported in part by grants from National Institutes of Health, National Institute of Drug Abuse DA032507, Office of Research Infrastructure Programs, P51 OD010425 (David Anderson, PI), DA014486, DA026430 and NS098777 (Nephi Stella, PI) and National Institute of Child Health and Human Development, HD083091 (Mike Guralnick, PI)
Abbreviations
- 2-AG
2-arachidonoyl glycerol
- 11-nor-THC-COOH
11-nor-tetrahydrocannabinol-9-caboxylic acid
- 11-OH-THC
11-hydroxy-tetrahydrocannabinol
- 8-OH-THC
8-hydroxy-tetrahydrocannabinol
- CB
cannabinoid
- CB1R
Type I cannabinoid receptor
- CB2R
Type II cannabinoid receptor
- CBD
cannabidiol
- Cmax
maximum concentration
- CNS
central nervous system
- CYP
cytochrome P450
- eCB
endocannabinoid
- GD
gestational day
- GPCR
G-protein-coupled receptor
- ip
intraperitoneal
- iv
intravenous
- MOA
mode of action
- phyto-CB
phyto-cannabinoids
- PND
postnatal day
- po
oral
- sc
subcutaneous
- THC
Δ9-tetrahydrocannabinol
- UGT
UDP-glucuronosyltransferase
Footnotes
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
References
- Abel EL (1984). Effects of ∆9-THC on pregnancy and offspring in rats. Neurobehavioral Toxicology and Teratology, 6(1), 29–32. [PubMed] [Google Scholar]
- Abood M, Sorensen R, & Stella N (Eds.). (2012). endoCANNABINOIDS: actions at Non-CB1/CB2 cannabinoid receptors (et al. ed.). Springer Science & Business Media. [Google Scholar]
- Abrams RM, Cook CE, Davis KH, Niederreither K, Jaeger MJ, & Szeto HH (1984). Plasma delta-9-tetrahydrocannabinol in pregnant sheep and fetus after inhalation of smoke from a marijuana cigarette. Alcohol and Drug Research, 6(5), 361–9. [PubMed] [Google Scholar]
- Aguado T, Monory K, Palazuelos J, Stella N, Cravatt B, Lutz B, Marsicano G, Kokaia Z, Guzmán M, & Galve-Roperh I (2005). The endocannabinoid system drives neural progenitor proliferation. The FASEB Journal, 19(12), 1704–1706. 10.1096/fj.05-3995fje [DOI] [PubMed] [Google Scholar]
- Aguado T, Palazuelos J, Monory K, Stella N, Cravatt B, Lutz B, Marsicano G, Kokaia Z, Guzmán M, & Galve-Roperh I (2006). The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. The Journal of Neuroscience, 26(5), 1551–1561. 10.1523/JNEUROSCI.3101-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed AIA, Van Den Elsen GAH, Colbers A, Kramers C, Burger DM, van der Marck MA, & Olde Rikkert MGM (2015). Safety, pharmacodynamics, and pharmacokinetics of multiple oral doses of delta-9-tetrahydrocannabinol in older persons with dementia. Psychopharmacology, 232(14), 2587–2595. 10.1007/s00213-015-3889-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpár A, Tortoriello G, Calvigioni D, Niphakis MJ, Milenkovic I, Bakker J, Cameron GA, Hanics J, Morris CV, Fuzik J, Kovacs GG, Cravatt BF, Parnavelas JG, Andrews WD, Hurd YL, Keimpema E, & Harkany T (2014). Endocannabinoids modulate cortical development by configuring Slit2/Robo1 signalling. Nature Communications, 5, 4421 10.1038/ncomms5421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Chemical Society. (2015). Legalizing marijuana and the new science of weed [Press Release; ]. Retrieved May 3, 2017, from https://www.acs.org/content/acs/en/pressroom/newsreleases/2015/march/legalizing-marijuana-and-the-new-science-of-weed video.html?_ga=1.115036224.695999133.1470076540 [Google Scholar]
- Asch RH, & Smith CG (1986). Effects of delta 9-THC, the principal psychoactive component of marijuana, during pregnancy in the rhesus monkey. The Journal of Reproductive Medicine, 31(12), 1071–1081. [PubMed] [Google Scholar]
- Astley SJ, & Little RE (1990). Maternal marijuana use during lactation and infant development at one year. Neurotoxicology and Teratology, 12(2), 161–168. 10.1016/0892-0362(90)90129-Z [DOI] [PubMed] [Google Scholar]
- Atwood BK, Huffman J, Straiker A, & MacKie K (2010). JWH018, a common constituent of “Spice” herbal blends, is a potent and efficacious cannabinoid CB1 receptor agonist. British Journal of Pharmacology, 160(3), 585–593. 10.1111/j.1476-5381.2009.00582.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azofeifa A, Mattson ME, Schauer G, McAfee T, Grant A, & Lyerla R (2016). National Estimates of Marijuana Use and Related Indicators - National Survey on Drug Use and Health, United States, 2002–2014. Morbidity and Mortality Weekly Report: Surveillance Summaries, 65(11), 1–28. https://doi.org/10.15585/mmwr.ss6511a1 [DOI] [PubMed] [Google Scholar]
- Bada HS, Reynolds EW, & Hansen WF (2006). Marijuana use, adolescent pregnancy, and alteration in newborn behavior: how complex can it get? The Journal of Pediatrics, 149(6), 742–5. 10.1016/j.jpeds.2006.10.049 [DOI] [PubMed] [Google Scholar]
- Bailey JR, Cunny HC, Paule MG, & Slikker W (1987). Fetal disposition of Δ9-tetrahydrocannabinol (THC) during late pregnancy in the rhesus monkey. Toxicology and Applied Pharmacology, 90(2), 315–321. 10.1016/0041-008X(87)90338-3 [DOI] [PubMed] [Google Scholar]
- Bellinger DC, Matthews-Bellinger JA, & Kordas K (2016). A developmental perspective on early-life exposure to neurotoxicants. Environment International, 94, 103–112. 10.1016/j.envint.2016.05.014 [DOI] [PubMed] [Google Scholar]
- Benevenuto SG, Domenico MD, Martins MAG, Costa NS, de Souza ARL, Costa JL, Tavares MFM, Dolhnikoff M, & Veras MM (2017). Recreational use of marijuana during pregnancy and negative gestational and fetal outcomes: An experimental study in mice. Toxicology, 1 (376) 94–101. 10.1016/j.tox.2016.05.020 [DOI] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urbán GM, … Harkany T (2007). Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science, 316(5828), 1212–6. 10.1126/science.1137406 [DOI] [PubMed] [Google Scholar]
- Blackard C, & Tennes K (1984). Human placental transfer of cannabinoids. New England Journal of Medicine, 311, 797. [DOI] [PubMed] [Google Scholar]
- Bland TM, Haining RL, Tracy TS, & Callery PS (2005). CYP2C-catalyzed delta(9)-tetrahydrocannabinol metabolism: Kinetics, pharmacogenetics and interaction with phenytoin. Biochemical Pharmacology, 70(7), 1096–1103. 10.1016/j.bcp.2005.07.007 [DOI] [PubMed] [Google Scholar]
- Borgen LA, Davis WM, & Pace HB (1973). Effects of Prenatal ∆9-Tetrahydrocannabinol on the Development of Rat Offspring. Pharmacology Biochemistry and Behavior, 1, 203–206. [Google Scholar]
- Bornheim LM, Lasker JM, & Raucy JL (1992). Human hepatic microsomal metabolism of ∆1-tetrahydrocannabinol. Drug Metabolism and Disposition, 20(2), 241–246. [PubMed] [Google Scholar]
- Boskovic R, Klein J, Woodland C, Karaskov T, & Koren G (2001). The role of the placenta in variability of fetal exposure to cocaine and cannabinoids: A twin study. Canadian Journal of Physiology and Pharmacology, 79(11), 942–945. 10.1139/y01-080 [DOI] [PubMed] [Google Scholar]
- Brake SC, Hutchings DE, Morgan B, Lasalle E, & Shi T (1987). Delta-9-tetrahydrocannabinol during pregnancy in the rat: II. Effects on ontogeny of locomotor activity and nipple attachment in the offspring. Neurotoxicology and Teratology, 9(1), 45–49. 10.1016/0892-0362(87)90069-9 [DOI] [PubMed] [Google Scholar]
- Brown QL, Sarvet AL, Shmulewitz D, Martins SS, Wall MM, & Hasin DS, (2016). Trends in Marijuana Use Among Pregnant and Nonpregnant Reproductive-Aged Women, 2002–2014. JAMA, 72(12), 1235–1242. 10.1001/jama.2016.17383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buser GL, Gerona RR, Horowitz BZ, Vian KP, Troxell ML, Hendrickson RG, Houghton DC, Rozansky D, Su SW, & Leman RF (2014). Acute kidney injury associated with smoking synthetic cannabinoid. Clinical Toxicology, 52(7), 664–673. 10.3109/15563650.2014.932365 [DOI] [PubMed] [Google Scholar]
- Calvigioni D, Hurd YL, Harkany T, & Keimpema E (2014). Neuronal substrates and functional consequences of prenatal cannabis exposure. European Child & Adolescent Psychiatry, 23(10), 931–941. 10.1007/s00787-014-0550-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campolongo P, Trezza V, Cassano T, Gaetani S, Morgese MG, Ubaldi M, Soverchia L, Antonelli T, Ferraro L, Massi M, Ciccocioppo R, & Cuomo V (2007). Perinatal exposure to delta-9-tetrahydrocannabinol causes enduring cognitive deficits associated with alteration of cortical gene expression and neurotransmission in rats. Addiction Biology, 12(3–4), 485–495. 10.1111/j.1369-1600.2007.00074.x [DOI] [PubMed] [Google Scholar]
- Campolongo P, Trezza V, Ratano P, Palmery M, & Cuomo V (2011). Developmental consequences of perinatal cannabis exposure: Behavioral and neuroendocrine effects in adult rodents. Psychopharmacology, 214(1), 5–15. 10.1007/s00213-010-1892-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli MP, Paola Piras A, D’Agostino A, Pibiri F, Perra S, Gessa GL, Maccarrone M, & Pistis M (2007). Dysregulation of the endogenous cannabinoid system in adult rats prenatally treated with the cannabinoid agonist WIN 55,212–2. European Journal of Pharmacology, 573(1–3), 11–9. 10.1016/j.ejphar.2007.06.047 [DOI] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, & Hashimotodani Y (2012). Endocannabinoid Signaling and Synaptic Function. Neuron, 76(1), 70–81. 10.1016/j.neuron.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Behavioral Health Statistics and Quality. (2016). Results from the 2015 National Survey on Drug Use and Health: Detailed Tables Rockville, Maryland: Retrieved from https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015.pdf [Google Scholar]
- Chambers RA, Taylor JR, & Potenza MN (2003). Developmental neurocircuitry of motivation in adolescence: A critical period of addiction vulnerability. American Journal of Psychiatry, 160(6),1041–52. 10.1176/appi.ajp.160.6.1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen HD, Freudenthal RI, Gidley JT, Rosenfeld R, Boegli G, Testino L, Brine DR, Pitt CG, & Wall ME (1971). Activity of ∆8- and ∆9-tetrahydrocannabinol and related compounds in the mouse. Science, 172(3979), 165–167. [DOI] [PubMed] [Google Scholar]
- Conner SN, Carter EB, Tuuli MG, Macones GA, & Cahill AG (2015). Maternal marijuana use and neonatal morbidity. American Journal of Obstetrics and Gynecology, 213(3), 422.e1–422.e4. 10.1016/j.ajog.2015.05.050 [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, & Lichtman AH (2001). Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proceedings of the National Academy of Sciences, 98(16), 9371–9376. 10.1073/pnas.161191698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews F, He J, & Hodge C (2007). Adolescent cortical development: A critical period of vulnerability for addiction. Pharmacology Biochemistry and Behavior, 86(2),189–99. 10.1016/j.pbb.2006.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristino L, & Di Marzo V (2014). Fetal cannabinoid receptors and the "dis-joint-ed" brain. The EMBO Journal, 33(7), 665–7. 10.1002/embj.201488086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl RE, Scher MS, Williamson DE, Robles N, & Day NL (1995). A Longitudinal Study of Prenatal Marijuana Use: Effects on Sleep and Arousal at Age 3 Years. Archives of Pediatrics & Adolescent Medicine, 149(2), 145 10.1001/archpedi.1995.02170140027004 [DOI] [PubMed] [Google Scholar]
- Day NL, Richardson GA, Geva D, & Robles N (1994). Alcohol, Marijuana, and Tobacco: Effects of Prenatal Exposure on Offspring Growth and Morphology at Age Six. Alcoholism: Clinical and Experimental Research, 18(4), 786–794. 10.1111/j.1530-0277.1994.tb00041.x [DOI] [PubMed] [Google Scholar]
- Day NL, Leech SL, & Goldschmidt L (2011). The effects of prenatal marijuana exposure on delinquent behaviors are mediated by measures of neurocognitive functioning. Neurotoxicology and Teratology, 33(1), 129–136. 10.1016/j.ntt.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Chiara V, Angelucci F, Rossi S, Musella A, Cavasinni F, Cantarella C, Mataluni G, Sacchetti L, Napolitano F, Castelli M, Caltagirone C, Bernardi G, Maccarrone M, Usiello A, & Centonze D (2010). Brain-derived neurotrophic factor controls cannabinoid CB1 receptor function in the striatum. Journal of Neuroscience, 30(24), 8127–8137. 10.1523/JNEUROSCI.1683-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De March Z, Zuccato C, Giampia C, Patassini S, Bari M, Gasperi V, De Ceballos ML, Bernardi G, Maccarrone M, Cattaneo E, & Fusco FR (2008). Cortical expression of brain derived neurotrophic factor and type-1 cannabinoid receptor after striatal excitotoxic lesions. Neuroscience, 152(3), 734–740. 10.1016/j.neuroscience.2007.11.044 [DOI] [PubMed] [Google Scholar]
- de Salas-Quiroga A, Díaz-Alonso J, García-Rincón D, Remmers F, Vega D, Gómez-Cañas M, Lutz B, Guzmán M, & Galve-Roperh I (2015). Prenatal exposure to cannabinoids evokes long-lasting functional alterations by targeting CB 1 receptors on developing cortical neurons. Proceedings of the National Academy of Sciences of the United States of America, 112(44), 1–6. 10.1073/pnas.1514962112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane WA, Dysarz III FA, Johnson MR, Melvin LS, & Howlett AC (1988). Determination and Characterization of a Cannabinoid Receptor in Rat Brain. Molecular Pharmacology, 34(5), 605–613. [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, & Piomelli D (1994). Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature, 372(6507), 686–691. 10.1038/372686a0 [DOI] [PubMed] [Google Scholar]
- Diamond A, & Lee K (2011). Interventions shown to aid executive function development in children 4 to 12 years old. Science, 333(6045), 959–64. 10.1126/science.1204529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Alonso J, Aguado T, Wu C-S, Palazuelos J, Hofmann C, Garcez P, Guillemot F, Lu HC, Lutz B, Guzmán M, & Galve-Roperh I (2012). The CB(1) cannabinoid receptor drives corticospinal motor neuron differentiation through the Ctip2/Satb2 transcriptional regulation axis. The Journal of Neuroscience, 32(47), 16651–65. 10.1523/JNEUROSCI.0681-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, & Piomelli D (2002). Brain monoglyceride lipase participating in endocannabinoid inactivation. Proceedings of the National Academy of Sciences of the United States of America, 99(16), 10819–24. 10.1073/pnas.152334899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Kathuria S, & Piomelli D (2004). RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Molecular Pharmacology, 66(5), 1260–4. 10.1124/mol.104.002071 [DOI] [PubMed] [Google Scholar]
- Dinis-Oliveira RJ (2016). Metabolomics of ∆9 -tetrahydrocannabinol: implications in toxicity. Drug Metabolism Reviews, 2532(1), 80–87. 10.3109/03602532.2015.1137307 [DOI] [PubMed] [Google Scholar]
- El Marroun H, Tiemeier H, Steegers EA, Jaddoe VW, Hofman A, Verhulst FC, van den Brink W, & Huizink AC (2009). Intrauterine cannabis exposure affects fetal growth trajectories: the Generation R Study. Journal of the American Academy of Child and Adolescent Psychiatry, 48(12), 1173–1181. 10.1016/S1090-798X(10)79394-2 [DOI] [PubMed] [Google Scholar]
- El Marroun H, Hudziak JJ, Tiemeier H, Creemers H, Steegers EA, Jaddoe VW, Hofman A, Verhulst FC, van den Brink W, & Huizink AC (2011). Intrauterine cannabis exposure leads to more aggressive behavior and attention problems in 18-month-old girls. Drug and Alcohol Dependence, 118(2–3), 470–474. 10.1016/j.drugalcdep.2011.03.004 [DOI] [PubMed] [Google Scholar]
- El Marroun H, Tiemeier H, Franken IHA, Jaddoe VWV, Van Der Lugt A, Verhulst FC, Lahey BB, & White T (2015). Prenatal Cannabis and Tobacco Exposure in Relation to Brain Morphology: A Prospective Neuroimaging Study in Young Children. Biological Psychiatry, 79(12), 1–9. 10.1016/j.biopsych.2015.08.024 [DOI] [PubMed] [Google Scholar]
- ElSohly MA, Mehmedic Z, Foster S, Gon C, Chandra S, & Church JC (2016). Changes in cannabis potency over the last 2 decades (1995–2014): Analysis of current data in the United States. Biological Psychiatry, 79(7), 613–619. 10.1016/j.biopsych.2016.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon M, Pichini S, Joya J, Pujadas M, Sanchez A, Vall O, García Algar O, Luna A, de la Torre R, Rotolo MC, & Pellegrini M (2012). Maternal hair testing for the assessment of fetal exposure to drug of abuse during early pregnancy: Comparison with testing in placental and fetal remains. Forensic Science International, 218(1–3), 92–96. 10.1016/j.forsciint.2011.10.022 [DOI] [PubMed] [Google Scholar]
- Fergusson DM, Horwood LJ, & Northstone K (2002). Maternal use of cannabis and pregnancy outcome. BJOG: An International Journal of Obstetrics and Gynaecology, 109(1), 21–27. [DOI] [PubMed] [Google Scholar]
- Frank DA, Kuranz S, Appugliese D, Cabral H, Chen C, Crooks D, Heeren T, Liebschutz J, Richardson M, & Rose-Jacobs R (2014). Problematic substance use in urban adolescents: Role of intrauterine exposures to cocaine and marijuana and post-natal environment. Drug and Alcohol Dependence, 142, 181–190. 10.1016/j.drugalcdep.2014.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal RI, Martin J, & Wall ME (1972). Distribution of Δ9-tetrahydrocannabinol in the mouse. British Journal of Pharmacology, 44(2), 244–249. 10.1111/j.1476-5381.1972.tb07260.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried PA (1976). Short and long-term effects of pre-natal Cannabis inhalation upon rat offspring. Psychopharmacology, 50(3), 285–291. 10.1007/BF00426846 [DOI] [PubMed] [Google Scholar]
- Fried PA (1980). Marihuana use by pregnant women: Neurobehavioral effects in neonates. Drug and Alcohol Dependence, 6(6), 415–424. 10.1016/0376-8716(80)90023-X [DOI] [PubMed] [Google Scholar]
- Fried PA (2002). The Consequences of Marijuana Use During Pregnancy: A Review of the Human Literature. Women and Cannabis: Medicine, Science, and Sociology, 2(3), 85–104. [Google Scholar]
- Fried PA, & O’Connell CM (1987). A comparison of the effects of prenatal exposure to tobacco, alcohol, cannabis and caffeine on birth size and subsequent growth. Neurotoxicology and Teratology, 9(2), 79–85. 10.1016/0892-0362(87)90082-1 [DOI] [PubMed] [Google Scholar]
- Fried PA, & Smith AM (2001). A literature review of the consequences of prenatal marihuana exposure - An emerging theme of a deficiency in aspects of executive function. Neurotoxicology and Teratology, 23(1), 1–11. 10.1016/S0892-0362(00)00119-7 [DOI] [PubMed] [Google Scholar]
- Fried PA, & Watkinson B (1988). 12- and 24-month neurobehavioural follow-up of children prenatally exposed to marihuana, cigarettes and alcohol. Neurotoxicology and Teratology, 10(4), 305–313. 10.1016/0892-0362(88)90032-3 [DOI] [PubMed] [Google Scholar]
- Fried PA, & Watkinson B (1990). 36-and 48-month neurobehavioral follow-up of children prenatally exposed to marijuana, cigarettes, and alcohol. Journal of Developmental & Behavioral Pediatrics, 11(2), 49–58. [PubMed] [Google Scholar]
- Fried PA, & Watkinson B (2001). Differential effects on facets of attention in adolescents prenatally exposed to cigarettes and marihuana. Neurotoxicology and Teratology, 23(5), 421–430. 10.1016/S0892-0362(01)00160-X [DOI] [PubMed] [Google Scholar]
- Fried PA, Watkinson B, Dillon RF, & Dulberg CS (1987). Neonatal neurological status in a low-risk population after prenatal exposure to cigarettes, marijuana, and alcohol. Journal of Developmental & Behavioral Pediatrics, 8(6), 318–326. https://doi.org/0196-206x/87/0806-0318$02.00/0 [PubMed] [Google Scholar]
- Fried PA, Buckingham M, & Von Kulmiz P (1983). Marijuana use during pregnancy and perinatal risk factors. American Journal of Obstetrics and Gynecology, 146(8), 992–994. 10.1016/0002-9378(83)90989-4 [DOI] [PubMed] [Google Scholar]
- Fried PA, O’Connell CM, & Watkinson B (1992a). 60- and 72-month follow-up of children prenatally exposed to marijuana, cigarettes, and alcohol: cognitive and language assessment. Journal of Developmental & Behavioral Pediatrics, 13(6), 383–91. [PubMed] [Google Scholar]
- Fried PA, Watkinson B, & Gray R (1992b). A follow-up study of attentional behavior in 6-year-old children exposed prenatally to marihuana, cigarettes, and alcohol. Neurotoxicology and Teratology, 14(5), 299–311. [DOI] [PubMed] [Google Scholar]
- Fried PA, Watkinson B, & Siegel LS (1997). Reading and language in 9- to 12-year olds prenatally exposed to cigarettes and marijuana. Neurotoxicology and Teratology, 19(3), 171–183. 10.1016/S0892-0362(97)00015-9 [DOI] [PubMed] [Google Scholar]
- Fried PA, Watkinson B, & Gray R (1998). Differential Effects on Cognitive Functioning in 9- to 12-Year Olds Prenatally Exposed to Cigarettes and Marihuana. Neurotoxicology and Teratology, 20(3), 293–306. 10.1016/S0892-0362(97)00091-3 [DOI] [PubMed] [Google Scholar]
- Fried PA, Watkinson B, & Gray R (1999). Growth from birth to early adolescence in offspring prenatally exposed to cigarettes and marijuana. Neurotoxicology and Teratology, 21(5), 513–525. 10.1016/S0892-0362(99)00009-4 [DOI] [PubMed] [Google Scholar]
- Fried PA, James DS, & Watkinson B (2001). Growth and pubertal milestones during adolescence in offspring prenatally exposed to cigarettes and marihuana. Neurotoxicology and Teratology, 23(5), 431–436. 10.1016/S0892-0362(01)00161-1 [DOI] [PubMed] [Google Scholar]
- Fried PA, Watkinson B, & Gray R (2003). Differential effects on cognitive functioning in 13- to 16-year-olds prenatally exposed to cigarettes and marihuana. Neurotoxicology and Teratology, 25(4), 427–436. 10.1016/S0892-0362(03)00029-1 [DOI] [PubMed] [Google Scholar]
- Fu J, Bottegoni G, Sasso O, Bertorelli R, Rocchia W, Masetti M, Guijarro A, Lodola A, Armirotti A, Garau G, Bandiera T, Reggiani A, Mor M, Cavalli A, & Piomelli D (2012). A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nature Neuroscience, 15(1), 64–9. 10.1038/nn.2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galve-Roperh I, Chiurchiù V, Díaz-Alonso J, Bari M, Guzmán M, & Maccarrone M (2013). Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Progress in Lipid Research, 52(4), 633–650. 10.1016/j.plipres.2013.05.004 [DOI] [PubMed] [Google Scholar]
- Galve-Roperh I, Palazuelos J, Aguado T, & Guzmán M (2009). The endocannabinoid system and the regulation of neural development: Potential implications in psychiatric disorders. European Archives of Psychiatry and Clinical Neuroscience 259(7):371–82. 10.1007/s00406-009-0028-y [DOI] [PubMed] [Google Scholar]
- Garcia-Gil L, Romero J, Ramos JA, & Fernandez-Ruiz JJ (1999). Cannabinoid receptor binding and mRNA levels in several brain regions of adult male and female rats perinatally exposed to delta9-tetrahydrocannabinol. Drug and Alcohol Dependence, 55(1–2), 127–136. [DOI] [PubMed] [Google Scholar]
- Garg M, Garrison L, Leeman L, Hamidovic A, Borrego M, Rayburn WF, & Bakhireva L (2016). Validity of Self-Reported Drug Use Information Among Pregnant Women. Maternal and Child Health Journal, 20(1), 41–47. 10.1007/s10995-015-1799-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett ER, & Hunt AC (1977). Pharmacokinetics of Δ9-Tetrahydrocannabinol in Dogs. Journal of Pharmaceutical Sciences, 66(3), 395–407.n 10.1002/jps.2600660322 [DOI] [PubMed] [Google Scholar]
- Gianutsos G, & Abbatiello ER (1972). The effect of pre-natal Cannabis Sativa on maze learning ability in the rat. Psychopharmacologia, 27(2), 117–122. 10.1007/BF00439370 [DOI] [PubMed] [Google Scholar]
- Ginsburg BC, Hruba L, Zaki A, Javors MA, & McMahon LR (2014). Blood levels do not predict behavioral or physiological effects of ∆9-tetrahydrocannabinol in rhesus monkeys with different patterns of exposure. Drug and Alcohol Dependence, 139, 1–8. 10.1016/j.drugalcdep.2014.02.696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaz-Sandberg A, Dietz L, Nguyen H, Oberwittler H, Aderjan R, & Mikus G (2007). Pharmacokinetics of 11-nor-9-Carboxy-∆9-Tetrahydrocannabinol (CTHC) After Intravenous Administration of CTHC in Healthy Human Subjects. Clinical Pharmacology and Therapeutics, 82(1), 63–9. 10.1038/sj.clpt.6100199 [DOI] [PubMed] [Google Scholar]
- Goldschmidt L, Day NL, & Richardson GA (2000). Effects of prenatal marijuana exposure on child behavior problems at age 10. Neurotoxicology and Teratology, 22(3), 325–336. 10.1016/S0892-0362(00)00066-0 [DOI] [PubMed] [Google Scholar]
- Goldschmidt L, Richardson GA, Cornelius MD, & Day NL (2004). Prenatal marijuana and alcohol exposure and academic achievement at age 10. Neurotoxicology and Teratology, 26(4), 521–532. 10.1016/j.ntt.2004.04.003 [DOI] [PubMed] [Google Scholar]
- Goldschmidt L, Richardson GA, Willford J, & Day NL (2008). Prenatal marijuana exposure and intelligence test performance at age 6. Journal of the American Academy of Child and Adolescent Psychiatry, 47(3), 254–63. 10.1097/CHI.0b013e318160b3f0 [DOI] [PubMed] [Google Scholar]
- Goldschmidt L, Richardson GA, Larkby C, & Day NL (2016). Early marijuana initiation: The link between prenatal marijuana exposure, early childhood behavior, and negative adult roles. Neurotoxicology and Teratology, 58,40–45. 10.1016/j.ntt.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant KS, & Rice DC (2008). Exposure to Environmental Chemicals and Developmental Risk. In Burbacher TM, Sackett GP, & Grant KS (Eds.), Primate Models of Children’s Health and Developmental Disabilities (pp. 377–419). Elsevier. [Google Scholar]
- Gray KA, Day NL, Leech S, & Richardson GA (2005). Prenatal marijuana exposure: Effect on child depressive symptoms at ten years of age. Neurotoxicology and Teratology, 27, 439–448. 10.1016/j.ntt.2005.03.010 [DOI] [PubMed] [Google Scholar]
- Grotenhermen F (2003). Pharmacokinetics and Pharmacodynamics of Cannabinoids. Clinical Pharmacokinetics, 42(4), 327–360. 10.2165/00003088-200342040-00003 [DOI] [PubMed] [Google Scholar]
- Grotenhermen F, Russo E, & Zuardi AW (2017). Even High Doses of Oral Cannabidol Do Not Cause THC-Like Effects in Humans: Comment on Merrick et al. Cannabis and Cannabinoid Research 2016;1(1):102–112; DOI: 10.1089/can.2015.0004 . Cannabis and Cannabinoid Research, 2(1), 1–4. 10.1089/can.2015.0004. Cannabis and Cannabinoid Research, 2(1), 1–4. https://doi.org/10.1089/can.2016.0036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkany T, Keimpema E, Barabás K, & Mulder J (2008a). Endocannabinoid functions controlling neuronal specification during brain development. Molecular and Cellular Endocrinology, 286(1–2), S84–S90. 10.1016/j.mce.2008.02.011 [DOI] [PubMed] [Google Scholar]
- Harkany T, Mackie K, & Doherty P (2008b). Wiring and firing neuronal networks: endocannabinoids take center stage. Current Opinion in Neurobiology, 18(3), 338–45. 10.1016/j.conb.2008.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huestis MA (2007). Human Cannabinoid Pharmacokinetics. Chemical Biodiversity, 4(8), 1770–1804. 10.1002/cbdv.200790152.Human [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt C, & Jones R (1980). Tolerance and disposition of tetrahydrocannabinol in man. Journal of Pharmacology and Experimental Therapeutics, 215(1), 35–44. 10.1017/CBO9781107415324.004 [DOI] [PubMed] [Google Scholar]
- Hurd YL, Wang X, Anderson V, Beck O, Minkoff H, & Dow-Edwards D (2005). Marijuana impairs growth in mid-gestation fetuses. Neurotoxicology and Teratology, 27(2), 221–229. 10.1016/j.ntt.2004.11.002 [DOI] [PubMed] [Google Scholar]
- Hutchings DE, & Dow-Edwards D (1991). Animal models of opiate, cocaine, and cannabis use. Clinics in Perinatology, 18(1), 1–22. [PubMed] [Google Scholar]
- Hutchings DE, Morgan B, Brake SC, Shi T, & Lasalle E (1987). Delta-9-tetrahydrocannabinol during pregnancy in the rat: I. Differential effects on maternal nutrition, embryotoxicity, and growth in the offspring. Neurotoxicology and Teratology, 9(1), 39–43. 10.1016/0892-0362(87)90068-7 [DOI] [PubMed] [Google Scholar]
- Hutchings DE, Martin BR, Gannagaris Z, Miller N, & Fico T (1989). Plasma Concentrations of delta-9-tetrahydrocannabinol in dams and fetuses following acute or multiple dosing in rats. Life Sciences, 44, 697–701. [DOI] [PubMed] [Google Scholar]
- Hutchings DE, Brake SC, Banks AN, Nero TJ, Dick LS, & Zmitrovich AC (1991a). Prenatal Delta-9-Tetrahydrocannabinol in the Rat: Effects on auditory startle in adulthood. Neurotoxicology and Teratology, 13(4), 413–416. 10.1016/0892-0362(91)90090-J [DOI] [PubMed] [Google Scholar]
- Hutchings DE, Fico TA, Banks AN, Dick LS, & Brake SC (1991b). Prenatal delta-9-tetrahydrocannabinol in the rat: Effects on postweaning growth. Neurotoxicology and Teratology, 13(2), 245–248. 10.1016/0892-0362(91)90018-R [DOI] [PubMed] [Google Scholar]
- Hutchings DE, Gamagaris Z, Miller N, & Fico TA (1989). The Effects of Prenatal Exposure to Delta-9-Tetrahydrocannabinol on the Rest-Activity Cycle of the Preweanling Rat. Neurotoxicology and Teratology, 11, 353–356. [DOI] [PubMed] [Google Scholar]
- Isoherranen N, & Thummel KE (2013). Drug Metabolism and Transport During Pregnancy: How Does Drug Disposition Change during Pregnancy and What Are the Mechanisms that Cause Such Changes? Drug Metabolism and Disposition, 41(2), 256–262. 10.1124/dmd.112.050245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaddoe VWV, Van Duijn CM, Van Der Heijden AJ, MacKenbach JP, Moll HA, Steegers EAP, Tiemeier H, Uitterlinden AG, Verhulst FC, & Hofman A (2008). The generation R study: Design and cohort update until the age of 4 years. European Journal of Epidemiology, 23(12), 801–811. 10.1007/s10654-008-9309-4 [DOI] [PubMed] [Google Scholar]
- Jaddoe VWV, Van Duijn CM, Van Der Heijden AJ, MacKenbach JP, Moll HA, Steegers EAP, Tiemeier H, Uitterlinden AG, Verhulst FC, & Hofman A (2010). The Generation R Study: Design and cohort update 2010. European Journal of Epidemiology, 25(11), 823–841. 10.1007/s10654-010-9516-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson M, Contreras JA, Hellman U, Tornqvist H, & Holm C (1997). cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. Journal of Biological Chemistry, 272(43), 27218–27223. 10.1074/jbc.272.43.27218 [DOI] [PubMed] [Google Scholar]
- Katona I, & Freund TF (2008). Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nature Medicine, 14(9), 923–30. 10.1038/nm.f.1869 [DOI] [PubMed] [Google Scholar]
- Katona I, & Freund TF (2012). Multiple functions of endocannabinoid signaling in the brain. Annual Review of Neuroscience, 35(1), 529–58. 10.1146/annurev-neuro-062111-150420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauert GF, Ramaekers JG, Schneider E, Moeller MR, & Toennes SW (2007). Pharmacokinetic properties of ∆9-tetrahydrocannabinol in serum and oral fluid. Journal of Analytical Toxicology, 31, 288–293. 10.1093/jat/31.5.288 [DOI] [PubMed] [Google Scholar]
- Keimpema E, Barabás K, Morozov YM, Tortoriello G, Torii M, Cameron G, Yanagawa Y, Watanabe M, Mackie K, & Harkany T (2010). Differential subcellular recruitment of monoacylglycerol lipase generates spatial specificity of 2-arachidonoyl glycerol signaling during axonal pathfinding. The Journal of Neuroscience, 30(42), 13992–4007. 10.1523/JNEUROSCI.2126-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keimpema E, Mackie K, & Harkany T (2011). Molecular model of cannabis sensitivity in developing neuronal circuits. Trends in Pharmacological Sciences, 32(9), 551–561. 10.1016/j.tips.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner HA, & Dingell JV (1971). The metabolism and excretion of Δ9-tetrahydrocannabinol in the rat. Life Sciences, 10, 49–59. 10.1016/0024-3205(71)90245-1 [DOI] [PubMed] [Google Scholar]
- Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, & Moran JH (2011). Severe toxicity following synthetic cannabinoid ingestion. Clinical Toxicology, 49(8), 760–4. 10.3109/15563650.2011.609822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly MEM, & Denovan-Wright EM (2015). Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. British Journal of Pharmacology, 172(20), 4790–4805. 10.1111/bph.13250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech SL, Larkby CA, Day R, & Day NL (2006). Predictors and correlates of high levels of depression and anxiety symptoms among children at age 10. Journal of the American Academy of Child and Adolescent Psychiatry, 45(2), 223–230. 10.1097/01.chi.0000184930.18552.4d [DOI] [PubMed] [Google Scholar]
- Lemberger L, Axelrod J, & Kopin IJ (1971). Metabolism and Disposition of Tetrahydrocannabinols in Naive Subjects and Chronic Marijuana Users. Annals of the New York Academy of Sciences, 191(1), 142–154. 10.1111/j.1749-6632.1971.tb13994.x [DOI] [Google Scholar]
- Leuschner J. T. a, Harvey DJ, Bullingham RES, & Paton WDM (1986). Pharmacokinetics of delta-9-tetrahydrocannabinol in rabbits following single or multiple intravenous doses. Drug Metabolism and Disposition, 14(2), 230–238. [PubMed] [Google Scholar]
- Lipari R, International R, Kroutil LA, & Pemberton MR (2015). Risk and Protective Factors and Initiation of Substance Use: Results from the 2014 National Survey on Drug Use and Health Retrieved from https://www.samhsa.gov/data/sites/default/files/NSDUH-DR-FRR4-2014rev/NSDUH-DR-FRR4-2014.pdf
- López-Gallardo M, López-Rodríguez AB, Llorente-Berzal Á, Rotllant D, Mackie K, Armario A, Nadal R, & Viveros MP (2012). Maternal deprivation and adolescent cannabinoid exposure impact hippocampal astrocytes, CB1 receptors and brain-derived neurotrophic factor in a sexually dimorphic fashion. Neuroscience, 204, 90–103. 10.1016/j.neuroscience.2011.09.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Guzmán M, Mackie K, Doherty P, & Harkany T (2014). Programming of neural cells by (endo)cannabinoids: from physiological rules to emerging therapies. Nature Reviews Neuroscience, 15(12), 786–801. 10.1038/nrn3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark K, Gryczynski J, Axenfeld E, Schwartz RP, & Terplan M (2017). Pregnant Womenʼs Current and Intended Cannabis Use in Relation to Their Views Toward Legalization and Knowledge of Potential Harm. Journal of Addiction Medicine, in press. [DOI] [PubMed] [Google Scholar]
- Marrs W, & Stella N (2007). 2-AG + 2 New Players = Forecast for Therapeutic Advances. Chemistry & Biology, 14(12), 1309–1311. 10.1016/j.chembiol.2007.12.004 [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, van der Stelt M, López-Rodriguez ML, Casanova E, Schütz G, Zieglgänsberger W, Di Marzo V, Behl C, & Lutz B (2003). CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science, 302(5642), 84–8. 10.1126/science.1088208 [DOI] [PubMed] [Google Scholar]
- Marsot A, Audebert C, Attolini L, Lacarelle B, Micallef J, & Blin O (2016). Comparison of Cannabinoid Concentrations in Plasma, Oral Fluid and Urine in Occasional Cannabis Smokers After Smoking Cannabis Cigarette. Journal of Pharmacy & Pharmaceutical Sciences, 19(3), 411 https://doi.org/10.18433/J3F31D [DOI] [PubMed] [Google Scholar]
- Martin BR, Dewey WL, Harris LS, & Beckner JS (1977). 3H-∆9-tetrahydrocannabinol distribution in pregnant dogs and their fetuses. Research Communications in Chemical Pathology and Pharmacology, 17(3), 457–470. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/897339 [PubMed] [Google Scholar]
- Mathews CA, Scharf JM, Miller LL, Macdonald-Wallis C, Lawlor DA, & Ben-Shlomo Y (2014). Association between pre- and perinatal exposures and tourette syndrome or chronic tic disorder in the alspac cohort. British Journal of Psychiatry, 204(1), 40–45. 10.1192/bjp.bp.112.125468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur A, Lichti CF, Prather PL, Zielinska AK, Bratton SM, Gallus-Zawada A, Finel M, Miller GP, Radomińska-Pandya A, & Moran JH (2009). Characterization of human hepatic and extrahepatic UDP-glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metabolism and Disposition: The Biological Fate of Chemicals, 37(7), 1496–1504. 10.1124/dmd.109.026898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckinney MK, & Cravatt BF (2005). Structure and Function of Fatty Acid Amide Hydrolase. Annual Reviews of Biochemistry, 74(1), 411–32. 10.1146/annurev.biochem.74.082803.133450 [DOI] [PubMed] [Google Scholar]
- McLemore GL, & Richardson KA (2016). Data from three prospective longitudinal human cohorts of prenatal marijuana exposure and offspring outcomes from the fetal period through young adulthood. Data in Brief, 9, 753–757. 10.1016/j.dib.2016.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu G, Fà M, Ferraro L, Cagiano R, Antonelli T, Tattoli M, Ghiglieri V, Tanganelli S, Gessa GL, & Cuomo V (2003). Prenatal exposure to a cannabinoid agonist produces memory deficits linked to dysfunction in hippocampal long-term potentiation and glutamate release. Proceedings of the National Academy of Sciences of the United States of America, 100(8), 4915–20. 10.1073/pnas.0537849100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metna-Laurent M, & Marsicano G (2015). Rising stars: Modulation of brain functions by astroglial type-1 cannabinoid receptors. Glia, 63(3), 353–364. 10.1002/glia.22773 [DOI] [PubMed] [Google Scholar]
- Miller AM, & Stella N (2008). CB2 receptor-mediated migration of immune cells: it can go either way. British Journal of Pharmacology, 153(2), 299–308. 10.1038/sj.bjp.0707523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno M, Trigo JM, Escuredo L, Rodriguez de Fonseca F, & Navarro M (2003). Perinatal exposure to ∆9 -tetrahydrocannabinol increases presynaptic dopamine D2 receptor sensitivity: a behavioral study in rats. Pharmacology Biochemistry and Behavior, 75(3), 565–575. 10.1016/S0091-3057(03)00117-5 [DOI] [PubMed] [Google Scholar]
- Mulder J, Aguado T, Keimpema E, Barabás K, Ballester Rosado CJ, Nguyen L, Monory K, Marsicano G, Di Marzo V, Hurd YL, Guillemot F, Mackie K, Lutz B, Guzmán M, Lu HC, Galve-Roperh I, & Harkany T (2008). Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proceedings of the National Academy of Sciences of the United States of America, 105(25), 8760–5. 10.1073/pnas.0803545105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musshoff F, & Madea B (2006). Review of biologic matrices (urine, blood, hair) as indicators of recent or ongoing cannabis use. Therapeutic Drug Monitoring, 28(2), 155–63. 10.1097/01.ftd.0000197091.07807.22 [DOI] [PubMed] [Google Scholar]
- Narimatsu S, Matsubara K, Shimonishi T, Watanabe K, Yamamoto I, & Yoshimura H (1988). Enzymatic oxidation of 7-hydroxylated delta 8-tetrahydrocannabinol to 7-oxo-delta 8-tetrahydrocannabinol by hepatic microsomes of the guinea pig. Drug Metabolism and Disposition: The Biological Fate of Chemicals, 16(1), 156–161. [PubMed] [Google Scholar]
- Narimatsu S, Watanabe K, Matsunaga T, Yamamoto I, Imaoka S, Funae Y, & Yoshimura H (1990). Cytochrome P-450 isozymes in metabolic activation of delta 9-tetrahydrocannabinol by rat liver microsomes. Drug Metabolism and Disposition, 18(6). [PubMed] [Google Scholar]
- Narimatsu S, Watanabe K, Matsunaga T, Yamamoto I, Imaoka S, Funae Y, & Yoshimura H (1992). Cytochrome P-450 isozymes involved in the oxidative metabolism of delta 9-tetrahydrocannabinol by liver microsomes of adult female rats. Drug Metabolism and Disposition, 20(1), 79–83. [PubMed] [Google Scholar]
- National Academies Press. (2017). The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. National Academies of Sciences Engineering and Medicine Washington, D.C. https://doi.org/10.17226/24625 [PubMed] [Google Scholar]
- Navarrete M, & Araque A (2010). Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron, 68(1), 113–126. 10.1016/j.neuron.2010.08.043 [DOI] [PubMed] [Google Scholar]
- Navarro M, Rubio P, & Rodriguez de Fonseca F (1995). Behavioral consequences of maternal exposure to natural cannabinoids in rats. Psychopharmacology, 122(1), 1–14. [DOI] [PubMed] [Google Scholar]
- Newsom RJ, & Kelly SJ (2008). Perinatal delta-9-tetrahydrocannabinol exposure disrupts social and open field behavior in adult male rats. Neurotoxicology and Teratology, 30(3), 213–219. 10.1016/j.ntt.2007.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noland JS, Singer LT, Short EJ, Minnes S, Arendt RE, Kirchner HL, & Bearer C (2005). Prenatal drug exposure and selective attention in preschoolers. Neurotoxicology and Teratology, 27, 429–438. 10.1016/j.ntt.2005.02.001 [DOI] [PubMed] [Google Scholar]
- Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MCG, Ward AM, Hahn YK, Lichtman AH, Conti B, & Cravatt BF (2011). Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science, 334(6057), 809–13. 10.1126/science.1209200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea M, & Mallet PE (2005). Impaired learning in adulthood following neonatal ∆9-THC exposure. Behavioural Pharmacology, 16(5–6), 455–61. https://doi.org/00008877-200509000-00019 [pii] [DOI] [PubMed] [Google Scholar]
- Ohlsson A, Lindgren JE, Wahlen A, Agurell S, Hollister LE, & Gillespie HK (1980). Plasma delta-9 tetrahydrocannabinol concentrations and clinical effects after oral and intravenous administration and smoking. Clinical Pharmacology and Therapeutics, 28(3), 409–416. 10.1038/clpt.1980.181 [DOI] [PubMed] [Google Scholar]
- Oliveira da Cruz JF, Robin LM, Drago F, Marsicano G, & Metna-Laurent M (2016). Astroglial type-1 cannabinoid receptor (CB1): A new player in the tripartite synapse. Neuroscience, 323, 35–42. 10.1016/j.neuroscience.2015.05.002 [DOI] [PubMed] [Google Scholar]
- Parikh N, Kramer WG, Khurana V, Smith CC, & Vetticaden S (2016). Bioavailability study of dronabinol oral solution versus dronabinol capsules in healthy volunteers. Clinical Pharmacology: Advances and Applications, 8, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG (2008). The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. British Journal of Pharmacology, 153(2), 199–215. 10.1038/sj.bjp.0707442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SPH, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, & Ross RA (2010). International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1 and CB2. Pharmacological Reviews, 62(4), 588–631. 10.1124/pr.110.003004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D (2003). The molecular logic of endocannabinoid signalling. Nature Reviews. Neuroscience, 4(11), 873–84. 10.1038/nrn1247 [DOI] [PubMed] [Google Scholar]
- Renard J, Norris C, Rushlow W, & Laviolette SR (2017). Neuronal and molecular effects of cannabidiol on the mesolimbic dopamine system: Implications for novel schizophrenia treatments. Neuroscience & Biobehavioral Reviews, 75, 157–165. 10.1016/j.neubiorev.2017.02.006 [DOI] [PubMed] [Google Scholar]
- Renard J, Rosen LG, Loureiro M, De Oliveira C, Schmid S, Rushlow WJ, & Laviolette SR (2017). Adolescent Cannabinoid Exposure Induces a Persistent Sub-Cortical Hyper-Dopaminergic State and Associated Molecular Adaptations in the Prefrontal Cortex. Cerebral Cortex, 27(2), 1297–1310. 10.1093/cercor/bhv335 [DOI] [PubMed] [Google Scholar]
- Richardson GA, Day NL, & Taylor PM (1989). The effect of prenatal alcohol, marijuana, and tobacco exposure on neonatal behavior. Infant Behavior and Development, 12(2), 199–209. 10.1016/0163-6383(89)90006-4 [DOI] [Google Scholar]
- Richardson GA, Day NL, & Goldschmidt L (1995). Prenatal alcohol, marijuana, and tobacco use: Infant mental and motor development. Neurotoxicology and Teratology, 17(4), 479–487. 10.1016/0892-0362(95)00006-D [DOI] [PubMed] [Google Scholar]
- Richardson GA, Ryan C, Willford J, Day NL, & Goldschmidt L (2002). Prenatal alcohol and marijuana exposure: Effects on neuropsychological outcomes at 10 years. Neurotoxicology and Teratology, 24(3), 309–320. 10.1016/S0892-0362(02)00193-9 [DOI] [PubMed] [Google Scholar]
- Rosenthaler S, Pöhn B, Kolmanz C, Nguyen Huu C, Krewenka C, Huber A, Kranner B, Rausch WD, & Moldzio R (2014). Differences in receptor binding affinity of several phytocannabinoids do not explain their effects on neural cell cultures. Neurotoxicology and Teratology, 46, 49–56. 10.1016/j.ntt.2014.09.003 [DOI] [PubMed] [Google Scholar]
- Ross RA (2009). The enigmatic pharmacology of GPR55. Trends in Pharmacological Sciences, 30(3),156–63. 10.1016/j.tips.2008.12.004 [DOI] [PubMed] [Google Scholar]
- Saurel-Cubizolles MJ, Prunet C, & Blondel B (2014). Cannabis use during pregnancy in France in 2010. BJOG: An International Journal of Obstetrics and Gynaecology, 121(8), 971–977. 10.1111/1471-0528.12626 [DOI] [PubMed] [Google Scholar]
- Scher MS, Richardson GA, Coble PA, Day NL, & Stoffer DS (1988). The Effects of Prenatal Alcohol and Marijuana Exposure: Disturbances in Neonatal Sleep Cycling and Arousal. Pediatric Research, 24(1), 101–105. [DOI] [PubMed] [Google Scholar]
- Schwilke EW, Schwope DM, Karschner EL, Lowe RH, William D, Kelly DL, Goodwin RS, Gorelick DA, & Huestis MA (2009). ∆9-Tetrahydrocannabinol (THC), 11-Hydroxy-THC, and 11-Nor-9-carboxy-THC Plasma Pharmacokinetics during and after Continuous High-Dose Oral THC. Clinical Chemistry, 55(12), 2180–2189. 10.1373/clinchem.2008.122119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharir H, & Abood ME (2010). Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacology and Therapeutics,126(3), 301–13. 10.1016/j.pharmthera.2010.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva L, Zhao N, Popp S, & Dow-Edwards D (2012). Prenatal tetrahydrocannabinol (THC) alters cognitive function and amphetamine response from weaning to adulthood in the rat. Neurotoxicology and Teratology, 34(1), 63–71. 10.1016/j.ntt.2011.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Fried PA, Hogan MJ, & Cameron I (2004). Effects of prenatal marijuana on response inhibition: An fMRI study of young adults. Neurotoxicology and Teratology, 26(4), 533–542. 10.1016/j.ntt.2004.04.004 [DOI] [PubMed] [Google Scholar]
- Smith AM, Fried PA, Hogan MJ, & Cameron I (2006). Effects of prenatal marijuana on visuospatial working memory: An fMRI study in young adults. Neurotoxicology and Teratology, 28(2), 286–295. 10.1016/j.ntt.2005.12.008 [DOI] [PubMed] [Google Scholar]
- Smith AM, Mioduszewski O, Hatchard T, Byron-Alhassan A, Fall C, & Fried PA (2016). Prenatal marijuana exposure impacts executive functioning into young adulthood: An fMRI study. Neurotoxicology and Teratology, 58, 53–59. 10.1016/j.ntt.2016.05.010 [DOI] [PubMed] [Google Scholar]
- Sonon KE, Richardson GA, Cornelius J, Kim KH, & Day NL (2015). Prenatal marijuana exposure predicts marijuana use in young adulthood. Neurotoxicology and Teratology, 47, 10–15. 10.1016/j.ntt.2014.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N (2010). Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia, 58(9), 1017–1030. 10.1002/glia.20983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N (2012). Neuroscience. Inflammation to rebuild a brain. Science, 338(6112), 1303–4. 10.1126/science.1232331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, & Piomelli D (2001). Receptor-dependent formation of endogenous cannabinoids in cortical neurons. European Journal of Pharmacology, 425(3), 189–196. 10.1016/S0014-2999(01)01182-7 [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, & Piomelli D (1997). A second endogenous cannabinoid that modulates long-term potentiation. Nature, 388(6644), 773–8. 10.1038/42015 [DOI] [PubMed] [Google Scholar]
- Stott C, White L, Wright S, Wilbraham D, & Guy G (2013). A Phase I, open-label, randomized, crossover study in three parallel groups to evaluate the effect of Rifampicin, Ketoconazole, and Omeprazole on the pharmacokinetics of THC/CBD oromucosal spray in healthy volunteers. SpringerPlus, 2(1), 236 10.1186/2193-1801-2-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BL, Levitt P, & Stanwood GD (2009). Prenatal exposure to drugs: effects on brain development and implications for policy and education. Nature Reviews Neuroscience, 10(4), 303–312. 10.1038/nrn2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd SM, & Arnold JC (2016). Neural correlates of interactions between cannabidiol and Δ(9) -tetrahydrocannabinol in mice: implications for medical cannabis. British Journal of Pharmacology, 173(1), 53–65. 10.1111/bph.13333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornqvist H, & Belfrage P (1976). Purification and some properties of a monoacylglycerol-hydrolyzing enzyme of rat adipose tissue. Journal of Biological Chemistry, 251(3), 813–9. [PubMed] [Google Scholar]
- Tortoriello G, Morris CV, Alpár A, Fuzik J, Shirran SL, Calvigioni D, Keimpema E, Botting CH, Reinecke K, Herdegen T, Courtney M, Hurd YL, & Harkany T (2014). Miswiring the brain: Δ9-tetrahydrocannabinol disrupts cortical development by inducing an SCG10/stathmin-2 degradation pathway. The EMBO Journal, 33(7), 668–85. 10.1002/embj.201386035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trezza V, Campolongo P, Cassano T, Macheda T, Dipasquale P, & Carratù MR (2008). Effects of perinatal exposure to delta-9-tetrahydrocannabinol on the emotional reactivity of the offspring: a longitudinal behavioral study in Wistar rats. Psychopharmacology, 198, 529–537. 10.1007/s00213-008-1162-3 [DOI] [PubMed] [Google Scholar]
- Trezza V, Campolongo P, Manduca A, Morena M, Palmery M, Vanderschuren LJMJ, & Cuomo V (2012). Altering endocannabinoid neurotransmission at critical developmental ages: impact on rodent emotionality and cognitive performance. Frontiers in Behavioral Neuroscience, 6, 1–12. 10.3389/fnbeh.2012.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner SE, Williams CM, Iversen L, & Whalley BJ (2017). Molecular Pharmacology of Phytocannabinoids. Progress in the Chemistry of Organic Natural Products, 103, 61–101. 10.1007/978-3-319-45541-9_3 [DOI] [PubMed] [Google Scholar]
- van Gelder MMHJ, Reefhuis J, Caton AR, Werler MM, Druschel CM, & Roeleveld N (2010). Characteristics of pregnant illicit drug users and associations between cannabis use and perinatal outcome in a population-based study. Drug and Alcohol Dependence, 109(1–3), 243–247. 10.1016/j.drugalcdep.2010.01.007 [DOI] [PubMed] [Google Scholar]
- Vitalis T, Lainé J, Simon A, Roland A, Leterrier C, & Lenkei Z (2008). The type 1 cannabinoid receptor is highly expressed in embryonic cortical projection neurons and negatively regulates neurite growth in vitro. European Journal of Neuroscience, 28(9), 1705–1718. 10.1111/j.1460-9568.2008.06484.x [DOI] [PubMed] [Google Scholar]
- Wall ME, Sadler BM, Brine D, Taylor H, & Perez-Reyes M (1983). Metabolism, disposition, and kinetics of delta-9-tetrahydrocannabinol in men and women. Clinical Pharmacology and Therapeutics, 34(3), 352–363. 10.1038/clpt.1983.179 [DOI] [PubMed] [Google Scholar]
- Warf B (2014). High Points: An Historical Geography of Cannabis. Geographical Review, 104(4), 414–438. 10.1111/j.1931-0846.2014.12038.x [DOI] [Google Scholar]
- Warner TD, Roussos-Ross D, & Behnke M (2014). It’s not your mother’s marijuana: Effects on maternal-fetal health and the developing child. Clinics in Perinatology, 41(4), 877–894. 10.1016/j.clp.2014.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshak C, Regan J, Moore B, Magner K, Kritzer S, & Hook J. Van. (2015). Association between marijuana use and adverse obstetrical and neonatal outcomes. Journal of Perinatology, 35(12), 991–995. 10.1038/jp.2015.120 [DOI] [PubMed] [Google Scholar]
- Watanabe K, Yamaori S, Funahashi T, Kimura T, & Yamamoto I (2007). Cytochrome P450 enzymes involved in the metabolism of tetrahydrocannabinols and cannabinol by human hepatic microsomes. Life Sciences, 80(15), 1415–1419. 10.1016/j.lfs.2006.12.032 [DOI] [PubMed] [Google Scholar]
- Westfall RE, Janssen PA, Lucas P, & Capler R (2009). Survey of medicinal cannabis use among childbearing women: Patterns of its use in pregnancy and retroactive self-assessment of its efficacy against “morning sickness.” Complementary Therapies in Clinical Practice, 15(4), 242–246. 10.1016/j.ctcp.2009.07.001 [DOI] [PubMed] [Google Scholar]
- Wise RA (2004). Dopamine, learning and motivation. Nature Reviews Neuroscience, 5(6), 483–494. 10.1038/nrn1406 [DOI] [PubMed] [Google Scholar]
- Wu C-S, Zhu J, Wager-Miller J, Wang S, O’Leary D, Monory K, Lutz B, Mackie K, & Lu HC (2010). Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. The European Journal of Neuroscience, 32(5), 693–706. 10.1111/j.1460-9568.2010.07337.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zammit S, Thomas K, Thompson A, Horwood J, Menezes P, Gunnell D, Hollis C, Wolke D, Lewis G, & Harrison G (2009). Maternal tobacco, cannabis and alcohol use during pregnancy and risk of adolescent psychotic symptoms in offspring. British Journal of Psychiatry, 195(4), 294–300. 10.1192/bjp.bp.108.062471 [DOI] [PubMed] [Google Scholar]
- Zuckerman B, Amaro H, & Cabral H (1989). Validity of self-reporting of marijuana and cocaine use among pregnant adolescents. The Journal of Pediatrics, 115 10.1016/S0022-3476(89)80668-7 [DOI] [PubMed] [Google Scholar]