

Abstract
In adults, treatment of hepatitis C virus (HCV) infection with ombitasvir (OBV)/paritaprevir (PTV)/ritonavir (r) with or without dasabuvir (DSV) and ±ribavirin (RBV) results in high rates of sustained virologic response (SVR). However, these regimens have not been investigated in adolescents. This ongoing, open‐label, phase 2/3 study evaluated the pharmacokinetics, safety, and efficacy of OBV/PTV/r+DSV±RBV treatment for 12 weeks in adolescents infected with HCV genotype (GT) 1 without cirrhosis (part 1) and the safety and efficacy of OBV/PTV/r±DSV±RBV treatment for 12 or 24 weeks in adolescents infected with GT1 or GT4 without cirrhosis or with compensated cirrhosis (parts 1 and 2). Patients were 12‐17 years of age and treatment naive or interferon experienced. Treatment regimens were based on HCV GT and cirrhosis status. Endpoints were SVR at posttreatment week 12 (SVR12), adverse events (AEs), and pharmacokinetic parameters. Thirty‐eight adolescents were enrolled, 66% were female patients, and 76% were White; 42%, 40%, and 18% of patients had HCV GT1a, GT1b, and GT4 infections, respectively. Median age was 15 years (range, 12‐17 years), and 1 patient had cirrhosis. The SVR12 rate was 100% (38/38; 95% confidence interval [CI], 90.8%‐100%). No treatment‐emergent grade 3 or 4 laboratory abnormalities were reported. No serious AEs occurred on treatment, and no AEs led to study drug discontinuation. The most common AEs were headache (21%), fatigue (18%), nasopharyngitis (13%), pruritus (13%), and upper respiratory tract infection (11%). Intensive pharmacokinetic results showed OBV, PTV, DSV, and ritonavir drug exposures were comparable to those seen in adults. Conclusion: Treatment with OBV/PTV/r±DSV±RBV was well tolerated and highly efficacious in adolescents with HCV GT1 or GT4 infection.
Abbreviations
- AE
adverse event
- ALT
alanine aminotransferase
- AUC
area under the curve
- CI
confidence interval
- Cmax
maximum plasma concentration
- Ctrough
trough concentration
- DAA
direct‐acting antiviral
- DSV
dasabuvir
- FDA
U.S. Food and Drug Administration
- GT
genotype
- HCV
hepatitis C virus
- IFN
interferon
- ITT
intention‐to‐treat
- LLOQ
lower limit of quantification
- NS5
nonstructural protein 5
- OBV
ombitasvir
- PTV
paritaprevir
- RBV
ribavirin
- r
ritonavir
- SVR
sustained virologic response
- SVR12/24
sustained virologic response at posttreatment week 12/week 24
- ULN
upper limit of normal
- ZIRCON
Study to Evaluate Treatment of Hepatitis C Virus Infection in Pediatric Subjects
Therapeutic options for the treatment of chronic HCV infection have advanced rapidly in recent years. Combination treatments comprising all‐oral, interferon (IFN)‐free, direct‐acting antiviral (DAA) drugs are now the standard of care for adult patients infected with any HCV GT.1, 2 These regimens are consistently associated with high rates of SVR and are well tolerated in most patients.1, 2
HCV infection is a global health problem with approximately 71 million people worldwide chronically infected with the virus.3, 4 Children and adolescents represent a small proportion of those infected with HCV; estimates in 2016 indicate that 4 million children (under 19 years of age) had HCV infection.5 However, a large proportion of pediatric HCV infections remain undiagnosed, likely owing to a lack of screening programs6 as well as the asymptomatic course of infection in younger individuals.7 The majority of children and adolescents newly infected with HCV will develop chronic infection, which typically progresses more slowly in younger individuals than in adults.8 Nevertheless, adolescents infected with HCV are at risk for complications associated with cirrhosis and advanced liver disease.9, 10
The standard of care treatment for children with chronic HCV infection was until recently pegylated IFN in combination and RBV.11, 12 In 2017, the European Medicines Agency and the U.S. Food and Drug Administration (FDA) approved the use of the nonstructural protein 5B (NS5B) polymerase inhibitor sofosbuvir with RBV for HCV GT2 and GT3 and the fixed‐dose combination of sofosbuvir plus the NS5A inhibitor ledipasvir for HCV GT1, GT4, GT5, and GT6 for the treatment of adolescents (12 years or older, or weighing at least 35 kg) with chronic HCV infection. Current guidance from the joint American Association for the Study of Liver Diseases and Infectious Diseases Society of America, the European Association for the Study of the Liver, and the Hepatology Committee of European Society of Pediatric Gastroenterology, Hepatology, and Nutrition no longer recommends pegylated IFN and RBV for the treatment of adolescents. For children 3 to 12 years of age, the guidance is to defer treatment until IFN‐free regimens are approved for this age group,1, 2, 13 whereas adolescents older than 12 years should receive sofosbuvir‐based regimens based on the recent approvals.1, 2, 13
The combination DAA treatment comprising OBV (an HCV NS5A inhibitor), PTV (an NS3/4A protease inhibitor), and the pharmacokinetic enhancer ritonavir is coadministered with or without DSV (a non‐nucleoside NS5B polymerase inhibitor) and with or without RBV (OBV/PTV/r±DSV±RBV) for up to 24 weeks in adult patients infected with GT1 or GT4 without cirrhosis or with compensated cirrhosis.14, 15, 16, 17 These all‐oral, IFN‐free, multitargeted regimens have shown high SVR12 rates and good tolerability in phase 3 studies for a broad range of adult patients infected with GT1 or GT4.18, 19, 20, 21, 22, 23 However, these regimens have not been studied in children or adolescents.
The ongoing Study to Evaluate Treatment of Hepatitis C Virus Infection in Pediatric Subjects (ZIRCON) was designed to evaluate the pharmacokinetics, safety, and efficacy of OBV/PTV/r±DSV±RBV in children and adolescents infected with HCV GT1 or GT4. Here, we report the results from ZIRCON parts 1 and 2 in adolescents 12‐17 years of age.
Patients and Methods
STUDY DESIGN
ZIRCON (NCT02486406) is an ongoing, open‐label, multinational study. Part 1 was a phase 2 study designed to evaluate the pharmacokinetics of OBV, PTV, ritonavir, and DSV to confirm the use of adult doses and formulation in adolescents and to evaluate the safety and efficacy of OBV/PTV/r+DSV±RBV in adolescents infected with HCV GT1, without cirrhosis, who were treatment naive. Part 2 was a phase 3 study conducted to evaluate the safety and efficacy of OBV/PTV/r±DSV±RBV in adolescents infected with GT1 or GT4, without cirrhosis or with compensated cirrhosis, who were treatment naive or had prior experience with IFN‐based therapies (Fig. 1).
Figure 1.
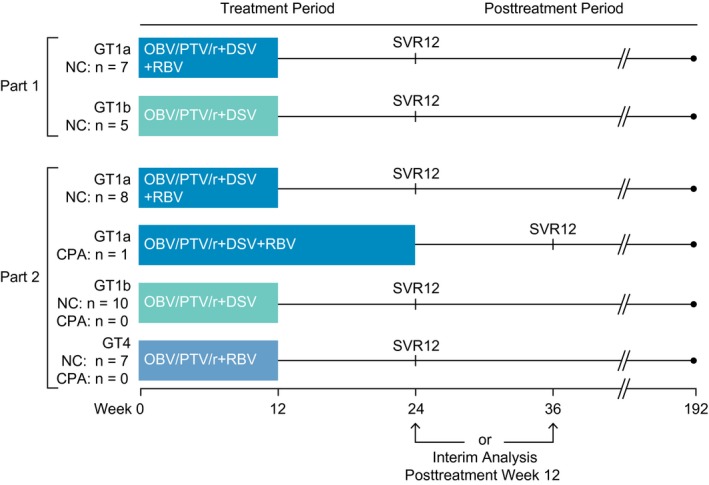
ZIRCON study design. Abbreviations: CPA, Child‐Pugh A cirrhosis; NC, no cirrhosis.
Parts 1 and 2 both comprised screening, treatment, and 24‐week posttreatment periods. At screening, patients without a history of cirrhosis and who did not have a liver biopsy or FibroScan (Echosens, Paris, France) within the 24 months before screening underwent FibroTest‐based (BioPredictive S.A., Paris, France) or FibroScan‐based transient elastography to determine cirrhosis status. Patients were considered to not have cirrhosis based on a liver biopsy within the 24 months before screening (Metavir score <4, Ishak score <5) or if a liver biopsy was not available, a historical FibroScan result of <14.6 kPa within the 24 months before screening.24 If neither historical result was available, a FibroTest score of <0.75 at screening was used to exclude cirrhosis.
Eligible patients were enrolled to receive 12 weeks of treatment in part 1 or either 12 or 24 weeks of treatment in part 2. Enrollment in part 2 commenced when dosing recommendations for the study drugs became available based on pharmacokinetic and clinical data from part 1. Patients in both parts received open‐label OBV/PTV/r (25 mg/150 mg/100 mg, once daily) ±DSV (250 mg, twice daily) ±weight‐based RBV (according to the local label). The treatment regimens and durations depended on genotype, subtype, and cirrhosis status (Fig. 1) and are consistent with label indications that have been approved by the FDA and European Medicines Agency for the treatment of chronic HCV infections in adults.14, 15, 16, 17
The study protocol and amendments were approved by the independent ethics committee or institutional review board for each study center. The study was designed and conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines and the ethical principles of the Declaration of Helsinki. Parents or legal guardians provided written informed consent, and patients gave assent as appropriate for age and country. All authors had access to study data and reviewed and approved the final manuscript.
STUDY PATIENTS
Adolescents 12‐17 years of age were eligible if they had a body weight ≥45 kg, were willing to swallow the adult formulation, had HCV infection (GT1 in part 1; GT1 or GT4 in part 2) defined as having positive anti‐HCV antibody and HCV RNA level >1000 IU/mL at screening, were without cirrhosis (parts 1 or 2) or with compensated cirrhosis (part 2), and were HCV treatment naive (parts 1 or 2) or IFN experienced (part 2; IFN‐based therapy with or without RBV must have been stopped ≥6 months before enrollment). Key exclusion criteria included coinfection with hepatitis B virus or human immunodeficiency virus; any concomitant liver disease; current or past clinical evidence of Child‐Pugh B or C (Child‐Pugh score ≥7) or clinical history of liver decompensation; radiographically confirmed hepatocellular carcinoma; previous or current use of any investigational or commercially available anti‐HCV drug other than IFN, pegylated IFN, or RBV; history of solid organ transplantation; or any of the following abnormal laboratory results: albumin <2.8 g/dL, hemoglobin <10 g/dL, platelets <25,000 cells/mm3, or total bilirubin >3.0 mg/dL. Full eligibility criteria are provided in the Supporting Appendix.
EFFICACY ASSESSMENTS
Blood samples were collected for measurement of HCV RNA by a central laboratory (Covance Central Laboratory Services) in parts 1 and 2 at screening, treatment day 1, and weeks 2, 4, 8, and 12 for patients who received a 12‐week regimen and additionally at treatment weeks 16, 20, and 24 for the patient who received a 24‐week regimen. Samples were also collected at posttreatment weeks 4, 12, and 24. HCV RNA concentration was quantified using the COBAS AmpliPrep/COBAS TaqMan HCV Test, v2.0 (lower limit of quantification [LLOQ] and detection were 15 IU/mL).
The primary efficacy endpoint in parts 1 and 2 was SVR12, which was defined as the percentage of patients who had HCV RNA concentrations less than the LLOQ at posttreatment week 12. Secondary efficacy endpoints in parts 1 and 2 included the percentage of patients who achieved SVR24 and the percentage of patients with normalization of alanine aminotransferase (ALT) concentrations during treatment, defined as ALT less than or equal to the central laboratory’s upper limit of normal (ULN; 34 U/L for female individuals and 43 U/L for male individuals) at the final treatment visit for patients with ALT >ULN at baseline. Additional efficacy endpoints were on‐treatment virologic failure (confirmed HCV RNA ≥LLOQ after HCV RNA was <LLOQ during treatment or confirmed increase from nadir in HCV RNA [two consecutive HCV RNA measurements >1 log10 IU/mL greater than nadir]) and posttreatment relapse (confirmed HCV RNA ≥LLOQ between end of treatment and 12 weeks after the last actual dose of active study drug [up to and including the SVR12 assessment time point] for a patient who completed treatment with HCV RNA <LLOQ).
SAFETY ASSESSMENTS
The following safety evaluations were performed during each study visit: AE assessments, vital sign assessments, physical examination, and clinical laboratory assessments. AEs that occurred from the time of first study drug administration until 30 days after study drug discontinuation were monitored by site investigators and coded according to MedDRA System Organ Class and Preferred Term (version 19.1). The relationship of the AE to the study drug and grade was assessed by site investigators according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE; version 4).
PHARMACOKINETIC ASSESSMENTS
Blood samples were collected in part 1 of the study at the week 2 visit (at 2, 4, 8, 12, and 24 hours after the dose) for intensive pharmacokinetic sampling of the study drugs. Plasma concentrations were determined using validated analytic methods at a central laboratory under the supervision of the Drug Analysis Department at AbbVie. The primary pharmacokinetic endpoints for each study drug were the maximum plasma concentration (Cmax) and the area under the plasma concentration–time curve (AUC) at 0 to 24 hours for OBV, PTV, and ritonavir or 0 to 12 hours for DSV, after dosing at week 2. In addition, the trough concentrations (Ctrough) for OBV, PTV, ritonavir, and DSV were obtained after dosing at week 2.
STATISTICAL ANALYSIS
For the primary and secondary efficacy analyses, the percentages of patients who achieved SVR12 across parts 1 and 2 were calculated for the intention‐to‐treat (ITT) population, which comprised all patients who received at least one dose of the study drug in part 1 or part 2 of the study. The 95% CI for any simple percentage was calculated using normal approximation to the binomial distribution if the percentage was not 0% or 100%; otherwise, Wilson's score method was used. All patients in parts 1 and 2 who received at least one dose of study drug were included in the safety analyses. Treatment‐emergent AEs were summarized. Demographics and baseline characteristics as well as mean changes from baseline in clinical laboratory tests were also summarized. All data were analyzed using the SAS software package (SAS Institute, Inc., Cary, NC).
A planned sample size of 12 adolescent patients in part 1 was deemed adequate to characterize the pharmacokinetics of the study drugs. Pharmacokinetic parameters for OBV, PTV, ritonavir, and DSV, including the Cmax, Ctrough, and AUC, were summarized for patients in part 1. The geometric means for each of the analytes (OBV, PTV, ritonavir, or DSV) were compared with the range of the geometric means from adult data from phase 3 clinical studies with intensive pharmacokinetic data.
Results
PATIENTS
Adolescent patients were recruited between November 24, 2015, and July 12, 2016. A total of 38 adolescent patients 12 to 17 years of age were enrolled at 18 sites in the following countries: Belgium, Germany, Spain, and the United States, including Puerto Rico. Twelve patients were enrolled in part 1, and 26 patients were enrolled in part 2. All patients received treatment with OBV/PTV/r±DSV±RBV for 12 or 24 weeks, as shown in Fig. 1.
Demographics and clinical characteristics at baseline are presented in Table 1. Baseline fibrosis stage data were available for 30 patients: 27 patients had F0‐F1 fibrosis and 1 patient each had F2 and F3 based on the FibroTest. One patient had F4 fibrosis based on the FibroScan (Child‐Pugh score of 5; GT1a infection). Of the 13 patients who had prior IFN‐based treatment experience, 8 patients were nonresponsive to prior treatment, 4 patients had experienced posttreatment virologic relapse, and 1 patient reported partial response to prior HCV treatment.
Table 1.
Baseline Demographics and Clinical Characteristics (ITT Population)
| Characteristic | Patients* N = 38 |
|---|---|
| Male | 13 (34) |
| Race | |
| White | 29 (76) |
| Black or African American | 5 (13) |
| Asian | 3 (8) |
| Multiple races | 1 (3) |
| Ethnicity | |
| Hispanic or Latino | 3 (8) |
| Median age, years [range] | 15 [12‒17] |
| Median weight, kg [range] | 66.4 [48.5‒118.7] |
| Body mass index ≥30 kg/m2 | 6 (16) |
| HCV genotype/subtype | |
| GT1a | 16 (42) |
| GT1b | 15 (40) |
| GT4 | 7 (18) |
| HCV RNA ≥800,000 IU/mL | 23 (61) |
| IL28B genotype, non‐CC | 29 (76) |
| Previous HCV treatment with IFN‐based regimens | |
| Naive | 25 (66) |
| Experienced | 13 (34) |
| Cirrhotic status | |
| No cirrhosis | 37 (97) |
| Compensated cirrhosis | 1 (3) |
*Data are n (%) unless stated otherwise.
EFFICACY
For the primary endpoint, all 38 patients had undetectable HCV RNA at posttreatment week 12 (100% SVR12; 95% CI, 90.8%‐100%; Fig. 2). Accordingly, no difference in response rates was observed based on prior HCV treatment experience or presence of cirrhosis. At treatment week 4, 95% of patients (36/38) had HCV RNA levels <LLOQ. No viral relapses occurred after posttreatment week 12; the SVR24 rate was 100%. Of the 24 patients who had elevated ALT concentrations (>ULN) at baseline, 21 patients (87.5%) had ALT normalization during treatment (ALT ≤ULN at the final treatment visit).
Figure 2.
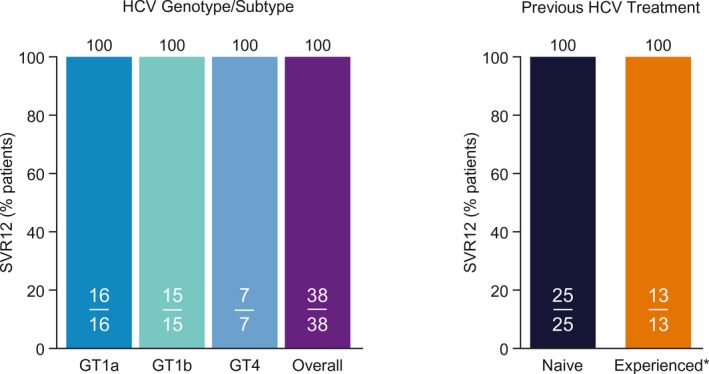
SVR rates by HCV genotype/subtype and previous HCV treatment (ITT population). *Interferon‐based treatment.
One patient had an HCV RNA level >LLOQ at treatment week 8 despite having had an HCV RNA level <LLOQ at treatment weeks 2 and 4. This patient was enrolled in part 1 of the study and received OBV/PTV/r+DSV+RBV but self‐discontinued the OBV/PTV/r tablets at treatment week 5 because of a stressful personal event. The investigator deemed that the viral rebound was caused by study drug interruption, and as per protocol, it was appropriate for this patient to receive retreatment within the protocol. Upon retreatment, this patient achieved an HCV RNA level <LLOQ at treatment week 2 and remained HCV RNA undetectable from treatment week 4 through posttreatment week 24.
SAFETY
Most patients (n = 32; 84%) experienced an AE during the study period (Table 2). No AE led to discontinuation of the study drugs, and no severe or serious AEs (CTCAE grade ≥3) or deaths occurred during treatment. The most common AEs were headache, fatigue, nasopharyngitis, pruritus, and upper respiratory tract infection.
Table 2.
Summary of Adverse Events
| Adverse Event | Patients N = 38 |
|---|---|
| Any AE, n (%) | 32 (84) |
| Any AE possibly related to DAAs,* n (%) | 15 (39) |
| Any AE possibly related to RBV,* n (%)† | 14 (61) |
| Any AE leading to discontinuation of study drug, n (%) | 0 |
| Any serious AE, n (%) | 0 |
| AEs in ≥10% of all adolescents, n (%) | |
| Headache | 8 (21) |
| Fatigue | 7 (18) |
| Nasopharyngitis | 5 (13) |
| Pruritus | 5 (13) |
| Upper respiratory tract infection | 4 (11) |
*As assessed by investigator. †23 patients were treated with RBV.
Post‐baseline laboratory abnormalities were infrequent, and no grade 3 or 4 abnormalities were reported (Table 3). One patient who received OBV/PTV/r+DSV+RBV had a grade 2 decrease in hemoglobin concentration (9.7 g/dL) at treatment week 12. One patient who received OBV/PTV/r+DSV+RBV had an alkaline phosphatase concentration >1.5 × ULN (585‐633 U/L) at baseline, treatment weeks 4 and 8, and posttreatment week 4. One patient who received OBV/PTV/r+RBV had a total bilirubin concentration ≥2 × ULN (2.7‐3.4 mg/dL) at treatment weeks 2, 4, 8, and 12 that was mainly driven by the elevation of indirect bilirubin (2.5‐3.2 mg/dL).
Table 3.
Laboratory Abnormalities
| Parameter | Patients N = 38 |
|---|---|
| Hemoglobin, n (%) | |
| <10 g/dL (grade 2) | 1 (3) |
| <8 g/dL (grade 3 or 4) | 0 |
| Alkaline phosphatase >1.5 × ULN elevation, n (%) | 1 (3) |
| Total bilirubin ≥2 × ULN elevation, n (%) | 1 (3) |
| Postnadir increase in ALT, n (%) | |
| >5‐20 × ULN (grade 3) | 0 |
| >20 × ULN (grade 4) | 0 |
| Creatinine clearance <50 mL/minute in patients with baseline >50 mL/minute, n (%) | 0 |
PHARMACOKINETICS
Of the 12 patients enrolled in part 1 of the study, 75% were female patients, 58% were White, and 58% had GT1a infection. The median age was 15.5 years (range, 12‐17). Per protocol, all patients in part 1 were treatment naive and none had cirrhosis (Supporting Table S1). The pharmacokinetic parameters of OBV, PTV, ritonavir, and DSV are shown in Table 4. The exposures for OBV, PTV, ritonavir, and DSV in the present study were generally comparable to exposures observed in previous phase 3 studies of adults who received similar formulations of these drugs (Table 4).
Table 4.
Preliminary Pharmacokinetic Parameters From Adolescents in ZIRCON Part 1 Versus Historical Data From Studies in Adults
| Drug |
Adolescents (N = 12)*
Geometric Mean (CV%) |
Adults (Historical, N = 10 to 22 per Study)†
Range of Geometric Means Across Studies (Range of CV% Across Studies) |
||||
|---|---|---|---|---|---|---|
|
Cmax
ng/mL |
AUC‡
ng·hour/mL |
Ctrough
ng/mL |
Cmax
ng/mL |
AUC‡
ng·hour/mL |
Ctrough
ng/mL |
|
| Ombitasvir | 75.4 (31) | 918 (23) | 19.0 (18) | 95.0‐114 (31‐45) | 1,350‐1,570 (31‐41) | 23.7‐30.2 (47‐54) |
| Paritaprevir | 738 (70) | 4,880 (52) | 19.4 (64) | 535‐1,350 (89‐124) | 4,760‐14,100 (98‐129) | 12.6‐57.6 (86‐237) |
| Ritonavir | 1,020 (38) | 7,700 (31) | 27.3 (60) | 894‐1,260 (32‐53) | 8,540‐13,400 (21‐56) | 31.3‐52.4 (44‐225) |
| Dasabuvir | 646 (49) | 4,460 (45) | 158 (43) | 666‐752 (44‐48) | 4,800‐5,220 (37‐54) | 119‐168 (53‐104) |
Abbreviation: CV, coefficient of variation.
*N = 11 for AUC and Ctrough of ombitasvir, paritaprevir, and ritonavir.
†Range of geometric means from three studies in adults infected with HCV who had intensive pharmacokinetic data(29) (and AbbVie data on file).
‡AUC0‐24hour for ombitasvir, paritaprevir, and ritonavir; AUC0‐12hour for dasabuvir.
Discussion
Hepatitis C virus infection acquired during childhood or adolescence is usually mild and typically follows a slower disease progression course than in patients who acquire infection during adulthood.8 However, if left untreated, chronic HCV infection can predispose pediatric patients to liver‐related complications, including cirrhosis and advanced liver disease.9, 10 Therefore, curative treatment of HCV infection should be considered for children and adolescents to mitigate the risk of liver disease progression, reduce the rates of transmission, and lessen the social stigma that can be associated with HCV infection.
Until recently, the standard of care for chronic HCV infection in pediatric patients was pegylated IFN in combination with RBV (GT2 and GT3, 24‐week treatment; HCV GT1 and GT4, 48‐week treatment).11, 12 In children infected with GT2 and GT3, the SVR rate was approximately 90%; with GT1 and GT4, the SVR rate was approximately 48%.13, 25 Treatment is associated with a high incidence of undesirable AEs, such as anemia, neutropenia, leukopenia, and depression.11, 25
The European Medicines Agency and the FDA have approved the use of sofosbuvir with RBV for adolescents with chronic GT2 or GT3 infection and the fixed‐dose combination of sofosbuvir plus ledipasvir for adolescents with chronic GT1, GT4, GT5, or GT6 infection.1, 2, 13 In clinical trials,26, 27 adult formulations of both sofosbuvir‐based regimens had plasma exposures in adolescent patients that were comparable to those in adults, with no severe AEs or laboratory abnormalities. In adolescents with GT1 infection and treated for 12 weeks with sofosbuvir plus ledipasvir, the SVR12 rate was 98%.26 In adolescents infected with GT2 or GT3 and treated for 12 or 24 weeks with sofosbuvir with RBV, respective SVR12 rates were 100% and 97%.27 Although the combination of sofosbuvir plus the NS5A inhibitor daclatasvir is not currently approved for treating pediatric patients, a 12‐week regimen achieved a SVR12 rate of 97% adolescents (12‐17 years) infected with GT4 .28 The safety and efficacy of more recently developed DAAs are currently being evaluated in clinical studies for children and adolescent patients (NCT03067129 and NCT03022981).
In part 1 of ZIRCON, intensive pharmacokinetic analyses following administration of the adult formulation of OBV/PTV/r+DSV±RBV to adolescents infected with HCV GT1 at the approved adult doses indicated that drug exposures were generally comparable to those observed in adult patients. In some cases, drug exposures were lower than the adult geometric mean ranges; however, this clearly did not impact overall efficacy. These findings suggest that dose adjustment of the adult regimen is not required in the adolescent population.
Treatment with OBV/PTV/r±DSV±RBV in patients infected with GT1 or GT4 in parts 1 and 2 was well tolerated, and no new safety signals were identified. This safety profile is consistent with that reported in previous studies in adult patients treated with these drug regimens,18, 19, 20, 21, 22, 23 suggesting that the adult regimen is suitable for treating adolescents without the need for additional clinical monitoring or dose adjustment.
Most importantly, 100% of 38 adolescent patients (12‐17 years) with HCV GT1 or GT4 infection enrolled in ZIRCON achieved SVR12 following treatment with OBV/PTV/r±DSV and ±RBV for 12 or 24 weeks. Furthermore, patient characteristics traditionally associated with a reduced response rate to HCV treatment, such as cirrhosis, nonresponse to prior IFN‐based therapies, and unfavorable interleukin 28B (IL28B) genotype, had no impact on virologic response rates. There were no posttreatment virologic relapses.
The results from the ZIRCON study further support the body of evidence demonstrating that adult formulations of existing DAA therapies are well tolerated and highly efficacious in adolescent patients. As guidance on HCV treatment recommendations continues to be updated, the availability of an increasing number of effective DAAs for adolescent patients will help practitioners to select the optimal treatment regimen for this underserved population.
The safety and efficacy of OBV/PTV/r±DSV±RBV in younger age groups are not yet known. However, ZIRCON is an ongoing study, and enrollment of pediatric patients 3‐11 years of age has been completed.
In conclusion, treatment with the all‐oral IFN‐free adult formulation of OBV/PTV/r±DSV±RBV for 12 or 24 weeks was well tolerated and highly efficacious in adolescent patients 12‐17 years of age who are infected with HCV GT1 or GT4.
Potential conflict of interest
Dr. Leung received grant/research support from Gilead, AbbVie, Bristol Myers Squibb, and Roche/Genentech and is a consultant for Merck. Dr. Wirth, Dr. Lobritto, Dr. Fortuny, Dr. Hsu, and Dr. Del Valle‐Segarra received grant/research support from AbbVie. Dr. Gonzalez‐Peralta received grant/research support from AbbVie, Gilead, and Merck and is an advisor/consultant for Genentech, Shire, and Kadmon. Dr. Jonas received grant support from Gilead, Bristol Myers Squibb, Echosens, Merck, AbbVie, and Roche and is a consultant for Gilead. Dr. Narkewicz received grant/research support from Vertex, AbbVie, and Gilead and is a consultant for Vertex. Dr. Sokal is Founder and CSO of Promethera Biosciences and consults for and owns intellectual property rights and stock in Promethera; he is an investigator for AbbVie, Alexion, Roche, Gilead, and Takeda and is a consultant for AbbVie and Alexion. Dr. Rosenthal received grant/research support from Gilead, AbbVie, Bristol Myers Squibb, and Roche; he is on the speakers’ bureau for Retrophin and is a consultant for Gilead, AbbVie, Intercept, Alexion, Retrophin, Albireo, and Audentes. Dr. Yao, Dr. Viani, Dr. Zha, Dr. Larsen, Dr. Liu, Dr. Shuster, and Dr. Cohen are current or former employees of AbbVie and own stock in AbbVie.
Author names in bold designate shared co‐first authorship.
Supporting information
Acknowledgment
Medical writing support was provided by Dr. Paul MacCallum of Fishawack Communications Ltd. Paritaprevir was identified by AbbVie and Enanta. We thank the patients who participated in this study and their families. We also thank the following ZIRCON study investigators: Cornelia Feiterna‐Sperling (Charité ‐ Universitätsmedizin Berlin, Berlin, Germany), Patrick Gerner (Universitäts Klinikum, Freiburg, Germany), Loreto Hierro (Hospital Universitario La Paz, Madrid, Spain), Christopher Jolley (University of Florida Health, Gainesville, FL), Begoña Polo (Hospital Universitari i Politècnic la Fe, Valencia, Spain), Jesus Quintero (University Hospital Vall d'Hebron, Barcelona, Spain), and Peter Witters (UZ Leuven, Leuven, Belgium).
SEE EDITORIAL ON PAGE http://doi.org/10.1002/hep4.1261
ClinicalTrials.gov ID: NCT02486406.
Supported by AbbVie Inc.; AbbVie also contributed to study design and participated in the collection, analysis, and interpretation of the data and in the writing, reviewing, and approval of the publication.
References
- 1. AASLD‐IDSA . HCV guidance: recommendations for testing, managing, and treating hepatitis C. https://www.hcvguidelines.org. Accessed June 21, 2018.
- 2. European Association for the Study of the Liver . EASL recommendations on treatment of hepatitis C 2018. J Hepatol 2018;69:461‐511. [DOI] [PubMed] [Google Scholar]
- 3. Polaris Observatory HCV Collaborators . Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. Lancet Gastroenterol Hepatol 2017;2:161‐176. [DOI] [PubMed] [Google Scholar]
- 4. WHO . Global hepatitis report 2017. Published 2017. https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/. Accessed June 21, 2018.
- 5. CDA Foundation . 52 million children suffering from hepatitis. https://cdafound.org/featured3/. Published January 5, 2018. Accessed June 21, 2018.
- 6. Delgado‐Borrego A, Smith L, Jonas MM, Hall CA, Negre B, Jordan SH, et al. Expected and actual case ascertainment and treatment rates for children infected with hepatitis C in Florida and the United States: epidemiologic evidence from statewide and nationwide surveys. J Pediatr 2012;161:915‐921. [DOI] [PubMed] [Google Scholar]
- 7. Bortolotti F, Verucchi G, Camma C, Cabibbo G, Zancan L, Indolfi G, et al. Italian Observatory for HCV Infection and Hepatitis C in Children. Long‐term course of chronic hepatitis C in children: from viral clearance to end‐stage liver disease. Gastroenterology 2008;134:1900‐1907. [DOI] [PubMed] [Google Scholar]
- 8. Garcia‐Monzon C, Jara P, Fernandez‐Bermejo M, Hierro L, Frauca E, Camarena C, et al. Chronic hepatitis C in children: a clinical and immunohistochemical comparative study with adult patients. Hepatology 1998;28:1696‐1701. [DOI] [PubMed] [Google Scholar]
- 9. Barshes NR, Udell IW, Lee TC, O'Mahony CA, Karpen SJ, Carter BA, et al. The natural history of hepatitis C virus in pediatric liver transplant recipients. Liver Transpl 2006;12:1119‐1123. [DOI] [PubMed] [Google Scholar]
- 10. Goodman ZD, Makhlouf HR, Liu L, Balistreri W, Gonzalez‐Peralta RP, Haber B, et al. Pathology of chronic hepatitis C in children: liver biopsy findings in the Peds‐C Trial. Hepatology 2008;47:836‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Genentech , Inc. PEGASYS (peginterferon alfa‐2a). US prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/103964s5204lbl.pdf. Published 2011. Accessed June 21, 2018.
- 12. Ghany MG, Strader DB, Thomas DL. Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 2009;49:1335‐1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Indolfi G, Hierro L, Dezsofi A, Jahnel J, Debray D, Hadzic N, et al. Treatment of chronic hepatitis C virus infection in children: a position paper by the Hepatology Committee of European Society of Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr 2018;66:505‐515. [DOI] [PubMed] [Google Scholar]
- 14. AbbVie Inc . VIEKIRA PAK (ombitasvir, paritaprevir, and ritonavir tablets; dasabuvir tablets) US prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/206619s017lbl.pdf. Published March 2017. Accessed June 21, 2018.
- 15. AbbVie Inc. VIEKIRAX (ombitasvir, paritaprevir, and ritonavir tablets) Summary of product characteristics. Available at: https://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003839/WC500183997.pdf. Accessed June 21, 2018.
- 16. AbbVie Inc. EXVIERA (dasabuvir tablets). Summary of product characteristics https://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003837/WC500182233.pdf. Accessed June 21, 2018.
- 17. AbbVie Inc. TECHNIVIE (ombitasvir, paritaprevir, and ritonavir tablets) US prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/207931s011lbl.pdf. Published March 2017. Accessed June 21, 2018.
- 18. Andreone P, Colombo MG, Enejosa JV, Koksal I, Ferenci P, Maieron A, et al. ABT‐450, ritonavir, ombitasvir, and dasabuvir achieves 97% and 100% sustained virologic response with or without ribavirin in treatment‐experienced patients with HCV genotype 1b infection. Gastroenterology 2014;147(359–365):e351. [DOI] [PubMed] [Google Scholar]
- 19. Feld JJ, Kowdley KV, Coakley E, Sigal S, Nelson DR, Crawford D, et al. Treatment of HCV with ABT‐450/r‐ombitasvir and dasabuvir with ribavirin. N Engl J Med 2014;370:1594‐1603. [DOI] [PubMed] [Google Scholar]
- 20. Ferenci P, Bernstein D, Lalezari J, Cohen D, Luo Y, Cooper C, et al. PEARL‐III Study; PEARL‐IV Study. ABT‐450/r‐ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med 2014;370:1983‐1992. [DOI] [PubMed] [Google Scholar]
- 21. Hezode C, Asselah T, Reddy KR, Hassanein T, Berenguer M, Fleischer‐Stepniewska K, et al. Ombitasvir plus paritaprevir plus ritonavir with or without ribavirin in treatment‐naive and treatment‐experienced patients with genotype 4 chronic hepatitis C virus infection (PEARL‐I): a randomised, open‐label trial. Lancet 2015;385:2502‐2509. [DOI] [PubMed] [Google Scholar]
- 22. Poordad F, Hezode C, Trinh R, Kowdley KV, Zeuzem S, Agarwal K, et al. ABT‐450/r‐ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med 2014;370:1973‐1982. [DOI] [PubMed] [Google Scholar]
- 23. Zeuzem S, Jacobson IM, Baykal T, Marinho RT, Poordad F, Bourliere M, et al. Retreatment of HCV with ABT‐450/r‐ombitasvir and dasabuvir with ribavirin. N Engl J Med 2014;370:1604‐1614. [DOI] [PubMed] [Google Scholar]
- 24. Ganne‐Carrie N, Ziol M, de Ledinghen V, Douvin C, Marcellin P, Castera L, et al. Accuracy of liver stiffness measurement for the diagnosis of cirrhosis in patients with chronic liver diseases. Hepatology 2006;44:1511‐1517. [DOI] [PubMed] [Google Scholar]
- 25. Druyts E, Thorlund K, Wu P, Kanters S, Yaya S, Cooper CL, et al. Efficacy and safety of pegylated interferon alfa‐2a or alfa‐2b plus ribavirin for the treatment of chronic hepatitis C in children and adolescents: a systematic review and meta‐analysis. Clin Infect Dis 2013;56:961‐967. [DOI] [PubMed] [Google Scholar]
- 26. Balistreri WF, Murray KF, Rosenthal P, Bansal S, Lin CH, Kersey K, et al. The safety and effectiveness of ledipasvir‐sofosbuvir in adolescents 12–17 years old with hepatitis C virus genotype 1 infection. Hepatology 2017;66:371‐378. [DOI] [PubMed] [Google Scholar]
- 27. Wirth S, Rosenthal P, Gonzalez‐Peralta RP, Jonas MM, Balistreri WF, Lin CH, et al. Sofosbuvir and ribavirin in adolescents 12–17 years old with hepatitis C virus genotype 2 or 3 infection. Hepatology 2017;66:1102‐1110. [DOI] [PubMed] [Google Scholar]
- 28. Yakoot M, El‐Shabrawi MH, AbdElgawad MM, Mahfouz AA, Helmy S, Abdo AM, et al. Dual sofosbuvir/daclatasvir therapy in adolescent patients with chronic hepatitis C infection. J Pediatr Gastroenterol Nutr 2018;67:86‐89. [DOI] [PubMed] [Google Scholar]
- 29. Lalezari J, Sullivan JG, Varunok P, Galen E, Kowdley KV, Rustgi V, et al. Ombitasvir/paritaprevir/r and dasabuvir plus ribavirin in HCV genotype 1‐infected patients on methadone or buprenorphine. J Hepatol 2015;63:364‐369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials