

Abstract
The treatment of HIV and AIDS was revolutionized by the introduction of peptidomimetic aspartyl protease inhibitors. One of the major limitations of this type of therapy is that higher therapeutic doses are necessary because of the presence of ‘peptide-like’ features in the drugs. Therefore, adequate supplies and cost effective syntheses of these drugs are of utmost importance. To date, there are six protease inhibitors approved by the United States Food and Drug Administration (FDA) for the treatment of HIV and AIDS. This review focuses on the published syntheses of currently FDA approved HIV protease inhibitor drugs, their isosteres and ligands.
Keywords: HIV, protease, inhibitor, isostere, synthesis
1. Introduction
Acquired immunodeficiency syndrome (AIDS), a degenerative disease of the immune system, is one of the most challenging problems in medicine. The Centers for Disease Control estimated that at the end of 2000, there were 36.1 million people living with the disease and an estimated 21.8 million have died since the beginning of the epidemic, 3 million in the year 2000 alone. Among various strategies to combat this devastating disease, therapeutic inhibition of the virally encoded HIV protease became an attractive target.1 During viral replication, gag and gagpol gene products are translated as precursor polyproteins. These proteins are processed by a virally encoded pro-tease to provide structural proteins (p 17, p 24, p 9, and p7) and essential viral enzymes (including protease, reverse transcriptase, and integrase).2 The virally encoded pro-tease has been characterized as a homodimeric endopeptidase of the aspartyl protease family.3
Based on the transition state mimetic concept utilizing various nonhydrolyzable hydroxyethylene and hydroxyethylamine isosteres, an impressive number of potent and selective HIV protease inhibitors have been developed.1,4 The clinical effectiveness of combination therapy consisting of protease inhibitors and reverse transcriptase inhibitors has now been well documented.5 These treatment regimens have changed the course of HIV management and the progression of AIDS.6 One of the major limitations of the current therapy is that higher therapeutic doses are necessary because of the presence of ‘peptide-like’ features in the drugs. Consequently, adequate supplies and cost effective syntheses of these drugs are of utmost importance. To date, there are six protease inhibitors approved by the United States Food and Drug Administration, (FDA) for the treatment of HIV and AIDS. All of these inhibitor drugs contain multiple stereocenters, complex heterocycles and rare functionalities. No doubt, large scale synthesis in enantiomerically pure form posed considerable challenges as well as opportunities for organic synthesis. An incredible effort has been carried out by academic and pharmaceutical laboratories in the area of design and synthesis of HIV protease inhibitors. It is beyond the scope of this review to cover all the synthetic work in this area. This review is intended to focus on the syntheses of current FDA approved inhibitor drugs, their isosteres and ligands. Herein, we report a full review of the published syntheses of these important therapeutics.
2. Saquinavir (Invirase®, Fortovase®, Ro 31–8959)
In December 1995, the FDA approved the first HIV pro-tease inhibitor for the treatment of AIDS, saquinavir (tradenames Invirase®, Fortovase®). Saquinavir (1, Figure 1), discovered by Hoffman-La Roche, displays an IC50 value of 2 nM and a Ki value at pH 5.5 of 0.12 nM against HIV-1; moreover, it is a very selective inhibitor of the HIV protease, showing less than 50% inhibition of the other aspartic proteases found in humans.7
Figure 1.
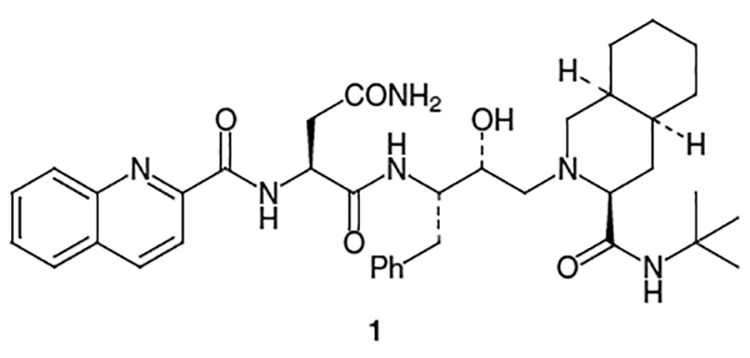
Structure of Saquinavir (Invirase®, Fortovase®, Ro 31–8959)
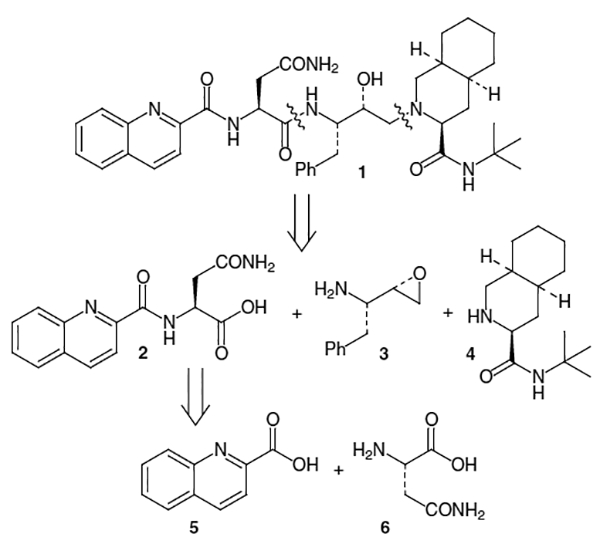
Most syntheses of saquinavir (1) utilize a convergent strategy based on the following disconnections: (i) carboxylic acid moiety 2, which can be derived from quinaldic acid (5) and l-asparagine (6), (ii) an electrophilic amino alcohol equivalent such as l-phenylalanine-based isostere 3 and (iii) decahydroisoquinoline derivative 4 (Figure 2).
Figure 2.
Retrosynthesis of Saquinavir
2.1. Synthesis of the Saquinavir Isostere
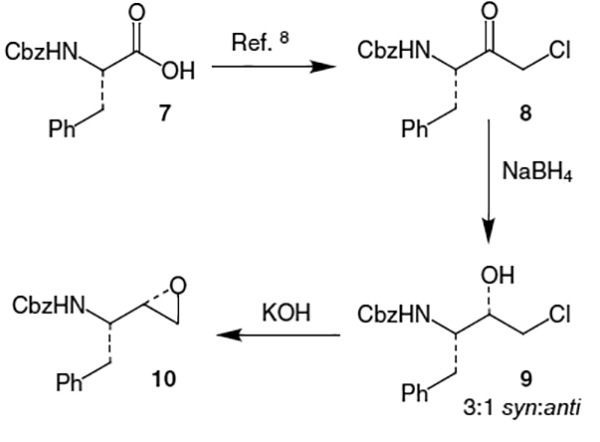
An early synthesis of the saquinavir isostere by scientists at Roche began with benzyloxycarbonyl (Cbz) protected l-phenylalanine (7) which was transformed into corresponding chloromethyl ketone 8 using established procedures (Scheme 1).8 Chloromethyl ketone 8 was then reduced with NaBH4 to afford a 3:1 mixture of diastereomers in favor of the desired (S)-alcohol (9) which was separated from the minor (R)-isomer. To complete the isostere synthesis, epoxide 10 was formed by the reaction of KOH with chloroalcohol 9.9
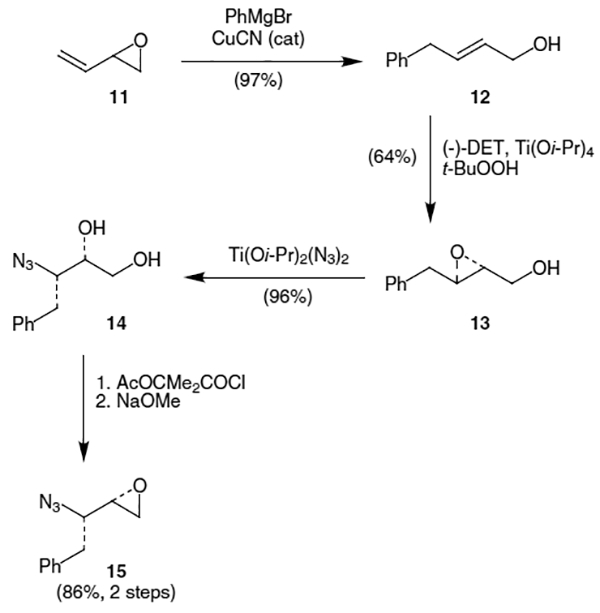
A number of other methods have been developed to produce an N-protected form of (2S,3S)-amino epoxide 10, with azidoepoxides being the most common targets. In 1993, Ghosh and co-workers published an efficient synthesis of azidoepoxide 15 utilizing Sharpless asymmetric epoxidation (SAE).10 Their first step was the copper cyanide-catalyzed addition of phenylmagnesium bromide to oxirane 11, which provided allylic alcohol 12 in 97% yield. This was followed by SAE using (−)-diethyl-D-tartrate [(−)-DET] which afforded epoxide 13 in 64% yield (Scheme 2). Regioselective epoxide ring opening with [Ti(Oi-Pr)2(N3)2], which was prepared in situ by refluxing Ti(Oi-Pr)4 and Me3SiN3 in benzene for 5 hours,11 provided azidodiol 14 in 96% yield. Finally, epoxide 15 was formed in 86% yield by treatment of 14 with 2-acetoxy-isobutyryl chloride followed by excess NaOMe.
An alternative synthesis published by Bennet et al. also utilizes SAE to introduce chirality into the molecule, but uses a slightly different procedure for forming the allylic alcohol.12 In their synthesis, phenylacetaldehyde (16) is subjected to Horner–Emmons homologation with triethylphosphonoacetate to afford E-unsaturated ester 17 which was subsequently reduced using diisobutylaluminum hydride (Dibal-H) to provide allylic alcohol 12 (Scheme 3). The corresponding azidodiol (14) was produced by a similar sequence of reactions as the Ghosh synthesis above. Reaction of 14 with p-toluenesulfonyl chloride (TsCl) followed by NaH produced azidoepoxide 15 in 70% yield.
Figure 3.
Structure and Retrosynthesis of Nelfinavir (Viracept®, AG1343)
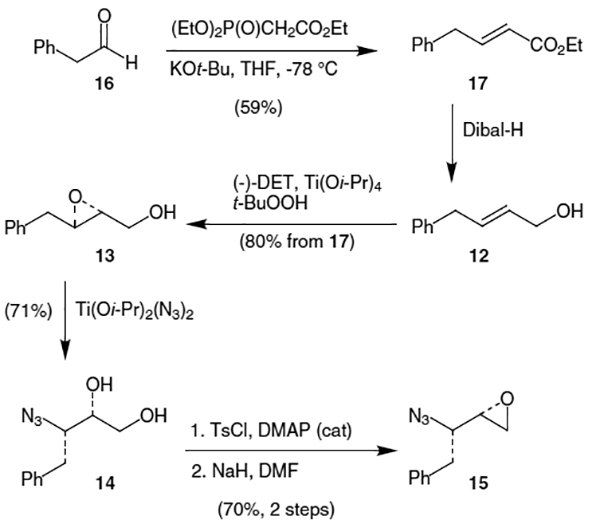
Ghosh and co-workers have also prepared azidoepoxide 15 starting instead from diethyl-D-tartrate.13 Thus, reduction of benzylidene protected (−)-DET (18)14 with lithium aluminum hydride (LAH) and AlCl3 gave the corresponding benzyl protected triol, which was protected as the corresponding acetonide with acetone and catalytic p-toluenesulfonic acid (TsOH) to afford alcohol 19 in 88% yield over 2 steps (Scheme 4).15 Epoxide 20 was produced in 77% yield by removal of the O-benzyl protecting group with Pd(OH)2 (Pearlman’s Catalyst) and H2 followed by Mitsunobu condensation of the resulting 1,2-diol with triphenylphosphine and diethylazodicarboxylate (DEAD). Copper cyanide-catalyzed addition of phenyl-magnesium bromide regioselectively produced alcohol 21 in 93% yield. Mitsunobu azidation of the resulting alcohol with PPh3, diphenylphosphoryl azide (DPPA) and DEAD afforded the corresponding protected azide, which was deprotected with 40% aqueous acetic acid to furnish azidodiol 14 in 64% yield over 2 steps.16 The desired azidoepoxide (15) was synthesized in 72% yield using 2-acetyoxyisobutyryl chloride followed by excess NaOMe, while the diastereomeric epoxide (22) was produced via successive treatment of 14 with benzoyl chloride (BzCl), MeSO2Cl (MsCl) and NaOMe in 66% yield.
Figure 4.
Structure and Retrosynthesis of Amprenavir (Agenerase®, VX-478, 141W94)
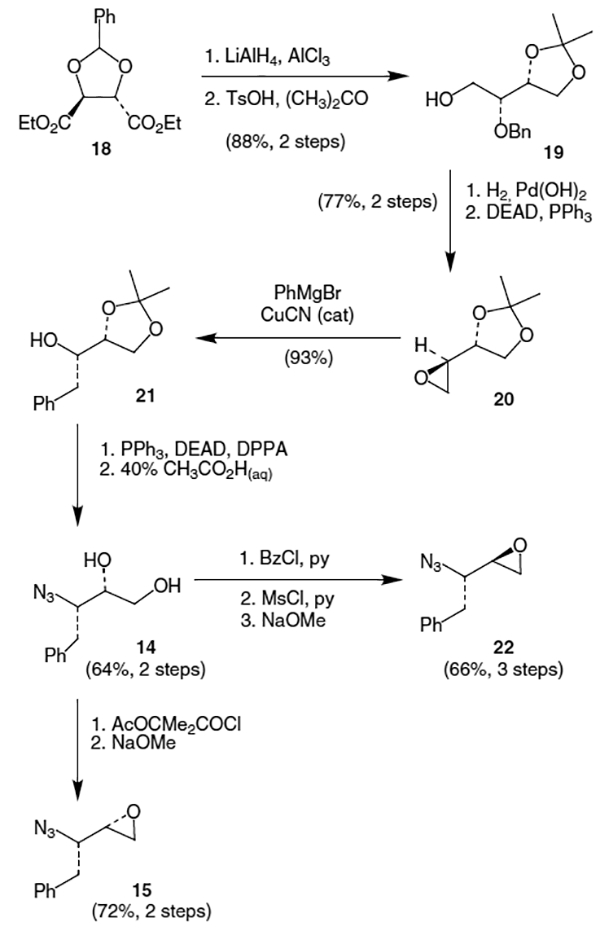
Several different synthetic equivalents of 15 have been developed with t-butoxycarbonyl (Boc) and benzyloxycarbonyl (Cbz) protected amino epoxides being quite common. As shown in Scheme 5, N-Boc-l-phenylalanine (23a) and N-Cbz-l-phenylalanine (23b) were treated with carbonyldiimidazole (CDI) followed by the magnesium enolate of malonic acid monoester to afford -keto esters 24a and 24b, respectively.17 Sodium borohydride reduction of 24a and 24b afforded 3:1 mixtures of diastereomers with the desired (R)-alcohols being the major products.9 After separation, protection as the corresponding acetonide using 2,2-dimethoxypropane (DMP) and TsOH was followed by NaOH hydrolysis to afford acids 25a and 25b. Treatment of the resulting acids with oxalyl chloride produced the corresponding acid chlorides. Conversion to bromides 26a and 26b was accomplished via the Barton decarboxylative bromination reaction utilizing 2-mercaptopyridine N-oxide sodium salt.18 Bromides 26a and 26b were then deprotected with glacial acetic acid and concentrated HCl followed by cyclization with methanolic KOH to afford epoxides 27a and 27b.
Figure 5.
Structure of Indinavir (Crixivan®, L-735,524, MK-639)
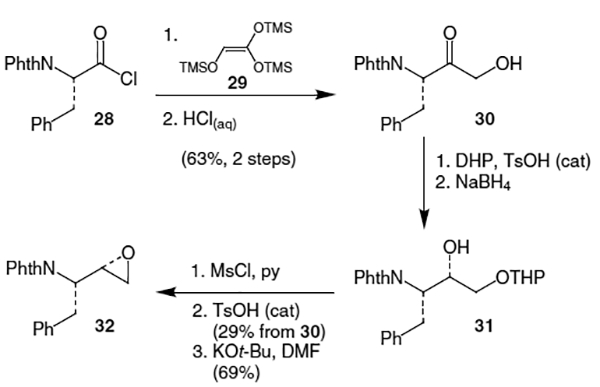
Another approach to N-protected epoxides involved the use of tris-((trimethylsilyl)oxy)ethene (29).19 In this synthesis,9 treatment of N-phthalyl protected acid chloride 28 with 2 equivalents of tris-((trimethylsilyl)oxy)ethene (29) at 95–100 °C followed by aqueous HCl produced α-hydroxymethyl ketone 30 in 63% yield (Scheme 6). Protection of the alcohol as a tetrahydropyranyl (THP) ether using dihydropyran (DHP) and a catalytic amount of TsOH followed by reduction of the ketone with NaBH4 produced alcohol 31. Mesylation with MsCl followed by removal of the THP group using TsOH gave the corresponding mesylate in 29% yield from 30. Epoxide ring formation was accomplished by treatment with t-BuOK, which afforded 32 in 69% yield.
Figure 6.
Retrosynthesis of Indinavir Fragments (Merck)
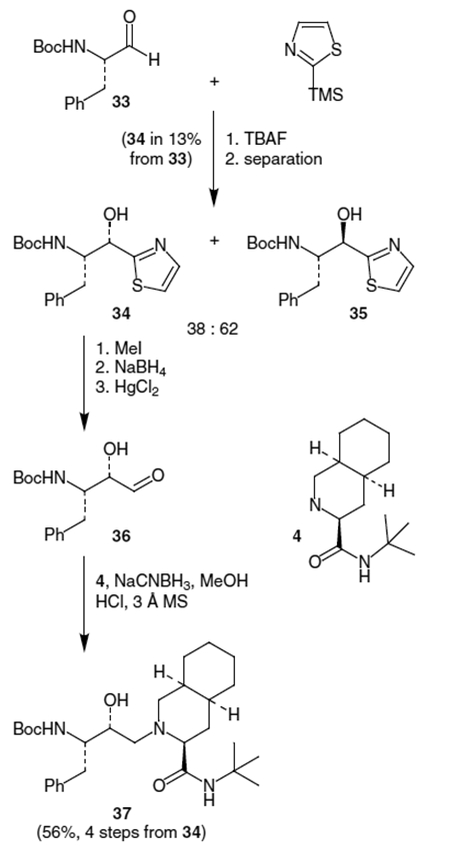
An alternative synthetic equivalent of 15 was produced by formation of an α-hydroxy aldehyde 36, which can then undergo a reductive coupling with decahydroisoquinoline derivative 4 using NaCNBH3.9 In the event, N-Boc-protected l-phenylalanal (33) was reacted with 2-(trimethylsilyl)thiazole followed by desilylation with tetrabutylammonium fluoride (TBAF) to give a mixture of diastereomeric alcohols (34) and (35) in a ratio of approximately 2:3 (Scheme 7). Methylation of the thiazole nitrogen followed by NaBH4 reduction and hydrolysis of the thiazolidine using HgCl2 gave α-hydroxy aldehyde 36. This compound was then coupled with 4 in the presence of NaCNBH3 to produce 37 in 56% overall yield from 34.
Figure 7.
Coupling of Epoxide for Indinavir Synthesis
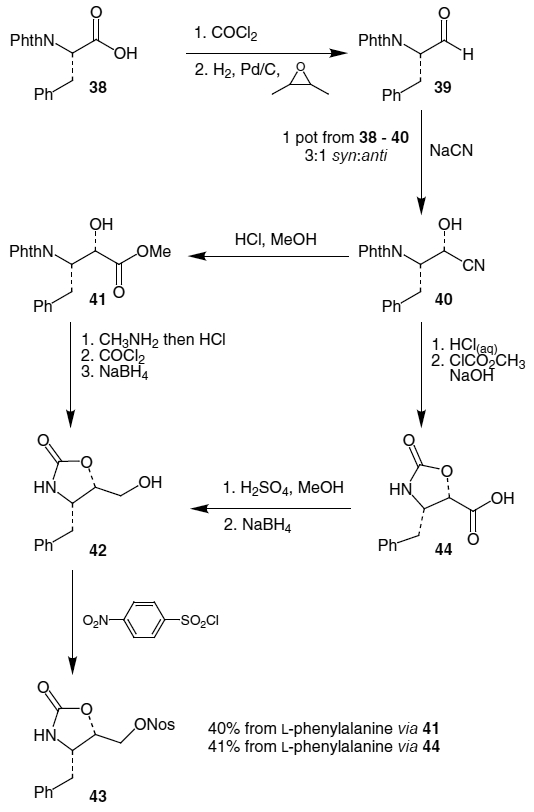
An improved method for preparing an electrophilic hydroxyamine equivalent starting from an acid chloride was published by Göhring et al. in 1996.20 Starting with N-phthalyl-l-phenylalanine (38), conversion to the acid chloride with phosgene followed by reduction with H2 and Pd/C produced aldehyde 39 (Scheme 8). Interestingly, the use of bases such as triethylamine and lutidine in the Rosenmund reaction led to high degrees of racemization, therefore, butylene oxide was used to neutralize the HCl produced during the reaction. Formation of cyanide 40 was accomplished using NaCN and led to a 3:1 mixture of diastereomers in favor of the (S)-alcohol which, when treated with HCl and methanol, yielded hydroxyester 41. After removing the phthalyl group using methylamine and HCl, oxazolidinone 42 was produced by reaction of the aminoalcohol with COCl2 followed by reduction of the ester using NaBH4. Reaction of oxizolidinone 42 with p-nitrobenzenesulfonyl chloride (NosCl) afforded nosylate 43 in 40% overall yield from phenylalanine.
Figure 8.
Structure of Ritonavir 167 (Norvir®, ABT-538) and Retrosynthesis of Isostere Fragment 168
An alternative procedure was developed which eliminated the need for phosgene and also reduced the number of steps required. In this synthesis (also found in Scheme 8), following transformation of cyanide 40 to the corresponding carboxylic acid using HCl and water, oxazolidinone 44 was produced by reaction with methyl chloroformate.20 Formation of the methyl ester with MeOH and H2SO4 followed by reduction with NaBH4 led to oxazolidinone 42 which was transformed into the nosylate as above, this time in 41% yield from phenylalanine.
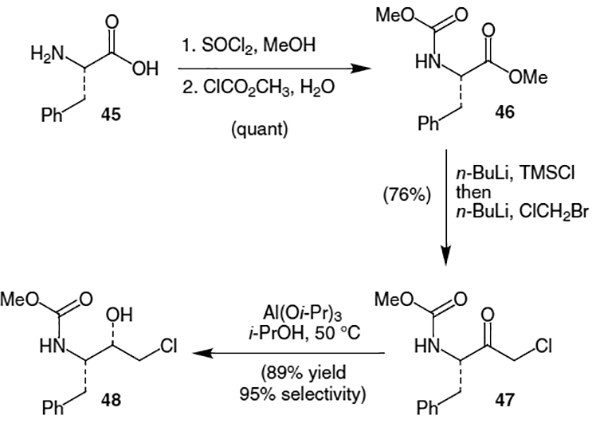
The most efficient synthesis of the central isostere containing portion of saquinavir and the one used to produce the large quantities necessary for clinical development, begins with l-phenylalanine (45).20 Construction of the methyl ester using thionyl chloride and methanol followed by treatment with methyl chloroformate resulted in quantitative formation of methyl ester 46 (Scheme 9). A one-pot sequence involving protection of the carbamate group using n-BuLi and TMSCl followed by reaction with chloromethyllithium (derived from n-BuLi and bromochloromethane) afforded chloromethyl ketone 47 in 76% yield after aqueous workup which also removed the NTMS group. Reduction selectivity was improved from the usual 3:1 obtained using NaBH4 to 19:1 by the use of Al(i-PrO)3. Therefore, Meerwein–Ponndorf–Verley reduction of chloromethyl ketone 47 using Al(i-PrO)3 and iso-propanol provided (S)-alcohol 48 in 89% yield and with 95% selectivity. Corey reduction of chloromethyl ketone 47 using a chiral oxazaborolidine catalyst also showed excellent selectivity (95:5) but required reagents deemed too expensive for large-scale production.
Figure 9.
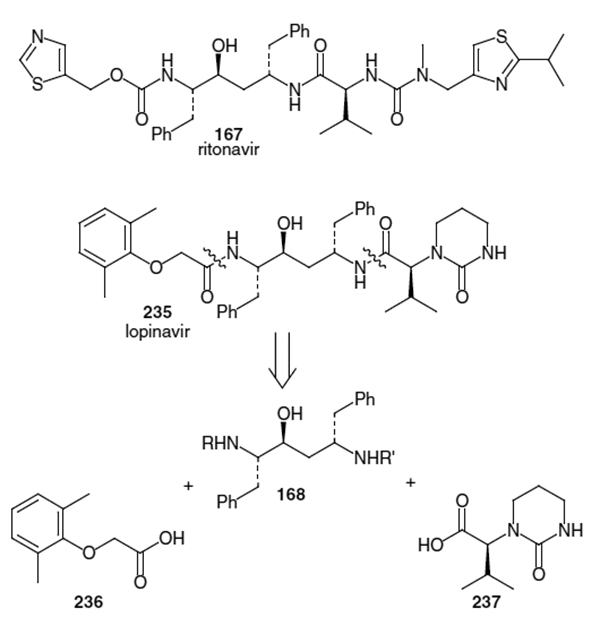
Structures of Ritonavir 167 (Norvir®, ABT-538), Lopinavir 235 (Aluviran®, ABT-378, Component of Kaletra®) and Retrosynthesis of Lopinavir
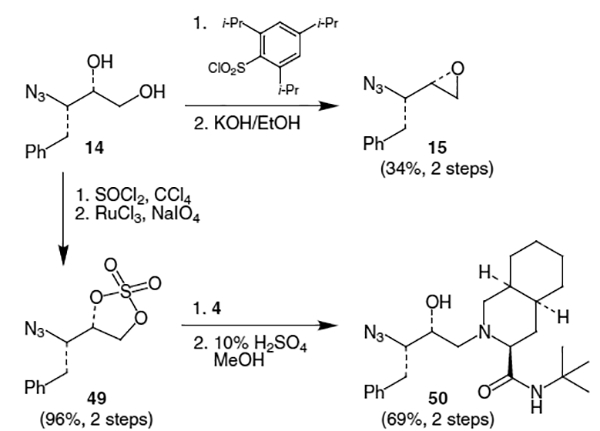
Several other approaches to synthetic equivalents of 15 have been used. Starting from azidodiol 14, reaction with triisopropylbenzenesulfonyl chloride gave the primary arylsulfonyl derivative which was cyclized with KOH to afford epoxide 15 in 34% yield for 2 steps (Scheme 10).9 Alternatively, azidodiol 14 was reacted with SOCl2 to afford the cyclic sulfite derivative, which was oxidized using RuCl3/NaIO4 to provide cyclic sulfate 49 in 96% yield over 2 steps. After coupling with decahydroisoquinoline derivative 4, the cyclic sulfate derivative was hydrolyzed using methanolic sulfuric acid. An advantage of the cyclic sulfate route is its higher reactivity in the coupling reaction with 4. While epoxides typically required elevated temperatures to react, 49 proceeded to give 69% of azidodecahydroisoquinoline 50 at room temperature.
Scheme 10.
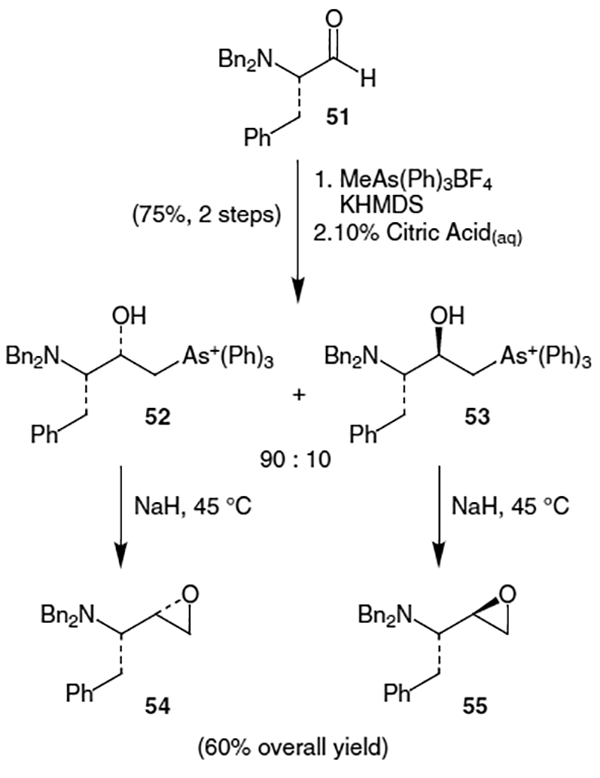
A method was developed by Reetz and Binder that provides good epoxide diastereoselectivity directly from α-amino aldehydes, which in turn can be derived from their corresponding amino acids.21 In their procedure it is essential that the nitrogen be doubly protected to afford the non-chelation controlled syn-addition product as the major isomer. Beginning with N,N-dibenzyl protected l-phenylalanal (51), addition of the arsonium ylide (derived from methyltriphenylarsonium tetrafluoroborate and potassium hexamethyldisilylazide (KHMDS)) provided a 90:10 mixture of syn:anti diastereomers (52) and (53) in 75% combined yield (Scheme 11). Reaction of these with NaH afforded the corresponding epoxides in 60% overall yield from aldehyde 51. While these results demonstrate an efficient diastereoselective synthesis of α-amino ep-oxides in 2 steps from their respective aldehydes, the conditions employed and toxicity of arsenic were thought by scientists at Hoffmann–La Roche to be too severe for application in large scale synthesis.9
Scheme 11.
2.2. Synthesis of the Decahydroisoquinoline Fragment of Saquinavir
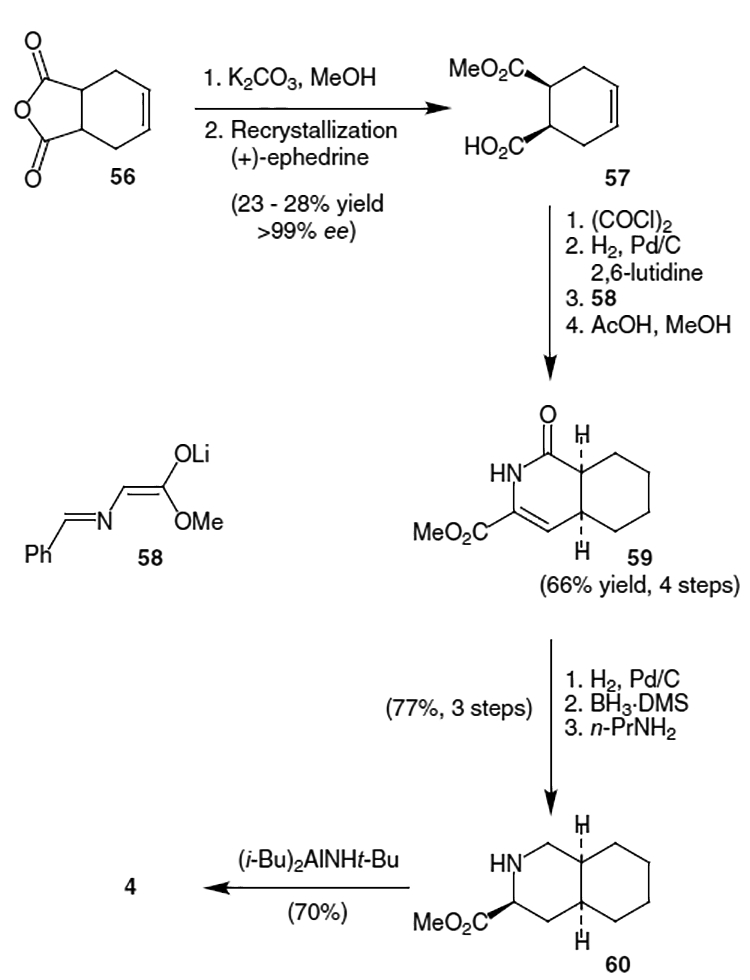
Several syntheses of the decahydroisoquinoline portion of saquinavir have been published. Houpis et al. published a synthesis in 1993 which begins with meso tetrahydrophthalic anhydride (56).22 Hydrolysis with K2CO3 and MeOH followed by recrystallization using (+)-ephedrine gave a single enantiomer of acid 57 in approximately 25% yield (2 steps) and >99% ee (Scheme 12). Formation of 59 was accomplished in 66% yield from 57 by treating the acid with oxalyl chloride, hydrogenating the resulting compound over Pd/C with 2,6-lutidine, addition of eno-late 58 to the resulting aldehyde, and cyclizing with acetic acid in methanol. Saturation of the conjugated alkene in 59 by hydrogenation over Pd/C in THF followed by reduction of the amide carbonyl using BH3 DMS and n-PrNH2 provided α-amino ester 60 in 77% yield from 59. Finally, the t-butyl amide was installed by reaction of 60 with the aluminum reagent derived from Al(i-Bu)3 and t-BuNH2 to afford 4 in 70% yield.
Scheme 12.
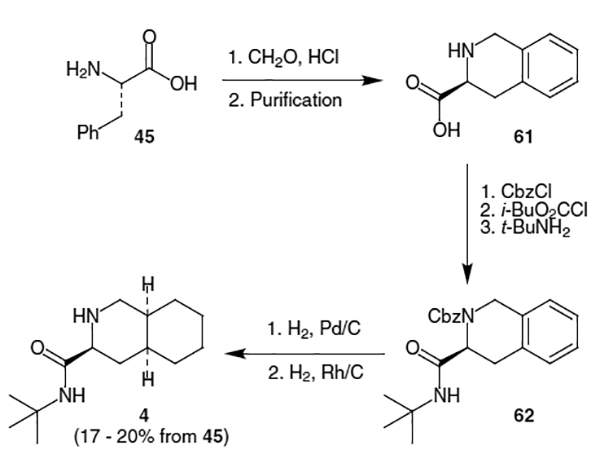
Another synthesis of the decahydroisoquinoline fragment begins with l-phenylalanine (45), which undergoes a Pictet–Spengler cyclization to produce acid 61 as shown in Scheme 13.20 Significant racemization took place during this reaction which required purification via the p-TsOH salt. Protection of the amine as the benzyl carbamate followed by displacement of the mixed carbonate (derived from iso-butyl chloroformate) with t-BuNH2 provided amide 62. Removal of the Cbz group using H2 and Pd/C followed by dearomatization with a Rh/C catalyst at elevated H2 pressure led to decahydroisoquinoline 4 in 17–20% overall yield from phenylalanine.
Scheme 13.
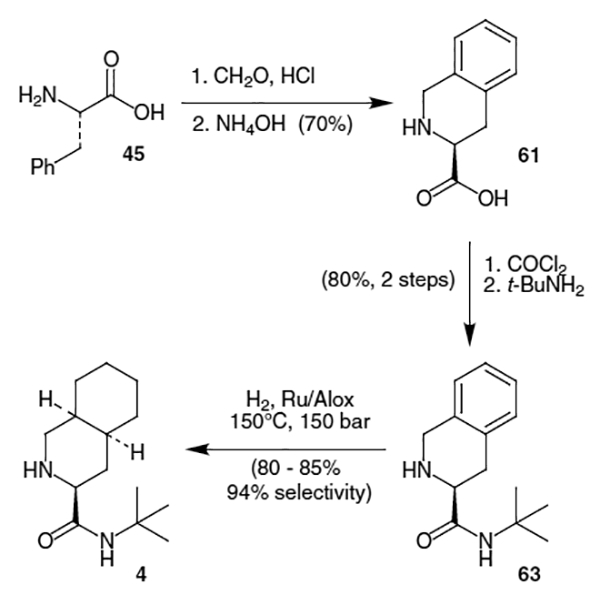
An improved synthesis of decahydroisoquinoline fragment 4 was developed by Hipert and co-workers at Hoffmann–La Roche which decreased the number of steps to 3 and increased the yield to 46%.20 Beginning with l-phenylalanine (45), Pictet–Spengler cyclization using highly concentrated HCl in formaldehyde proceeded with very little racemization to provide acid 61 in 70% yield after recrystallization of the free amino acid to assure enantiopurity (Scheme 14). Amide 63 was formed in 80% yield by reaction of 61 with COCl2 followed by t-BuNH2. Finally, hydrogenation at high temperature and pressure with a Ru catalyst afforded 4 in up to 85% yield and with 94% selectivity to complete the synthesis of the decahydroisoquinoline fragment.
Scheme 14.
2.3. Synthesis of Saquinavir
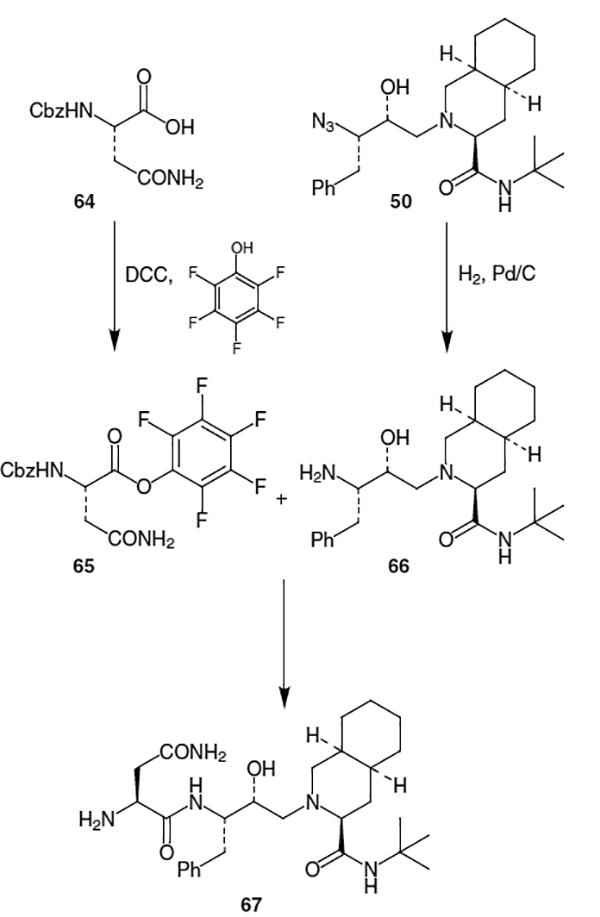
The remaining portion of saquinavir, fragment 2, is usually constructed from quinaldic acid (5) and l-asparagine (6). An early synthesis of saquinavir completed this portion by beginning with Cbz-protected l-asparagine (64) and coupling it to pentafluorophenol using dicyclohexylcarbodiimide (DCC) to provide 65 (Scheme 15).20 Reaction of pentafluorobenzene ester 65 with 66 gave compound 67 (Scheme 15), which subsequently was coupled to 68 (produced by the activation of quinaldic acid (5) using N-hydroxysuccinimide (NHS) and DCC, Scheme 16), to produce saquinavir (1).
Scheme 15.
Scheme 16.
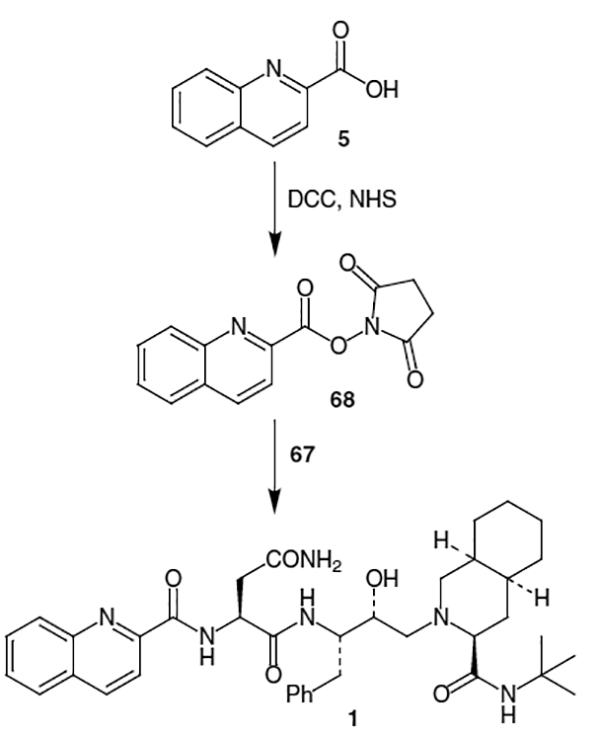
A more convergent approach, which also decreased the number of steps has been developed.20 In this synthesis, quinaldic acid (5) was coupled to l-asparagine via activation with N-hydroxysuccinimide to provide 2 in 82–85% yield for the 4 step coupling procedure (Scheme 17). The quinargine thus produced was reacted with 67 and DCC along with a catalytic amount of N-hydroxypyridone to afford saquinavir mesylate (69) in 81% yield after recrystallization of the mesylate salt.
Scheme 17.
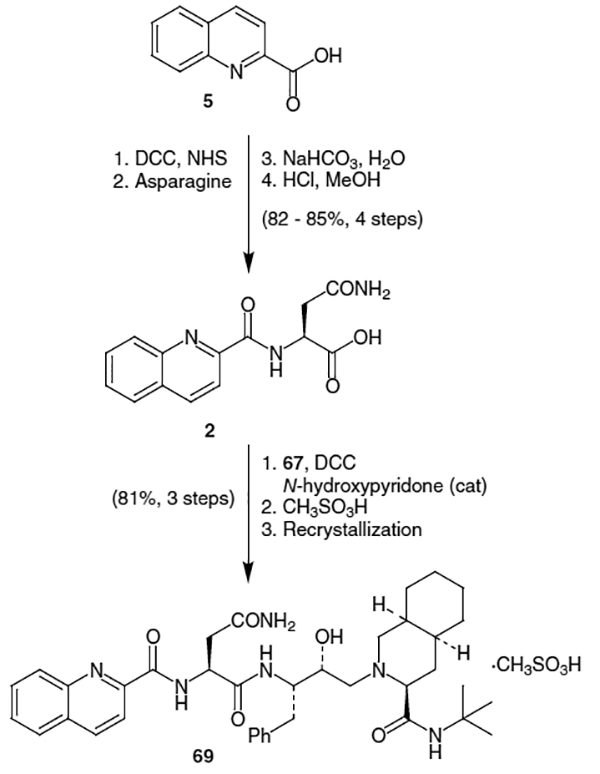
An alternative procedure for the coupling of decahydroisoquinoline derivative 4 with azidoepoxide 15 in good (75%) yield, which does not require heating has been published.23 In this scheme, silica gel is added to a chloroform solution of amine 4 and epoxide 15, and after removal of the solvent under reduced pressure, allowed to stand for 3 days (Scheme 18). Azidodecahydroisoquinoline 50 can be isolated by simply adding the solid to a silica gel column and purifying. This method may have advantages when using more temperature sensitive substrates.
Scheme 18.
3. Nelfinavir (Viracept®, AG1343)
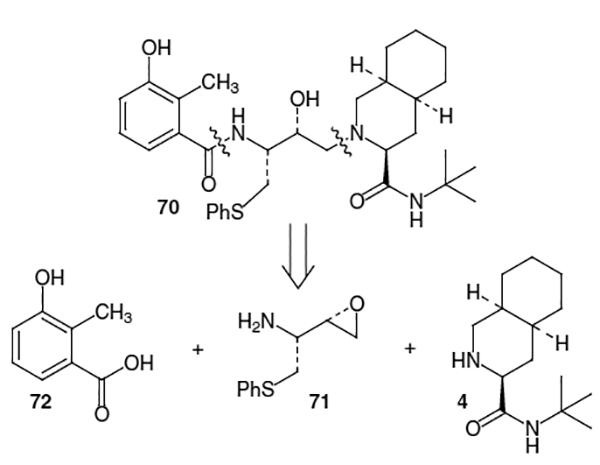
Nelfinavir mesylate, Viracept®, approved in March, 1997 is another potent inhibitor of the HIV protease enzyme, which contains a hydroxyethylamine isostere. Marketed by Agouron Pharmaceuticals, nelfinavir (70, Figure 3) exhibits an ED50 value of 14 nm.24
As seen in Figure 3, nelfinavir (70) can be disconnected into three units. Fragment 4 is the same decahydroisoquinoline contained in saquinavir. The transition state mimic in nelfinavir, 71, is a hydroxyethylamine isostere with an S-phenyl substituent, which replaces the phenyl group found in the saquinavir isostere. Finally, the isostere is coupled with 3-hydroxy-2-methylbenzoic acid (72), or a suitable equivalent.
3.1. Synthesis of the Nelfinavir Isostere
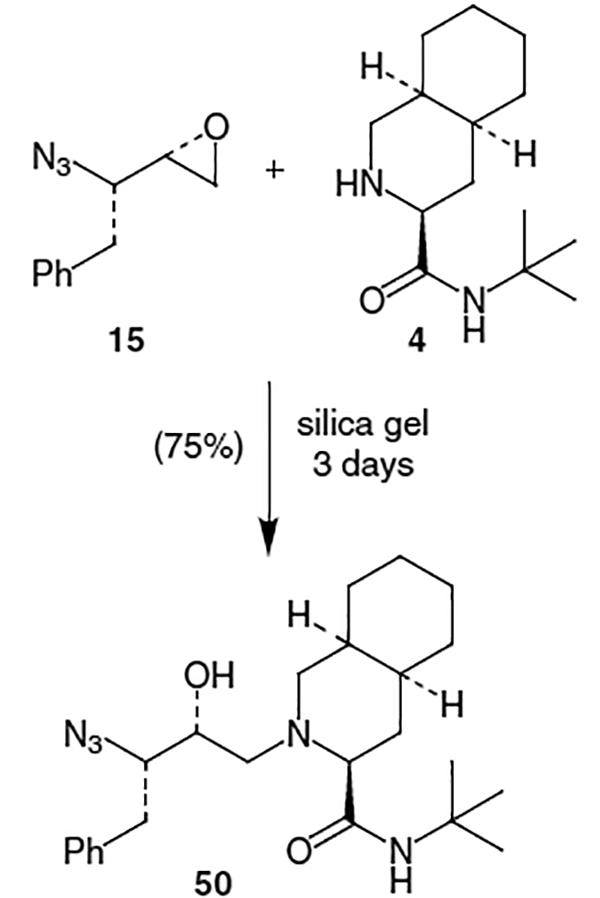
The main challenge in the synthesis of nelfinavir involves construction of the unusual hydroxyethylyamine isostere (71). One approach to the nelfinavir isostere utilizes 1,3-dithiane.25 Reiger’s synthesis begins with N-Cbz-S-phenyl-l-cysteine (73), produced by a previously described method.26 Synthesis of the N,N-dimethyl amide proceeded by exposure of 73 to pivaloyl chloride followed by in situ displacement of the mixed anhydride by dimethylamine and afforded amide 74 in 98% yield (Scheme 19). Addition of the anion derived from the treatment of 1,3-dithiane with n-BuLi to dimethyl amide 74 followed by reduction with NaBH4 afforded alcohol 75 in 70% isolated yield from a 4.5:1 mixture of diastereomers. Removal of the dithiane group was effected by treatment with Hg(ClO4)2 3H2O to give unstable aldehyde 76 which was immediately used in the following reductive amination step. Thus, treatment of 76 with 4, molecular sieves, and NaBH4 yielded 77 in 68% yield and 83% ee from 75. Amino alcohol 77 could then be converted to nelfinavir (70) by established procedures.27
Scheme 19.
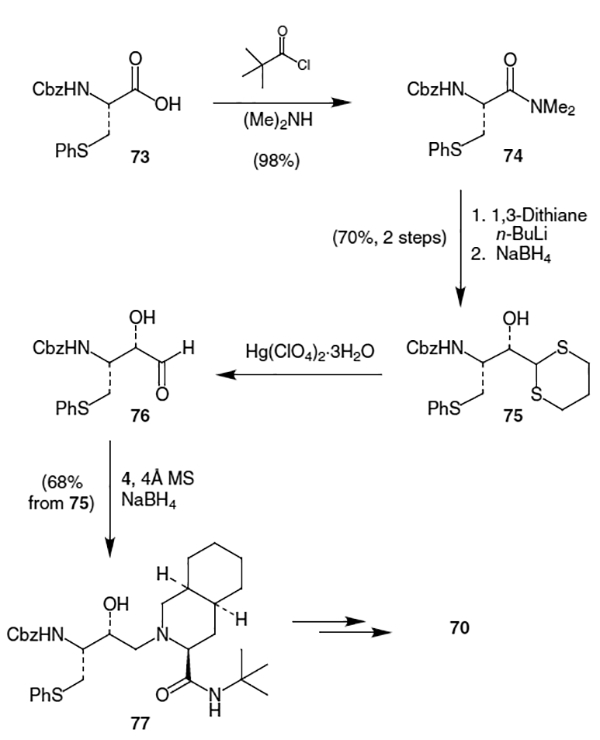
In another approach, a chiral 2-amino-1,3,4-butanetriol was used as a building block in the synthesis of nelfinavir.28 This synthesis began with readily available 4,4-dimethyl-3,5,6-trioxa-bicyclo[5.1.0]octane (78), which underwent asymmetric ring opening using a chiral Ti catalyst to provide amino alcohol 79 in nearly quantitative yield and >97% de (Scheme 20).29 Amino alcohol 79 was then transformed into protected amino triol benzoic acid salt 80 in 82% yield by the following one pot procedure: conversion to the more thermodynamically stable dioxolane using 2,2-dimethoxypropane (DMP) and MsOH in acetone followed by debenzylation with Pd/C and H2 in the presence of benzoic acid. Amine 80 was protected as the benzyl carbamate using CbzCl and K2CO3. Replacement of the free alcohol by an S-phenyl group was accomplished by first forming the mesylate with MsCl and TEA followed by displacement using thiophenol/NaOH/Bu4NBr which afforded 81. The acetonide was then removed from 81 by refluxing with HCl in MeOH/H2O and the resulting primary alcohol was selectively protected as the p-nitrobenzyl (PNB) ether using PNBCl and 2-picoline to provide alcohol 82. Reaction of alcohol 82 with MsCl provided the corresponding mesylate, which was then epoxidized by in situ removal of the PNB protecting group using KOH to afford 83 in 82% yield from 80, thus completing the isostere synthesis.
Scheme 20.
There have been many reports of syntheses of unnatural S-phenyl cysteine in the literature. Some of the methods described include: opening of a serine derived -lactone intermediate with a thiophenol based nucleophile,26,30 displacement of a serine derived tosylate by a thiolate anion,31 opening of an aziridinecarboxylic acid by thiophenol,32 Michael addition of thiophenol to a chiral nickel complex incorporating an electrophilic alanine-type substituent,33 and an enzymatic process using cysteine desulfhydrase.34
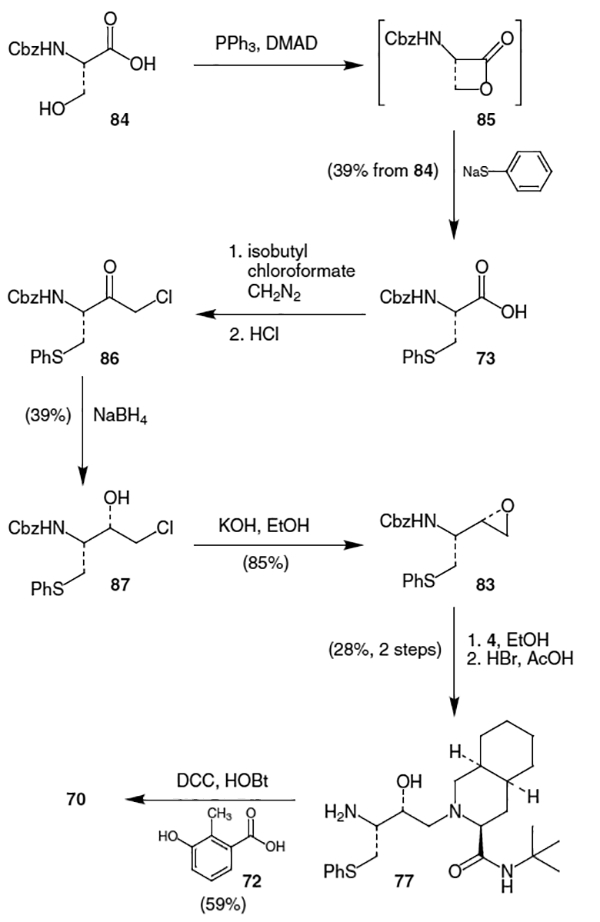
One synthesis of nelfinavir created the isostere via a -lactone ring opening using a thiophenol nucleophile. In the synthesis by Kaldor et al. in 1997, treatment of Cbz protected l-serine (84) with PPh3 and dimethylazidodicarboxylate (DMAD) produced lactone 85, which was then reacted in situ with the thiolate anion derived from thiophenol and NaH to afford Cbz protected S-phenyl cysteine (73) in 39% overall yield (Scheme 21).24 Reaction of 73 with iso-butylchloroformate followed by CH2N2 generated the diazoketone in 73% yield, which was subsequently treated with HCl to provide chloromethyl ketone 86. Sodium borohydride reduction of ketone 86 produced chloro alcohol 87 in 39% isolated yield from a mixture of diastereomers. Base induced cyclization of alcohol 87 using KOH/EtOH afforded epoxide 83 in 85% yield to complete the synthesis of the isostere portion of nelfinavir.
Scheme 21.
3.2. Synthesis of Nelfinavir
Construction of nelfinavir then proceeded via coupling of epoxide 83 with decahydroisoquinoline derivative 4 in refluxing ethanol followed by Cbz deprotection with HBr/AcOH to afford 77 in 28% yield from 83 (Scheme 21). Finally, reaction of 77 with 3-hydroxy-2-methylbenzoic acid (72) in the presence of DCC and 1-hydroxybenzotriazole (HOBt) provided nelfinavir (70) in 59% yield.
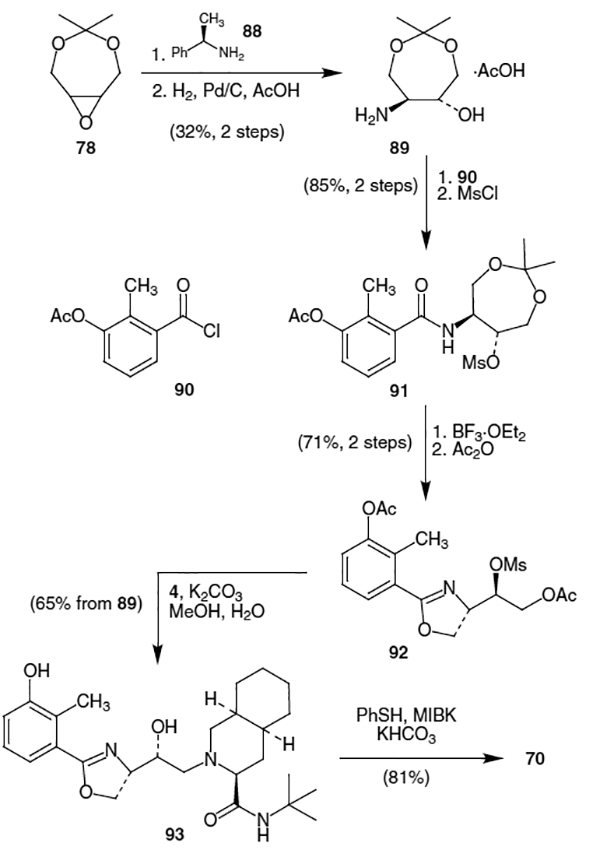
A different approach to the synthesis of nelfinavir proceeded through an oxazoline intermediate.35 Thus, epoxide 78 was opened with (R)-methylbenzylamine (88) to afford the trans amino alcohol in 39% yield, which was subsequently hydrogenated with H2, Pd/C and AcOH to give amino alcohol acetate salt 89 (Scheme 22). Reaction of 89 with 3-acetoxy-2-methylbenzoyl chloride (90) gave the corresponding amido alcohol, which, upon treatment with MsCl, yielded mesylate 91. Addition of BF3 OEt2 to a solution of 91 followed by quenching with acetic anhydride produced oxazoline 92 in 71% yield. Heating a solution of 92, K2CO3 and amine 4 in methanol at 50 °C afforded 93 in 65% yield from 89. Oxazoline 93 was then converted to nelfinavir (70) in 81% yield by reaction with thiophenol using KHCO3 as base and methyl iso-butylketone (MIBK) as solvent. It is worth noting that the reaction conditions employed were critical. The use of ethylene glycol as solvent with no base for example, led to product formation in a ratio of 18:82 (nelfinavir–regioisomer) while the conditions stated above led to a 92:8 product distribution.
Scheme 22.
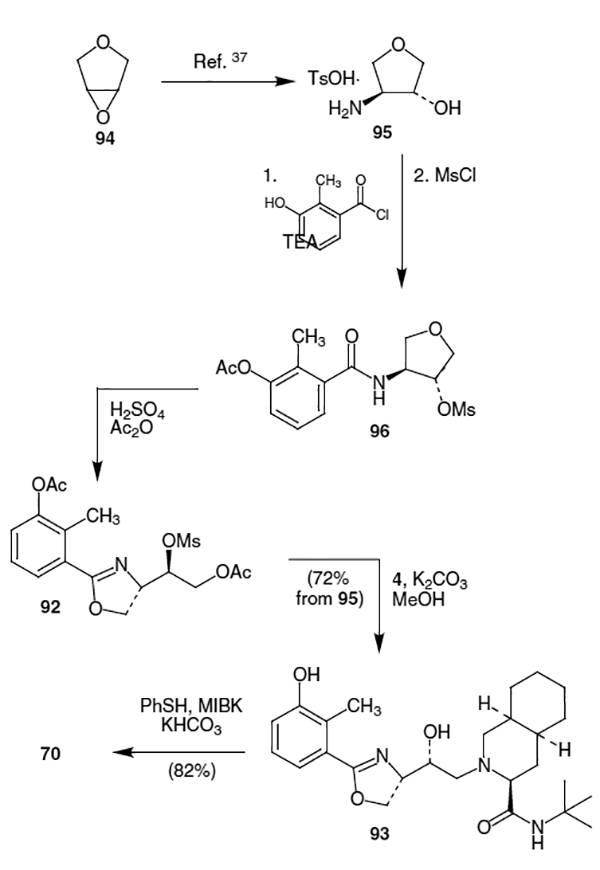
A similar approach to nelfinavir that also goes through an oxazoline intermediate was published by Zook et al. in 2000.36 Beginning with epoxide 94, catalytic asymmetric ring opening with azidotrimethylsilane produced the corresponding azide in 96% yield and >99% ee.37 The corresponding product was subsequently deprotected, hydrogenated and converted to the corresponding tosylate salt (95) (Scheme 23). Treatment of 95 with commercially available 3-acetoxy-2-methylbenzoyl chloride and triethylamine (TEA) gave the corresponding amide, which was then reacted with MsCl to afford mesylate 96. Without any purification, the freshly prepared mesylate was reacted with excess acetic anhydride and concentrated H2SO4 to provide oxazoline 92. The oxazoline thus produced was coupled with amine 4 using MeOH and K2CO3 to afford 93 in 72% yield from tosylate salt 95. Finally, reaction of 93 with thiophenol and KHCO3 in MIBK produced nelfinavir (70) in 82% yield to complete the synthesis.
Scheme 23.
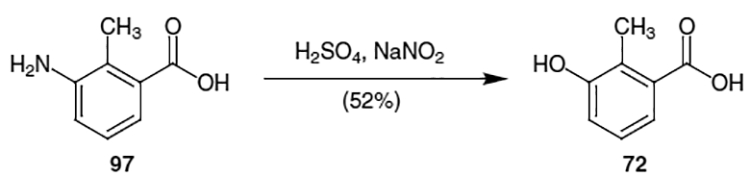
Moiety 72 and the corresponding acid chloride are both commercially available, however 3-hydroxy-2-methylbenzoic acid can also be prepared as in Scheme 24. Treatment of commercially available 3-amino-2-methylbenzoic acid (97) with concentrated H2SO4 and sodium nitrite afforded 72 in 52% yield.24
Scheme 24.
4. Amprenavir (Agenerase®, VX-478, 141W94)
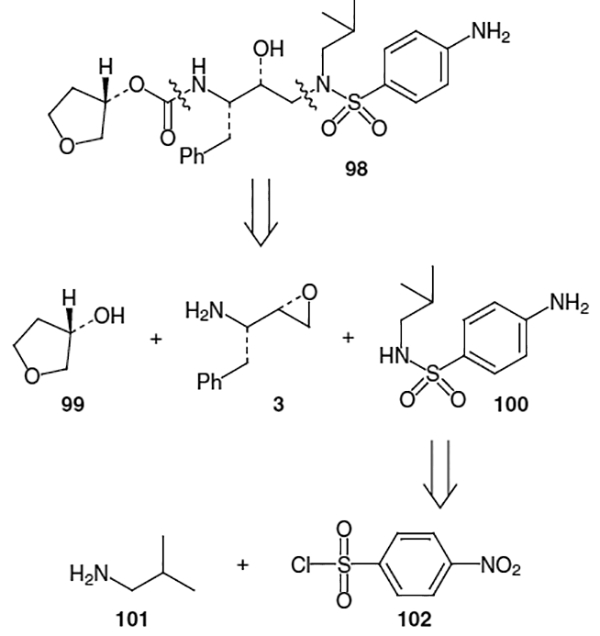
One of the more recently approved HIV protease inhibitors is amprenavir, discovered by scientists at Vertex Pharmaceuticals and currently marketed as Agenerase® by Glaxo Wellcome. This molecule resulted from the desire to find smaller weight protease inhibitors, which it was hoped, would enhance the oral bioavailability while still maintaining or increasing potency. Amprenavir, approved in April 1999 by the FDA, has a Ki value of 0.6 nM against HIV-1 and, with a molecular weight of 505.64, is the smallest of the 6 currently approved HIV protease inhibitors.38
As can be seen in Figure 4, amprenavir (98) can be disconnected into 3 fragments of which 3 is the common benzyl substituted amino epoxide which can be used to form the central hydroxyethylamine isostere. Moiety 99 is commercially available (S)-3-hydroxytetrahydrofuran, which can be activated to form the P1 carbamate ligand. The remaining portion of amprenavir (100) can be synthesized from iso-butylamine (101) and an activated benzenesulfonyl derivative such as p-nitrobenzenesulfonyl chloride (102).
4.1. Synthesis of the Amprenavir Isostere
Ghosh and Fidanze have developed a flexible synthesis which allows access to the hydroxyethylamine isostere found in amprenavir, saquinavir and nelfinavir, as well as the hydroxyethylene isostere, which occurs in ritonavir, lopinavir, and indinavir.39 Their approach utilizes asymmetric syn- and anti-aldol chemistry using N-tosyl-cis-aminoindanol as a chiral auxiliary. In the synthesis leading to amprenavir, N-tosyl-cis-aminoindanol (103) is coupled to 3-phenylpropionic acid using 4-dimethyl aminopyridine (DMAP) and DCC to afford ester 104 in 85% yield (Scheme 25). Aldol reaction between the titanium enolate of 104 (generated using TiCl4 and Hünig’s base) and benzyloxy-acetaldehyde provided syn-aldol product 105 as a single isomer in 97% yield. Removal of the chiral auxiliary by treatment with hydrogen peroxide and LiOH occurred in 82% yield. Exposure of the resulting acid to diphenylphosphorylazide (DPPA) and TEA initiated a Curtius rearrangement, which resulted in formation of oxazolidinone 106 in 75% yield. Hydrolysis of 106 by KOH followed by protection of the free amine as the t-butyloxycarbonyl, (Boc) derivative produced alcohol 107 in 87% yield from oxazolidinone 106. Removal of the O-benzyl protecting group by hydrogenation over Pearlman’s catalyst followed by reaction with PPh3 and DEAD afforded the desired amino epoxide (108) in 61% yield from 107.
Scheme 25.
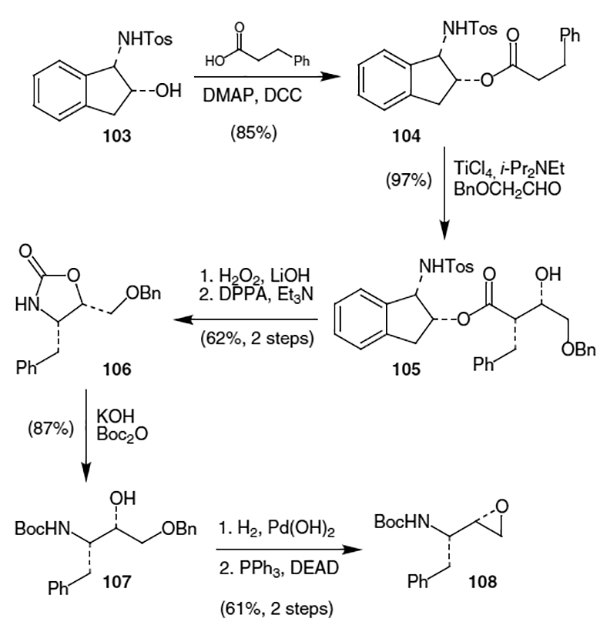
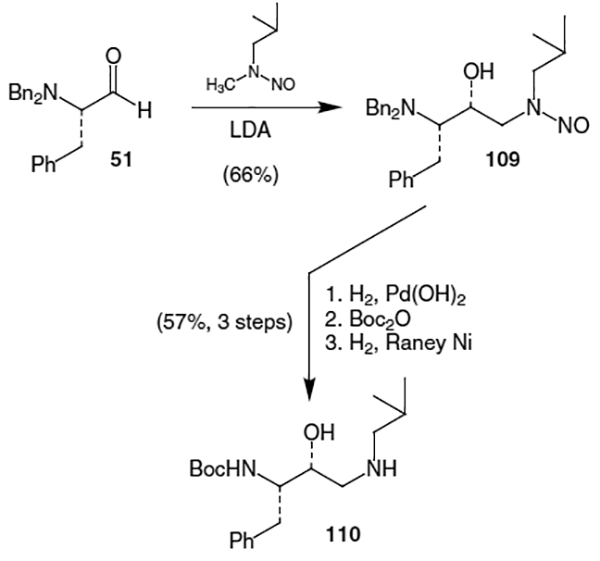
A remarkably straightforward synthesis of the amprenavir isostere is outlined in Scheme 26 and involves the diastereoselective addition of an amino carbanion to protected phenylalanal.40 Beginning with N,N-dibenzyl protected l-phenylalanal (51), reaction with the anion derived from lithium diisopropylamine and N-methyl-N-nitrosoisobutylamine gave 109 as a 4:1 mixture of syn:anti diastereomers in 83% combined yield. Removal of the benzyl protecting groups with H2 and Pearlman’s catalyst followed by reprotection with Boc2O proceeded in 89% yield. Finally, treatment of the nitroso derivative with H2 and Raney nickel afforded iso-butyl amine 110 in 64% yield which could be further elaborated to furnish amprenavir (98).
Scheme 26.
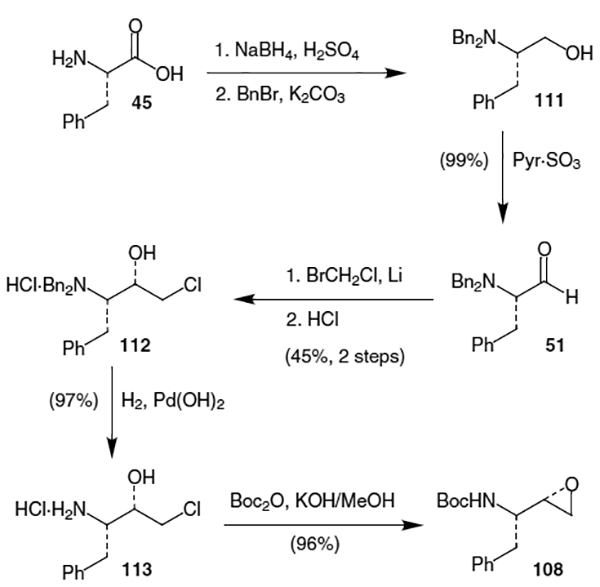
Another approach to the hydroxyethylamine isostere was developed by Beaulieu and co-workers.41 The synthesis begins with l-phenylalanine (45). Reduction of the acid to the alcohol took place using NaBH4 and H2SO4, followed by protection as the N,N-dibenzyl derivative using BnBr and K2CO3, afforded alcohol 111 (Scheme 27). Oxidation using sulfur trioxide provided dibenzyl protected l-phenylalanal (51) in 99% yield. Reaction of this aldehyde with bromochlororomethane and Li metal (15 equiv) at – 65 °C gave the analogous epoxide, which was immediately treated with 6 N HCl to afford hydrochloride salt 112 in 45% yield from phenylalanol. Removal of the benzyl protecting groups was accomplished by hydrogenating over Pearlman’s catalyst to afford 113 in 97% yield. To complete the isostere synthesis, 113 was reprotected with Boc2O and subsequently cyclized with methanolic KOH to provide 108 in 96% yield.
Scheme 27.
4.2. Synthesis of Amprenavir
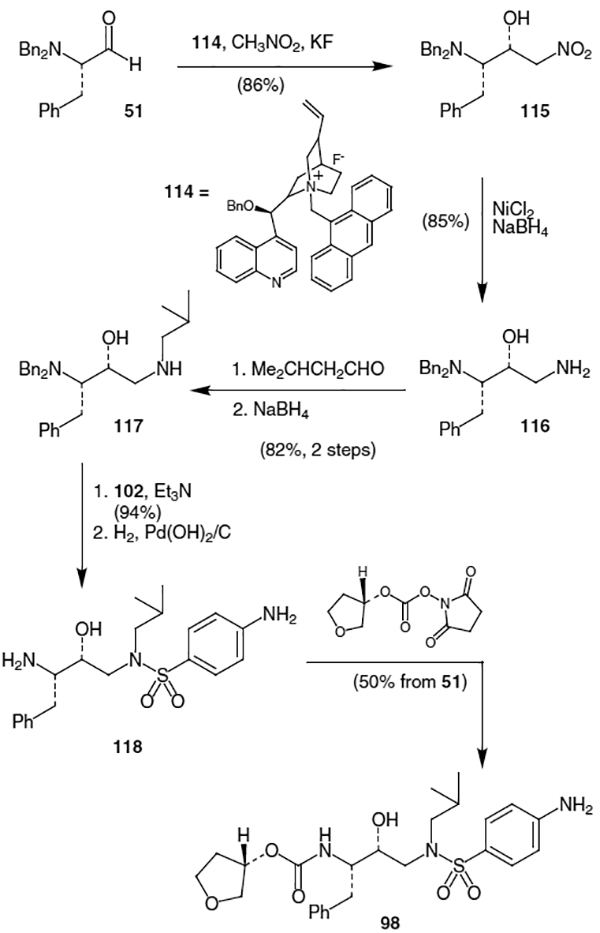
There have been very few total syntheses of amprenavir in the literature. Corey and Zhang have published one such synthesis which uses a chiral quaternary ammonium salt to promote a diastereoselective nitroaldol reaction.42 Reaction of dibenzyl protected l-phenylalanal (51) with 10 mol% of chiral ammonium salt 114, nitromethane, and excess KF in THF at −10 °C afforded corresponding nitro alcohol 115 in 86% yield as a 17:1 ratio of diastereomers in favor of 115 (Scheme 28). Treatment of 115 with NiCl2 and a large excess of NaBH4 provided amino alcohol 116 in 85% yield. Formation of the corresponding imine by reaction of amine 116 with iso-butyraldehyde followed by reduction using NaBH4 produced amino alcohol 117 in 82% yield. Reaction of 117 with p-nitrobenzenesulfonyl chloride (102) gave the corresponding sulfonamide in 94% yield, which was deprotected with H2 and Pearl-man’s catalyst to afford 118. Finally, coupling of amine 118 with the N-oxysuccinimidyl carbonate of (S)-3-hydroxytetrahydrofuran produced amprenavir (98) in 50% overall yield from 51.
Scheme 28.
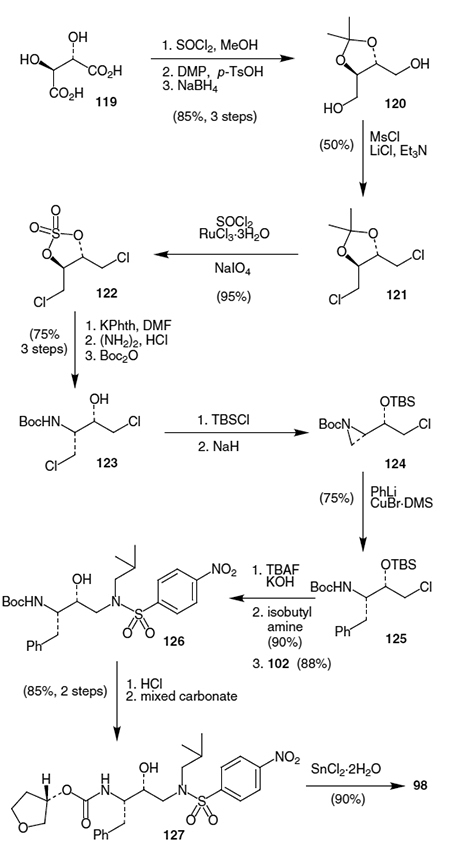
Another total synthesis was recently published by Kim et al., which is especially noteworthy because it allows easy access to protease inhibitors that use hydroxyethylamine isosteres not derived from naturally occurring amino acids.43 The synthesis begins with D-tartaric acid (119) which is first esterified with SOCl2 and MeOH followed by protection of the diol as the acetonide with 2,2-dimethoxypropane (DMP) and p-TsOH and subsequent reduction to diol 120 using NaBH4 in ~85% overall yield (Scheme 29). Transformation of diol 120 to the corresponding dichloride (121) was completed in 50% yield by treatment with MsCl, LiCl and TEA. Conversion to the cyclic sulfate was accomplished by treating 121 SOCl2 followed by oxidation using RuCl3 with 3H2O and sodium periodate to afford 122 in 95% yield. The Boc-protected nitrogen was introduced into the molecule by opening the cyclic sulfate with potassium phthalimide (KPhth) in DMF, removal of the phthayl group by treatment with hydrazine followed by HCl, and reprotection as the t-butyl carbamate using Boc2O, which afforded 123 in 75% yield from cyclic sulfate 122. Protection of the free alcohol as the t-butyldimethylsilyl (TBS) ether was accomplished by reaction with TBSCl. Next the aziridine was formed by treatment with NaH to furnish 124 in near quantitative yield, which completed the carbon skeleton of the isostere portion of amprenavir. Introduction of the benzyl side chain of the amprenavir isostere was accomplished by treating 124 with phenyllithium and CuBr DMS, which selectively opened the aziridine to afford 125 in 75% yield. The epoxide was then formed by deprotecting 125 with tetrabutylammonium fluoride (TBAF), and subsequent treatment with KOH in methanol. Opening the epoxide with iso-butyl amine proceeded in 90% yield. The corresponding alcohol was subsequently reacted with p-nitrobenzenesulfonyl chloride (102) to afford 126 in 88% yield. Removal of the Boc group with HCl gave the free amine, which was then reacted with the active carbonate of (S)-3-hydroxytetrahydrofuran to provide 127 in 85% yield. Lastly, reduction of the nitro group in 127 was accomplished by treatment with SnCl2 2H2O to afford amprenavir (98) in 90% yield.
Scheme 29.
5. Indinavir (Crixivan®, l-735,524, MK-639)
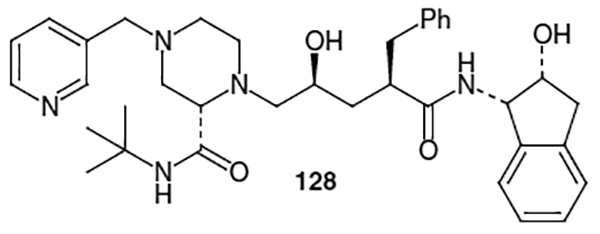
Crixivan® (128, Figure 5) is the trade name for indinavir sulfate. The FDA approved indinavir sulfate for the treatment of HIV and AIDS in March of 1996.44 This inhibitor exhibits an IC50 value of 0.56 nM ± 0.2 nM and a CIC95 value of 25–100 nM.
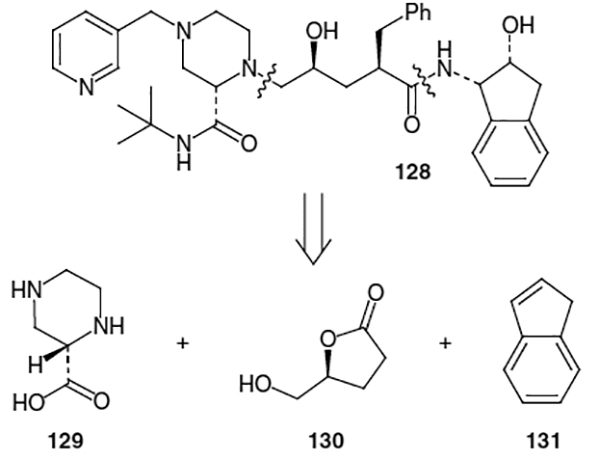
The structure of this inhibitor can be disconnected into three fragments: the piperazine moiety, the Phe-Gly hydroxyethylene isostere and the (−)-cis-(1S,2R)-1-aminoindan-2-ol [(−)-CAI] moiety, as shown in Figure 6. The first total synthesis of indinavir was published by Merck Research Laboratories in 1994.45 They used a convergent approach for the construction of indinavir where the piperazine moiety was derived from (S)-2-piperazinecarboxylic acid (129),46 the hydroxyethylene isostere was derived from commercially available, optically active (S)-(+)-dihydro-5-(hydroxymethyl)-2(3H)-furanone (130) and the (−)-cis-(1S,2R)-1-aminoindan-2-ol moiety was derived from indene (131).47
5.1. Synthesis of the Indinavir 2-Piperazine Fragment
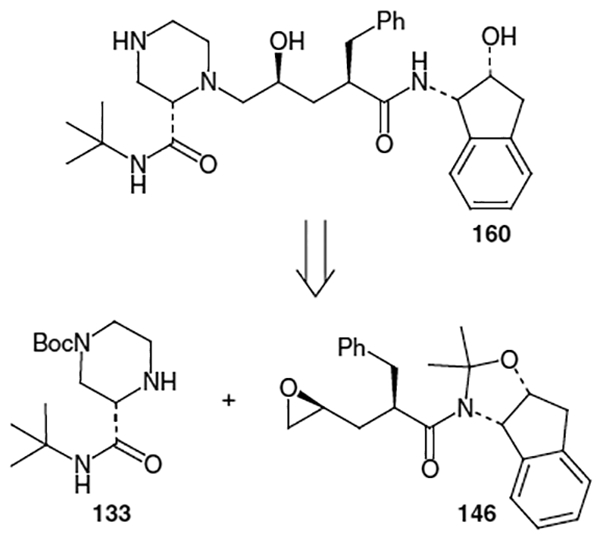
The synthetic route began with (S)-2-piperazinecarboxylic acid bis[(S)-(+)-camphorsulfonic acid] salt (132, Scheme 30). This starting material can be produced via the method of Felder et al., which entails the hydrogenation of pyrazinecarboxylic acid followed by resolution through crystallization with (S)-(+)-camphorsulfonic acid.46 The synthetic route towards indinavir continued with sequential protection of the amine functionalities as t-butylcarbamate and benzyl carbamate in 96% yield followed by amidation with t-butylamine. Removal of the Cbz protecting group was accomplished using hydrogenation over 10% Pd/C in methanol to afford intermediate 133 in 96% yield.
Scheme 30.
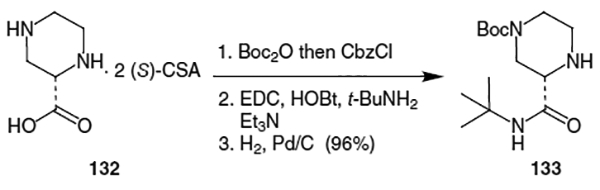
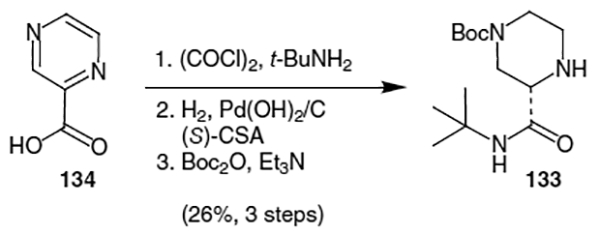
This piperazine fragment can also be produced by amidation of 2-pyrazinecarboxylic acid (134) with oxalyl chloride and t-butylamine followed by hydrogenation using Pearlman’s catalyst and (S)-camphorsulfonic acid [(S)-CSA] to afforded the corresponding optically active salt (Scheme 31).48 Selective protection of N4 of the resulting product using Boc2O and TEA afforded optically active t-butyl amide 133 in 26% overall yield.
Scheme 31.
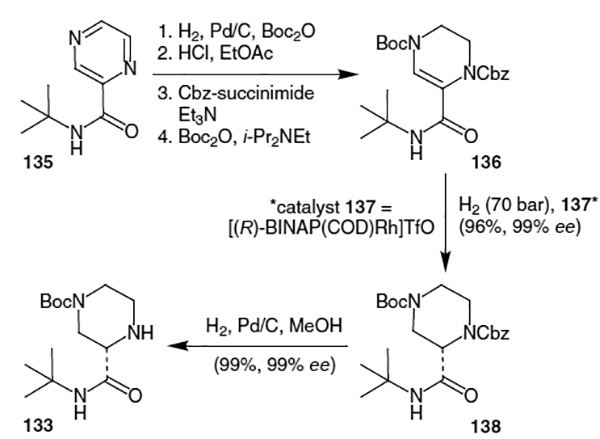
A modified preparation of the piperazine fragment of indinavir was developed by Rossen et al.49 It proceeds from the t-butylamide derived from 2-pyrazinecarboxylic acid (135, Scheme 32). Partial hydrogenation of the pyrazine ring followed by sequential protection of the resulting amine moieties produced intermediate 136 in 62% overall yield. Catalytic hydrogenation of 136 with 2% [(R)-BINAP(COD)Rh]TfO in methanol afforded 138 in 96% yield with 99% ee. Removal of the Cbz group was accomplished by hydrogenation over Pd/C in methanol to afford the desired piperazine derivative (133) in >99% ee.
Scheme 32.
5.2. Synthesis of the Indinavir Isostere
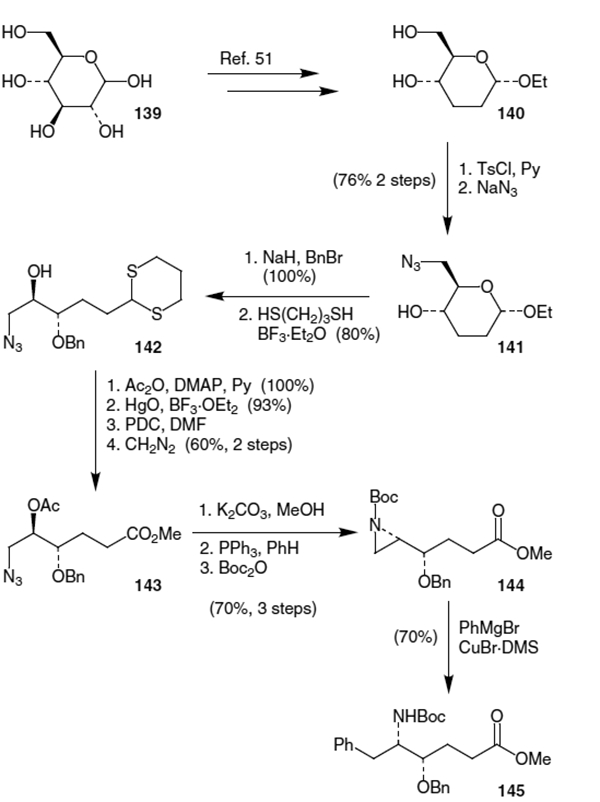
Chakraborty and Gangakhedkar have devised a synthesis of the Phe-Gly hydroxyethylene isostere starting from d-glucose (139).50 As shown in Scheme 33, d-glucose (139) was converted into protected derivative 140 using known carbohydrate transformations.51 Tosylation of the primary alcohol was followed by displacement using sodium azide and afforded azidoalcohol 141. Benzyl protection of the remaining alcohol followed by thioacetal protection of the ring-open form of the pyranoside afforded 142 in 80% yield over two steps. Acetate protection of the resulting alcohol followed by deprotection of the dithioacetal, oxidation of the resulting aldehyde, and esterification of the acid afforded ester 143 in 56% yield (4 steps). Deprotection and activation of the resulting alcohol with triphenyl phosphine led to aziridine formation affording the desired isomer (144) in 70% isolated yield after Boc protection. Regioselective opening of the aziridine was accomplished using phenyl Grignard in the presence of CuBr DMS. This produced the desired Phe-Gly hydroxyethylene isostere (145) in 70% yield.
Scheme 33.
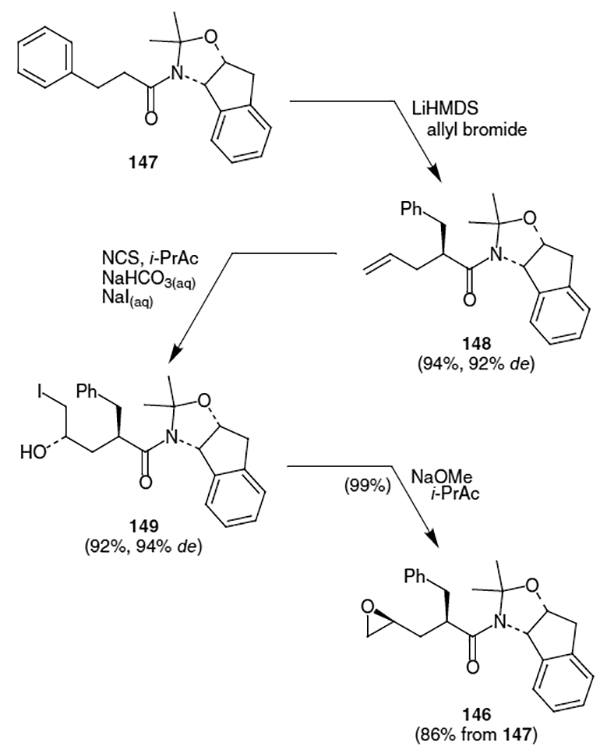
Maligres et al. published an efficient synthesis of epoxide 146 which contains the epoxide equivalent of the indinavir isostere.52 The key steps of this synthesis are the stereoselective allylation of 147 and the stereoselective epoxidation of the resulting olefin. Thus, as shown in Scheme 34, aminoindanol derivative 147 was treated with LiHMDS and allyl bromide to afford a 96:4 ratio of the corresponding olefins in 94% yield (major diastereomer shown, 148). The resulting major product was converted into iodohydrin 149 via treatment with N-chlorosuccinimide (NCS) in a mixture of i-propylacetate and aqueous sodium bicarbonate followed by an aqueous solution of sodium iodide. These conditions afforded primary iodohydrin 149 in 92% yield (94% de). Addition of a 25% solution of sodium methoxide to iodohydrin 149 in i-propylacetate afforded epoxide 146 in near quantitative yield. This resulted in formation of the key epoxide in an overall yield of 86% from 147.53
Scheme 34.
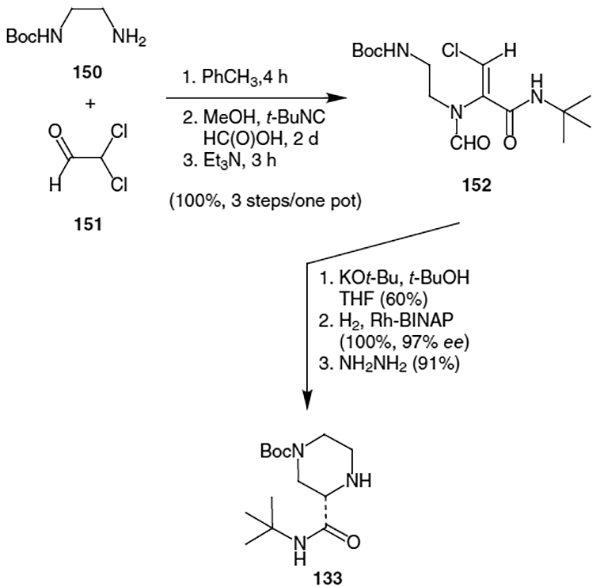
5.3. Synthesis of Indinavir
Rossen and co-workers’ total synthesis of indinavir at Merck began by improving the efficiency of the construction of the piperazine fragment as shown in Scheme 35.54 Thus, condenstaion of N-Boc-ethylenediamine (150), dichloroacetaldehyde (151), t-butylisocyanideformic acid followed by treatment with triethylamine led to the formation of intermediate 152 in quantitative yield. Deprotonation with t-BuOK led to cyclization (60% yield), which was followed by catalytic hydrogenation using Rh-BINAP (quantitative yield, 97% ee). The formyl protecting group was cleanly removed using hydrazine to afford the desired piperazine fragment (133) in 91% yield without racemization of the chiral center (98% ee).
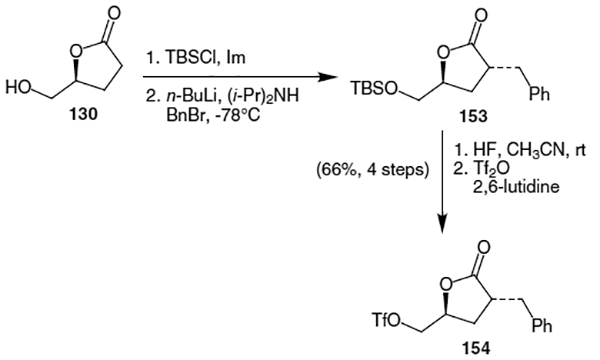
Scheme 35.
The synthesis of indinavir continued by coupling piperazine 133 with the triflate derivative of furanone 130. This triflate derivative (154) was constructed via alkylation of the corresponding protected lactone (Scheme 36). This alkylation was accomplished by protection of (S)-(+)-dihydro-5-(hydroxymethyl)-2(3H)-furanone (130) with t-butyldimethylsilyl chloride (TBSCl) followed by treatment with n-BuLi, diisopropylamine, and benzyl bromide to yield alkylated lactone 153. Deprotection of the silyl group with HF in acetonitrile followed by conversion of the resulting alcohol to a triflate yielded derivative 154 in 66% yield over four steps.
Scheme 36.
Having two of the three fragments for indinavir completed, Merck Laboratories turned to the synthesis of the third fragment, (−)-cis-(1S,2R)-1-aminoindan-2-ol. Merck’s synthesis of (−)-cis-(1S,2R)-1-aminoindan-2-ol begins with the conversion of indene (131) to optically active indene oxide (156), as shown in Scheme 37.47,55 This was accomplished using Jacobsen’s epoxidation catalyst, S,S-(salen)Mn(III)Cl (155), 4-(3-phenylpropyl)pyridine N-oxide (P3NO) and aqueous sodium hypochlorite and afforded indene oxide in 89% yield with an enantiomeric excess of 88%. Indene oxide was then converted into methyl oxazoline 157 by treatment with oleum in acetonitrile. Hydrolysis of the oxazoline followed by crystallization with l-tartaric acid yielded (−)-cis-(1S,2R)-1-aminoindan-2-ol (158) in 50% overall yield with greater then 99% enantiomeric excess.
Scheme 37.
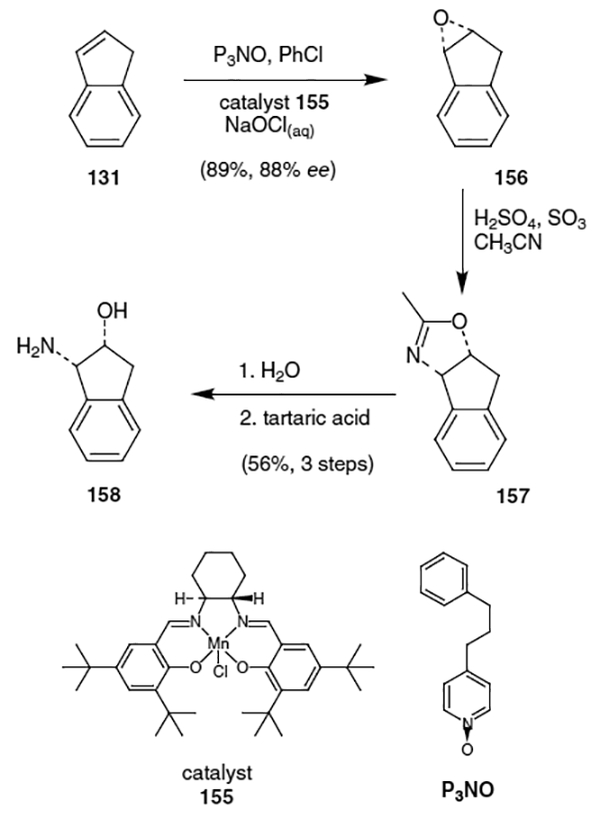
Completion of the synthesis of indinavir was accomplished by the coupling of triflate derivative 154 with piperazine 133 using diisoproplylethylamine in iso-propanol (Scheme 38). This afforded the corresponding coupled product in 83% yield. Hydrolysis of the lactone using lithium hydroxide yielded the hydroxyethylene isostere, which was protected as a t-butyldimethylsilyl ether in 96% yield over two steps. Coupling of the resulting acid with (−)-cis-(1S,2R)-1-aminoindan-2-ol (158) using 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) and 1-hydroxybenzotriazole (HOBt) resulted in intermediate 159. The completion of the synthesis of indinavir was accomplished by removal of the silyl and Boc protecting groups using hydrochloric acid and finally, coupling of the piperazine with 3-picolyl chloride. The synthesis provided indinavir in ten steps with an overall yield of 35%. A separate synthetic route to indinavir was devised using coupling of epoxide 146 with piperazine fragment 133, as shown in Figure 7. This synthesis is different from the previous in that it creates the isostere hydroxy group via opening of an epoxide.
Scheme 38.
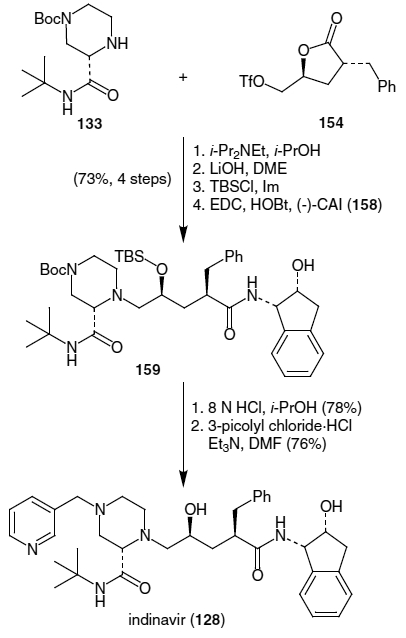
This route begins with the synthesis of the (−)-cis-(1S,2R)-1-aminoindan-2-ol fragment. This was achieved by N-alkylation of (−)-cis-(1S,2R)-1-aminoindan-2-ol (158) with 3-phenylpropionyl chloride (161) followed by protection of the resulting amidoalcohol as an acetonide (Scheme 39).56 The resulting amide derivative (162) was then functionalized at the α-carbon using LiHMDS and commercially available (S)-(+)-glycidyl tosylate (163). This led to coupled product 146 in 72% yield as a 98:2 ratio of epimers at the α-carbon (major diastereomer is shown). Completion of the synthesis of indinavir was accomplished by coupling of epoxide 146 with piperazine fragment 133, synthesized as shown in Scheme 32. Coupling of the two fragments along with concomitant deprotection of the acetonide and Boc moieties with HCl afforded the corresponding diol. This intermediate was then coupled with 3-picolyl chloride to afford indinavir in 71% yield (from 146).
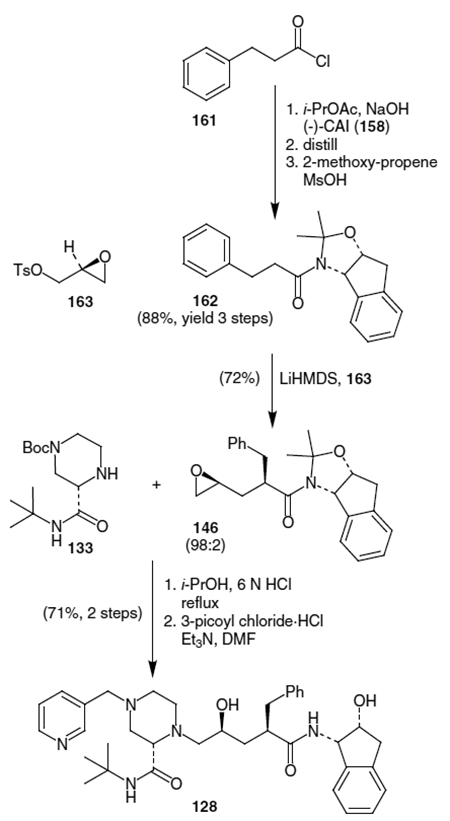
Scheme 39.
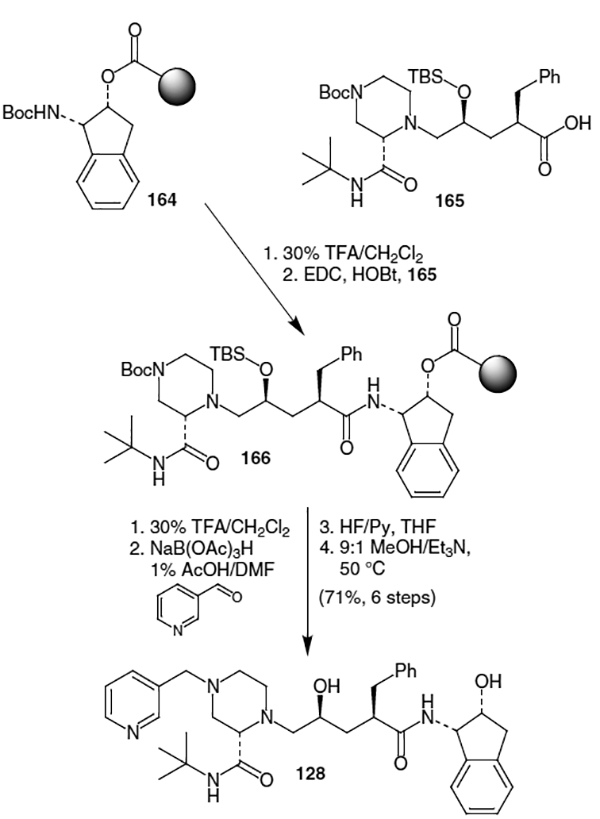
A solid phase synthesis of indinavir was developed by Cheng et al. and it began with linking N-Boc-(−)-cis-(1S,2R)-1-aminoindan-2-ol to the Rapp TentaGel S COOH resin via an ester linkage (Scheme 40).57 This coupling was accomplished using EDC and dimethylaminopyridine (DMAP) in a mixture of dimethylformamide and dichloromethane to yield linked N-Boc-(−)-cis-(1S,2R)-1-aminoindan-2-ol 164. Removal of the Boc protecting group with trifluoracetic acid (TFA) followed by coupling with 16545 under standard conditions (EDC/HOBt) afforded intermediate 166. Boc deprotection (TFA) followed by reductive amination yielded protected indinavir. The silyl group was removed with hydrogen fluoride in pyridine and indinavir was cleaved from the resin using a 9:1 mixture of methanol–TEA at 50 °C. This synthesis provided the target molecule in 71% yield with an optical purity of >95%.
Scheme 40.
6. Ritonavir (Norvir®, ABT-538)
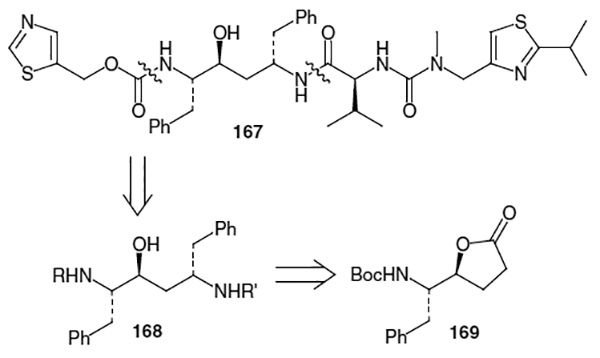
Norvir® (167, Figure 8) is the trade name for ritonavir, an FDA approved (March, 1996) HIV protease inhibitor from Abbott Laboratories. This inhibitor exhibits an EC50 value of 0.022–0.13 μM versus HIV-1 and an EC50 value of 0.16 μM versus HIV-2.58
6.1. Synthesis of the Ritonavir Isostere
This molecule was synthesized at Abbott Laboratories by Kempf et al.59 The route began with the construction of the Phe-Phe hydroxyethylene isostere moiety. As shown in Figure 8, the isostere can be derived from (S,S,S)-diaminoalcohol 168. This hydroxyethylene isostere has been prepared via several different routes over the years. Many of the methods used for its construction proceed using an alkylation of lactone 169 as a means of introducing the third stereocenter into the isostere.
Evans et al. published an early synthesis of this type of lactone starting from N-Boc-phenylalanine, which they converted into N-Boc phenylalanal (33, Scheme 41).60 This aldehyde was subjected to Corey–Chaykovsky epoxidation, which afforded epoxide derivative 170 as a mixture (ca. 1:1) of diastereomers. The epoxide moiety was then opened using the anion of diethylmalonate, and after in situ lactonization and chromatography, the corresponding lactone was isolated in 32% yield from 170. The α-carbon of the lactone was then alkylated using NaOEt and benzyl bromide. The resulting compound was treated with base, acid and heated to afford a mixture of epimers of the Phe-Phe isostere of ritonavir in 78% yield.
Scheme 41.
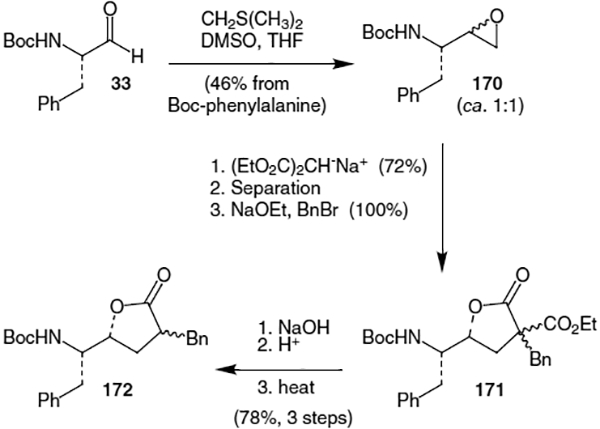
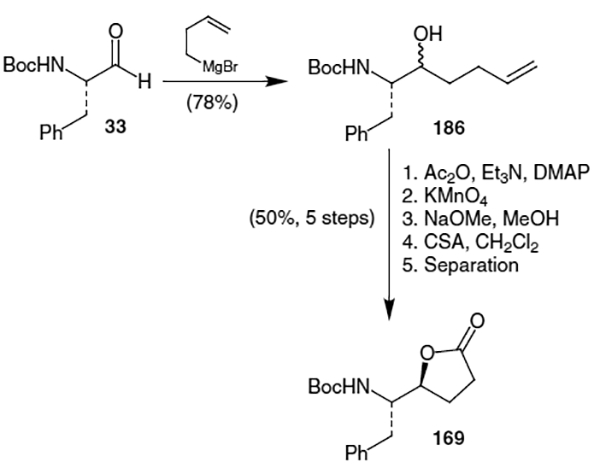
DeCamp et al. completed the synthesis of lactone 169 using addition of a homoenolate to Boc-protected phenylalanal (33).61 Thus, as shown in Scheme 42, addition of the appropriate titanium homoenolate to N-Boc phenylalanal (33) provided the corresponding alcohol in a 16:1 ratio of diastereomers (major isomer shown, 173). Refluxing the alcohol with acetic acid in toluene afforded lactone 169 containing two of the stereocenters for the Phe-Phe isostere of ritonavir.
Scheme 42.
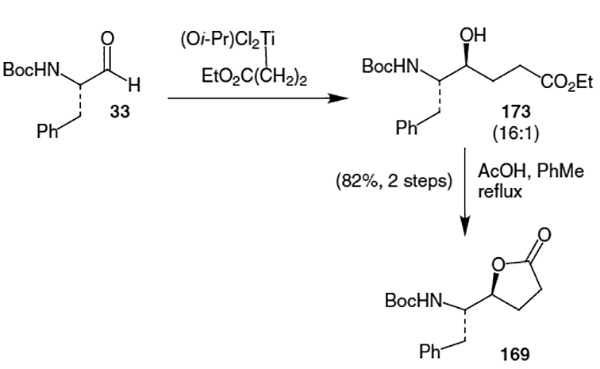
A synthesis of an unsaturated version of lactone 169 was accomplished in 1991 by Harding et al.62 As shown in Scheme 43, this synthetic route began with compound 174 and proceeded via stereoselective addition of 2-(trimethylsilyloxy)furan (175) in the presence of BF3 OEt2. This afforded the corresponding Cbz-protected lactone (176) in 70% yield and 74% diastereomeric excess.
Scheme 43.
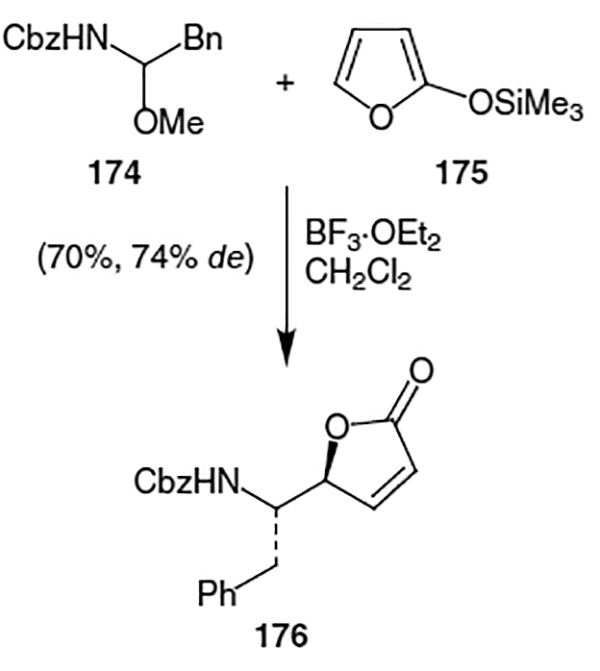
Also in 1991, Chu et al. devised a synthesis of lactone 169 starting from D-mannose (177).63 Protection followed by elimination and isomerization of the resulting olefin afforded dihydrofuran derivative 179 (Scheme 44). Hydrogenation and deprotection of the acetonide moiety were followed by formation of an epoxide via elimination of the primary tosylate. Phenyl cuprate was used to open the epoxide and afforded alcohol 181. Inversion of the resulting alcohol with diphenylphosphoryl azide afforded azide 182 in 92% yield. Lactonization with mCPBA followed by hydrogenation of the azide and Boc protection of the resulting amine led to lactone 169. Chu et al. proceeded to alkylate lactone 169 using LiHMDS and benzyl iodide followed by hydrolysis using trimethyl aluminum and benzyl amine to yield isostere 183. However, this isostere contains an incorrect stereocenter for incorporation into ritonavir.
Scheme 44.
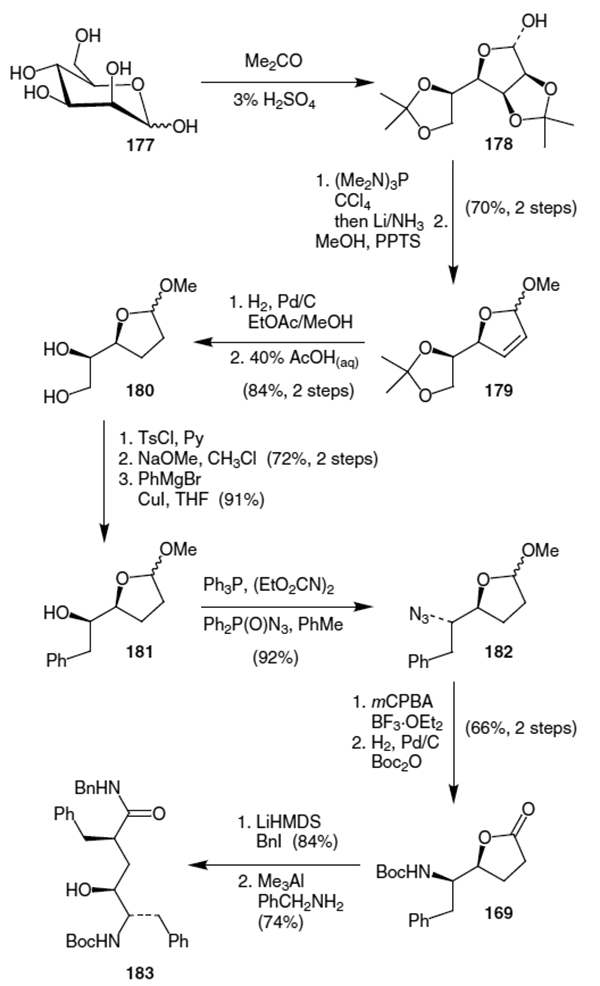
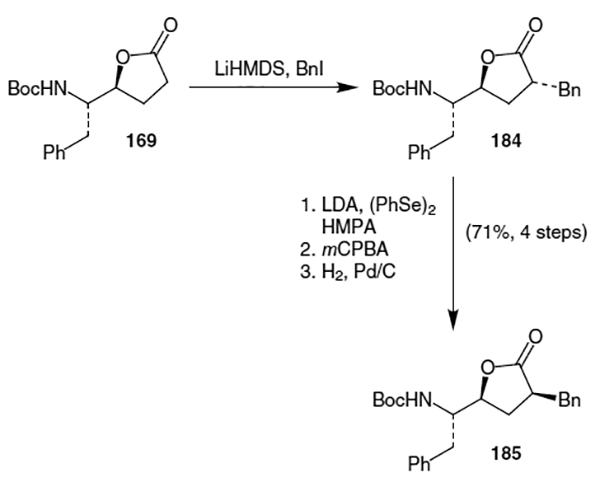
Ghosh and co-workers published an improved alkylation procedure in 1993.64 This proceeded from common lactone 169, as shown in Scheme 45. Thus, alkylation using LiHMDS and benzyl iodide provided lactone 184. This was followed by isomerization of the newly created stereocenter using a sequence of: (i) conversion to the corresponding phenyl selenide, (ii) elemination of the selenide using mCPBA and (iii) stereoselective hydrogenation of the resulting olefin. This sequence yielded the correct isomer of the alkylated lactone in a combined yield of 71% over the four steps. Ghosh et al. completed their synthesis of the isostere by hydrolyzing the lactone and using a Curtius rearrangement to convert the resulting carboxylic acid moiety to an amide.
Scheme 45.
Yet another synthesis of lactone 169 was reported in 1992 by Dreyer et al. (Scheme 46).65 Grignard addition to N-Boc phenylalanal (33) provided unsaturated alcohol 186 in 78% yield as a mixture of isomers. Acetate protection of the alcohol was followed by permanganate oxidation of the olefin, deprotection of the acetate group and lactonization to afford lactone 169 in 50% yield after chromatography.
Scheme 46.
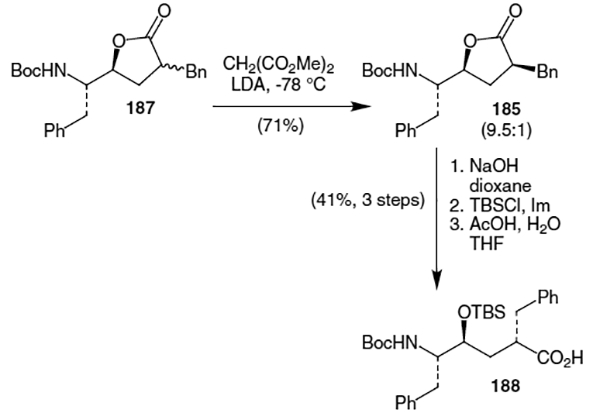
Baker and Pratt published a synthesis of the hydroxyethylene isostere of ritonavir starting form alkylated lactone 187.66 As shown in Scheme 47, kinetic protonation of lactone 187 using LDA and dimethylmalonate at −78 °C afforded a stereochemically enriched mixture of lactones(9.5:1 for the isomer shown, 185). Hydrolysis using NaOH followed by silyl protection of the resulting alcohol afforded the ritonavir hydroxyethylene isostere (188) containing the correct stereochemistry at all three chiral centers.
Scheme 47.
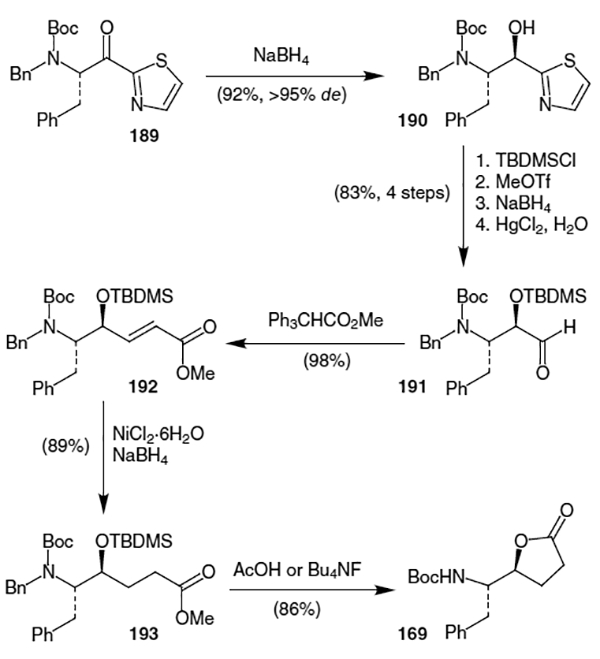
Stereoselective reduction of thiazole derivative 189 (Scheme 48) allowed Dondoni and co-workers access to lactone 169.67 Thus, treatment of 189, made from 2-(trimethylsilyl)thiazole addition to activated phenylalanine,68 with NaBH4 afforded the corresponding alcohol (190) in 92% yield (>95% de). After silyl protection of the alcohol, a three step procedure was used to convert the thiazole to an aldehyde. The resulting aldehyde (191) was subjected to a Wittig reaction to yield the corresponding unsaturated ester (192) in 98% yield. Reduction of the double bond was followed by deprotection and lactonization to give 169 in 76% yield over two steps.
Scheme 48.
Ghosh and co-workers contributed to this body of work again in 1998 with their anti-aldol approach to the isostere39 and in 1999 with their dihydroxylation approach.69 The anti-aldol approach produced aldol 194 (Scheme 49). The chiral auxiliary (N-tosyl-cis-aminoindanol, 103) of aldol 194 was hydrolyzed followed by conversion of the resulting acid to an amine via a Curtius rearrangement and Boc protection to afford allylic alcohol 195. Ozonolysis of the olefin was followed by Horner– Emmons–Wittig reaction of the resulting aldehyde, hydrogenation of the double bond and acidic lactonization to provide 169. Alkylation and hydrolysis in the presence of benzylamine completed the isostere synthesis.
Scheme 49.
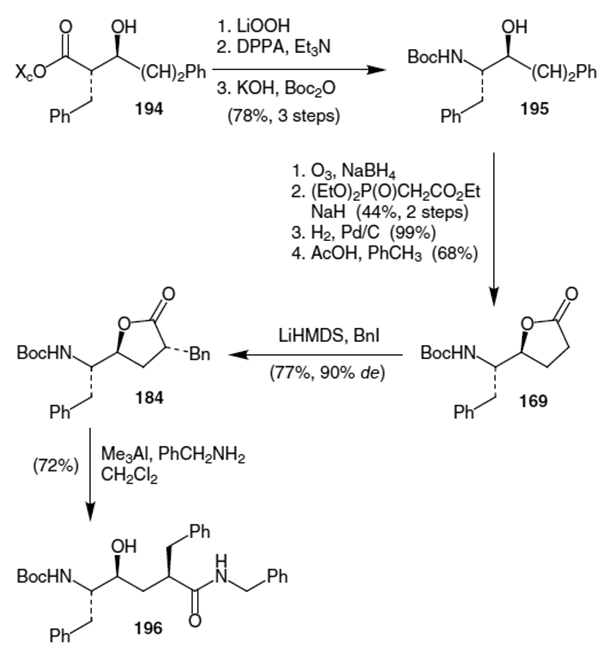
As shown in Scheme 50, Ghosh’s dihydroxylation approach proceeded by treatment of unsaturated ester 197 with AD-mix-β and methanesulfonamide to afford lactone 198 in 87% yield.69 Inversion of the hydroxy stereochemistry was achieved by conversion to the mesylate and substitution using sodium azide. Upon reduction of the azide and protection using Boc anhydride, the lactone was alkylated using LiHMDS and benzyl iodide to afford 200.
Scheme 50.
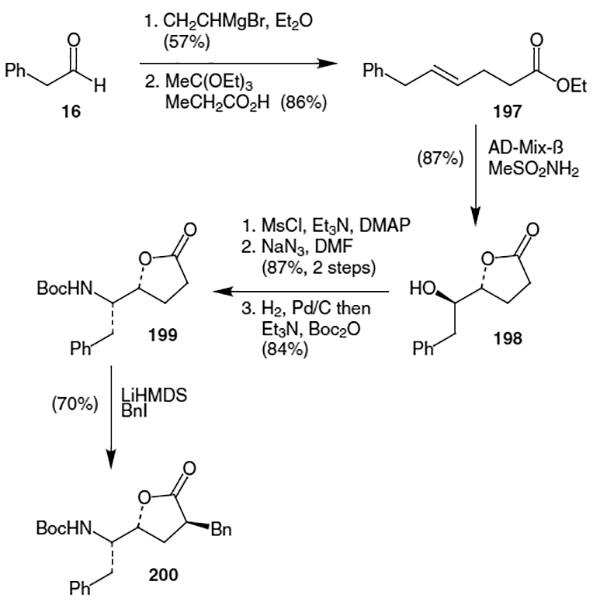
Finally, as shown in Scheme 51, inversion of the lactone oxygen was accomplished by, (after hydrolysis and several protection/deprotection steps) conversion of 201 to the oxazolidinone using SO2Cl2 (73% yield). Hydrolysis of the oxazolidinone and protection of the resulting amines with Boc anhydride yielded the hydroxyethylene isostere (202) in 52% yield (2 steps).
Scheme 51.
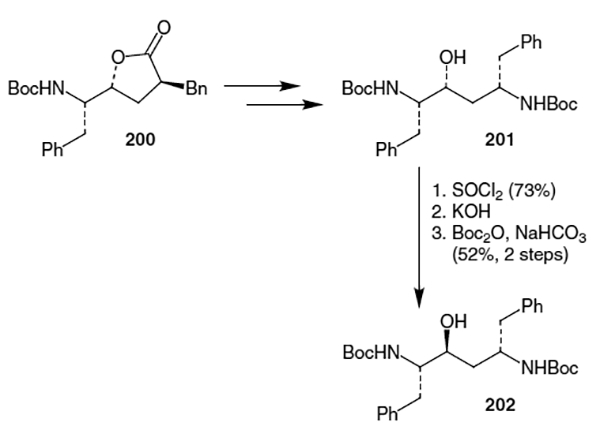
A different approach to the isostere was taken by Hanessian et al. in which alkylation of lactone 169 was not necessary.70 Thus, as shown in Scheme 52, addition of (2-bromomethyl)acrylate to protected phenylalanal 203 in the presence of zinc dust afforded a 90% yield of olefin 204. Acidic lactonization was followed by phenyl cuprate addition to give lactone 206 in 86% diastereomeric excess. Isomerization of the hydroxy stereocenter after a deprotection/protection sequence yielded lactone 184 containing one incorrect stereocenter for the ritonavir isostere.
Scheme 52.
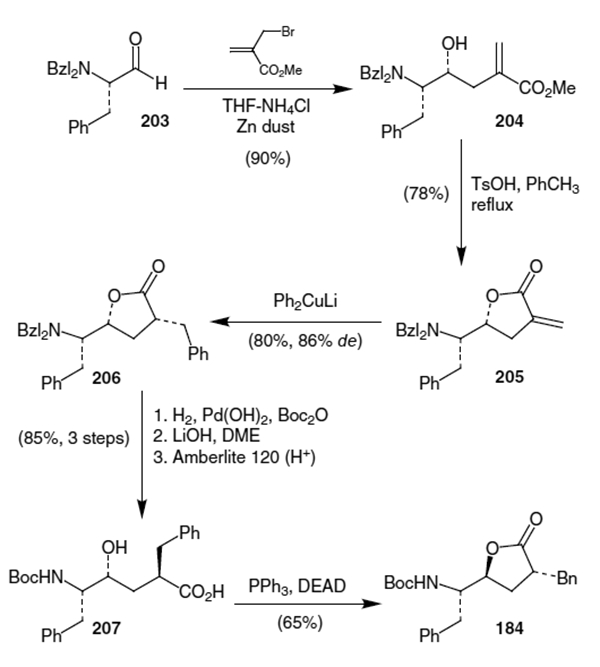
Another synthesis of the Phe-Phe hydroxyethylene isostere that does not proceed through lactone 169 was published by Kempf and co-workers.71 They originally constructed the hydroxyethylene isostere for ritonavir starting from Cbz-phenylalanol (208, Scheme 53). Vanadium-mediated coupling of two molecules of the corresponding aldehyde of 208 proceeded to give an 8:1:1 mixture of diols in favor of 209.
Scheme 53.
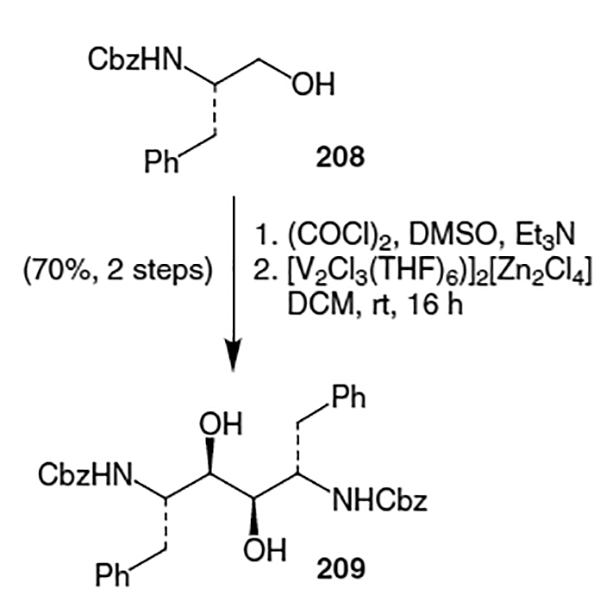
Kempf et al. continued by dehydration of diol 209.72 Thus, treatment of 209 with α-acetoxyisobutyryl bromide yielded the corresponding bromoacetate (210, Scheme 54). Reductive debromination followed by hydrolysis of the protecting groups afforded isostere 211 in 81% yield (3 steps).
Scheme 54.
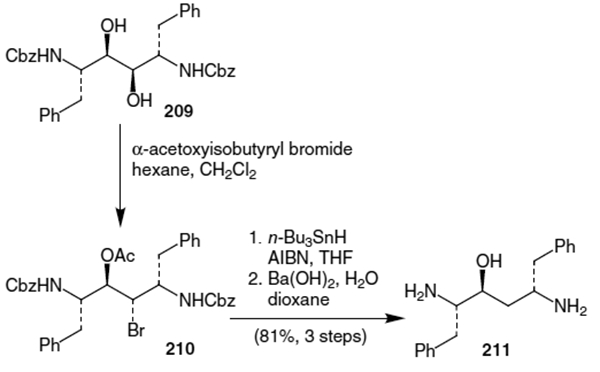
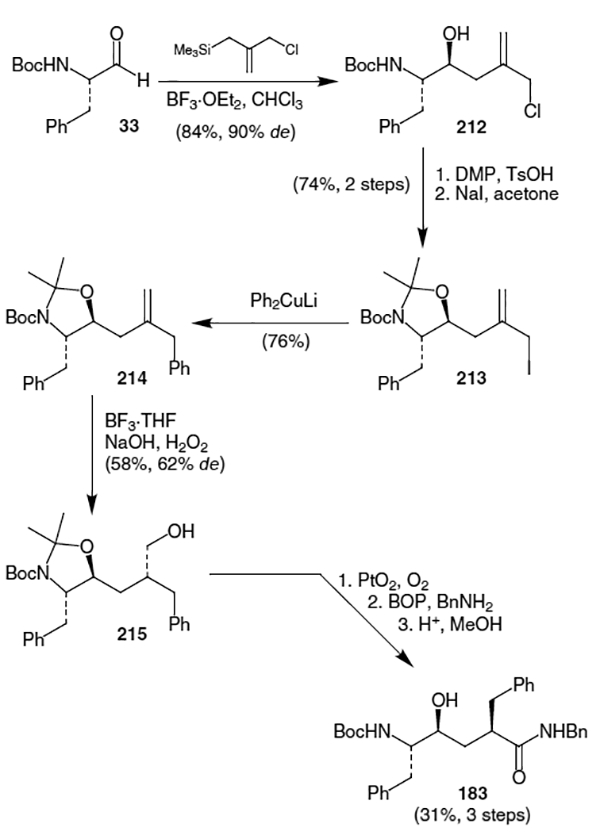
D’Aniello et al. published another route to the hydroxyethylene isostere of ritonavir.73 This route, shown in Scheme 55, began with the addition of 2-(chloromethyl)-3-(trimethylsilyl)-1-propene to N-Boc phenylalanal (33). This afforded homoallylic alcohol 212 in 84% yield and 90% diastereomeric excess. After acetonide protection, halogen exchange and treatment with phenyl cuprate afforded a 76% yield of compound 214. Hydroboration of the olefin of 214 resulted in a 58% yield (62% de) of alcohol 215. Oxidation to the acid followed by amidation and acidic deporotection of the acetonide yielded isostere 183. This isostere, however, contains an incorrect stereocenter for ritonavir.
Scheme 55.
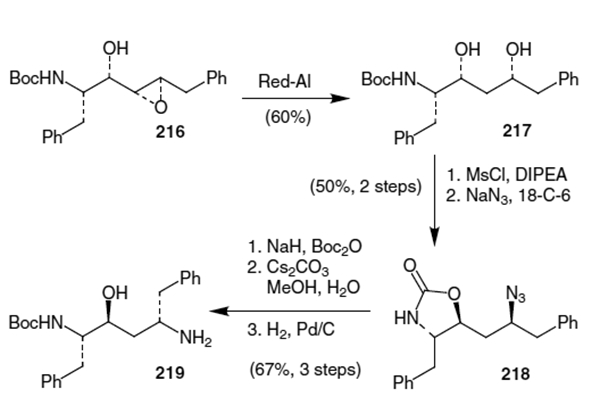
Benedetti et al. elaborated epoxide 216,74 shown in Scheme 56, into the hydroxyethylene isostere of ritonavir.75 This was accomplished starting with reduction of the epoxide with Red-Al in THF (60% yield). Mesylation of the diol resulted in spontaneous oxazolidinone formation and the remaining mesylate was displaced using sodium azide to afford 218. Protection of the carbamate nitrogen with Boc2O followed by hydrolysis of the oxazolidinone and hydrogenation of the azide afforded isostere 219 in 67% yield over three steps.
Scheme 56.
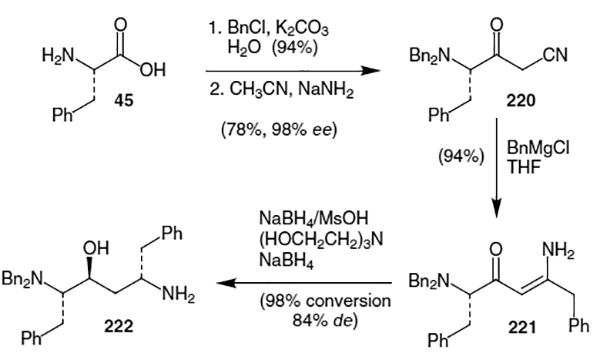
The actual isostere synthesis reported by Abbott Laboratories is based on the reduction of an enaminone.76 Thus phenylalanine (45) was treated with benzyl chloride in the presence of K2CO3 to yield the corresponding protected benzyl ester (Scheme 57) in 94% yield. An acetonitrile anion was added to the benzyl ester to afford 220 in a yield of at least 78% after recrystallization (>98% ee). Benzyl Grignard addition to the resulting nitrile afforded key enaminone 221 in greater than 99.5% enantiomeric excess and 94% yield after recrystallization. Selective reduction of the enaminone was accomplished by treatment with a complex of NaBH4 and methanesulfonic acid (MsOH) followed by a second reduction step with NaBH4 in the presence of triethanolamine. This provided greater than 98% conversion of 221 to 222 with a diastereomeric excess of 84%. Compound 222 can easily be converted into the desired diamine (211) via hydrogenation.
Scheme 57.
6.2. Synthesis of Ritonavir
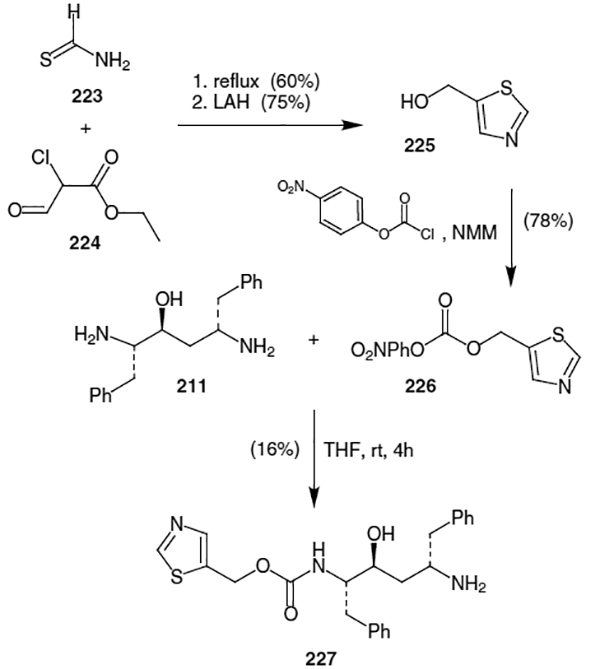
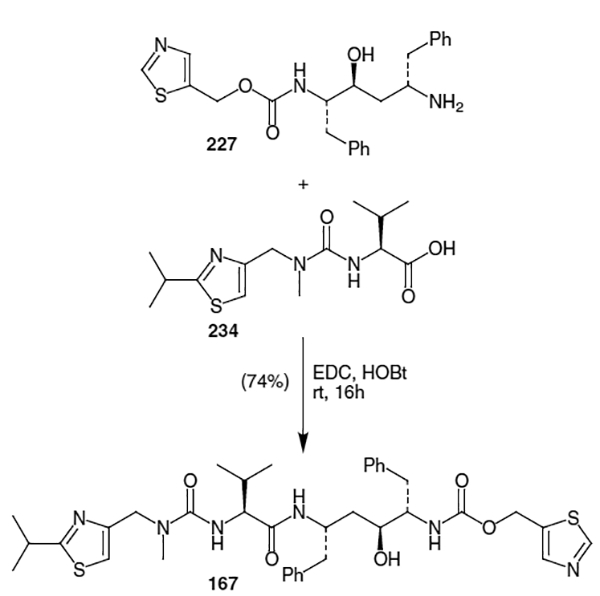
With the isostere (211) in hand, Abbott’s synthesis turned to construction of the remaining fragments and coupling to form ritonavir.59 The thiazolyl carbamate fragment of ritonavir was elaborated as shown in Scheme 58. Thus, condensation of thioformamide (223) and ethyl-2-chloro-2-formylacetate (224) followed by reduction of the resulting ester afforded 5-(hydroxymethyl)thiazole (225) in 45% yield (2 steps). Conversion of this thiazole to the corresponding (p-nitrophenyl)carbonate was followed by coupling with diamine 211. This resulted in carbamate 227 in 16% isolated yield.
Scheme 58.
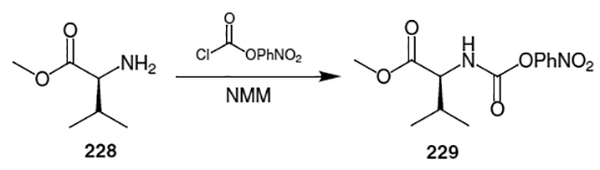
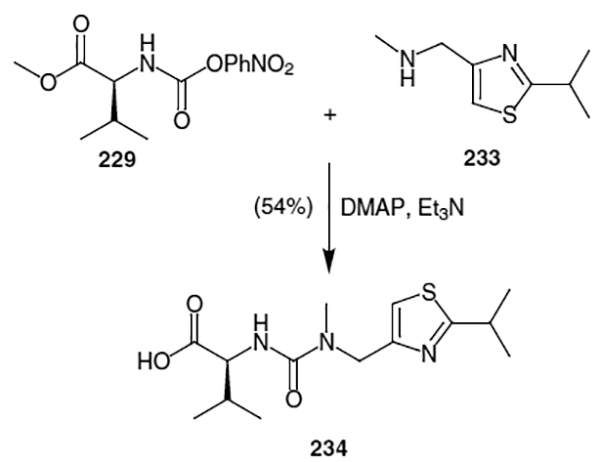
Elaboration of the remaining fragment of ritonavir was accomplished by coupling valine methyl ester (228) with N- methyl-N-[(2-isopropyl-4-thiazolyl)-methyl]amine. Thus, as shown in Scheme 59, valine methyl ester (228) was activated as a (p-nitrophenyl)carbamate.
Scheme 59.
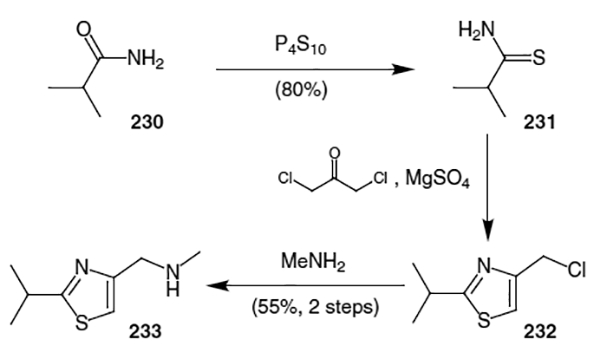
N-methyl-N-[(2-isopropyl-4-thiazolyl)-methyl]amine (233) was constructed through conversion of iso-butyramide (230) to 2-methylpropane thioamide (231) via treatment with P4S10 (Scheme 60). This was followed by condensation with 1,3-dichloroacetone and methylamine to afford N-methyl-N-[(2-isopropyl-4-thiazolyl)-methyl]amine (233) in 55% yield over two steps.
Scheme 60.
Coupling of N-methyl-N-[(2-isopropyl-4-thiazolyl)-methyl]amine (233) and valine derivative 229 was accomplished through treatment with TEA and DMAP. This resulted in the final fragment (234) of ritonavir in 54% yield.
Coupling of fragments 227 and 234 proceeded under standard conditions (EDC/HOBt) to afford ritonavir in 74% yield.
7. Lopinavir (Aluviran®, ABT-378, Component of Kaletra®)
Lopinavir (235, Figure 9) is an FDA approved (September, 2000) HIV protease inhibitor from Abbott Laboratories. This therapeutic is contained in a protease inhibitor formulation (Kaletra®) that includes ritonavir.77 Ritonavir is known to inhibit cytochrome P-450 3A, the enzyme responsible for metabolism of lopinavir,78 therefore the combination allows for increased plasma levels of lopinavir. Both ritonavir and lopinavir contain the same Phe-Phe hydroxyethylene isostere subunit. The retrosynthetic disconnections for lopinavir can be seen in Figure 9. This shows that construction of lopinavir can be accomplished via coupling of isostere fragment 168 with acids 236 and 237.
7.1. Synthesis of the Lopinavir Cyclic Urea Fragment
The synthesis of lopinavir from Abbott Laboratories began by preparation of acid 237.79 As shown in Scheme 63, valine (238) was coupled with phenylchloroformate to afford carbamate derivative 239 in 92% yield. This carbamate was treated with 3-chloropropylamine and NaOH followed by t-BuOK to afford the target acid (237) in 77% yield.
Scheme 63.
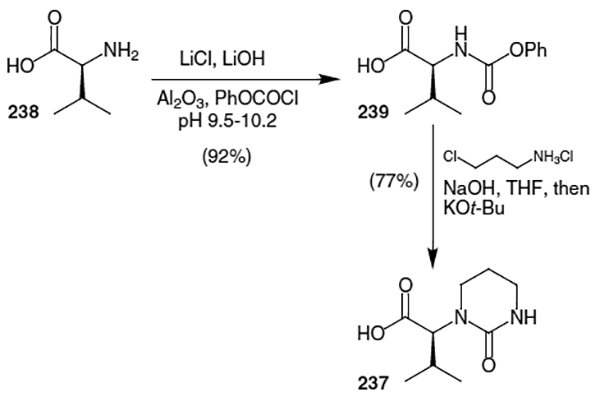
7.2. Synthesis of Lopinavir
Acid 237 was then transformed into the corresponding acid chloride for coupling with the hydroxyethylene isostere (Scheme 64). This transformation was accomplished using thionyl chloride in THF allowing for isolation of the acid chloride (240) in quantitative yield. Coupling of 240 with isostere 24180 was accomplished using three equivalents of imidazole in ethyl acetate and dimethylformamide. This coupling proceeded in quantitative yield along with the subsequent debenzylation using transfer hydrogenation conditions. Crystallization of crude 243 with (S)-2-pyrrolidone-5-carboxylic acid (l-pyroglutamic acid, 242) afforded an 80% yield of pure 243.
Scheme 64.
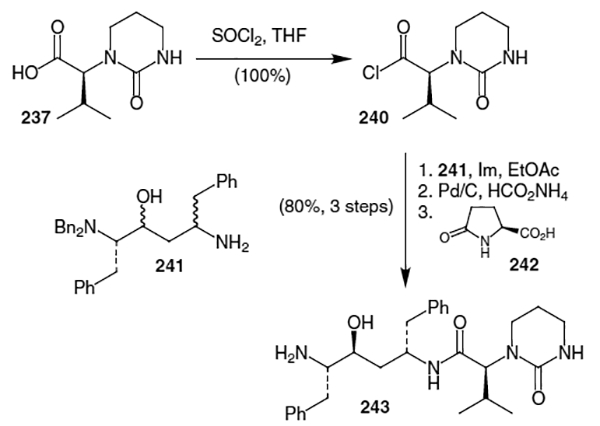
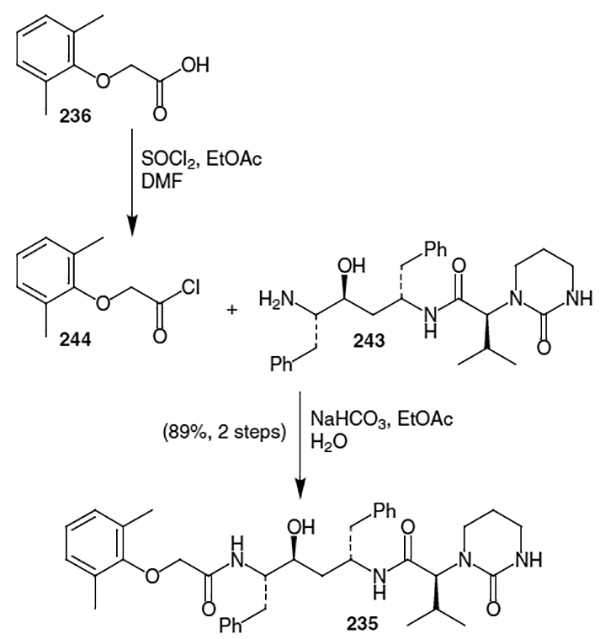
Acid 23681 was then converted into acid chloride 244 for use in coupling with fragment 243. This was accomplished, as shown in Scheme 65, by treatment with SO2Cl2 in ethyl acetate and dimethylformamide. This acid chloride was coupled with fragment 243 in a buffered mixture of ethyl acetate and water. This afforded lopinavir (235) in 89% yield and greater than 99% purity after recrystallization. This reaction sequence allowed for the production of lopinavir in 58% overall yield form the isostere fragment and greater than 99% diastereomeric excess.
Scheme 65.
8. Conclusion
This review reports the synthesis of currently approved HIV protease inhibitor drugs. All of these inhibitors contain multiple chiral centers, therefore the synthesis of hundreds of thousands of kilograms of inhibitor drugs in optically pure form is a tribute to asymmetric synthesis. While the current therapies have helped in the fight against HIV and AIDS, the epidemic is still raging. Approximately 95% of people with HIV live in developing countries. The current therapies are very expensive and possess other limitations as well. These include: (i) low oral bioavailability, (ii) necessity of higher therapeutic doses, (iii) inability to cross the blood-brain barrier, and most concerning, (iv) the development of viral resistance to drugs. Thus, the search for less expensive, more effective, new protease inhibitors that are not cross resistant to current therapies is of critical importance. There are currently three experimental protease inhibitors undergoing clinical evaluations: tipranavir (Boehringer Ingelheim), atazanavir (Bristol-Myers Squibb) and mozenavir (Triangle Pharmaceuticals). These, along with other therapeutics currently in development will add to the arsenal against the current HIV epidemic. This timely application of powerful organic synthetic methodologies to meet today’s challenges may serve as an inspiration to deal with other complex human ailments. We hope that this review will stimulate further research and inspiration in the development of protease inhibitors and other peptidomimetic drugs.
Scheme 1.
Scheme 2.
Scheme 3.
Scheme 4.
Scheme 5.
Scheme 6.
Scheme 7.
Scheme 8.
Scheme 9.
Scheme 61.
Scheme 62.
Acknowledgment
Financial support for this work was provided by the National Institutes of Health (GM 53386).
Biographical Sketches
Arun K. Ghosh was born in India in 1958. He obtained his Ph.D. from the University of Pittsburgh with Professor Alan P. Kozikowski (1985). After his postdoctoral studies with Professor E. J. Corey at Harvard University (1985–1988), he joined Merck Research Laboratories (West Point) as a Senior Research Chemist. In 1994, he moved to the University of Illinois at Chicago where he is currently Professor of Chemistry. His current research interests include: development of asymmetric synthetic methodologies; asymmetric catalysis; total synthesis of biologically important natural products; design and synthesis of molecular probes for biological systems; peptidomimetic design and molecular modeling.

Geoffrey Bilcer was born in 1973 and raised near Chicago, Illinois. He received his B.A. in chemistry from Northwestern University in Evanston, Illinois in 1995. After working in industry for two years, he began his graduate studies at the University of Illinois at Chicago. Geoff is working towards his Ph.D. under Professor Arun Ghosh, focusing on the areas of bioactive natural product synthesis and the design and synthesis of various peptidomimetic protease inhibitors. He was awarded a National Science Foundation fellowship for the 2000–2001 academic year. Geoff anticipates completion of his Ph.D. studies in early 2002.

Gary Schiltz was born in Bismarck, N.D. in 1974 and grew up near Chicago, Illinois. He received a B.S. in chemistry from the University of Illinois at Urbana-Champaign in 1997. In 1999 he enrolled at the University of Illinois at Chicago and subsequently joined Professor Ghosh’s research group. He is currently working towards his Ph.D. with his research focusing on design and synthesis of selective aspartyl protease inhibitors.

References
- (1).(a) Cihlar T; Bischofberger N Ann. Rep. Med. Chem 2000, 35, 177. [Google Scholar]; (b) Darke PL; Huff JR Advances in Pharmacology, 25; August JT; Anders MW; Murad F, Eds.; Academic Press: San Diego, 1994, 399–454. [DOI] [PubMed] [Google Scholar]; (c) Clare M Drug Discovery and Design 1993, 1, 49. [Google Scholar]; (d) Debouck C Human Retroviruses 1992, 8, 153. [DOI] [PubMed] [Google Scholar]; (e) Martin JA Antiviral Res 1992, 17, 265. [DOI] [PubMed] [Google Scholar]; (f) Huff JR J. Med. Chem 1991, 34, 2305. [DOI] [PubMed] [Google Scholar]; (g) Norbeck DW; Kempf DJ Annu. Rep. Med. Chem 1991, 26, 141. [Google Scholar]; (h) Tomasselli AG; Howe WJ; Sawyer TK; Wlodawer A; Heinrikson RL Chimicaoggi 1991, 6. [Google Scholar]
- (2).(a) Mervis RJ; Ahmad N; Lillehoj EP; Raum MG; Salazar FHR; Chan HW; Venkatesan S J. Virol 1988, 62, 3993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Farmerie WG; Leob DD; Cassavant NC; Hutchinson CA III; Edgell MH; Swanstrom R Science 1987, 236, 305. [DOI] [PubMed] [Google Scholar]
- (3).(a) Wlodawer A; Erickson JW Annu. Rev. Biochem 1993, 62, 543. [DOI] [PubMed] [Google Scholar]; (b) Navia MA; Fitzgerald PMD; McKeever BM; Leu C-T; Heimbach JC; Herber WK; Sigal IS; Darke PL; Springer JP Nature 1989, 337, 615. [DOI] [PubMed] [Google Scholar]; (c) Wlodawer A; Miller M; Jaskolski M; Sathyanarayana BK; Baildwin E; Weber IT; Selk LM; Clawson L; Schneider J; Kent SBH Science 1987, 245, 616. [DOI] [PubMed] [Google Scholar]
- (4).(a) Kempf DJ Methods in Enzymology, 241; Kuo LC; Shafer JA, Eds.; Academic Press: New York, 1994, 334.7854187 [Google Scholar]; (b) Vacca JP Methods in Enzymology, 241; Kuo LC; Shafer JA, Eds.; Academic Press: New York, 1994, 311. [DOI] [PubMed] [Google Scholar]
- (5).Flexner CN Engl. J. Med 1998, 338, 1281. [DOI] [PubMed] [Google Scholar]
- (6).(a) Cohen J Science 1996, 272, 1882. [DOI] [PubMed] [Google Scholar]; (b) Moyle G; Gazzard B Drugs 1996, 5, 701. [DOI] [PubMed] [Google Scholar]; (c) Mascolini M Int. Assoc. Physicians AIDS Care 1995, 11. [PubMed] [Google Scholar]
- (7).Roberts NA; Marti D; Kinchington AV; Broadhurst JC; Craig JC; Duncan IB Science 1990, 248, 358. [DOI] [PubMed] [Google Scholar]
- (8).Shaw E Methods in Enzymology; Hirs CHW, Ed.; Academic Press: New York, 1967, 677. [Google Scholar]
- (9).Parkes KEB; Bushnell DJ; Crazkett PH; Dundson SJ; Freeman AC; Gunn MP; Hopkins RA; Lambert RW; Martin JA; Merrett JH; Redshaw S; Spurden WC; Thomas GJ J.Org. Chem 1994, 59, 3656. [Google Scholar]
- (10).Ghosh AK; Thompson WT; Holloway KM; McKee SP; Duong TT; Lee HY; Munson PM; Smith AM; Wai JM; Darke PL; Zugay JA; Emini EA; Schleif WL; Huff JR; Anderson PS J. Med. Chem 1993, 36, 2300. [DOI] [PubMed] [Google Scholar]
- (11).Caron M; Carlier K; Sharpless KB J. Org. Chem 1988, 53, 5185; and references cited therein. [Google Scholar]
- (12).Bennet F; Girijavallabhan VM; Patel N J. Chem. Soc. Chem. Comm 1993, 737. [Google Scholar]
- (13).Ghosh AK; McKee SP; Lee HY; Thompson WT J. Chem. Soc. Chem. Comm 1992, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Wenger RM Helv. Chim. Acta 1983, 66, 2308. [Google Scholar]; (b) Curtis DW; Laidler DA; Stoddart JF; Jones GH J. Chem. Soc. Chem. Comm 1975, 833. [DOI] [PubMed] [Google Scholar]
- (15).Seebach D; Hüngerbuhler E Modern Synthetic Methods; Scheffold R, Ed.; Otto Salle–Sauerländer: Frankfurt-Aarau, 1980, 152. [Google Scholar]
- (16).A 3:1 mixture of azidodiol 22 and elimination product was produced where the elimination product was carried through to the deprotection stage before being separated.
- (17).Maibaum J; Rich DH J. Org. Chem 1988, 53, 869. [Google Scholar]
- (18).Barton DH; Crich D; Motherwell WB Tetrahedron Lett 1983, 24, 4979. [Google Scholar]
- (19).Wissner AJ J. Org. Chem 1979, 44, 4617. [Google Scholar]
- (20).Göhring W; Gokhale S; Hilpert H; Roessler F; Schlageter M; Vogt P Chimia 1996, 11, 532. [Google Scholar]
- (21).Reetz MT; Binder J Tetrahedron Lett 1989, 30, 5425. [Google Scholar]
- (22).Houpis IN; Molina A; Reamer RA; Lynch JE; Volante RP; Reider PJ Tetrahedron Lett 1993, 34, 2593. [Google Scholar]
- (23).Bennett F; Patel NM; Girijavallabhan VM; Ganguly AK Synlett 1993, 703. [Google Scholar]
- (24).Kaldor SW; Kalish VJ; Davies JF; Shetty BV; Fritz JE; Appelt K; Burgess JA; Campanale KM; Chirgadze NY; Clawson DK; Dressman BA; Hatch SD; Khalil DA; Kosa MB; Lubbehusen PP; Muesing MA; Patick AK; Reich SH; Su KS; Tatlock JH J. Med. Chem 1997, 40, 3979. [DOI] [PubMed] [Google Scholar]
- (25).Reiger DL J. Org. Chem 1997, 62, 8546. [DOI] [PubMed] [Google Scholar]
- (26).Marzoni G; Kaldor SW; Trippe AJ; Shamblin BM; Fritz JE Synth. Commun 1995, 25, 2475. [Google Scholar]
- (27).Dressman BA; Fritz JE; Hammond M; Hornback WJ; Kaldor SW; Kalish VJ; Munroe JE; Reich SH; Tatlock JH; Shepherd TA; Rodriguez MJ U.S. Patent 5 484–926, 1994; (Agouron Pharmaceuticals, Inc.). [Google Scholar]
- (28).Inaba T; Yamada Y; Abe H; Sagawa S; Cho H J. Org. Chem 2000, 65, 1623. [DOI] [PubMed] [Google Scholar]
- (29).Sagawa S; Abe H; Hase Y; Inaba T J. Org. Chem 1999, 65, 4962. [DOI] [PubMed] [Google Scholar]
- (30).Arnold LD; Kalantar TH; Vederas JC J. Am. Chem. Soc 1985, 107, 7105. [Google Scholar]
- (31).Sasaki NA; Hashimoto C; Potier P Tetrahedron Lett 1987, 28, 6069. [Google Scholar]
- (32).Hata Y; Watanabe M Tetrahedron 1987, 17, 3881. [Google Scholar]
- (33).Belokon YN; Sagyan SM; Djamgaryan VI; Belikov VM Tetrahedron 1988, 17, 5507. [Google Scholar]
- (34).Yamada H; Kumagai H Pure Appl. Chem 1978, 50, 1117. [Google Scholar]
- (35).Inaba T; Birchler AG; Yamada Y; Sagawa S; Yokota K; Ando K; Uchida I J. Org. Chem 1998, 63, 7582. [Google Scholar]
- (36).Zook SE; Busse JK; Borer BC Tetrahedron Lett 2000, 41, 7017. [Google Scholar]
- (37).Schaus SE; Larrow JF; Jacobsen EN J. Org. Chem 1997, 62, 4197. [Google Scholar]
- (38).Kim EE; Baker CT; Dwyer MD; Murcko MA; Rao BG; Tung RD; Navia MA J. Am. Chem. Soc 1995, 117, 1181. [Google Scholar]
- (39).Ghosh AK; Fidanze S J. Org. Chem 1998, 63, 6146. [DOI] [PubMed] [Google Scholar]
- (40).Shibata N; Katoh T; Terashima S Tetrahedrom Lett 1997, 38, 619. [Google Scholar]
- (41).(a) Beaulieu PL; Wernic D; Duceppe J; Guindon Y Tetrahedron Lett 1995, 36, 3317. [Google Scholar]; (b) Beaulieu PL; Wernic D J. Org. Chem 1996, 61, 3635. [DOI] [PubMed] [Google Scholar]
- (42).Corey EJ; Zhang F Angew. Chem. Int. Ed 1999, 38, 1931. [DOI] [PubMed] [Google Scholar]
- (43).Kim BM; Bae SJ; So SM; Yoo HT; Chang SK; Lee JH; Kang J Org. Lett 2001, 3, 2349. [DOI] [PubMed] [Google Scholar]
- (44).Ohta Y; Shinkai I Bioorg. Med. Chem 1997, 5, 463. [DOI] [PubMed] [Google Scholar]
- (45).Dorsey BD; Levin RB; McDaniel SL; Vacca JP; Guare JP; Darke PL; Zugay JA; Emini EA; Schleif WA; Quintero JC; Lin JH; Chen I-W; Holloway MK; Fitzgerald PM; Axel MG; Ostovic D; Anderson PS; Huff J J. Med. Chem 1994, 37, 3443. [DOI] [PubMed] [Google Scholar]
- (46).Felder E; Maffei S; Pietra S; Pitre D Helv. Chim. Acta 1960, 888. [Google Scholar]
- (47).For an excellent review on the synthesis of aminoindanol see: Ghosh AK; Fidanze S; Senanayake CH Synthesis 1998, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kajiro H; Mitamura S-I; Mori A; Hiyama T Synlett 1998, 51. [Google Scholar]; (c) Buckland BC; Drew SW; Connors NC; Chartrain MM; Lee C; Salmon PM; Gbewonyo K; Zhou W; Gailliot P; Singhvi R; Olewinski RC Jr; Sun WJ; Reddy J; Zhang J; Jackey B; Taylor C; Goklen KE; Junker B; Greasham RL Metab. Eng 1999, 1, 63. [DOI] [PubMed] [Google Scholar]; (d) Kajiro H; Mitamura S-I; Mori A; Hiyama T Bull. Chem. Soc. Jpn 1999, 72, 1093. [Google Scholar]; (e) Demir AS; Hamamci H; Doganel F; Ozgul E J. Mol. Catal. B: Enzym 2000, 9, 157. [Google Scholar]
- (48).Askin D; Eng KK; Rossen K; Purick RM; Wells KM; Volante RP; Reider PJ Tetrahedron Lett 1994, 35, 673. [Google Scholar]
- (49).Rossen K; Weissman SA; Sager J; Reamer RA; Askin D; Volante RP; Reider PJ Tetrahedron Lett 1995, 36, 6419. [Google Scholar]
- (50).(a) Chakraborty TK; Gangakhedkar KK Synth. Comm 1996, 26, 2045. [Google Scholar]; (b) Chakraborty TK; Gangakhedkar KK Tetrahedron Lett 1991, 32, 1897. [Google Scholar]
- (51).(a) Roth W; Pigman W Methods Carbohydr. Chem 1963, 121, 405. [Google Scholar]; (b) Stamatatos L; Sinay P; Pougny J-R Tetrahedron 1984, 40, 1713. [Google Scholar]
- (52).(a) Maligres PE; Upadhyay V; Rossem K; Cianciosi SJ; Purick RM; Eng KK; Reamer RA; Askin D; Volante RP; Reider PJ Tetrahedron Lett 1995, 36, 2195. [Google Scholar]; (b) Maligres PE; Weissman SA; Upadhyay V; Cianciosi SJ; Reamer RA; Purick RM; Sager J; Rossen K; Eng KK; Askin D; Volante RP; Reider PJ Tetrahedron 1996, 52, 3327. [Google Scholar]
- (53).For an overview of the epoxidation route used as well as the syntheses of the various fragments see: Reider PJ Chimia 1997, 51, 306. [Google Scholar]
- (54).Rossen K; Pye PJ; DiMichele LM; Volante RP; Reider PJ Tetrahedron Lett 1998, 39, 6823. [Google Scholar]
- (55).(a) Senanayake CH; Smith GB; Ryan KM; Fredenburgh LE; Liu J; Roberts F; Hughes DL; Larsen RD; Verhoeven TR; Reider PJ Tetrahedron Lett 1996, 37, 3271. [Google Scholar]; (b) Senanayake CH; Roberts FE; DiMichele LM; Ryan KM; Liu J; Fredenburgh LE; Foster BS; Douglas AW; Larsen RD; Verhoeven TR; Reider PJ Tetrahedron Lett 1995, 36, 3993. [Google Scholar]
- (56).Askin D; Eng KK; Rossen K; Purick RM; Wells KM; Volante RP; Reider PJ Tetrahedron Lett 1994, 35, 673. [Google Scholar]
- (57).Cheng Y; Lu Z; Chapman KT; Tata JR J. Comb. Chem 2000, 2, 445. [DOI] [PubMed] [Google Scholar]
- (58).Kempf DJ; Marsh KC; Denissen JF; McDonald E; Vasavanonda S; Flentge CA; Green BE; Fino L; Park CH; Kong X-P; Wideburg NE; Saldivar A; Ruiz L; Kati WM; Sham HL; Robins T; Stweart KD; Hsu A; Plattner JJ; Leonard JM; Norbeck DW Proc. Natl. Acad. Sci. USA 1995, 92, 2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Kempf DJ; Sham HL; Marsh KC; Flentge CA; Betebenner D; Green BE; McDonald E; Vasavanonda S; Saldivar A; Wideburg NE; Kati WM; Ruiz L; Zhao C; Fino L; Patterson J; Molla A; Plattner JJ; Norbeck DW J. Med. Chem 1998, 41, 602. [DOI] [PubMed] [Google Scholar]
- (60).Evans BE; Rittle KE; Homnick CF; Springer JP; Hirshfield J; Veber DF J. Org. Chem 1985, 50, 4615. [Google Scholar]
- (61).DeCamp AE; Kawaguchi AT; Volante RP; Shinkai I Tetrahedron Lett 1991, 32, 1867. [Google Scholar]
- (62).Harding KE; Coleman MT; Liu LT Tetrahedron Lett 1991, 32, 3795. [Google Scholar]
- (63).Chu CK; Beach JW; Jeong LS; Choi BG; Comer FI; Alves AJ; Schinazi RF J. Org. Chem 1991, 56, 6503. [Google Scholar]
- (64).Ghosh AK; McKee SP; Thompson WJ; Darke PL; Zugay JC J. Org. Chem 1993, 58, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Dreyer GB; Lambert DM; Meek TD; Carr TJ; Tomaszek TA Jr.; Fernandez AV; Bartus H; Cacciavillani E; Hassell AM; Minnich M; Petteway SR Jr.; Metcalf BW Biochemistry 1992, 31, 6646. [DOI] [PubMed] [Google Scholar]
- (66).Baker WR; Pratt JK Tetrahedron 1993, 49, 8739. [Google Scholar]; The lactone was prepared in racemic form following the procedure of: Kempf DJ J. Org. Chem 1986, 51, 3921. [Google Scholar]
- (67).Dondoni A; Perrone D; Semola MT J. Org. Chem 1995, 60, 7927. [Google Scholar]
- (68).(a) Dondoni A; Fantin G; Fogagnolo M; Pedrini P J. Org. Chem 1990, 55, 1439. [Google Scholar]; (b) Dondoni A; Perrone D; Merino P J. Org. Chem 1995, 60, 8074. [Google Scholar]
- (69).Ghosh AK; Shin D; Mathivanan P Chem. Comm 1999, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Hanessian S; Park H; Yang R-Y Synlett 1997, 351. [Google Scholar]
- (71).Kempf DJ; Sowin TJ; Doherty EM; Hannick SM; Codavoci L; Henry RF; Green BE; Spanton SG; Norbeck DW J. Org. Chem 1992, 57, 5692. [Google Scholar]
- (72).Kempf DJ; Marsh KC; Fino LC; Bryant P; Craig-Kennard A; Sham HL; Zhao C; Vasavanonda S; Kohlbrenner WE; Wideburg NE; Saldivar A; Green BE; Herrin T; Norbeck DW Bioorg. Med. Chem 1994, 2, 847. [DOI] [PubMed] [Google Scholar]
- (73).(a) D’Aniello F; Taddei M J. Org. Chem 1992, 57, 5247. [Google Scholar]; (b) D’Aniello F; Mann A; Mattii D; Taddei M J. Org. Chem 1994, 59, 3762. [Google Scholar]
- (74).Benedetti F; Miertus S; Norbedo S; Tossi A; Zlatoidsky P J. Org. Chem 1997, 62, 9348. [Google Scholar]
- (75).Benedetti F; Norbedo S Chem. Comm 2001, 2, 203. [Google Scholar]
- (76).(a) Stuk TL; Haight AR; Scarpetti D; Allen MS; Menzia JA; Robbins TA; Parekh SI; Langridge DC; Tien J-H; Pariza RJ; Kerdesky FAJ J. Org. Chem 1994, 59, 4040. [Google Scholar]; (b) Haight AR; Stuk TL; Allen MS; Bhagavatula L; Fitzgerald M; Hannick SM; Kerdesky FAJ; Menzia JA; Parekh SI; Robbins TA; Scarpetti D; Tien J-H Org. Proc. Res. Dev 1999, 3, 94. [Google Scholar]
- (77).For syntheses of ritonavir, see section 3.2.
- (78).Kumar GN; Dykstra J; Roberts EM; Jayanti VK; Hickman D; Uchic J; Yao Y; Surber B; Thomas S; Granneman GR Drug Metab. Dispos 1999, 27, 902. [PubMed] [Google Scholar]
- (79).(a) Stoner EJ; Stengel PJ; Cooper AJ Org. Process Res. Dev 1999, 3, 145. [Google Scholar]; (b) Stoner EJ; Cooper AJ; Dickman DA; Kolaczkowski L; Lallaman JE; Liu J-H; Oliver-Schaffer PA; Patel KM; Paterson JB Jr.; Plata DJ; Riley DA; Sham HL; Stengel PJ; Tien J-H J. Org. Process Res. Dev 2000, 4, 264. [Google Scholar]
- (80).The coupling was performed using a mixture of diastereomers of isostere 222. See Section 6.1 of this review for the preparation of isostere 222.
- (81).Prepared from 2,6-dimethylphenol following procedure of: Organic Syntheses Collect Vol 5; Baumgarten HE, Ed.; Wiley: New York, 1973, 251. [Google Scholar]