

Abstract
Niacin (vitamin B3) is available as a prescription medication and over‐the‐counter supplement. Although it is well known for its vasodilatory effect, it has also been associated with mild hepatotoxicity and, rarely, acute liver failure. We present the case of a 74‐year‐old Hispanic woman who developed acute liver failure (anicteric encephalopathy and coagulopathy) after her home dose of immediate‐release niacin was replaced with an extended‐release formulation during an inpatient hospital stay. This is the first reported case of niacin toxicity associated with a histopathologic finding of diffuse microvesicular steatosis. This unique phenotype strongly implicates mitochondrial impairment as a mechanism of niacin‐induced hepatotoxicity.
Abbreviations
- DILI
drug‐induced liver injury
- ER
extended release
- IR
immediate release
- SR
sustained release
Drug‐induced liver injury (DILI) usually exhibits transient liver test abnormalities with or without jaundice that reverses after medication discontinuation. In its severe form, however, DILI can result in acute liver failure. Typically, we classify DILI‐causing agents into direct‐acting and idiosyncratic (usually adaptive immune mediated). Acetaminophen is the major direct acting toxin with a well‐established body of literature that has elucidated its mechanism of injury. Niacin also induces direct hepatotoxicity, but little is known about its mechanism. Niacin has traditionally been used as a lipid‐lowering agent and, recently, as treatment for blistering skin disorders like bullous pemphigoid. Here, we present the first reported case of acute hepatic failure secondary to high‐dose niacin extended‐release (ER) with a histological finding of diffuse microvesicular steatosis that is characteristic of targeted mitochondrial impairment.
Case Presentation
A 74‐year‐old Hispanic woman presented with a chief complaint of progressive weakness and diarrhea over 4 weeks. Her past medical history included bullous pemphigoid, essential hypertension, and type 2 diabetes mellitus. She had no history of liver disease nor reported history of alcohol consumption, illicit drug use, or herbal or nutritional supplement use. Relevant home medications included prednisone 25 mg every other day, mycophenolate mofetil 1.5 g twice daily, and niacin immediate‐release (IR) 500 mg three times daily. She was admitted to the general medicine ward for suspected adrenal insufficiency secondary to rapid steroid tapering following a previously treated bullous pemphigoid flare. On admission, her home niacin was replaced with niacin ER 500 mg three times daily; other home medications were continued.
She was started on 25 mg of daily prednisone on day 1, which was increased to 40 mg on day 2. On day 5 she developed acute encephalopathy, coinciding with an elevation in her international normalized ratio to 3.75 (normal, 0.9‐1.11) from 1.4 on admission. The Liver Consult service was then called to see her and found a frail, chronically ill‐appearing female with a body weight of 84.7 kg and body mass index of 33.9 kg/m2; vital signs were normal. She was lethargic and disoriented to time with asterixis, a diffusely mildly tender abdomen, pitting edema in her bilateral lower extremities, and multiple superficial erosions and bullae over her body. Other laboratory tests revealed a white blood cell count of 18.2 103/cumm (normal, 4.5‐10.0), hemoglobin of 9 g/dL (normal, 12.0‐14.6), serum sodium of 126 mmol/L (normal, 135‐145), serum potassium of 5.1 mmol/L (normal, 3.5‐5.1), serum bicarbonate of 15 mmol/L (normal, 20‐30), serum glucose of 87 mg/dL (normal, 65‐99), and alkaline phosphatase of 322 U/L (normal, 30‐100). Results of the sequential liver test are given in Table 1. There were no recent hypotensive or ischemic events and no evidence of sepsis. Imaging was negative; acetaminophen level was less than 15 µg/mL; hepatitis A, B, and C serologies were negative; ammonia level was 49 µmol/L (normal, 11‐48); lactate dehydrogenase was 1111 U/L (normal, 90‐220); creatine kinase was 52 U/L (normal, 25‐145 U/L); and transthoracic echocardiogram demonstrated an ejection fraction of 65%.
Table 1.
Liver Tests Over the Course of Admission
| Hospital Day |
ALP (U/L) (normal, 30‐100) |
AST (U/L) (normal, 10‐40) |
ALT (U/L) (normal, 5‐40) |
TB (mg/dL) (normal, < 1.0) |
INR (normal, 0.9‐1.11) |
|---|---|---|---|---|---|
| 1 | — | — | — | — | — |
| 2 | 199 | 31 | 24 | 0.3 | 1.06 |
| 3 | 201 | 38 | 27 | 0.5 | 1.15 |
| 4 | 208 | 51 | 31 | 0.7 | 1.4 |
| 5 | 322 | 428 | 176 | 0.9 | 3.75 |
| 6 | 236 | 1091 | 523 | 1.8 | 10.1 |
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; INR, international normalized ratio; TB, tuberculosis.
Her clinical condition was consistent with anicteric hepatic failure, likely secondary to niacin administration given the temporal relationship between drug exposure and symptom onset. Continuation of outpatient mycophenolate, insulin, furosemide, spironolactone, and metronidazole was started on day 3 for fecal C. difficile; none were considered to have caused hepatotoxicity. She was transferred to the intensive care unit for close monitoring and started on N‐acetylcysteine. She was also given a vitamin K challenge for her coagulopathy. Serial laboratory testing demonstrated progressively worsening liver tests (Table 1). Niacin ER had been discontinued on day 4 and replaced with her home formulation of niacin IR. Niacin IR was discontinued on day 5. She became hypotensive late on day 5, requiring vasopressor support, and subsequently developed acute respiratory distress and died approximately 12 hours later.
Autopsy revealed liver weight of 750 g (normal, 1200‐1800 g) with smooth and glistening capsule. Cut sections showed the liver to be soft and uniformly tan‐yellow to light red without regenerative nodule formation or other lesions. Microscopic examination showed a normal architectural pattern without fibrosis. The portal tracts were unremarkable. The parenchyma showed diffuse, predominantly small, droplet microvesicular steatosis without lobular inflammation or liver cell ballooning (Figure 1). Perivenular early ischemic necrosis of hepatocytes was present (most likely due to terminal hypotension). No other contributing cause of her clinical condition or death was identified.
Figure 1.
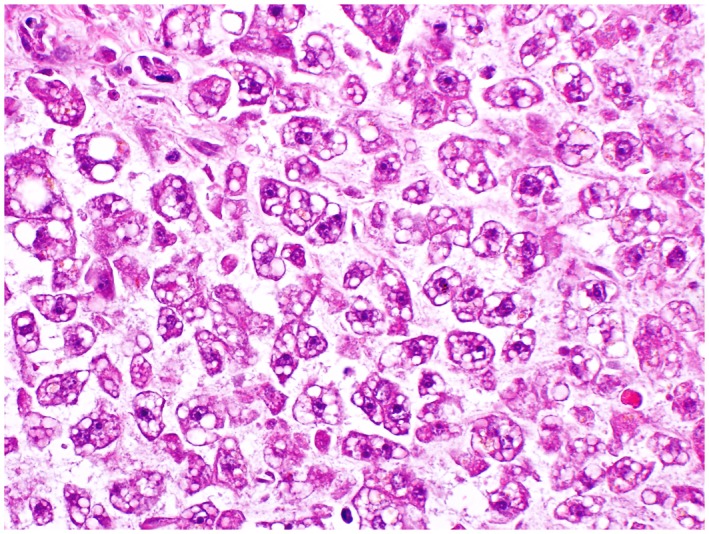
Hepatic microvesicular steatosis. A routine section of liver from the autopsy shows diffuse small droplet microvesicular steatosis without accompanying inflammation (hematoxylin and eosin, 40).
Discussion
Niacin traditionally has been used to lower low‐density lipoprotein and raise high‐density lipoprotein cholesterol levels. Niacin IR, also termed plain or crystalline niacin, was first introduced in the mid‐1950s. However, due to poor compliance from cutaneous flushing, a slower release formulation was introduced as niacin sustained‐release (SR) in the 1960s. Although it did not gain Food and Drug Administration approval because of suspected hepatotoxicity, it remains available as an over‐the‐counter dietary supplement. Niacin ER, with intermediate kinetics between the IR and SR formulations, was subsequently introduced in 1997 with Food and Drug Administration approval for the management of dyslipidemia.1 Although niacin is no longer commonly used in treating dyslipidemia, it remains in use in conditions such as bullous pemphigoid, where it is believed to inhibit chemotaxis of neutrophils and eosinophils.2
Niacin undergoes first‐pass metabolism in the liver through the high‐affinity, low‐capacity amidation pathway or the low‐affinity, high‐capacity glycine conjugation pathway. The conjugation pathway results in production of nicotinuric acid. Although it was previously believed that nicotinuric acid is responsible for flushing, it is now known that flushing results when the precursor compound, nicotinic acid, binds with the GPR109A receptor in epidermal Langerhans cells, triggering prostaglandin D2 release. The amidation pathway results in the formation of nicotinamide and pyrimidine metabolites and has been implicated in causing hepatotoxicity, although the precise mechanism remains unclear.3 The dissolution and absorption rates of the different niacin formulations determines the principle pathway for metabolism. Niacin IR, with its rapid absorption and dissociation, saturates the amidation pathway and is thereby metabolized primarily through the conjugation pathway. In contrast, niacin SR and ER, with their slower absorption and dissociation rates, are metabolized primarily through the amidation pathway.1
All formulations of niacin have been implicated in causing hepatotoxicity. Table 2 summarizes previous reports of niacin‐induced liver injury. Niacin IR typically causes hepatotoxicity at doses above 3 g/day.4, 5, 6, 7, 8 Although usually reversible, at least one documented case resulted in acute liver failure, requiring emergent liver transplantation.8 Earlier cases of niacin‐induced liver injury were most frequently seen in middle‐aged individuals taking niacin for dyslipidemia.4, 5 In more recent years, however, there have been reports of young adults ingesting high doses of niacin IR for purported sports performance enhancement or drug screen masking, with resultant liver injury and even acute liver failure.7, 8, 9, 10 Given the widespread availability of niacin in dietary supplements and energy drinks, greater awareness of its toxicity is warranted.
Table 2.
Niacin‐Induced Hepatotoxicity
| References | Age | Sex | Form | Dose (g/d) | Duration | ALP (U/L) | TB (mg/dL) | AST (U/L) | ALT (U/L) | PT (s)/INR | Histology |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Winter and Boyer4 | 35 | M | IR | 9 | Several days | 62 | 4.9 | 2,072 | 2,640 | 14.9/— | Increase in portal fibrosis; mild proliferation of bile ductules; sparse portal inflammatory infiltrate consisting of chronic inflammatory cells; centrilobular parenchymal cell swelling with cytoplasmic vacuolization; no cell necrosis or lobular inflammation; few mitotic figures and canalicular bile plugs |
| Patterson et al.6 | 41 | M | IR | 4.5 | — | 542 | 8.0 | 2,660 | 3,300 | 28.5/— | Distorted architecture with massive and submassive lobular collapse and increased fibrosis in a few areas of collapse; marked cholestasis with canalicular bile plugging; many hepatocytes with swollen vacuolated cytoplasm; normal intralobular ducts |
| Clementz and Holmes5 | 46 | M | IR | 3 | 10 weeks | 239 | 6.6 | 14,140 | 6,220 | 90/— | — |
| Mullin et al.21 | 44 | M | SR | 6 | 3 days | 384 | 9.82 | 6,181 | 5,162 | 64.6/— | Massive hepatic necrosis with collapse of the architecture; pigment‐laden macrophages within areas of collapse; cholestasis within remaining, viable hepatic parenchyma; minimal fatty change in some remaining hepatocytes |
| Henkin et al.14 | 62 | M | SR | 4 | 5 days | 70 | 0.82 | 800 | 700 | — | — |
| 50 | F | SR | 2 | Several weeks | 2,325 | 7.07 | 227 | 870 | — | — | |
| 47 | M | SR | 2 | 2 months | 85 | 0.53 | 160 | 155 | — | — | |
| Fischer22 | 56 | M | SR | 2 | 2 months | 270 | 6.0 | 2,010 | 2,567 | — | Massive and submassive lobular collapse with increased fibrosis in some areas of collapse; pronounced cholestasis with canalicular bile plugging; vacuolated cytoplasm |
| Etchason et al.12 | 51 | M | SR | 2 | 3 weeks | 293 | 1.5 | 3,170 | — | — | — |
| 60 | F | SR | 3 | 3 days | — | — | 65 | 50 | — | — | |
| 54 | M | SR | 2 | 7 weeks | — | — | 59 | — | — | — | |
| 34 | M | SR | 2.5 | 2 days | — | — | 132 | — | — | — | |
| 61 | F | SR | 1.5 | 10 months | 285 | — | 208 | — | — | — | |
| Dearing et al.18 | 55 | M | SR | 2 | 3 months | — | 1.0 | 55 | 86 | 17.2/— | — |
| 47 | M | SR | 2 | — | — | 0.6 | 65 | 56 | 19.2/— | — | |
| 44 | M | SR | 3 | 5 weeks | — | 0.7 | 73 | 56 | 17.7/— | — | |
| Dalton and Berry13 | 67 | F | SR | — | 2 days | 252 | 0.5 | 363 | 105 | 15.8/— | — |
| Lawrence et al.23 | 37 | F | SR | 4 | 1 month | 503 | 0.8 | 63 | 58 | — | — |
| Coppola et al.19 | 58 | M | SR | 2.5 | 5 months | 136 | 1.7 | 86 | 86 | 16.6/— | — |
| 52 | F | SR | 1.5 | 3 months | 70 | 0.6 | 209 | 208 | — | — | |
| 44 | M | SR | 2.25 | — | 151 | 1.1 | 57 | — | — | Mild focal intracellular cholestasis and fatty liver | |
| 62 | M | SR | 2 | — | 70 | 1.2 | 26 | 34 | — | — | |
| Ali et al.17 | 61 | M | ER | 2 | 4 years | — | 1.4 | 52 | 44 | 16.6/1.8 | — |
| Eswaran et al.7 | 20 | M | IM | 3.75 | 2 days | — | — | 4,070 | 3,832 | 4.95/— | — |
| 23 | M | IM | 1 | — | — | — | 2,903 | 4,087 | 4.3/— | — | |
| Ellsworth et al.9 | 17 | M | IM | — | — | — | 8.4 | 295 | 284 | 32.3/4.8 | — |
| Schaffellner et al.8 | 22 | F | IM | 20 | 1 day | 165 | 3.6 | 14,985 | 12,594 | —/> 8.9 | Panlobular parenchymal necrosis with congestion |
| Mittal et al.10 | 17 | F | — | 2.5 | 2 days | 89 | 1.3 | 35 | 20 | 23.1/3.8 | — |
| 14 | M | SR | 5.5 | 1 day | — | — | 193 | 344 | 18.8/1.74 | — |
Abbreviations: F, female; M, male.
Time‐release formulations of niacin have comparatively higher potential for causing liver injury. Up to 52% of patients taking niacin SR may develop dose‐dependent elevations in serum aminotransferases, with symptoms usually occurring with doses above 2 g/day.11 Hepatotoxicity can develop in as early as 2 days, but can occur even after months of exposure.12 Drug‐induced hepatitis has also been observed within days of converting from niacin IR to SR, with no evidence of liver injury after niacin IR rechallenge. Although our patient received a somewhat lower dose (1.5 g/day) compared with most of the reported cases of ER and SR DILI (> 2.0 g/day), we hypothesize that her complex underlying medical problems, such as bullous pemphigoid, steroid use and diabetes, may have increased the susceptibility to a somewhat lower dose of niacin ER than typically associated with niacin DILI.
Niacin ER has been associated with elevations in serum aminotransferases with doses above 2 g/day, though these elevations were not clinically significant.15, 16 Only 1 other report of niacin ER‐related liver injury was identified.17 This may not necessarily suggest lower risk of hepatotoxicity, but rather reflect the general decline in niacin’s use in clinical practice for the management of dyslipidemia since the introduction of other lipid lowering agents, namely statins.
Although marked elevations in serum aminotransferases are usually seen, impaired hepatic synthesis may be the only indication of niacin‐induced liver injury. Coagulopathy secondary to clotting factor deficiency with minimal elevations in transaminases has been observed with both niacin SR and ER.17, 18, 19 Low serum albumin levels have also been observed, with subsequent improvement after discontinuation of niacin.20 As previous studies have used serum aminotransferases as markers of liver injury, it is possible that impaired hepatic function as a manifestation of liver injury may be missed and underestimate the incidence of niacin‐induced hepatotoxicity.
Niacin has been associated with focal fatty infiltration of the liver. Focal fat appears as an area24 of increased echogenicity on ultrasound and lucency on computed tomography, which represents macrovesicular fatty change in sporadic foci, a benign phenomenon that resolves when niacin is discontinued.19, 23
Histopathologic features that have previously been described in association with niacin‐induced liver injury include necrosis, cholestasis, fibrosis, and/or hepatocyte vacuolization (Table 2).4, 8, 21, 22 This is the first demonstration of diffuse microvesicular steatosis, a feature seen in alcoholic foamy fatty liver disease, Reye’s syndrome, acute fatty liver disease of pregnancy, and nucleoside drug‐induced liver injury.24 Microvesicular steatosis is a marker of mitochondrial injury, presenting with coagulopathy, encephalopathy, and minimal hyperbilirubinemia. We believe that mitochondrial targeting may be frequently missed in such patients, especially because histology is often not available. However, the striking encephalopathy and coagulopathy that have been described in other cases of niacin hepatotoxicity with minimal hyperbilirubinemia are important clues to mitochondrial impairment. Although the exact pathogenesis of niacin‐induced mitochondrial injury is unknown, it may be due to imbalance in the mitochondrial redox status of nicotinamide adenine dinucleotide (NAD)/reduced nicotinamide adenine dinucleotide or nicotinamide adenine dinucleotide phosphate/reduced form of nicotinamide adenine dinucleotide phosphate NAD‐dependent sirtuin activation favoring excessive deacetylation, adenosine diphosphate ribosylation, or dose‐dependent metabolism of niacin to an unknown toxic intermediate. In conclusion, we describe a case of fatal anicteric acute liver failure with microvesicular hepatic steatosis as the only pathologic finding. We believe that this clinical scenario as a result of ER or SR niacin may be underrecognized.
Author Contributions
K.L. prepared and edited the manuscript. Z.C. and G.K. provided histopathologic images and descriptions. M.Q. and N.K. edited the manuscript.
Potential conflicts of interest: Nothing to report.
References
- 1. Pieper JA. Overview of niacin formulations: differences in pharmacokinetics, efficacy, and safety. Am J Health‐Syst Pharm 2003;60:S9‐S14. [DOI] [PubMed] [Google Scholar]
- 2. Hornschuh B, Hamm H, Wever S, Hashimoto T, Schröder U, Bröcker E‐B, et al. Treatment of 16 patients with bullous pemphigoid with oral tetracycline and niacinamide and topical clobetasol. J Am Acad Dermatol 1997;36:101‐103. [DOI] [PubMed] [Google Scholar]
- 3. Stern RH. The role of nicotinic acid metabolites in flushing and hepatotoxicity. J Clin Lipidol 2007;1:191‐193. [DOI] [PubMed] [Google Scholar]
- 4. Winter SL, Boyer JL. Hepatic toxicity from large doses of vitamin B3 (nicotinamide). N Engl J Med 1973;289:1180‐1182. [DOI] [PubMed] [Google Scholar]
- 5. Clementz GL, Holmes AW. Nicotinic acid‐induced fulminant hepatic failure. J Clin Gastroenterol 1987;9:582‐584. [DOI] [PubMed] [Google Scholar]
- 6. Patterson DJ, Dew EW, Gyorkey F, Graham DY. Niacin hepatitis. South Med J 1983;76:239‐240. [DOI] [PubMed] [Google Scholar]
- 7. Eswaran S, Alvey N, Fayek S, Nikunj S, Chan E. Niacin, the internet, and urine drug testing: a cause of acute liver failure. J Clin Toxicol 2013;3:1‐3. [Google Scholar]
- 8. Schaffellner S, Stadlbauer V, Sereinigg M, Mìller H, Högenauer C, Fickert P, et al. Niacin‐associated acute hepatotoxicity leading to emergency liver transplantation. Am J Gastroenterol 2017;112:1345‐1346. [DOI] [PubMed] [Google Scholar]
- 9. Ellsworth MA, Anderson KR, Hall DJ, Freese DK, Lloyd RM. Acute liver failure secondary to niacin toxicity. Case Rep Pediatr 2014;2014:1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mittal MK, Florin T, Perrone J, Delgado JH, Osterhoudt KC. Toxicity from the use of niacin to beat urine drug screening. Ann Emerg Med 2007;50:587‐590. [DOI] [PubMed] [Google Scholar]
- 11. McKenney JM, Proctor JD, Harris S, Chinchili VM. A comparison of the efficacy and toxic effects of sustained‐ vs immediate‐release niacin in hypercholesterolemic patients. JAMA 1994;271:672‐677. [PubMed] [Google Scholar]
- 12. Etchason JA, Miller TD, Squires RW, Allison TG, Gau GT, Marttila JK, et al. Niacin‐induced hepatitis: a potential side effect with low‐dose time‐release niacin. Mayo Clin Proc 1991;66:23‐28. [DOI] [PubMed] [Google Scholar]
- 13. Dalton TA, Berry RS. Hepatotoxicity associated with sustained‐release niacin. Am J Med 1992;93:102‐104. [DOI] [PubMed] [Google Scholar]
- 14. Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug‐induced hepatitis from sustained‐release niacin. JAMA 1990;264:241‐243. [PubMed] [Google Scholar]
- 15. Goldberg AC. Clinical trial experience with extended‐release niacin (Niaspan): dose‐escalation study. Am J Cardiol 1998;82:35U‐38U. [DOI] [PubMed] [Google Scholar]
- 16. Capuzzi DM, Guyton JR, Morgan JM, Goldberg AC, Kreisberg RA, Brusco OA, et al. Efficacy and safety of an extended‐release niacin (Niaspan): a long‐term study. Am J Cardiol 1998;82:74U‐81U. [DOI] [PubMed] [Google Scholar]
- 17. Ali EH, McJunkin B, Jubelirer S, Hood W. Niacin induced coagulopathy as a manifestation of occult liver injury. W V Med J 2013;109:12‐14. [PubMed] [Google Scholar]
- 18. Dearing BD, Lavie CJ, Lohmann TP, Genton E. Niacin‐induced clotting factor synthesis deficiency with coagulopathy. Arch Intern Med 1992;152:861‐863. [PubMed] [Google Scholar]
- 19. Coppola A, Brady PG, Nord HJ. Niacin‐induced hepatotoxicity. South Med J 1994;87:30‐32. [DOI] [PubMed] [Google Scholar]
- 20. Tatò F, Vega GL, Grundy SM. Effects of crystalline nicotinic acid‐induced hepatic dysfunction on serum low‐density lipoprotein cholesterol and lecithin cholesteryl acyl transferase. Am J Cardiol 1998;81:805‐807. [DOI] [PubMed] [Google Scholar]
- 21. Mullin GE, Greenson JK, Mitchell MC. Fulminant hepatic failure after ingestion of sustained‐release nicotinic acid. Ann Intern Med 1989;111:253‐255. [DOI] [PubMed] [Google Scholar]
- 22. Fischer DJ, Knight LL, Vestal RE. Fulminant hepatic failure following low‐dose sustained‐release niacin therapy in hospital. West J Med 1991;155:410‐412. [PMC free article] [PubMed] [Google Scholar]
- 23. Lawrence SP. Transient focal hepatic defects related to sustained‐release niacin. J Clin Gastroenterol 1993;16:234‐236. [DOI] [PubMed] [Google Scholar]
- 24. Ress C, Kaser S. Mechanisms of intrahepatic triglyceride accumulation. World J Gastroenterol 2016;22:1664‐1673. [DOI] [PMC free article] [PubMed] [Google Scholar]