

Abstract
Bile acids (BAs) activate various dedicated receptors, including the farnesoid X receptor (FXR) and the Takeda G protein‐coupled receptor 5 (TGR5). The FXR agonist obeticholic acid (OCA) is licensed for the treatment of primary biliary cholangitis and has shown promising results in NASH patients, whereas TGR5 agonists target inflammation and metabolism. We hypothesized that FXR and/or TGR5 agonists may be therapeutic in early alcoholic liver disease (ALD) in mice, in which hepatic inflammation plays a major role. OCA, INT‐777, and INT‐767 are BA derivatives with selective agonist properties for FXR, TGR5, or both, respectively. These compounds were tested in two mouse models (3‐day binge model and prolonged Lieber DeCarli diet for 12 days) of early ALD. Serum alanine aminotransferase and liver histology were used to assess liver injury, Oil Red O staining of liver sections to assess steatosis, and real‐time polymerase chain reaction to assess changes in gene expression. In the ethanol binge model, treatment with OCA and INT‐777 decreased hepatic macrovesicular steatosis and protected from ethanol‐induced liver injury. After prolonged ethanol administration, mice treated with OCA, INT‐767, or INT‐777 showed decreased hepatic steatosis, associated with reduced liver fatty acid synthase protein expression, and protection from liver injury. Treatment with BA receptor agonists in both models of ethanol administration modulated lipogenic gene expression, and decreased liver interleukin‐1β mRNA expression associated with increased ubiquitination of NLRP3 inflammasome through cyclic adenosine monophosphate–induced activation of protein kinase A. Conclusion: OCA, INT‐767, or INT‐777 administration is effective in reducing acute and chronic ethanol‐induced steatosis and inflammation in mice, with varying degrees of efficacy depending on the duration of ethanol administration, indicating that both FXR and TGR5 activation can protect from liver injury in ALD models.
Abbreviations
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- ASH
alcoholic steatohepatitis
- BA
bile acids
- cAMP
cyclic adenosine monophosphate
- Casp‐1
caspase‐1
- EtOH
ethanol
- FASN
fatty acid synthase
- FXR
farnesoid X receptor
- H&E
hematoxylin and eosin
- IL
interleukin
- NLRP3
NACHT domain‐, leucine‐rich repeat‐, and PYD‐containing protein 3
- OCA
obeticholic acid
- PF
pair‐fed
- PKA
protein kinase A
- SREBP
sterol regulatory element–binding protein
- TGR5
Takeda G protein‐coupled receptor 5
- and WT
wild type
Excessive alcohol consumption leads to alcoholic liver disease (ALD), which is a cause of considerable morbidity and mortality in the Western world. ALD consists of a spectrum of stages, ranging from simple steatosis to alcoholic steatohepatitis (ASH) to fibrosis, cirrhosis, and ultimately hepatocellular carcinoma.( 1 ) In its initial phase, it is characterized by hepatic lipid accumulation and innate immune system activation leading to chronic inflammation.( 2 ) Activation of resident liver macrophages, the Kupffer cells (KCs), plays a critical role in the initiation and development of ALD, and the NLRP3 inflammasome has been found to be activated in KCs of ethanol‐fed mice.( 3 ) Elevated liver expression of inflammasome components (interleukin [IL]‐1β, IL‐18, caspase‐1 [Casp‐1]), correlated with liver injury, has also been found in patients with ALD, confirming the important role of innate immune system activation in the pathogenesis of ALD. However, despite significant advances in understanding ALD pathogenesis, very few treatment options are still available.( 4 )
Bile acids (BAs) act as signaling molecules, activating two dedicated receptors: farnesoid X receptor (FXR), a member of the nuclear receptor superfamily of transcription factors, and transmembrane G protein‐coupled receptor 5 (TGR5).( 5 ) FXR (NR1H4), highly expressed in liver, gut, kidney and adrenal glands, regulates BA synthesis, lipid, glucose and energy homeostasis, as well as inflammation and fibrosis, all of which are important components in the pathogenesis of NASH and ASH.( 6, 7, 8, 9 ) TGR5 (also known as G‐protein coupled bile acid receptor 1) is more broadly expressed than FXR and induces gallbladder filling, exerts potent anti‐inflammatory effects, enhances energy expenditure, prevents obesity, and decreases insulin resistance.( 9 ) Therefore, the two BA receptors play complementary roles in the regulation of several metabolic and inflammatory pathways. Recent reports have highlighted the potential of synthetic agonists for these receptors in various liver disease models and in the treatment of different chronic liver diseases.( 10, 11 ) The FXR agonist obeticholic acid (OCA, also known as INT‐747) has been recently approved for the treatment of primary biliary cholangitis, a chronic autoimmune liver disease.( 8 ) This drug has also shown promising results in the treatment of NASH in a phase II trial,( 12 ) demonstrating anti‐inflammatory and antifibrotic effects in the liver, and is currently in a phase III clinical trial in NASH patients (ClinicalTrials.gov identifier, NCT02548351; sponsored by Intercept Pharmaceuticals).
Although ASH and NASH have distinct triggers, chronic alcohol consumption and obesity‐associated lipotoxicity, respectively, both liver diseases share overlapping pathogenic mechanisms, including steatosis and altered hepatocyte lipid metabolism, hepatic stellate cell activation, innate immune system activation with subsequent inflammation, fibrosis, and liver injury.( 13 ) However, molecular mechanisms can differ, as indicated by the predominant involvement of inflammasome activation and IL‐1β signaling in ASH.( 14 )
Here, we have tested the hypothesis that FXR and/or TGR5 agonists may be therapeutic in the treatment of early ASH in mice. The results indicate that both FXR and TGR5 activation can protect from liver injury in ALD models.
1. Materials and Methods
1.1. Animal Studies
Six‐ to 8‐week‐old female C57BL/6 wild‐type (WT) mice (Jackson Laboratory, Bar Harbor, ME) were used. OCA,( 15 ) INT‐777,( 15 ) and INT‐767( 16 ) are BA derivatives with selective agonist properties for FXR, TGR5, or both, respectively (Supporting Information Fig. S1A). These bile acid receptor agonists were generously provided by Intercept Pharmaceuticals. These compounds were tested in two mouse models of early alcohol steatohepatitis. In the binge model, an ethanol gavage (20% vol/vol ethanol) was administered for 3 days to mice concurrently treated with either agonist (30 mg/kg/day) or vehicle (0.5% methylcellulose) through oral gavage (Supporting Information Fig. S1B). In the prolonged ethanol model, mice were given a Lieber DeCarli diet for 12 days (7 days of 5% vol/vol ethanol after a 5‐day ramp‐up), with oral administration of vehicle or agonist at 30 mg/kg every day during ramp‐up period and for 4 days spread throughout the remaining 7 days as we previously described (Supporting Information Fig. S1C).( 17 ) All animals received proper care in agreement with animal protocols approved by the Institutional Animal Use and Care Committee of the University of Massachusetts Medical School.
1.2. Biochemical Assays
Serum alanine aminotransferase (ALT) was determined using a kinetic method (D‐Tek LLC., Bensalem, PA).
1.3. RNA Analysis
RNA was purified using the RNeasy kit (Qiagen Sciences) and on‐column DNA digestion. Complementary DNA was transcribed with the Reverse Transcription System (Promega Corp.). Quantitative PCR (qPCR) was performed using Sybr‐Green and iCycler from BioRad (Bio‐Rad Laboratories Inc.).
1.4. Histopathological Analysis
Liver sections were stained with hematoxylin and eosin (H&E) or Oil Red O and analyzed by microscopy, and positive staining was quantified using ImageJ (NIH) as previously described.( 17 )
1.5. Protein Analysis
Whole‐cell lysates were extracted from liver, and 20 µg of liver extracts were separated on polyacrylamide gel and transferred to a nitrocellulose membrane. Target proteins were detected by western blot and immunostaining with specific primary antibody, followed by horseradish peroxidase–labeled secondary antibody. The specific immunoreactive bands of interest were detected by chemiluminescence (Bio‐Rad). Digital system (ImageQuant LAS 4000; GE Healthcare) was used for image acquisition. Antibodies were specific for Fatty Acid Synthase (3180S, Cell Signaling Technology), Phospho‐(Ser/Thr) PKA Substrate (9621S, Cell Signaling Technology), CYP7A1 (MABD42, EMD Millipore), and Casp1p10 (sc‐514, Santa Cruz Technology).
1.6. Statistical Analysis
Two‐sided t tests or analysis of variance and Dunnett’s multiple comparison posttest were used to compare the means of groups as appropriate. Data are shown as mean ± SEM and were considered statistically significant at P < 0.05 (* P < 0.05 versus baseline; ** P ≤ 0.001 versus baseline; # P ≤ 0.05 versus vehicle‐treated ethanol [EtOH] condition). We used SPSS 19.0 (IBM SPSS, Chicago, IL) and GraphPad Prism 7.0c (San Diego, CA) for calculations.
2. Results
2.1. Increased CYP7A1 Expression in ALD
Ethanol exposure is associated with increased bile acid signaling,( 18 ) with increasing evidence to suggest that an altered bile acid homeostasis may be responsible for some pathogenic changes observed in ALD.( 19, 20 ) It has been previously reported that CYP7A1 mRNA is expressed by primary human hepatocytes after stimulation with ethanol.( 21 ) For this reason, we explored signs of altered bile acid signal transduction in human livers from patients with acute alcoholic hepatitis. Remarkably, we found increased CYP7A1 protein expression in the three human livers with alcoholic hepatitis tested, compared with three healthy controls (Fig. 1A). It should be noted that CYP7A1 expression was hepatocyte‐specific, with connective tissue in these fibrotic livers negative for CYP7A1 staining. We wondered whether these findings would also extrapolate to a shortened 12‐day ALD murine Lieber DeCarli model of prolonged ethanol administration. These mouse livers showed increased CYP7A1 protein expression compared with their pair‐fed (PF) controls (Fig. 1B). These data indicate that in humans and mice prolonged ethanol exposure results in increased CYP7A1 expression, suggesting altered bile acid signaling.
Figure 1.

Increased CYP7A1 expression in human and mouse ALD. (A) Three human livers from patients with severe alcoholic hepatitis and three healthy controls were probed for CYP7A1 protein expression through immunohistochemistry. Images are shown at 100‐times magnification, with inset shown at 200‐times magnification. Numbers indicate patient identification number. WT mice were given a Lieber DeCarli ethanol diet for 12 days. (B) Mice were sacrificed and livers analyzed for CYP7A1 protein expression through western blot. Densitometry of ethanol‐fed group compared with the PF, calorie‐matched control group was assessed using ImageJ software (n = 5 for PF group, n = 8 for ethanol‐fed group).
We hypothesized that reducing CYP7A1 expression may be therapeutic, and thus BA receptor agonists may represent a suitable treatment in mice with ALD. We first tested whether delivery of BA receptor agonists in a short‐term and long‐term alcohol feeding model would be sufficient to target CYP7A1 protein expression, independent of ethanol administration. Thus, we administered three different BA receptor agonists over a 3‐day regimen (short term) and a 12‐day regimen (prolonged term) of daily alcohol oral gavage. We used OCA, INT‐767, and INT‐777, which differentially target BA receptors. OCA is an FXR agonist, INT‐777 is a TGR5 agonist, and INT‐767 is a dual FXR/TGR5 agonist (Supporting Information Fig. S1A). We found that in both short‐term and prolonged models, daily administration of OCA and INT‐767, both of which target FXR, significantly reduced CYP7A1 protein expression in mouse livers (Fig. 2A). Conversely, CYP7A1 expression was not affected by the TGR5 agonist INT‐777, as expected. We then administered WT mice with a 12‐day ethanol feeding and found that, similarly, ethanol‐treated mice expressed reduced levels of CYP7A1 protein in the liver when simultaneously treated with FXR agonists OCA or INT‐767, compared with their PF controls, but not with the TGR5 agonist INT‐777 (Fig. 2C). Interestingly, INT‐767 showed the greatest reduction of liver CYP7A1 expression, possibly due to the three‐fold higher potency in FXR activation of INT‐767 compared with OCA or to the reinforcing effect of the crosstalk between TGR5 and FXR.( 22 )
Figure 2.
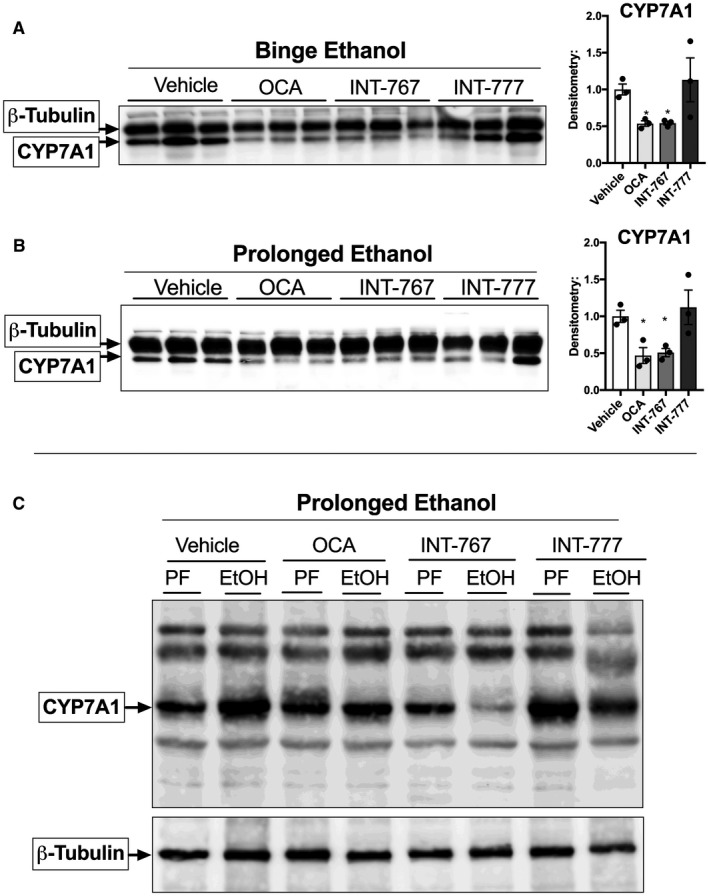
(A) FXR agonists reduce CYP7A1 protein expression. In the short‐term, 3‐day model, WT mice were treated with either agonist (30 mg/kg/day) or vehicle (0.5% methylcellulose) for 3 days through oral administration. (B) In the prolonged, 12‐day model, mice were given a Lieber DeCarli diet for 12 days, with oral administration of vehicle (0.5% methylcellulose) or BA receptor agonist at 30 mg/kg every day during ramp‐up period and for 4 days spread throughout the remaining 7 days (Supporting Information Fig. S1). (A,B) CYP7A1 protein expression was assessed in whole‐cell liver lysates. (C) In the prolonged model, changes in CYP7A1 protein expression were assessed in whole‐cell liver lysates from ethanol‐treated mice dosed with either agonist or vehicle, compared with its PF controls.
2.2. BA Receptor Agonists Attenuate Liver Injury and Microvesicular Steatosis After Acute Ethanol Administration
In the binge ethanol injury model, treatment with OCA and INT‐777 decreased hepatic microvesicular steatosis detected by H&E histology, whereas the FXR/TGR5 dual agonist, INT‐767, resulted only in mild improvement of these histological features (Fig. 3A). We then analyzed liver lipid accumulation through Oil Red O staining and found reduced levels in the percent of positive Oil Red O area (Fig. 3B,C) with each BA receptor agonist treatment, albeit only OCA showed a statistically significant reduction compared with vehicle treatment. In the acute binge ethanol model, mice treated with OCA showed significant protection from liver injury, as measured by serum ALT, compared with ethanol‐binged mice treated with vehicle (Fig. 3D). Treatment with OCA resulted in slight attenuation in liver‐to‐body‐weight ratio induced by acute ethanol administration (Supporting Information Fig. S2A), whereas BA receptor agonist treatment had no effect on serum ethanol levels (Supporting Information Fig. S2B).
Figure 3.

Bile acid receptor agonists attenuate liver injury and microvesicular steatosis after acute ethanol injury. WT mice treated with bile acid receptor agonist or vehicle were exposed to ethanol or sugar binges. Liver injury and microvesicular steatosis was assessed by H&E histology on liver sections (A‐C), and serum ALT was evaluated (D). Steatosis was assessed by Oil Red O staining (B), and percent‐positive staining was quantified by ImageJ software from the average of five random images per sample (C). *P < 0.05 compared with baseline; # P < 0.05 compared with vehicle‐treated ethanol group (n = 5 for PF, 10 for ethanol, per treatment).
2.3. BA Receptor Agonists Attenuate Liver Injury and Macrovesicular Steatosis in Early ALD
We similarly analyzed mouse liver sections through H&E histology and Oil Red O staining after prolonged ethanol administration, resulting in liver injury and steatosis compared with PF mice (Fig. 4A,B) (Table 2). Mice treated with vehicle plus ethanol showed substantial accumulation of macrovesicular steatosis (Fig. 4A), a phenotype not present in the acute liver injury model (Fig. 3A). Furthermore, H&E staining showed that mice treated with OCA, INT‐767, or INT‐777 had decreased hepatic macrovesicular steatosis in the liver compared with the vehicle‐treated ethanol group (Table 1). Likewise, Oil Red O staining showed a significant decrease in steatosis after treatment with each of the three BA receptor agonists (Fig. 4B,C).
Figure 4.

Bile acid receptor agonists attenuate liver injury and macrovesicular steatosis in early ALD. WT mice treated with bile acid agonist or vehicle were exposed to 12 days of prolonged ethanol or PF liquid diet. Liver injury and microvesicular steatosis was assessed by H&E histology on liver sections (A‐C), and serum ALT was evaluated (D). Steatosis was assessed by Oil Red O staining (B), and percent‐positive staining was quantified by ImageJ software from the average of five random images per sample (C). * P < 0.05 compared with baseline; ** P < 0.001 compared with baseline; # P < 0.05 compared with vehicle‐treated ethanol group (n = 5 for PF, 10 for ethanol, per treatment).
Table 2.
Summary of Attenuation in Serum ATL, Steatosis, and Induction of Liver IL‐1β mRNA After Treatment With BA Receptor Agonists After Prolonged Ethanol
| Prolonged Ethanol | |||
|---|---|---|---|
| ALT | Steaotosis | mRNA IL‐1β | |
| No Treatment | 100% | 100% | 100% |
| OCA | 93% | 35% | 57% |
| INT‐767 | 52% | 33% | 51% |
| INT‐777 | 88% | 42% | 53% |
Percentages indicate the amount of increase compared with its own baseline, where vehicle‐treated (No Treatment) is shown as highest increase (100%).
Table 1.
Summary of Attenuation in Serum ALT, Steatosis, and Induction of Liver IL‐1β mRNA After Treatment With BA Receptor Agonists in Mice Administered With Binge Ethanol
| Binge Ethanol | |||
|---|---|---|---|
| ALT | Steaotosis | mRNA IL‐1β | |
| No Treatment | 100% | 100% | 100% |
| OCA | 39% | 24% | 38% |
| INT‐767 | 85% | 31% | 55% |
| INT‐777 | 73% | 36% | 64% |
Percentages indicate the amount of increase compared with its own baseline, in which vehicle‐treated (“No Treatment”) is shown as the highest increase (100%).
To test whether changes in serum ALT correlated with changes in liver structure, we performed liver histology. Prolonged ethanol administration resulted in liver injury, as indicated by elevated serum ALT (Fig. 4D). Interestingly, OCA treatment did not attenuate liver damage after 12 days of chronic ethanol administration. In this prolonged alcohol feeding model that causes features of early ALD, only treatment with the FXR/TGR5 dual agonist INT‐767 reduced serum ALT levels significantly compared with the vehicle‐treated ethanol group (Fig. 4D). BA receptor agonist treatment did not result in changes in serum ethanol levels or body‐to‐weight ratios (Supporting Information Fig. S1C,D).
2.4. Bile Acid Receptor Agonists Decrease Steatosis Through Fatty Acid Synthase Downregulation
The role of FXR in lipid and glucose metabolism has been explored extensively explored.( 23, 24, 25, 26 ) FXR activation lowers plasma and liver triglyceride levels by enhancing plasma triglyceride clearance( 27 ) and repressing sterol regulatory element–binding protein (SREBP) 1c‐mediated hepatic lipogenesis.( 28 ) Therefore, it is likely that BA receptor agonists directly modulate fatty acid synthesis (FASN) pathways. Based on the fact that ethanol administration led to an increase in steatosis, but more importantly, having shown that each of these compounds lead to a decrease in steatosis, we analyzed protein expression changes of FASN.
We found no change in expression of FASN in mouse livers after short acute binge ethanol or after prolonged ethanol administration when treated with vehicle (Fig. 5A). Although it has been previously shown that ethanol induces an increase in FASN through SREBP‐1, an effect likely mediated through acetaldehyde,( 29 ) the absence of change from PF to ethanol‐treated groups might be due to the fact that the PF controls receive either calorie‐matched sugar binge or iso‐caloric Lieber DeCarli diet for the short term and prolonged term models, respectively, both of which may increase FASN protein expression compared with chow‐fed mice. We found a statistically significant reduction in FASN protein expression in the acute binge model after treatment with OCA. More importantly, in the prolonged model, in which steatosis is greater than in the short‐term model, treatment with all tested BA receptor agonists effectively reduced ethanol‐induced FASN protein expression (Fig. 5B‐D). The lipogenic and lipolytic changes correlate with previously described reversal in pathology associated with the development of ALD.( 30, 31 )
Figure 5.

Bile acid receptor agonists decrease steatosis through FASN downregulation. WT mice treated with bile acid agonist or vehicle were exposed to ethanol or sugar binges, or to 12 days of prolonged ethanol or PF liquid diet. FASN expression was assessed by probing for FASN in liver lysate by western blot in the acute (A) and prolonged ethanol feeding model (B). Individual western blots are shown in (C), where acute is shown first followed by prolonged ethanol administration, listed in groups according to treatment. Quantification was done using ImageJ software. * P < 0.05 compared with baseline; ** P < 0.001 compared with vehicle‐treated group (n = 4‐5 for PF, 9‐10 for ethanol, per treatment).
2.5. BA Receptor Agonists Regulate NLRP3 Inflammasome Through Protein Kinase A
To assess the effect of BA receptor agonists on ethanol‐induced inflammation, we analyzed changes in pro‐inflammatory signaling, First, we found that BA receptor agonist treatment consistently reduced expression of IL‐1β (Supporting Information Fig. S3A,B), a known driver of ethanol‐induced inflammation in the liver. Based on these results, we hypothesized that BA receptor agonists were inhibiting inflammasome signaling.
To test this possibility, we first probed for Casp‐1 cleavage in the liver, indicative of inflammasome activity. We found increased Casp‐1 cleavage (Casp‐1p10) in both vehicle‐treated acute and prolonged ethanol models (Fig. 6A,B). This increase was slightly reduced following treatment with OCA and INT‐767, but not INT‐777 (Fig. 6A,B). To gain a better understanding of the mechanism by which these BA receptor agonists could modulate inflammasome signaling, we tested whether inflammasome inhibition involved cyclic adenosine monophosphate–induced activation of protein kinase A (PKA), resulting in ubiquitination and potential subsequent degradation of NLRP3.( 32 )
Figure 6.
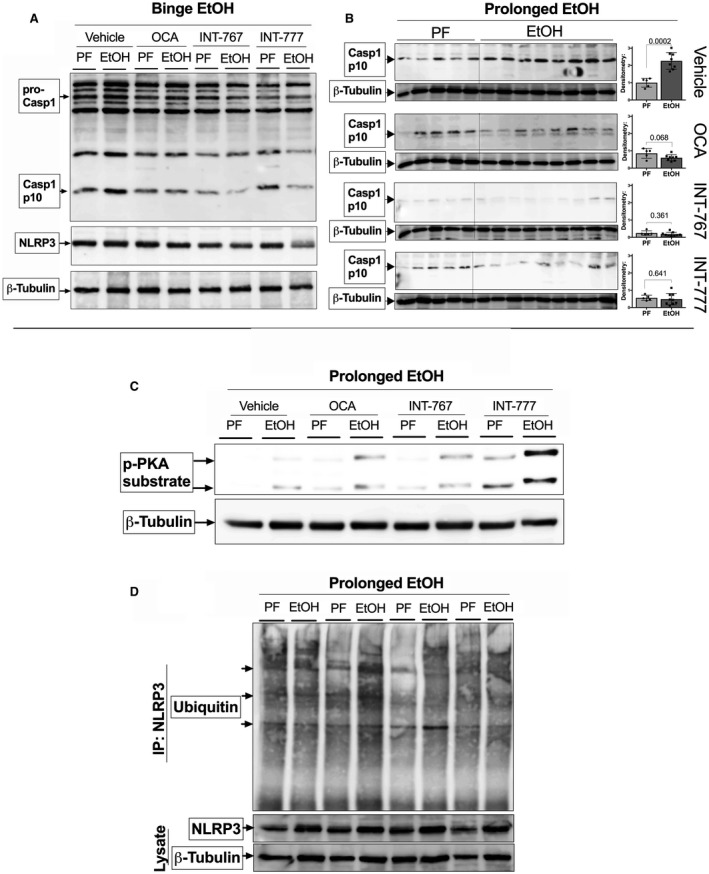
BA agonists regulate NLRP3 inflammasome through PKA. WT mice treated with BA agonist or vehicle were exposed to ethanol or sugar binges, or to 12 days of prolonged ethanol or PF liquid diet. Effects on inflammasome activity were assessed by probing for Casp‐1 cleavage in liver lysate by western blot in the acute (A) and prolonged ethanol feeding model (B). PKA activity in the prolonged ethanol model was assessed by probing for phospho‐PKA substrate in liver lysate (C). Ubiquitination of NLRP3 was assessed by immunoprecipitation of NLRP3, followed by probing of ubiquitin protein by western blot (D).
Our lab has previously shown that the NLRP3 sensor is a key driver of inflammation in ALD through activation from uric acid and adenosine triphosphate (ATP), released by damaged hepatocytes.( 17, 33 ) In ALD models, we found that both FXR and TGR5 agonists increase phosphorylated PKA substrate, suggesting PKA activity, especially after treatment with TGR5 agonists (Fig. 6C). To test whether PKA activity resulted in increased ubiquitination of NLRP3, we performed immunoprecipitation of NLRP3 and probed for ubiquitin, observing an increase in ubiquitin‐tagged NLPR3, especially following INT‐767 treatment (Fig. 6D). Consistent with increased NLRP3 ubiquitination, both FXR and TGR5 agonists inhibited expression of IL‐1β transcripts in the prolonged ALD model (Table 2)(Supporting Information Fig. S3B), whereas they were significantly inhibited only by OCA in the acute ALD model (Table 1) (Supporting Information Fig. S3A). These data are further supported by the previously reported role of TGR5‐cyclic adenosine monophosphate (cAMP) signaling in Kupffer cells in being able to reduce lipopolysaccharide‐induced expression of pro‐inflammatory cytokines such as IL‐1β and tumor necrosis factor α.( 34 ) Taken together, our results indicate that BA receptor agonists lower steatosis and inflammation, and improve liver pathology in ALD models by targeting NLRP3 inflammasome signaling as well as reducing FASN‐induced steatosis.
3. Discussion
Despite significant advances in understanding the pathogenesis of ALD, very few treatment options are currently available.( 4, 35 ) Here, we have tested the therapeutic potential of BA receptor agonists in two mouse models of alcoholic liver injury. Our data show that FXR and TGR5 agonists inhibit liver CYP7A1 expression, which is increased in ALD, reduce liver damage, and blunt hepatic steatosis through FASN downregulation. FXR agonists inhibit NLRP3 inflammasome signaling, as detected by reduced Casp‐1 cleavage. In the two ALD models tested, FXR and TGR5 agonists increase phosphorylated PKA, resulting in increased ubiquitination of NLRP3 inflammasome, and leading to decreased expression of IL‐1β transcripts.
FXR and TGR5 play key roles in ALD by modulating BA metabolism, lipid and glucose metabolism, as and well as inflammation and liver regeneration.( 36 ) Alcohol consumption induces hepatic lipid accumulation and immune cell infiltration, and increases oxidative stress.( 4, 35 ) More importantly, chronic alcohol consumption results in increased BA pool and decreased excretion of BA, suggesting that alcohol consumption may affect the enterohepatic circulation.( 37 ) However, acute alcohol exposure has been reported to induce BA synthesis in humans and primary hepatocytes.( 21, 38 ) Hence, we chose to test both acute and prolonged alcohol exposure models (Supporting Information Fig. S1). Treatment with OCA, INT‐767, or INT‐777 was effective in reducing ethanol‐induced steatosis after acute or chronic ethanol administration. However, only mice treated with the selective FXR agonist, OCA, showed protection from acute binge ethanol‐induced liver injury, whereas mice treated with the dual FXR‐TGR5 agonist, INT‐767, showed liver protection after chronic ethanol exposure. Our findings demonstrate that treatment with FXR and/or TGR5 agonists results in improved biochemical and histological features of early ALD, with varying degrees of efficacy depending on the mode of ethanol administration.
Ethanol‐induced CYP7A1 signaling in hepatocytes may represent a therapeutic target through which steatosis and inflammation can be attenuated. Consistent with a previous report,( 21 ) we show here that ethanol exposure increases CYP7A1 production in the liver, which catalyzes the rate‐limiting step in BA biosynthesis from cholesterol. BA receptor agonists downregulate expression of CYP7A1, and the resulting negative feedback loop, decreasing bile acid synthesis, may indirectly result in decreased steatosis. FXR agonist treatment induces expression of small heterodimer partner 1, an atypical member of the nuclear receptor family,( 39 ) which represses expression of CYP7A1 by inhibiting the activity of liver receptor homolog 1, an orphan nuclear receptor known to positively regulate CYP7A1 expression.( 40 ) In our ALD models, treatment with OCA and INT‐767, both of which signal through FXR (but not INT‐777), decreases CYP7A1 expression, confirming that administration of these FXR agonists induces the expected effect in the liver of mice undergoing alcohol‐induced liver injury.
Our study, based on two murine models of ethanol administration, is supported by a report showing that OCA treatment results in decreased hepatic steatosis and oxidative stress in a model of liver injury induced by ethanol combined with a low‐protein diet.( 41 ) This study suggests that reduced lipid accumulation is mediated by FXR‐induced regulation of FASN and SREBP1c expression.( 42 ) In addition, FXR activation by the specific agonist WAY‐362450 has been shown to protect mice from ALD development.( 43 ) Confirming and extending these results, our data show that in both models of ethanol‐induced liver injury, and in particular in the prolonged model in which steatosis is greater, treatment with all BA receptor agonists tested reduces ethanol‐induced FASN protein expression, providing an explanation for the beneficial effects of FXR and TGR5 activation on ALD‐induced liver pathology.( 30, 31 )
Pathogen‐associated molecular patterns from bacterial fragments drive TLR4‐mediated inflammation.( 44 ) Specifically relevant to ALD is the NLRP3‐inflammasome, a multiprotein complex primed through TLR4 signaling and activated by release of endogenous damage‐associated molecular patterns, such as hepatocyte‐derived ATP and uric acid.( 17, 33 ) We have previously shown that not only is inflammation a driver of ALD, but its effects are long lasting, even after ethanol exposure has stopped, by decreasing the liver’s intrinsic ability to regenerate and recover.( 45 )
Our results show that both FXR and TGR5 activation contribute to ameliorate liver pathology in ALD models. Although TGR5 is expressed in many tissues, including intestine, endocrine glands, adipocytes, muscle, gallbladder, brain and enteric nervous system, its hepatic expression is limited to Kupffer cells, macrophages, sinusoidal endothelial cells, cholangiocytes, gallbladder epithelial cells, and gallbladder smooth muscle cells, but is absent in hepatocytes.( 34, 46 ) TGR5 activation in macrophages by INT‐777 potently inhibits inflammation by targeting cAMP‐ and nuclear factor (NF) kB signaling,( 47 ) and bile acids inhibit LPS‐induced proinflammatory cytokine expression in macrophages( 46 ) and in Kupffer cells( 34 ) through TGR5‐cAMP‐dependent pathways.( 34 ) Therefore, TGR5 agonists may inhibit inflammation and steatosis in ALD also by targeting Kupffer cells–mediated inflammasome activation. Moreover, FXR activation inhibits NF‐kB signaling in the liver( 48 ) and in the intestine,( 49 ) leading to marked inhibition of inflammatory responses.( 50 ) When we assessed the effect of FXR/TGR5 agonists in protecting against alcohol‐induced inflammation, we found that IL‐1β mRNA expression and cleavage of Casp‐1 were consistently reduced in both models of ethanol administration following administration of all three tested BA receptor agonists. This phenotype was driven, more clearly in the case of INT‐767, through an increase in PKA activity and subsequent NLRP3 ubiquitination, suggesting that bile acid receptor agonists play a role in repression of inflammasome signaling, a key driver of inflammation in ALD.( 33 ) This finding is further supported by a recent report showing that bile acids negatively regulate the NLRP3 inflammasome.( 32 )
In conclusion, our findings demonstrate that FXR/TGR5 activation ameliorates murine ALD, providing the first evidence for the therapeutic potential of both FRX and TGR5 agonists to treat ALD. Although the models tested primarily represent early alcoholic steatohepatitis and do not fully replicate the complexity of human drinking patterns, our results provide a sound rationale for the clinical translation of bile acid receptor activation in ALD patients, and suggest that a combined treatment with both FXR and TGR5 agonists may be most effective. Given the limited efficacy of current standard‐of‐care with steroids in alcoholic hepatitis, there is a significant need for new effective and well‐tolerated therapies to increase survival and quality of life in patients with alcoholic hepatitis. Thus, FXR and TGR5 agonists deserve further investigations in human clinical trials in alcoholic hepatitis.
4. Potential conflict of interest
Dr. Adorini consults for and owns stock in Intercept.
5.
Supporting information
Supported by Intercept Pharmaceuticals and the National Institute on Alcohol Abuse and Alcoholism (NIAAA) (AA017729 to G.S. and PA‐12‐149 to A.I.‐V., as a supplement to grant 1R01AA020744‐02 to G.S. and F31AA025545 to A.I.‐V.).
*These authors contributed equally to this work.
Author names in bold designate shared co‐first authorship.
References
- 1. Chacko KR, Reinus J. Spectrum of alcoholic liver disease. Clin Liver Dis 2016;20:419‐427. [DOI] [PubMed] [Google Scholar]
- 2. Mandrekar P, Szabo G. Signalling pathways in alcohol‐induced liver inflammation. J Hepatol 2009;50:1258‐1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL‐1 receptor antagonist ameliorates inflammasome‐dependent alcoholic steatohepatitis in mice. J Clin Invest 2012;122:3476‐3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stickel F, Datz C, Hampe J, Bataller R. Pathophysiology and management of alcoholic liver disease: update 2016. Gut Liver 2017;11:173‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 2014;11:55‐67. [DOI] [PubMed] [Google Scholar]
- 6. Neuschwander‐Tetri BA. Farnesoid x receptor agonists: what they are and how they might be used in treating liver disease. Curr Gastroenterol Rep 2012;14:55‐62. [DOI] [PubMed] [Google Scholar]
- 7. Chiang JY. Bile acid metabolism and signaling. Compr Physiol 2013;3:1191‐1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016;375:631‐643. [DOI] [PubMed] [Google Scholar]
- 9. Guo C, Chen WD, Wang YD. TGR5, not only a metabolic regulator. Front Physiol 2016;7:646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ali AH, Carey EJ, Lindor KD. Recent advances in the development of farnesoid X receptor agonists. Ann Transl Med 2015;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Halilbasic E, Fuchs C, Traussnigg S, Trauner M. Farnesoid X receptor agonists and other bile acid signaling strategies for treatment of liver disease. Dig Dis 2016;34:580‐588. [DOI] [PubMed] [Google Scholar]
- 12. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Greuter T, Malhi H, Gores GJ, Shah VH. Therapeutic opportunities for alcoholic steatohepatitis and nonalcoholic steatohepatitis: exploiting similarities and differences in pathogenesis. JCI. Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011;54:133‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, et al. 6alpha‐ethyl‐chenodeoxycholic acid (6‐ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem 2002;45:3569‐3572. [DOI] [PubMed] [Google Scholar]
- 16. Rizzo G, Passeri D, De Franco F, Ciaccioli G, Donadio L, Rizzo G, et al. Functional characterization of the semisynthetic bile acid derivative INT‐767, a dual farnesoid X receptor and TGR5 agonist. Mol Pharmacol 2010;78:617‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Petrasek J, Iracheta‐Vellve A, Saha B, Satishchandran A, Kodys K, Fitzgerald KA, et al. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross‐talk between hepatocytes and immune cells in alcoholic liver disease. J Leukoc Biol 2015;98:249‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Gut microbiota, cirrhosis, and alcohol regulate bile acid metabolism in the gut. Dig Dis 2015;33:338‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hartmann P, Hochrath K, Horvath A, Chen P, Seebauer CT, Llorente C, et al. Modulation of the intestinal bile acid‐FXR‐FGF15 axis improves alcoholic liver disease in mice. Hepatology 2018;67:2150‐2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie G, Zhong W, Li H, Li Q, Qiu Y, Zheng X, et al. Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. FASEB J 2013;27:3583‐3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nilsson LM, Sjovall J, Strom S, Bodin K, Nowak G, Einarsson C, et al. Ethanol stimulates bile acid formation in primary human hepatocytes. Biochem Biophys Res Commun 2007;364:743‐747. [DOI] [PubMed] [Google Scholar]
- 22. Pathak P, Liu H, Boehme S, Xie C, Krausz KW, Gonzalez F, Chiang JYL. Farnesoid X receptor induces Takeda G‐protein receptor 5 cross‐talk to regulate bile acid synthesis and hepatic metabolism. J Biol Chem 2017;292:11055‐11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li S, Hsu DD, Li B, Luo X, Alderson N, Qiao L, et al. Cytoplasmic tyrosine phosphatase Shp2 coordinates hepatic regulation of bile acid and FGF15/19 signaling to repress bile acid synthesis. Cell Metab 2014;20:320 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 2009;89:147‐191. [DOI] [PubMed] [Google Scholar]
- 25. Fuchs C, Claudel T, Trauner M. Bile acid‐mediated control of liver triglycerides. Semin Liver Dis 2013;33:330‐342. [DOI] [PubMed] [Google Scholar]
- 26. Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med 2015;21:159‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Claudel T, Inoue Y, Barbier O, Duran‐Sandoval D, Kosykh V, Fruchart J, et al. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology 2003;125:544‐555. [DOI] [PubMed] [Google Scholar]
- 28. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP‐1c. J Clin Invest 2004;113:1408‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element‐binding protein (SREBP). J Biol Chem 2002;277:29342‐29347. [DOI] [PubMed] [Google Scholar]
- 30. Bukong TN, Iracheta‐Vellve A, Gyongyosi B, Ambade A, Catalano D, Kodys K, et al. Therapeutic benefits of spleen tyrosine kinase inhibitor administration on binge drinking‐induced alcoholic liver injury, steatosis, and inflammation in mice. Alcohol Clin Exp Res 2016;40:1524‐1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bukong TN, Iracheta‐Vellve A, Saha B, Ambade A, Satishchandran A, Gyongyosi B, et al. Inhibition of spleen tyrosine kinase activation ameliorates inflammation, cell death, and steatosis in alcoholic liver disease. Hepatology 2016;64:1057‐1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 2016;45:944. [DOI] [PubMed] [Google Scholar]
- 33. Iracheta‐Vellve A, Petrasek J, Satishchandran A, Gyongyosi B, Saha B, Kodys K, et al. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J Hepatol 2015;63:1147‐1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun 2008;372:78‐84. [DOI] [PubMed] [Google Scholar]
- 35. Petrasek J, Szabo G. Treatment of alcoholic liver disease including emerging therapies, novel targets, and liver transplantation In: Chalasani N, Szabo G, eds. Alcoholic and Non‐Alcoholic Fatty Liver Disease. Volume 1 Switzerland: Springer International Publishing; 2016:291‐312. [Google Scholar]
- 36. Manley S, Ding W. Role of farnesoid X receptor and bile acids in alcoholic liver disease. Acta Pharm Sin B 2015;5:158‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lefevre AF, DeCarli LM, Lieber CS. Effect of ethanol on cholesterol and bile acid metabolism. J Lipid Res 1972;13:48‐55. [PubMed] [Google Scholar]
- 38. Axelson M, Mork B, Sjovall J. Ethanol has an acute effect on bile acid biosynthesis in man. FEBS Lett 1991;281:155‐159. [DOI] [PubMed] [Google Scholar]
- 39. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP‐1, and LRH‐1 represses bile acid biosynthesis. Mol Cell 2000;6:517‐526. [DOI] [PubMed] [Google Scholar]
- 40. Abrahamsson A, Gustafsson U, Ellis E, Nilsson LM, Sahlin S, Bjorkhem I, et al. Feedback regulation of bile acid synthesis in human liver: importance of HNF‐4alpha for regulation of CYP7A1. Biochem Biophys Res Commun 2005;330:395‐399. [DOI] [PubMed] [Google Scholar]
- 41. Livero FA, Stolf AM, Dreifuss AA, Bastos‐Pereira AL, Chicorski R, de Oliveira LG, et al. The FXR agonist 6ECDCA reduces hepatic steatosis and oxidative stress induced by ethanol and low‐protein diet in mice. Chem Biol Interact 2014;217:19‐27. [DOI] [PubMed] [Google Scholar]
- 42. Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol 2015;16:678‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu X, Xue R, Ji L, Zhang X, Wu J, Gu J, et al. Activation of farnesoid X receptor (FXR) protects against fructose‐induced liver steatosis via inflammatory inhibition and ADRP reduction. Biochem Biophys Res Commun 2014;450:117‐123. [DOI] [PubMed] [Google Scholar]
- 44. Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, et al. The critical role of toll‐like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008;48:1224‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Iracheta‐Vellve A, Petrasek J, Gyogyosi B, Bala S, Csak T, Kodys K, et al. Interleukin‐1 inhibition facilitates recovery from liver injury and promotes regeneration of hepatocytes in alcoholic hepatitis in mice. Liver Int 2017;37:968‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein‐coupled receptor responsive to bile acids. J Biol Chem 2003;278:9435‐9440. [DOI] [PubMed] [Google Scholar]
- 47. Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab 2011;14:747‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008;48:1632‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gadaleta RM, Oldenburg B, Willemsen EC, Spit M, Murzilli S, Salvatore L, et al. Activation of bile salt nuclear receptor FXR is repressed by pro‐inflammatory cytokines activating NF‐kappaB signaling in the intestine. Biochim Biophys Acta 2011;1812:851‐858. [DOI] [PubMed] [Google Scholar]
- 50. Zhu Y, Liu H, Zhang M, Guo GL. Fatty liver diseases, bile acids, and FXR. Acta Pharm Sin B 2016;6:409‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials