

Abstract
West Nile virus (WNV) is a member of the flavivirus genus belonging to the Flaviviridae family. The viral serine protease NS2B/NS3 has been considered an attractive target for the development of anti-WNV agents. Although several NS2B/NS3 protease inhibitors have been described so far, most of them are reversible inhibitors. Herein, we present a series of α-aminoalkylphosphonate diphenyl esters and their peptidyl derivatives as potent inhibitors of the NS2B/NS3 protease. The most potent inhibitor identified was Cbz-Lys-Arg-(4-GuPhe)P(OPh)2 displaying Ki and k2/Ki values of 0.4 µM and 28 265 M−1s−1, respectively, with no significant inhibition of trypsin, cathepsin G, and HAT protease.
Keywords: NS2B/NS3 protease, aminophosphonates, serine proteases, enzyme inhibitors, West Nile virus
Introduction
The West Nile virus (WNV) belongs to the flavivirus genus (Flaviviridae family) and is a mosquito-borne human pathogen of global occurrence. WNV was first isolated from humans in 1937 in the West Nile district of Uganda1. In 1953, it was identified in birds of the Nile delta region. Until 1997, WNV was not considered pathogenic to birds when a more virulent strain appeared in Israel and caused fatal disease with signs of encephalitis and paralysis in various bird species. In 1999, a pathogenic WNV strain was transferred to New York leading to its rapid spread throughout the USA, Canada and in the following years, the virus further spread, reaching northern countries of South America2. The virus also became a relevant human pathogen in Eurasia, causing large outbreaks in Greece, Israel, Romania, and Russia3–6. Although the lifecycle of WNV involves the transmission of viruses between birds and mosquitoes, various mammalian species, including humans, and horses, are susceptible to the virus. However, mammals are generally dead-end hosts, being infected through the bites of infected mosquitoes7. Although infections with WNV are mainly asymptomatic, one-fifth of the infected humans develops symptoms of the milder West Nile fever or more severe neuroinvasive diseases (meningitis and encephalitis). Unfortunately, no vaccine or effective antiviral therapy against WNV is available8.
The flaviviral genome is a positive-sense single strand RNA. The viral replication process occurs in the cytoplasm where the RNA serves as a template for production of a large polyprotein, which is further processed by host and viral proteases. This proteolytic maturation yields structural (C, prM, and E) and non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). NS3 plays a key role during the polyprotein processing. This protein is composed of an N-terminal protease domain (1–179 amino acids) and a C-terminal helicase domain (residues 180–618). It has been demonstrated that inactivation of NS2B/NS3 protease catalytic centre blocks viral replication8. To become fully functional, the NS3 segment requires a short co-factor, NS2B. The WNV protease contains the classical serine protease catalytic triad Asp-His-Ser. The protease binding site exists as a shallow groove composed of 7 subsites (S4-S3’, according to the Schechter and Berger nomenclature)9. An analysis of the substrate preference of WNV NS2B/NS3 protease revealed that the natural substrates contain a highly conserved arginine residue in the P1 position. Further studies showed that basic amino acids were also preferred in P2 as well as in the P3 positions10,11.
Until now the most potent inhibitors of NS2B/NS3 protease have been reported by Stoermer et al.11. These compounds are tripeptide aldehydes (1,2) with a modified N-capping group (Figure 1). Although inhibitors 1 and 2 displayed low Ki values of 6 and 9 nM, respectively, due to the high reactivity of an aldehyde group, low stability and tendency to form hemiaminals, their application as potential therapeutics is limited12. Hammamy et al. presented a series of decarboxylated substrate analogues containing chlorophenylacetyl (3) or phenylacetyl moiety as an N-capping group which are one of the most potent reversible NS2B/NS3 inhibitors reported thus far13. Recently, Bastos et al. presented an interesting group of novel peptide-hybrids reversible inhibitors based on 2,4-thiazolidinedione scaffold (4)14. An interesting reversible inhibitor of NS2B/NS3 was described by Behnam et al.15 compound 5 containing a benzyloxyphenylglycine residue at P1 position showed a significant reduction of Dengue and WNV titres in cell-based assays of virus replication (EC50 = 15.5 µM).
Figure 1.
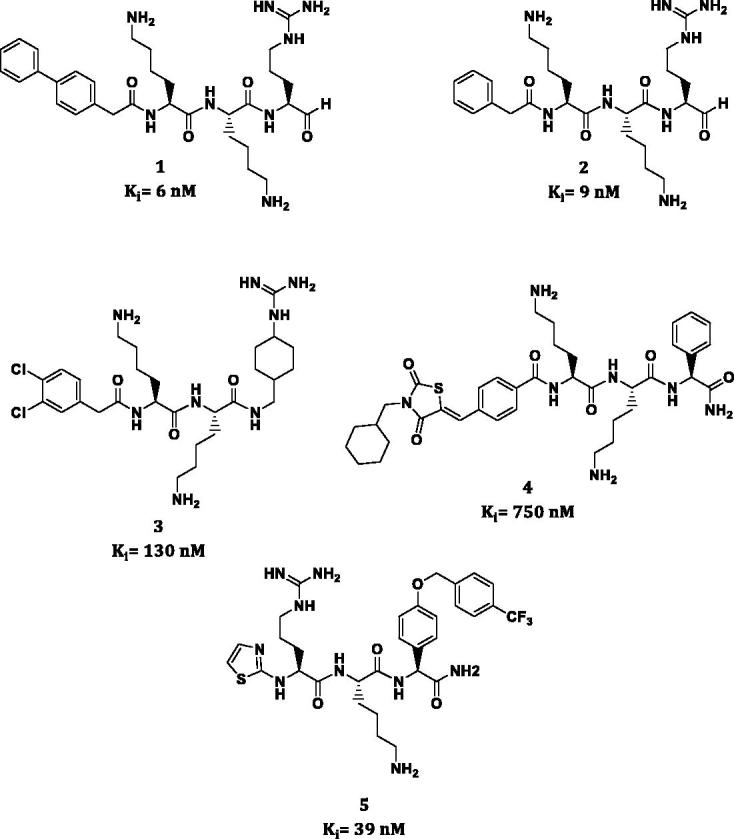
Inhibitors of the West Nile virus NS2B/NS3 protease.
Herein, we present the synthesis and application of α-aminoalkylphosphonates and their peptidyl derivatives as NS2B/NS3 WNV protease inhibitors. These compounds belong to a class of irreversible inhibitors that specifically and exclusively react with the active site serine residue leading to the formation of a slow hydrolysing protease-inhibitor complex (Figure 2)16. One of the major advantages of α-aminoalkylphosphonate diphenyl esters is their lack of reactivity with cysteine, aspartyl, and metalloproteases as well as good stability in buffer and human plasma17,18. In this work, we present a series of lysine, arginine, and peptidyl diphenylphosphonate derivatives which could be considered as starting templates for further structure optimisation studies as NS2B/NS3 protease inhibitors.
Figure 2.

Mechanism of serine proteases inhibition by α-aminoalkylphosphonate diphenyl esters. Residue numbering according to the West Nile virus NS2B/NS3 protease.
Chemistry
Chemical reagents
(Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP), CbzLys(Boc)-OH, Fmoc-Arg(Pbf)-OH, 2-chlorotrityl resin, trifluoroacetic acid, triisopropylsilane (TIPS), N,N-diisopropylethylamine (DIPEA), and di-tert-butyl dicarbonate (Boc2O) were purchased from IRIS Biotech (Marktredwitz, Germany). All other reagents, catalysts and solvents were purchased either from Sigma-Aldrich (Poznań, Poland), Merck (Warszawa, Poland), or Alfa Aesar (Karlsruhe, Germany).
Inhibitors synthesis
The synthesis of simple Cbz-protected α-aminoalkylphosphonate diphenyl esters (6–27) was performed as previously reported19,20. For the overall synthetic strategy as well as the spectroscopic data of the obtained key intermediates and new products, see the Supplementary material associated with the manuscript. The spectroscopic data for already known compounds fully agreed with the literature data. Briefly, the synthesis of ornithine (9) and lysine (6) diphenylphosphonates started with the preparation of N-phthalimide-protected amino aldehydes, which were further used in the α-amidoalkylation reaction with benzyl carbamate and triphenyl phosphite (Scheme S1.A and S1.B)20. The phthalimide group of resulting orthogonally protected derivatives was removed with hydrazine hydrate, which led to the target compounds21. Their subsequent guanidinylation with N,N’-di-Boc-S-ethyl isothiourea in the presence of HgCl2 and triethylamine followed by Boc deprotection (50% TFA in DCM) resulted in diphenylphosphonate analogues of arginine (7) and homoarginine (10)22. In order to synthesise phosphonate analogue of thio-arginine (8), 4-chlorobutylaldehyde obtained under Swern conditions from 4-chlorobutane-1-ol was used in the amidoalkylation reaction with benzyl carbamate and triphenyl phosphite with Cu(CF3SO3)2 as a catalyst (Scheme S2)23. The obtained product was treated with thiourea in refluxing ethanol yielding target compound which crystallised as white solid from diethyl ether. The synthesis of diphenylphosphonate glutamine from 4-oxo-N-tritylbutanamide followed the procedure Ewa et al. (Scheme S3)24. The synthesis of 4-amino (12) and 4-guanidine (13) derivatives of diphenylphosphonate phenylalanine started from the esterification of 4-nitrophenylacetic acid with methanol followed by the reduction of nitro group (Scheme S4). After Boc protection of the amino group, the ester function was reduced to alcohol and then oxidised, by means of the Swern method. Subsequently, α-amidoalkylation with benzyl carbamate and triphenyl phosphite under mild conditions with Cu(CF3SO3)2 as catalyst produced Cbz-(4-N-Boc)PheP(OPh)2. Boc deprotection with 50% TFA in DCM gave 12, which was further transformed into 13 as described above (for derivatives 7 and 10)25. A similar approach was applied for the synthesis of diphenylphosphonate phenylglycine derivatives (14–19) with the exception of nitrobenzaldehyde that was used for phosphonates synthesis, followed by nitro group reduction with SnCl2 prior to the guanylation step (Scheme S5)26. The synthesis of amidines 21 and 23 from the corresponding nitriles (20 and 22) followed the protocol developed by Oleksyszyn et al. (Scheme S6 and S7)20,27. Heterocyclic derivatives of diphenylphosphonate phenylglycine analogue (24–27) were obtained via the original α-amidoalkylation reaction procedure with substituted benzaldehydes obtained from 4-fluorobenzaldehyde and appropriate heterocyclic secondary amine (Scheme S8): pyrazole (24), benzimidazole (25), N-methylpiperazine (26), or morpholine (27)20,28.
For the Cbz-protected derivatives that are most active against NS2B/NS3 protease, we extended their structure with a dipeptidyl Cbz-Lys-Arg fragment. The synthetic strategy, outlined in Figure 3, started with the removal of the Cbz protecting group in orthogonally protected phosphonates via hydrogenation over 10% Pd/C, yielding target derivatives (32–35) containing a free amino group. In parallel, the Cbz-Lys(Boc)-Arg(Pbf)-OH was synthesised using solid phase peptide synthesis approach on 2-chlorotrityl resin. Next, the obtained phosphonate analogues of arginine were coupled to Cbz-Lys(Boc)-Arg(Pbf)-OH using PyBOP as the coupling agent in the presence of DIPEA. The reaction was performed in DMF for 12 h. The reaction mixture was then diluted five times with ethyl acetate and washed with 5% citric acid, 5% NaHCO3, and brine. The organic phase was dried over anhydrous MgSO4, filtered and evaporated to dryness. The obtained crude product was treated with cleavage solution (95% TFA, 2.5% TIPS, 2.5% H2O; v/v/v) for 2 h at room temperature prior to the precipitation of deprotected phosphonate peptides with diethyl ether. The final compounds were purified on the HPLC (Varian ProStar 210 with a dual λ absorbance detector system equipped with the Discovery® BIO Wide Pore C8 HPLC Column 250 mm × 212 mm, 10 µm) with a 15 ml/min flow rate using a gradient 5–95% (0.05% TFA/acetonitrile) in (0.05% TFA/H2O) over 15 min (Method A) or Discovery® BIO Wide Pore C8 HPLC Column (250 mm × 46 mm, 10 µm) with a 0.9 ml/min flow rate using a gradient 0 − 100% (0.05% TFA/acetonitrile) in (0.05% TFA/H2O) over 15 min (Method B)). The nuclear magnetic resonance spectra (1H and 31P) were recorded on either a Bruker Avance DRX-300 (300.13 MHz for 1H NMR, 121.50 MHz for 31P NMR) or Bruker Avance 600 MHz (600.58 MHz for 1H NMR, 243.10 MHz for 31P NMR) spectrometer. Chemical shifts are reported in parts per million (ppm) relative to a tetramethylsilane internal standard. High-resolution mass spectrometry was acquired on Waters Acquity Ultra Performance LC, LCT Premier, XE.
Figure 3.

Synthesis of peptidyl α-aminoalkylphosphonate diphenyl esters (35–38) of general formula Cbz-Lys-Arg-AaaP(OPh)2, where AaaP is a phosphonate ester analogue of arginine.
In vitro NS2B/NS3 protease inhibition
For the initial screening, Cbz-protected aminophosphonates were assayed in 96 well microplates (Nunc™ F96 MicroWell™ White Polystyrene Plate) in the following protease buffer: 50 mM Tris, 1 mM Chaps, 20% glycerol, pH 8.5. WNV NS2B/NS3 protease (AnaSpec, Liege, Belgium; 20 nM) was pre-incubated with tested inhibitor (100 µM) in the protease buffer at 37 °C for 10 min prior an addition of the fluorescent substrate Pyr-RTKR-AMC (AnaSpec, Liege, Belgium; 20 µM). The progress of the reaction was monitored continuously (λex = 354 nm, λem = 442 nm) at 37 °C on a Spectra Max Gemini XPS spectrofluorometer (Molecular Devices, Sunnyvale, CA) for 30 min. For compounds which exhibited more than 50% of inhibition, the Ki and k2/Ki values were calculated. In 96 well microplates narrowed concentration of inhibitors and constant substrate concentration (Pyr-RTKR-AMC, C = 20 µM, KM = 59 µM) were prepared. The enzyme solution was added and enzymatic reaction was monitored (Figure S1). Using a model for irreversible inhibition, in which the first order inactivation rate constant kobs is hyperbolic dependent from the inhibitor concentration Ki and k2/Ki values were calculated (equations (1–4) in supplementary material)29. Control progress curves in the absence of inhibitor were linear. The standard deviation for the presented values was calculated using the mean of two independent experiments and did not exceed 10%.
Control proteases inhibition assay
Bovine β-trypsin (AppliChem, Łódź, Poland), human cathepsin G (Biocentrum, Kraków, Poland) and Human Airway Trypsin-like Protease (HAT) (R&D Systems, Minneapolis, MN) were used as control proteases to screen the activity of NS2B/NS3 protease inhibitors (36–39) obtained in this study. Inhibitors were assayed in 96-well microplates in the 0.1 M HEPES, 0.5 M NaCl, 0.03%, pH 7.5 Triton X-100 (for bovine β-trypsin and cathepsin G). Bovine β-trypsin (15 nM), human cathepsin G (150 nM) or HAT protease (0.001 µg) was pre-incubated with tested inhibitor (25 µM) in the protease buffer at 37 °C for 10 min prior to the addition of the fluorescent substrate: Cbz-Arg-AFC (synthesised in-house according to the procedure described by Bissell30; 50 µM; λex = 400 nm, λem = 505 nm, for bovine β-trypsin); MeO-Suc-Ala-Ala-Pro-Val-AMC (Bachem, Bubendorf, Switzerland; 40 µM, λex = 340 nm, λem = 440 nm, for human cathepsin G) or Phe-Ser Arg-AMC (Bachem, Bubendorf, Switzerland; 40 µM, λex = 340 nm, λem = 440 nm, for HAT protease). The progress of the reactions was monitored continuously at 37 °C on a Spectra Max Gemini XPS spectrofluorometer for 20 min. Control curves in the absence of inhibitor were linear. The rate of the tested protease inhibition was calculated from the linear range of the plot.
Molecular docking
In order to evaluate the binding mode of the obtained phosphonate inhibitors into the NS2B/NS3 active site, we have performed a molecular docking simulation (AutoDock Vina 1.1.2) using the WNV NS2B/NS3 protease (2fp7.pdb) as a receptor31. The coordinates of the Bz-Nle-Lys-Arg-Arg-H inhibitor molecule as well as water molecules were removed from the structure (Figure 4)32. Since numerous studies have shown that α-aminoalkylphosphonates inhibitors complex serine proteases as phosphonic acids, we docked energy minimised (MM2 force field) inhibitor 38 in the chemical form of (Cbz-Lys-Arg-(4-GuPhe)P(OH)2)33,34. The centre of the grid box was defined at the catalytic Ser hydroxyl oxygen with the grid box size 100 × 100 × 100 Å. Pictures were prepared in Pymol35.
Figure 4.

(A) Docking conformation of Cbz-Lys-Arg-(4-GuPhe)P(OH)2 (38, green) in the binding site of NS2B/NS3 protease. The peptidyl inhibitor is shown in red (Bz-Nle-Lys-Arg-Arg-H) is present in the original crystal structure of WNV protease (2fp7.pdb). (B) Interaction of Cbz-Lys-Arg-(4GuPhe)P(OH)2 (38, orange) with the NS2B/NS3 active site. The hydrogen bond network is indicated with yellow dashed lines.
Results and discussion
Inhibitor P1 position screening
Compounds presented in this study are categorised into three groups: (I) compounds 6–11 are simple diphenyl phosphonate analogues of lysine, arginine, glutamine, ornithine, homoarginine, and thioarginine; (II) compounds 12–23 are aromatic analogues bearing a basic moiety, and (III) derivatives of diphenylphosphonatec phenylglycine (24–27) with different heterocyclic substituents. From all tested simple Cbz-N-capped derivatives, the highest potency of action toward NS2B/NS3 protease was observed for compounds 6, 7, 13, and 16 (Table 1). The most potent compound was observed to be 13 with k2/Ki value of 200 M−1s−1. Replacing the guanidine moiety in 13 with amino group (12) resulted in a dramatic drop in the inhibitory activity (11% of inhibition at 100 µM). In general, derivatives substituted at the meta position showed weaker inhibition levels as compared to their analogues substituted at para position of the phenyl ring (14, 15, 16 vs. 17, 18, 19). Compounds with heterocycles (24–27) showed very weak (1–5%) inhibitionagainst NS2B/NS3 protease when used at 100 µM concentration. This was probably because the heterocyclic group could not fit into the P1 binding pocket. The phenyl (21) and naphthyl (23) amidines were slightly more active against the tested protease. Nevertheless, among the tested series of α-aminoalkylphosphonate diphenyl esters we selected the most active compounds for further modifications. In summary, we identified lysine (6) and arginine (7) as the most favourable P1 residues, whereas for non-proteinogenic amino acid analogues we selected guanidine derivatives (13) and p (16)23.
Table 1.
Activities of simple Cbz N-capped phosphonates against the NS2B/NS3 WNV protease.a
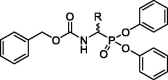 | |||
| No |
R |
Ki [μM]b |
k2/Ki [M−1s−1] |
| 6 | 22 ± 2 μM | 80 | |
| 7 |  |
13 ± 1 μM | 154 |
| 8 |  |
22% | |
| 9 | 12% | ||
| 10 | 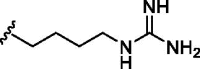 |
12% | |
| 11 | 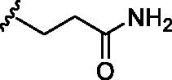 |
15% | |
| 12 | 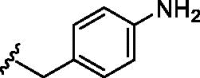 |
11% | |
| 13 | 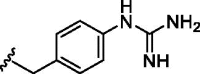 |
4 ± 0.3 μM | 200 |
| 14 | 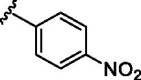 |
3% | |
| 15 | 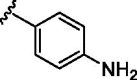 |
16% | |
| 16 | 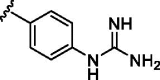 |
10 ± 1 μM | 87 |
| 17 | 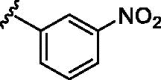 |
4% | |
| 18 | 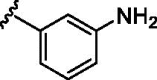 |
8% | |
| 19 | 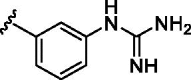 |
11% | |
| 20 | 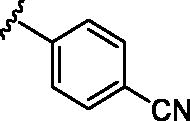 |
8% | |
| 21 | 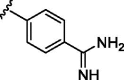 |
22% | |
| 22 | 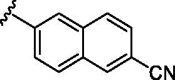 |
2% | |
| 23 | 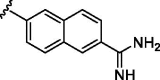 |
12% | |
| 24 | 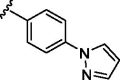 |
5% | |
| No |
R |
Ki [μM]b |
k2/Ki [M−1s−1] |
| 25 | 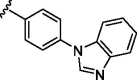 |
4% | |
| 26 | 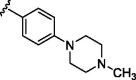 |
3% | |
| 27 | 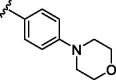 |
1% | |
Mean values ± standard deviation of two experiments conducted in duplicates.
percent of inhibition was calculated for compounds which displayed low activity toward NS2B/NS3 protease after 30 min incubation at 37 °C; substrate used: Pyr-RTKR-AMC (C = 20 µM, KM = 59 µM). Bold values indicate the most active compounds.
Influence of peptide chain elongation
The structure of the most active inhibitors identified in the initial screening step was elongated with a P2 Arg and P3 Lys. The resultant compounds (36–39) showed significantly increased inhibitory potencies against NS2B/NS3 protease (Table 2). The introduction of the additional two residues into the structure resulted in a similar (69-fold) improvement in their inhibitory potency, leading to 36 (k2/Ki = 5 520 M−1s−1) and 37 (k2/Ki = 10 725 M−1s−1). The most potent NS2B/NS3 protease inhibitor identified in the presented studies was compound 38, which displayed a k2/Ki value of 28 265 M−1s−1. The highest (∼290-fold) increase of inhibitory potencies was observed for the peptidyl derivative of the phosphonate analogue of 4-guanidinephenylglycine (39) which showed a k2/Ki value of 24 890 M−1s. The inhibition data observed for 36–39 is in agreement with the reported X-ray structure of NS2B/NS3 protease, thus highlighting the significant role of P2 and P3 residues in binding molecules (substrates and inhibitors) to the enzyme32. As reference compound we synthesised peptide aldehyde inhibitor (40). This reversible inhibitor showed Ki lower (∼4 times) than our most potent phosphonate inhibitor. However, it is difficult to compare the Ki values between reversible and irreversible inhibitors. Noteworthy, all of the obtained compounds were tested as diastereoisomeric mixtures thus their separation into single isomers will lead to significantly more potent inhibitors as observed previously36. Further investigation into the design and synthesis of phosphonate inhibitors of NS2B/NS3 protease might lead to discovering more potent and selective inhibitors. Future work should involve structure-activity relationship studies of the P2 and P3 residues as well as ring substituents. The next challenge is a more comprehensive structure-activity relationship study aiming to optimise the peptidyl fragment of the inhibitor as well as the structure of the aromatic ring substituent prior to the in vivo studies of most potent derivatives.
Table 2.
Activities of the peptide phosphonates against the NS2B/NS3 WNV proteasea
| No. | Compound | Ki (μM) | k2/Ki (M−1s−1) |
|---|---|---|---|
| 36 | Cbz-Lys-Arg-LysP(OPh)2 | 8 ± 0.9 | 5 520 |
| 37 | Cbz-Lys-Arg-ArgP(OPh)2 | 3 ± 0.3 | 10 725 |
| 38 | Cbz-Lys-Arg-(4-GuPhe)P(OPh)2 | 0.4 ± 0.03 | 28 265 |
| 39 | Cbz-Lys-Arg-(4-GuPhg)P(OPh)2 | 0.7 ± 0.2 | 24 890 |
| 40 | Cbz-Lys-Arg-Arg-H | 0.12 ± 0.02 | Not determined |
Mean values þ standard deviation of two experiments conducted in duplicates. Bold value indicates the most active compound.
Protease selectivity assay
The selectivity of inhibitors 36–39 were determined by means of serine proteases of a similar substrate recognition pattern such as bovine β-trypsin, human cathepsin G, and HAT protease. The results clearly showed that the inhibition levels observed for all of the investigated compounds at a concentration of 25 µM did not exceed 10% after a 30 min incubation period (Table S2). These results indicate that the obtained peptidyl inhibitors are not significantly active against members of proteases with a trypsin-like activity. However, the selectivity toward other members of this family will be further examined.
Molecular docking
The analysis of inhibitor 38 docked to the active site of NS2B/NS3 protease revealed docking conformation to be very similar to the one observed for Bz-Nle-Lys-Arg-Arg-H present in the crystal structure reported by Erbel et al. (2fp7.pdb)32. The reactive phosphonate warhead of the inhibitor is in close proximity (1.6 Å) to the catalytic serine residue allowing the formation of a covalent bond between the protease and inhibitor (Figure 4(A), Figure S3). The basic side chains of the amino acids in P1 and P3 positions are responsible for the formation of an extensive hydrogen bonding network with the protease (Figure 4(B)) including the P1 4-guanidinephenylalanine electrostatic interaction with the side chain of Asp129; P2 arginine with the carbonyl oxygen of Gly83, and Asp82 and the side chain of Asn84; and lysine in P3 position with Phe85 carbonyl oxygen. Additionally, two tyrosine residues (Tyr150, Tyr161) could be responsible for interaction with aromatic ring of 4-guanidinephenylalanine inhibitor residue (Figure 4(A), Figure S4). This interaction provides explanation for the improved activity of inhibitors 38 and 39 over simple lysine and arginine analogues (36,37).
Conclusions
In this work, we present a series of phosphonate diphenyl esters with low micromolar inhibitory activities against the WNV NS2B/NS3 protease. This class of inhibitors has never been reported to inhibit the NS2B/NS3 protease. The rigid 4-guanidinephenylalanine and 4-guanidinephenylglycine moieties at the P1 position were found to be more potent than a P1 arginine. Future work should involve more structure-activity relationship studies at the P2 and P3 residues and changing the phosphonate ester moieties.
Supplementary Material
Funding Statement
This work was supported by the Ministry of Science and Higher Education’s Iuventus Plus Programme [IP2012 0556 72]. The funder had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. MS and JO are grateful to Wroclaw University of Technology for support [Statute Funds 0401/0195/17]. MS is grateful to National Science Centre [UMO-2013/09/N/ST5/02439]. AM and KP are grateful to National Science Centre [UMO-2016/21/B/NZ6/01307]. Project supported by Wroclaw Centre of Biotechnology, programme The Leading National Research Centre (KNOW) for years 2014–2018.
Acknowledgements
The authors would like to thank Dr. Ewa Burchacka and Maciej Walczak for the kind gift of Cbz-GlnP(OPh)2 and Dr. Renata Grzywa for guidance in the inhibitors docking analysis.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Smithburn KC, Hughes TP, Burke AW, Paul JH. A neurotropic virus isolated from the blood of a native of Uganda. Am J Trop Med Hyg 1940;s1–20:471–92. [Google Scholar]
- 2.Lanciotti RS, Roehrig JT, Deubel V, et al. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 1999;286:2333–7. [DOI] [PubMed] [Google Scholar]
- 3.McMullen AR, May FJ, Li L, et al. Evolution of new genotype of West Nile virus in North America. Emerging Infect Dis 2011;17:785–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention CDC ) [cited 2018 Sept 5]. Available from: www.cdc.gov/westnile/index.html
- 5.Sejvar JJ. West Nile virus: an historical overview. Ochsner J 2003;5:6–10. [PMC free article] [PubMed] [Google Scholar]
- 6.Colpitts TM, Conway MJ, Montgomery RR, Fikrig E. West Nile virus: biology, transmission, and human infection. Clin Microbiol Rev 2012;25:635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holbrook MR. Historical perspectives on flavivirus research. Viruses 2017;9:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suthar MS, Diamond MS, Gale M. West Nile virus infection and immunity. Nat Rev Microbiol 2013;11:115–28. [DOI] [PubMed] [Google Scholar]
- 9.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 1967;27:157–62. [DOI] [PubMed] [Google Scholar]
- 10.Chappell KJ, Stoermer MJ, Fairlie DP, Young PR. West Nile virus NS2B/NS3 protease as an antiviral target. Curr Med Chem 2008;15:2771–84. [DOI] [PubMed] [Google Scholar]
- 11.Stoermer MJ, Chappell KJ, Liebscher S, et al. Potent cationic inhibitors of West Nile virus NS2B/NS3 protease with serum stability, cell permeability and antiviral activity. J Med Chem 2008;51:5714–21. [DOI] [PubMed] [Google Scholar]
- 12.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol 2001;8:739–58. [DOI] [PubMed] [Google Scholar]
- 13.Hammamy MZ, Haase C, Hammami M, et al. Development and characterization of new peptidomimetic inhibitors of the West Nile virus NS2B-NS3 protease. ChemMedChem 2013;8:231–41. [DOI] [PubMed] [Google Scholar]
- 14.Bastos LA, Behnam MA, El Sherif Y, et al. Dual inhibitors of the dengue and West Nile virus NS2B-NS3 proteases: synthesis, biological evaluation and docking studies of novel peptide-hybrids. Bioorg Med Chem 2015;23:5748–55. [DOI] [PubMed] [Google Scholar]
- 15.Behnam MAM, Graf D, Bartenschlager R, et al. Discovery of nanomolar dengue and West Nile virus protease inhibitors containing a 4-benzyloxyphenylglycine residue. J Med Chem 2015;58:9354–70. [DOI] [PubMed] [Google Scholar]
- 16.Oleksyszyn J, Powers JC. Irreversible inhibition of serine proteases by peptidyl derivatives of alpha-aminoalkylphosphonate diphenyl esters. Biochem Bioph Res Co 1989;161:143–9. [DOI] [PubMed] [Google Scholar]
- 17.Oleksyszyn J, Powers JC. Irreversible inhibition of serine proteases by peptide derivatives of (alpha-aminoalkyl)phosphonate diphenyl esters. Biochemistry 1991;30:485–93. [DOI] [PubMed] [Google Scholar]
- 18.Sienczyk M, Oleksyszyn J. Irreversible inhibition of serine proteases – design and in vivo activity of diaryl alpha-aminophosphonate derivatives. Curr Med Chem 2009;16:1673–87. [DOI] [PubMed] [Google Scholar]
- 19.Oleksyszyn J, Powers JC. Amino acid and peptide phosphonate derivatives as specific inhibitors of serine peptidases Methods in Enzymology 1994;244:423–441. [DOI] [PubMed] [Google Scholar]
- 20.Oleksyszyn J, Subotkowska L, Mastalerz P. Diphenyl 1-aminoalkanephosphonates. Synthesis 1979;1979:985–6. [Google Scholar]
- 21.Hamilton R, Walker BJ, Walker B. A convenient synthesis of N-protected diphenyl phosphonate ester analogues of ornithine, lysine and homolysine. Tetrahedron Lett 1993;34:2847–50. [Google Scholar]
- 22.Peterlin-Mašič L, Kikelj D. Arginine mimetics. Tetrahedron 2001;57:7073–105. [Google Scholar]
- 23.Van der Veken P, El Sayed I, Joossens J, et al. Lewis acid catalyzed synthesis of N-protected diphenyl 1-aminoalkyl-phosphonates. Synthesis 2004;2005:634–8. [Google Scholar]
- 24.Ewa B, Maciej W, Marcin S, et al. The development of first Staphylococcus aureus SplB protease inhibitors: phosphonic analogues of glutamine. Bioorg Med Chem Lett 2012;22:5574–8. [DOI] [PubMed] [Google Scholar]
- 25.Joossens J, Van der Veken P, Lambeir AM, et al. Development of irreversible diphenyl phosphonate inhibitors for urokinase plasminogen activator. J Med Chem 2004;47:2411–3. [DOI] [PubMed] [Google Scholar]
- 26.Sienczyk M, Oleksyszyn J. A convenient synthesis of new alpha-aminoalkylphosphonates, aromatic analogues of arginine as inhibitors of trypsin-like enzymes. Tetrahedron Lett 2004;45:7251–4. [Google Scholar]
- 27.Oleksyszyn J, Boduszek B, Kam CM, Powers JC. Novel amidine-containing peptidyl phosphonates as irreversible inhibitors for blood coagulation and related serine proteases. J Med Chem 1994;37:226–31. [DOI] [PubMed] [Google Scholar]
- 28.Magdolen P, Meciarova M, Toma S. Ultrasound effect on the synthesis of 4-alkyl-(aryl)aminobenzaldehydes. Tetrahedron 2001;57:4781–5. [Google Scholar]
- 29.Stein RL, Trainor DA. Mechanism of inactivation of human leukocyte elastase by a chloromethyl ketone: kinetic and solvent isotope effect studies. Biochemistry 1986;25:5414–9. [DOI] [PubMed] [Google Scholar]
- 30.Bissell ER, Mitchell AR, Smith RE. Synthesis and chemistry of “7-amino-4-(trifluoromethyl)coumarin and its amino-acid and peptide derivatives. J Org Chem 1980;45:2283–7. [Google Scholar]
- 31.Trott O, Olson AJ. Software news and update autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2009;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erbel P, Schiering N, D’Arcy A, et al. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat Struct Mol Biol 2006;13:372–3. [DOI] [PubMed] [Google Scholar]
- 33.Hof P, Mayr I, Huber R, et al. The 1.8 angstrom crystal structure of human cathepsin G in complex with Suc-Val-Pro-Phe(P)-(OPh)(2): a janus-faced proteinase with two opposite specificities. Embo J 1996;15:5481–91. [PMC free article] [PubMed] [Google Scholar]
- 34.Lechtenberg BC, Kasperkiewicz P, Robinson H, et al. The elastase-PK101 structure: mechanism of an ultrasensitive activity-based probe revealed. ACS Chem Biol 2015;10:945–51. [DOI] [PubMed] [Google Scholar]
- 35.DeLano WL. The PyMOL molecular graphics system. San Carlos (CA): DeLano Scientific; 2002. [Google Scholar]
- 36.Winiarski L, Oleksyszyn J, Sienczyk M. Human neutrophil elastase phosphonic inhibitors with improved potency of action. J Med Chem 2012;55:6541–53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.