

Abstract
Adverse clinical events in primary sclerosing cholangitis (PSC) happen too slowly to capture during clinical trials. Surrogate endpoints are needed, but no such validated endpoints exist for children with PSC. We evaluated the association between gamma glutamyltransferase (GGT) reduction and long‐term outcomes in pediatric PSC patients. We evaluated GGT normalization (< 50 IU/L) at 1 year among a multicenter cohort of children with PSC who did or did not receive treatment with ursodeoxycholic acid (UDCA). We compared rates of event‐free survival (no portal hypertensive or biliary complications, cholangiocarcinoma, liver transplantation, or liver‐related death) at 5 years. Of the 287 children, mean age of 11.4 years old, UDCA was used in 81% at a mean dose of 17 mg/kg/day. Treated and untreated groups had similar GGT at diagnosis (314 versus 300, P= not significant [NS]). The mean GGT was reduced at 1 year in both groups, with lower values seen in treated (versus untreated) patients (99 versus 175, P= 0.002), but 5‐year event‐free survival was similar (74% versus 77%, P= NS). In patients with GGT normalization (versus no normalization) by 1 year, regardless of UDCA treatment status, 5‐year event‐free survival was better (91% versus 67%, P< 0.001). Similarly, larger reduction in GGT over 1 year (> 75% versus < 25% reduction) was also associated with improved outcome (5‐year event‐free survival 88% versus 61%, P= 0.005). Conclusion:A GGT < 50 and/or GGT reduction of > 75% by 1 year after PSC diagnosis predicts favorable 5‐year outcomes in children. GGT has promise as a potential surrogate endpoint in future clinical trials for pediatric PSC.
Abbreviations
- AIH
autoimmune hepatitis
- ALP
alkaline phosphatase
- APRI
AST to platelet ratio index
- GGT
gamma glutamyltransferase
- IBD
inflammatory bowel disease
- PSC
primary sclerosing cholangitis
- UDCA
ursodeoxycholic acid
Primary sclerosing cholangitis (PSC) is a rare progressive disease of the liver characterized by cholestasis and ongoing destruction of the bile ducts.1 Compared with adult patients, children with PSC more commonly present with co‐occurring autoimmune hepatitis (AIH), have fewer dominant strictures, and have a lower incidence of cholangiocarcinoma. Long‐term outcomes are poor. Within 10 years of diagnosis, 30% of children will require liver transplantation and 50% of children will have developed complications of biliary stricturing or portal hypertension.2 There is no known effective treatment to stop the progression of biliary and liver fibrosis.3 PSC is recognized as having one the largest unmet needs in hepatology.4
The slow progression of PSC hinders attempts at pharmaceutical trials. With only 5% of children experiencing an adverse clinical outcome event annually,2 most patients will not reach clinical endpoints during the timeframe of a clinical trial. Adequate statistical power to show a 50% reduction in adverse clinical events over 2 years in a theoretical randomized, placebo‐controlled clinical trial of an investigational drug for PSC would require enrollment of at least 750 children, approximately every newly diagnosed child in North America for 3 to 4 consecutive years. Due to obvious cost and logistical concerns, such a study is impossible. The validation of biomarkers as surrogate endpoints is needed, but presently there is no surrogate endpoint that reliably predicts clinical outcomes in PSC.
Ursodeoxycholic acid (UDCA) therapy has been studied extensively in adult patients with PSC and has not been shown to improve rates of cirrhosis, liver transplantation, or death across a range of doses.5, 6, 7, 8, 9 High‐dose UDCA (25‐30 mg/kg/day) in particular was found to be harmful,8 especially for patients with early‐stage disease.10 The American Association for the Study of Liver Diseases practice guideline for PSC currently discourages the use of UDCA in adults.11 There is a paucity of data in the literature to make recommendations in pediatrics, and UDCA continues to be administered in at least 80% of pediatric PSC cases.2, 12 Data from pediatric PSC case series have shown that treatment with UDCA improves liver biochemistries and cholestatic parameters13, 14; however, the impact of UDCA on long‐term clinical outcomes has not been studied.
Adult studies show that serum alkaline phosphatase (ALP) is of prognostic importance in PSC.15, 16 The incidence of clinically important endpoints such as development of liver cirrhosis and cholangiocarcinoma has been found to be higher in PSC patients with persistently elevated serum ALP levels compared with those who achieve normalization of serum ALP.15, 16, 17 Because the serum ALP levels widely fluctuate in children due to bone growth, serum gamma glutamyltransferase (GGT) is used commonly in children as a marker for cholestasis.18 Serum GGT levels have been found to be of prognostic importance in other pediatric cholestatic diseases, including biliary atresia,19 total parenteral nutrition‐related cholestasis,20 idiopathic neonatal hepatitis,21, 22 and neonatal sepsis‐related cholestasis.23 Elevated serum GGT is the most consistent biochemical abnormality in pediatric patients with PSC, and it appears to be a more sensitive indicator for bile duct injury in PSC compared with ALP and bilirubin.24, 25, 26, 27, 28, 29, 30, 31 GGT at diagnosis of PSC in children correlates with long‐term outcomes, but ALP does not.2 GGT response in adult PSC patients paralleled other markers including ALP in a clinical trial of norursodeoxycholic acid.32 Larger elevations in GGT have been associated with more extensive liver fibrosis in adults with PSC.33 The aim of this study was to compare biochemical responses (using GGT as a biomarker) with long‐term outcomes, in response to UDCA therapy in the largest cohort of children with PSC worldwide.
Methods
We retrospectively reviewed medical records on all known PSC patients with disease onset prior to 18 years of age at 36 different institutions throughout Europe, North America, the Middle East, and Asia,2 including 11 non‐transplant referral centers, 13 large, tertiary referral liver transplant centers, and 12 centers with population‐based data (capturing all patients in a defined geographic region). For each patient, we collected basic demographics and laboratory data at liver disease diagnosis (including complete blood counts, serum chemistries, coagulation, serum globulins, and autoantibody titers). Midway through enrolling new centers for this project, we began additionally collecting laboratory studies at 1 year. These data were requested and available from a subset of the last 17 centers submitting data, which are noted in Appendix I. We included laboratory data present within 6 weeks of the date 1 year after PSC diagnosis. Because of the wide range of normal ALP values in children at different ages due to bone turnover and growth, we normalized all values for age using Mayo Medical Laboratories reference values.34 We collected the presence and type of associated inflammatory bowel disease (IBD), the presence of AIH, large‐duct versus small‐duct PSC phenotype, and the use of UDCA. UDCA was started within 1 month of PSC diagnosis. We observed that all cases of ulcerative colitis refractory to oral aminosalicylate monotherapy, all cases of Crohn’s disease, and all cases of AIH received systemic immunosuppression. Physicians generally followed established practice guidelines for these diseases.35, 36, 37 Patients with PSC generally did not receive immunosuppression outside of that used for coexisting IBD or AIH. Individual therapeutic regimens were not recorded, however. Data were abstracted and de‐identified at local study sites and reviewed and stored centrally.
The diagnosis of PSC was based on a cholestatic biochemical profile with either cholangiography showing multifocal stricturing and segmental dilations of the biliary tree or liver histopathology showing periductal, concentric fibrosis, fibro‐obliterative cholangitis, or primary ductular involvement.11 Patients with abnormal cholangiograms were labeled as large‐duct PSC. Patients with normal cholangiograms but with liver biopsy consistent with PSC were labeled as small‐duct PSC. AIH was diagnosed in patients who met the simplified AIH criteria that have been validated in children,38 based on histopathology, positive autoantibodies, elevated serum globulins, and exclusion of viral hepatitis. All patients with a “probable” or “definite” score were labeled with AIH.
We created a retrospective cohort of all patients and followed them from date of PSC diagnosis to the date of several clinical endpoints: (1) the development of portal hypertensive complications (ascites, hepatic encephalopathy, or esophageal varices with or without bleeding); (2) biliary complications (a clinical picture of biliary obstruction as evidenced by a biliary stricture requiring an intervention in the form of endoscopic or percutaneous stenting, balloon dilation, or drainage); (3) cholangiocarcinoma; (4) liver transplantation; or (5) death from liver disease. A composite outcome of any of these criteria was termed “event‐free survival.” Patients were censored at the date of last known follow‐up. We excluded patients who presented with portal hypertensive or biliary complications within 3 months of PSC diagnosis from this analysis, as their baseline laboratory studies were likely reflective of the outcomes of interest already being present. We used the Kaplan‐Meier method to calculate annual outcome probabilities. The log‐rank test assessed survival differences between groups.
To assess whether improvement in GGT (absolute level less than 50 U/L at 1 year) was associated with lower rates of long‐term adverse outcomes, we categorized patients into four groups: (1) no treatment, GGT at 1 year normalized; (2) UDCA treatment, GGT at 1 year normalized; (3) no treatment, GGT at 1 year not normalized; and (4) UDCA treatment, GGT at 1 year not normalized. We excluded patients who had a GGT < 50 at diagnosis. We performed survival analysis stratified by these groupings. We used previously identified risk cutoffs based on GGT and AST‐to‐platelet‐ratio Index (APRI) at PSC diagnosis to compare response to UDCA in patients who were low‐risk (GGT < 309, APRI < 1.33) versus high‐risk (GGT ≥ 309, APRI ≥ 1.33)2 of experiencing adverse clinical outcomes. We assessed GGT improvement over 1 year among these risk groupings. To assess whether UDCA treatment was effective for PSC phenotypes with IBD (versus none), AIH (versus none), or large duct disease (versus small), we repeated all calculations across these individual subgroups. AppendixII details which patients were included for each analysis. We used Stata version 13 (StataCorp, College Station, TX). All research was approved by the institutional review board of each participating center.
Results
A total of 287 patients had adequate data for analysis. The resulting cohort was 40% female, aged 11.4 years at PSC diagnosis, with PSC + AIH overlap present in 39%, IBD present in 84% (ulcerative colitis in 85% of cases and Crohn’s disease in 15%), and large duct involvement in 74%. UDCA therapy was used in 81% at a mean dose of 16.5 mg/kg/day.
UDCA‐treated patients were younger and had higher APRI values than untreated patients but were otherwise similar at PSC diagnosis (Table 1). After 1 year of UDCA therapy, UDCA‐treated patients had larger reductions in, and lower mean levels of GGT, ALT and APRI, compared with untreated patients. Rates of adverse liver outcomes were similar in treated and untreated groups (Fig. 1). Because median APRI was the only parameter that was different between treatment groups at baseline, we analyzed outcomes on and off treatment by patients in high and low APRI risk groups in Appendix III. Subgroup analyses for outcomes of UDCA therapy in patients with or without IBD or AIH, or with small‐duct versus large‐duct disease are detailed in Table 2. Patients had similar outcomes regardless of phenotype.
Table 1.
Demographics, Biochemical Response, and Clinical Outcomes of Treated and Untreated Patients
|
No Treatment (n = 51) |
UDCA (n = 236) |
PValue | |
|---|---|---|---|
| Demographics | |||
| Age at PSC diagnosis | 12.6 | 11.1 | 0.026 |
| Inflammatory bowel disease | 84% | 84% | 0.941 |
| Ulcerative colitis phenotype | 88% | 84% | 0.479 |
| Crohn’s disease phenotype | 12% | 16% | 0.479 |
| Autoimmune hepatitis overlap | 45% | 38% | 0.327 |
| Large duct involvement | 69% | 75% | 0.383 |
| Laboratory studies at PSC diagnosis | |||
| GGT (U/L) | 290 | 269 | 0.948 |
| ALP (x ULN) | 1.2 | 1.3 | 0.583 |
| ALT (U/L) | 175 | 238 | 0.162 |
| APRI | 0.6 | 1.1 | 0.014 |
| Total bilirubin (mg/dL) | 0.5 | 0.6 | 0.795 |
| Laboratory studies at 1 year | |||
| GGT (U/L) | 115 | 43 | < 0.001 |
| ALP (x ULN) | 0.8 | 0.6 | 0.017 |
| ALT (U/L) | 96 | 62 | 0.003 |
| APRI | 0.5 | 0.4 | 0.031 |
| Total bilirubin (mg/dL) | 0.5 | 0.6 | 0.235 |
| Percentage decrease in laboratory studies from diagnosis to 1 year | |||
| GGT (U/L) | 60% | 75% | 0.002 |
| ALP (x ULN) | 12% | 44% | < 0.001 |
| ALT (U/L) | 22% | 73% | < 0.001 |
| APRI | 22% | 58% | < 0.001 |
| Total bilirubin (mg/dL) | 0% | 9% | 0.322 |
| Rate of liver outcomes within 5 years of diagnosis | |||
| Portal hypertensive complications | 18% | 19% | 0.814 |
| Biliary stricture requiring procedural intervention | 8% | 6% | 0.698 |
| Liver transplantation | 12% | 11% | 0.807 |
| Cholangiocarcinoma | 0% | 1% | 0.511 |
| Death | 0% | 1% | 0.511 |
| Any adverse outcome | 25% | 25% | 0.945 |
Abbreviation: ULN, upper limit of normal.
Figure 1.
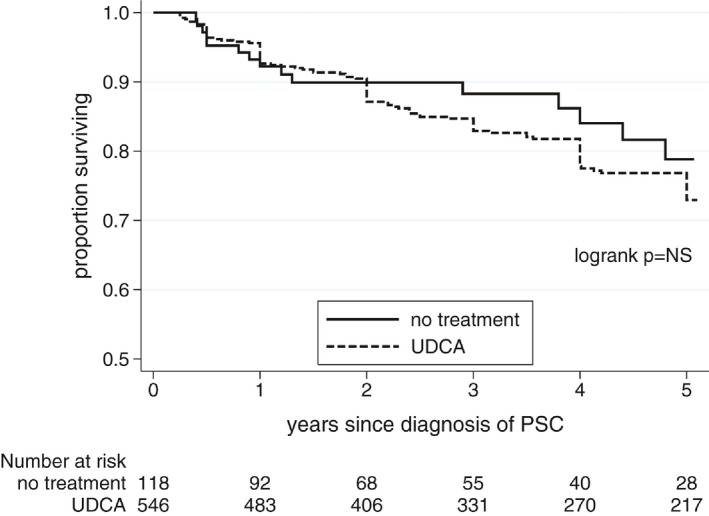
Event‐free survival based on UDCA treatment status.
Table 2.
Clinical Outcomes in UDCA‐Treated Patients Based on Phenotype
|
Portal Hypertensive Complications (5 Years) |
Biliary Complications (5 Years) |
Survival With Native Liver (5 Years) |
Event‐Free Survival (5 Years) |
|
|---|---|---|---|---|
| PSC without IBD |
30% (95% CI 15‐33) |
10% (95% CI 3‐29) |
81% (95% CI 48‐94) |
66% (95% CI 41‐82) |
| PSC‐IBD |
20% (95% CI 14‐27) |
6% (95% CI 3‐11) |
89% (95% CI 81‐93) |
80% (95% CI 72‐85) |
| Pvalue | 0.460 | 0.541 | 0.849 | 0.186 |
| PSC without AIH |
24% (95% CI 17‐33) |
8% (95% CI 4‐15) |
85% (95% CI 74‐91) |
74% (95% CI 64‐81) |
| PSC + AIH |
16% (95% CI 9‐28) |
4% (95% CI 1‐12) |
94% (95% CI 84‐98) |
84% (95% CI 72‐91) |
| Pvalue | 0.203 | 0.348 | 0.175 | 0.074 |
| Small‐duct PSC |
19% (95% CI 10‐34) |
0% (95% CI 0‐0) |
85% (95% CI 65‐94) |
82% (95% CI 65‐91) |
| Large‐duct PSC |
22% (95% CI 16‐30) |
8% (95% CI 5‐15) |
89% (95% CI 80‐94) |
76% (95% CI 67‐83) |
| Pvalue | 0.933 | * | 0.788 | 0.235 |
Abbreviation: CI, confidence interval.
By definition, patients with small‐duct PSC could not have cholangiographic abnormalities, so a statistical comparison is not listed.
Overall, patients who showed normalization of GGT over 1 year fared well, and patients who did not fared poorly, regardless of UDCA treatment status (Fig. 2). Normal GGT at 1 year was present in 53% (112 of 210) versus 29% (14 of 48) of UDCA‐treated versus untreated patients, respectively. Ignoring treatment status and pooling treated and untreated patients together, patients with 1‐year GGT < 50 had a 5‐year event‐free survival of 91% compared with 67% in patients with 1‐year GGT ≥ 50 (P< 0.0001) (Fig. 3). A breakdown of specific events that occurred in each group is included in Appendix IV. Outcomes were best in patients who achieved the most dramatic GGT reductions over 1 year: 5‐year event‐free survival of 88% versus 78%, versus 61% in patients who achieved a 1‐year GGT reduction (compared with GGT at diagnosis) of ≥ 75%, versus 26%‐74% versus ≤ 25%, respectively (Fig. 4).
Figure 2.
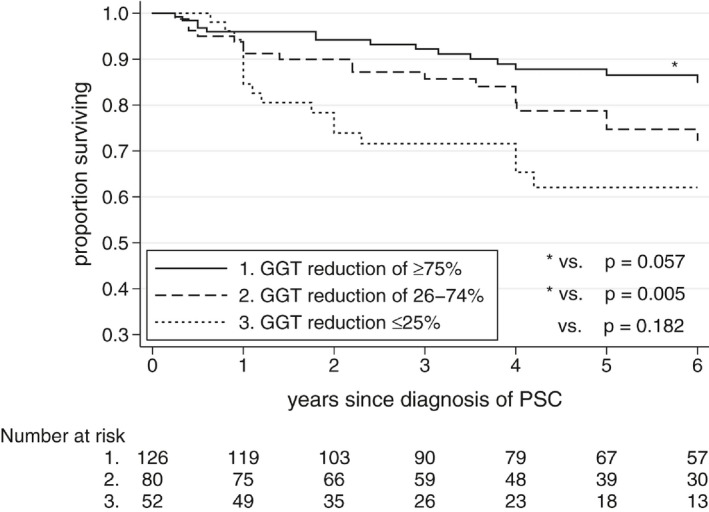
Event‐free survival based on percentage reduction in GGT over first year after diagnosis of PSC.
Figure 3.
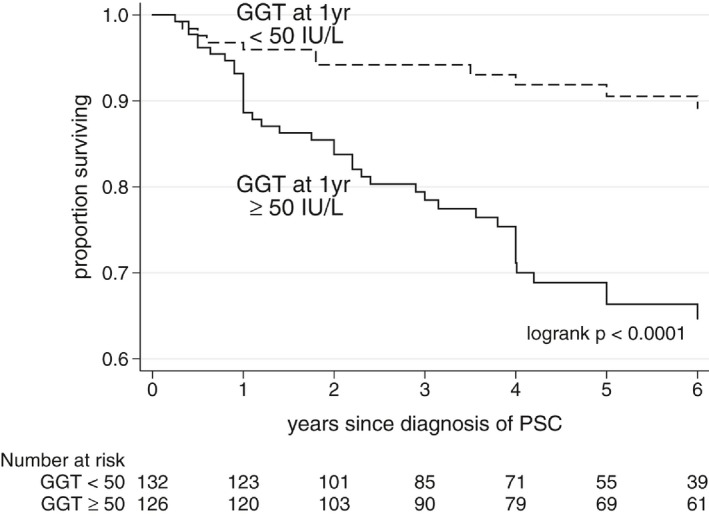
Event‐free survival based on normalization of GGT by 1 year after diagnosis of PSC.
Figure 4.
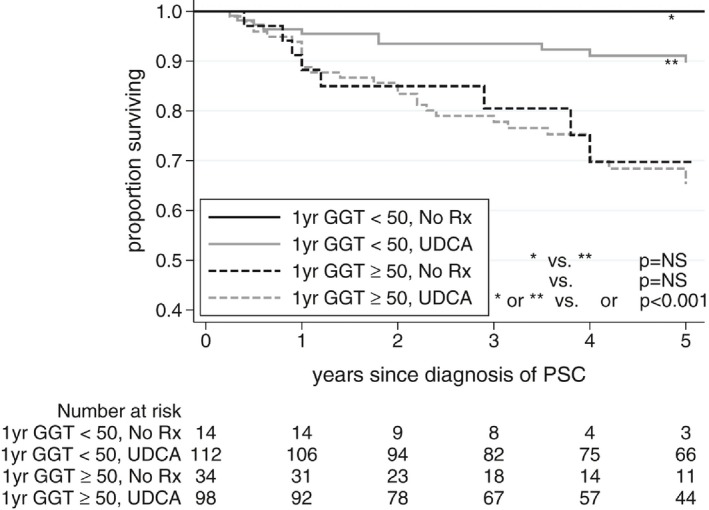
Event‐free survival based on UDCA treatment status and GGT at 1 year.
Analysis of biochemical response in patients stratified by baseline risk groups showed that most of the improvement in GGT observed in UDCA compared with no treatment arose in low‐risk patients who were unlikely to experience adverse outcomes anyway. In contrast, in patients at high baseline risk of adverse outcomes, UDCA‐treated and UDCA‐untreated patients showed a similar decline in GGT over 1 year (Table 3).
Table 3.
Percentage GGT Reduction in 1 Year, Stratified by Baseline Risk of Clinical Liver Complications
| Risk Group |
No Treatment (n = 51) |
UDCA (n = 236) |
PValue |
|---|---|---|---|
| Based on GGT at diagnosis | |||
| Low risk (< 309 IU/L) |
38% reduction n = 37 |
64% reduction n = 148 |
0.003 |
| High risk (≥ 309 IU/L) | 85% reduction | 90% reduction | 0.443 |
| n = 14 | n = 88 | ||
| Based on APRI at diagnosis | |||
| Low risk (< 1.33) |
60% reduction n =38 |
75% reduction n = 143 |
0.007 |
| High risk (≥ 1.33) |
73% reduction n = 13 |
78% reduction n = 93 |
0.199 |
Discussion
We assessed the biochemical and clinical response to UDCA therapy in the largest cohort of children with PSC ever reported in the literature. Regardless of UDCA treatment status, when a child with PSC had a normalized or dramatically reduced GGT at 1 year, outcomes were favorable, suggesting that GGT reduction may be a suitable surrogate endpoint for future clinical trials in children. We showed that UDCA therapy resulted in a larger reduction in liver biochemistry tests compared with no therapy, but that long‐term adverse liver outcomes were similar in treated and untreated patients. This was true across a range of outcome metrics and across PSC phenotypes.
The relationship between biochemical response to UDCA and long‐term liver outcomes seems paradoxical: (1) UDCA‐treated patients had lower liver biochemistry levels on average compared with patients who were not treated; (2) lower liver biochemistry levels generally predicted a more favorable outcome; but (3) UDCA treatment did not result in improved outcomes compared with untreated patients. Analogous results have been seen in clinical trials involving adult patients: Lower or normal ALP levels were associated with improved outcomes,15, 16, 39, 40 regardless of whether UDCA was used or if the labs normalized without therapy.16, 17
These seemingly discordant effects may be explained by several observations. Although the overall mean percentage GGT reduction was statistically superior in UDCA‐treated compared with untreated patients (75% versus 60%), it was not clinically superior, as both groups showed a large reduction. The difference in GGT reduction is further clarified when patients are stratified by baseline risk group. GGT reduction on UDCA therapy was superior to no treatment only in patients at low baseline risk of progressive liver disease, who had lower levels of ductular inflammation (lower baseline GGT), and lower levels of hepatic fibrosis (lower baseline APRI), and who were unlikely to experience clinical events regardless. Conversely, in the highest‐risk patients, with greater degrees of ductular inflammation (higher baseline GGT) and more extensive hepatic fibrosis (higher baseline APRI), most at risk for adverse clinical events and most in need of effective therapy, GGT reduction was similar in the treated and untreated groups.
The biochemical changes observed in untreated patients show that most patients undergo a spontaneous reduction in various parameters of hepatobiliary inflammation in the year following diagnosis. We speculate that PSC in children may generally be detected at an earlier, waxing and waning inflammatory stage at the transition of prediagnostic, subclinical inflammation when enzymes are normal, and the chronic, unrelenting inflammation and extensive biliary changes most characteristic of adults of PSC. ALP is known to wax and wane over years in PSC,3 and it is likely that the natural history of GGT is similar. A regression to the mean phenomenon likely clouds the role of UDCA in enzymatic improvement. Clinicians who follow a patient’s liver enzymes over time are biased to perform diagnostic testing when the enzymes spike upward. A subsequent reduction in enzymes is likely spontaneous in at least some patients and may be attributable to the natural history of the disease rather than to a treatment effect. UDCA treatment may have reduced hepatobiliary inflammation and normalized GGT in a subset of patients who otherwise would not have done so on their own, but this effect is likely too small to be of clinical benefit, as it could not be detected in this cohort of hundreds of children with PSC.
The strength of this study was its large size and inclusion of patients from a diverse mix of centers. Patients were drawn from tertiary care referral centers, population‐based cohorts, and small single centers, eliminating a referral bias. There are weaknesses to this study. The retrospective design prevented a standardized diagnostic and therapeutic algorithm. We did not have data on compliance with prescribed UDCA, which may have been poor especially in teenage patients.
The present data show convincingly that when GGT is lower, patients do better. Prospective data are needed to validate GGT as a proven surrogate endpoint in PSC, but such a study could take a decade or more. Patients are in dire need of clinical trials to identify an effective therapy. The U.S. Food and Drug Administration (FDA) recognizes the need for surrogate endpoints for clinical trials in slowly progressive diseases like PSC. In situations in which there is insufficient prospective data to validate a surrogate, but where there is convincing rationale from epidemiologic studies such as this one, they define a surrogate endpoint as “reasonably likely.”41 Clinical trials may progress on the basis of reasonably likely surrogate endpoints in the FDA’s Accelerate Approval program, with future prospective validation taking place after drug approval.42 We believe that the present data support GGT as a reasonably likely surrogate endpoint, at least for phase II clinical trials in children.
Conclusions
A GGT < 50 and/or GGT reduction of > 75% by 1 year after PSC diagnosis predicts favorable 5‐year outcomes in children, regardless of whether UDCA treatment is used. GGT is a useful biomarker in PSC and is clearly correlated with clinical liver outcomes. Phase II trials could proceed in children if the trials are designed around sustained GGT normalization as a reasonably likely surrogate endpoint, but prospective studies over several years are ultimately needed to prove the relationship between GGT response and clinical outcome.
Potential conflicts of interest
Dr. DiGuglielmo received grants from Synergy Pharmaceuticals. Dr. Kamath consults for Retrophin. Dr. Miloh consults, advises, and is on the speakers’ bureau for Alexion. Dr. Mohan received grants from Gilead. Dr. Vos consults for ARMA, Target Pharmasolutions, and Intercept; she received grants from Resonance Health and Gemphire.
Author Contributions
All authors participated in all phases of the study design, data collection, data analysis, and manuscript preparation and review.
1.
Pediatric PSC Consortium Centers Participating in the 1‐Year GGT Data Analysis (in Alphabetical Order)
Children’s National Medical Center, Washington, DC
Emory University, Atlanta, GA
Medical College of Wisconsin, Milwaukee, WI
Palacky University, Olomouc, Czech Republic
Sapienza University of Rome, Rome, Italy
Tel‐Aviv University, Tel‐Aviv, Israel
Texas Children’s Hospital, Houston, TX
University of Athens, Athens, Greece
University of Colorado, Aurora, CO
University of Helsinki, Helsinki, Finland
University of Naples Federico II, Naples, Italy
University of Manitoba, Winnipeg, Manitoba, Canada
University of Pittsburgh Medical Center, Pittsburgh, PA
University of Rochester, Rochester, NY
University of Toronto, Toronto, Ontario, Canada
University of Utah, Salt Lake City, UT
Yale University, New Haven, CT
Flow Diagram Detailing Patient Inclusion and Exclusion in Each Analysis
(Fig. 5)
Figure 5.
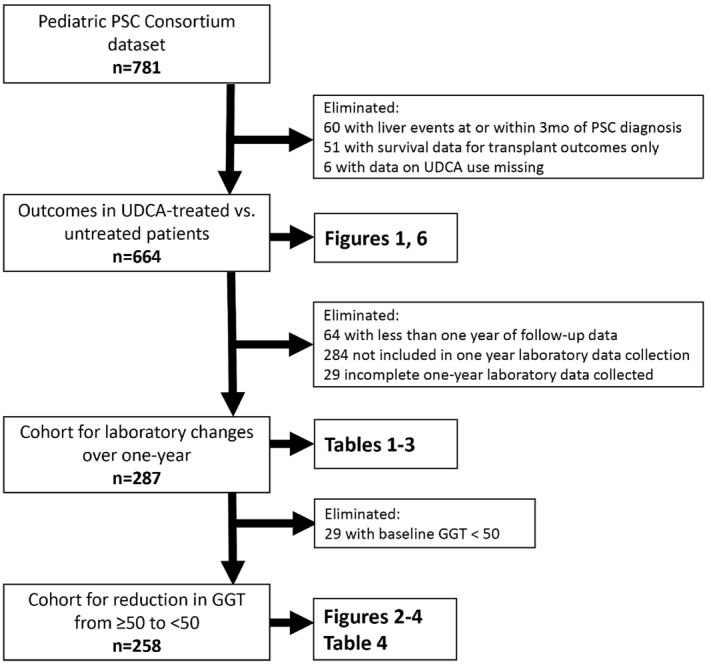
Flow diagram detailing patient inclusion and exclusion in each analysis.
Outcomes of UDCA Treatment Versus No Treatment, Stratified by APRI
Because the median APRI at diagnosis of PSC was different between treated versus untreated patients (1.1 versus 0.6, P= 0.014), we performed a survival analysis from diagnosis of PSC to any liver event, stratified by patients in the highest tertile of APRI (≥ 1.33) and lowest two tertiles of APRI (< 1.33). Clinical outcomes remained similar in treated and untreated patients in both the low‐risk and high‐risk APRI groups (Fig. 6).
Figure 6.
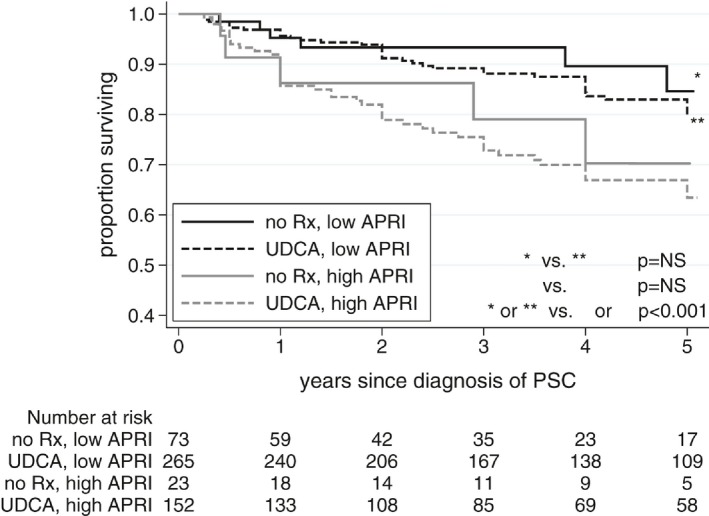
Event‐free survival based on UDCA treatment status, stratified by APRI risk group.
Clinical Events in Patients With GGT ≥ 50 Versus < 50 at 1 Year
(Table 4)
Table 4.
Clinical Events in Patients With GGT ≥ 50 Versus < 50 at 1 Year
|
1‐Year GGT ≥ 50 n = 126 |
1‐Year GGT < 50 n = 132 |
PValue | |
|---|---|---|---|
| Portal hypertensive complications | 38 (30%) | 12 (9%) | < 0.001 |
| Biliary stricture requiring intervention | 10 (8%) | 7 (5%) | 0.394 |
| Liver transplantation | 25 (20%) | 5 (4%) | < 0.001 |
| Cholangiocarcinoma | 1 (< 1%) | 1 (<1%) | 0.974 |
| Death | 1 (< 1%) | 1 (<1%) | 0.974 |
Columns listed as number (%); some patients had events in multiple categories.
Funding
Supported by the Primary Children’s Hospital Foundation and the National Center for Advancing Translational Sciences of the National Institutes of Health (KL2TR001065).
References
- 1. Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet 2013;382:1587‐1599. [DOI] [PubMed] [Google Scholar]
- 2. Deneau MR, El‐Matary W, Valentino PL, Abdou R, Alqoaer K, Amin M, et al. The natural history of primary sclerosing cholangitis in 781 children: a multicenter, international collaboration. Hepatology 2017;66:518‐527. [DOI] [PubMed] [Google Scholar]
- 3. Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis—a comprehensive review. J Hepatol 2017;67:1298‐1323. [DOI] [PubMed] [Google Scholar]
- 4. Dyson JK, Webb G, Hirschfield GM, Lohse A, Beuers U, Lindor K, et al. Unmet clinical need in autoimmune liver diseases. J Hepatol 2015;62:208‐218. [DOI] [PubMed] [Google Scholar]
- 5. Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis‐Ursodeoxycholic Acid Study Group. N Engl J Med 1997;336:691‐695. [DOI] [PubMed] [Google Scholar]
- 6. Cullen SN, Rust C, Fleming K, Edwards C, Beuers U, Chapman RW. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. J Hepatol 2008;48:792‐800. [DOI] [PubMed] [Google Scholar]
- 7. Olsson R, Boberg KM, de Muckadell OS, Lindgren S, Hultcrantz R, Folvik G, et al. High‐dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5‐year multicenter, randomized, controlled study. Gastroenterology 2005;129:1464‐1472. [DOI] [PubMed] [Google Scholar]
- 8. Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, et al. High‐dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009;50:808‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wunsch E, Trottier J, Milkiewicz M, Raszeja‐Wyszomirska J, Hirschfield GM, et al. Prospective evaluation of ursodeoxycholic acid withdrawal in patients with primary sclerosing cholangitis. Hepatology 2014;60:931‐940. [DOI] [PubMed] [Google Scholar]
- 10. Imam MH, Sinakos E, Gossard AA, Kowdley KV, Luketic VA, Edwyn Harrison M, et al. High‐dose ursodeoxycholic acid increases risk of adverse outcomes in patients with early stage primary sclerosing cholangitis. Aliment Pharmacol Ther 2011;34:1185‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Schneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010;51:660‐678. [DOI] [PubMed] [Google Scholar]
- 12. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single‐center longitudinal cohort study. J Pediatr Gastroenterol Nutr 2016;63:603‐609. [DOI] [PubMed] [Google Scholar]
- 13. Feldstein AE, Perrault J, El‐Youssif M, Lindor KD, Freese DK, Angulo P. Primary sclerosing cholangitis in children: a long‐term follow‐up study. Hepatology 2003;38:210‐217. [DOI] [PubMed] [Google Scholar]
- 14. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A retrospective single‐center review of primary sclerosing cholangitis in children. Clin Gastroenterol Hepatol 2009;7:239‐245. [DOI] [PubMed] [Google Scholar]
- 15. Al Mamari S, Djordjevic J, Halliday JS, Chapman RW. Improvement of serum alkaline phosphatase to xxaaa1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol 2013;58:329‐334. [DOI] [PubMed] [Google Scholar]
- 16. Lindstrom L, Hultcrantz R, Boberg KM, Friis‐Liby I, Bergquist A. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2013;11:841‐846. [DOI] [PubMed] [Google Scholar]
- 17. Stanich PP, Bjornsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis 2011;43:309‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cabrera‐Abreu JC, Green A. Gamma‐glutamyltransferase: value of its measurement in paediatrics. Ann Clin Biochem 2002;39:22‐25. [DOI] [PubMed] [Google Scholar]
- 19. Freeman AJ, Ng VL, Harpavat S, Hrycko A, Apted Z, Bulut P, et al. Level of gamma‐glutamyltransferase in 2‐year‐old children with biliary atresia associates with progression of portal hypertension. Clin Gastroenterol Hepatol 2017;15:1133‐1135. [DOI] [PubMed] [Google Scholar]
- 20. Spagnuolo MI, Iorio R, Vegnente A, Guarino A. Ursodeoxycholic acid for treatment of cholestasis in children on long‐term total parenteral nutrition: a pilot study. Gastroenterology 1996;111:716‐719. [DOI] [PubMed] [Google Scholar]
- 21. Wang J, Wang ZL, Wang XH, Zhu QR, Zheng S. The prognostic value of serum gamma glutamyltransferase activity in Chinese infants with previously diagnosed idiopathic neonatal hepatitis. HK J Paediatr (new series) 2008;13:39‐45. [Google Scholar]
- 22. Wang JS, Tan N, Dhawan A. Significance of low or normal serum gamma glutamyl transferase level in infants with idiopathic neonatal hepatitis. Eur J Pediatr 2006;165:795‐801. [DOI] [PubMed] [Google Scholar]
- 23. Oswari H, Widjaja RK, Rohsiswatmo R, Cleghorn G. Prognostic value of biochemical liver parameters in neonatal sepsis‐associated cholestasis. J Paediatr Child Health 2013;49:E6‐E11. [DOI] [PubMed] [Google Scholar]
- 24. Classen M, Gotze H, Richter HJ, Bender S. Primary sclerosing cholangitis in children. J Pediatr Gastroenterol Nutr 1987;6:197‐202. [DOI] [PubMed] [Google Scholar]
- 25. Debray D, Pariente D, Urvoas E, Hadchouel M, Bernard O. Sclerosing cholangitis in children. J Pediatr 1994;124:49‐56. [DOI] [PubMed] [Google Scholar]
- 26. el‐Shabrawi M, Wilkinson ML, Portmann B, Mieli‐Vergani G, Chong SKF, Williams R,, et al. Primary sclerosing cholangitis in childhood. Gastroenterology 1987;92:1226‐1235. [DOI] [PubMed] [Google Scholar]
- 27. Johnson DA, Cattau EL Jr, Hancock JE. Pediatric primary sclerosing cholangitis. Dig Dis Sci 1986;31:773‐777. [DOI] [PubMed] [Google Scholar]
- 28. Kagalwalla AF, Altraif I, Shamsan L, Omojola M, Khan H, Kagalwalla YA. Primary sclerosing cholangitis in Arab children: report of four cases and literature review. J Pediatr Gastroenterol Nutr 1997;24:146‐152. [DOI] [PubMed] [Google Scholar]
- 29. Sisto A, Feldman P, Garel L, Seidman E, Brochu P, Morin CL, et al. Primary sclerosing cholangitis in children: study of five cases and review of the literature. Pediatrics 1987;80:918‐923. [PubMed] [Google Scholar]
- 30. Werlin SL, Glicklich M, Jona J, Starshak RJ. Sclerosing cholangitis in childhood. J Pediatr 1980;96:433‐435. [DOI] [PubMed] [Google Scholar]
- 31. Wilschanski M, Chait P, Wade JA, Davis L, Corey M, St. Louis P, et al. Primary sclerosing cholangitis in 32 children: clinical, laboratory, and radiographic features, with survival analysis. Hepatology 1995;22:1415‐1422. [PubMed] [Google Scholar]
- 32. Fickert P, Hirschfield GM, Denk G, Marschall HU, Altorjay I, Färkkilä M, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol 2017;67:549‐558. [DOI] [PubMed] [Google Scholar]
- 33. Corpechot C, El Naggar A, Poujol‐Robert A, Ziol M, Wendum D, Chazouilléres O, et al. Assessment of biliary fibrosis by transient elastography in patients with PBC and PSC. Hepatology 2006;43:1118‐1124. [DOI] [PubMed] [Google Scholar]
- 34. Mayo Medical Laboratories . Alkaline phosphatase, total and isozymes, serum: clinical and interpretive. https://www.mayomedicallaboratories.com/test-catalog/Clinical+and+Interpretive/8340. Accessed August 31, 2018.
- 35. Ruemmele FM, Veres G, Kolho KL, Griffiths A, Levine A, Escher JC, et al. Consensus guidelines of ECCO/ESPGHAN on the medical management of pediatric Crohn's disease. J Crohns Colitis 2014;8:1179‐1207. [DOI] [PubMed] [Google Scholar]
- 36. Manns MP, Czaja AJ, Gorham JD, Krawitt EL, Mieli‐Vergani G, Vergani D, et al. Diagnosis and management of autoimmune hepatitis. Hepatology 2010;51:2193‐2213. [DOI] [PubMed] [Google Scholar]
- 37. Turner D, Levine A, Escher JC, Griffiths AM, Russell RK, Dignass A, et al. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence‐based consensus guidelines. J Pediatr Gastroenterol Nutr 2012;55:340‐361. [DOI] [PubMed] [Google Scholar]
- 38. Mileti E, Rosenthal P, Peters MG. Validation and modification of simplified diagnostic criteria for autoimmune hepatitis in children. Clin Gastroenterol Hepatol 2012;10(417–421):e1‐e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rupp C, Rossler A, Halibasic E, Sauer P, Weiss KH, Friedrich K, et al. Reduction in alkaline phosphatase is associated with longer survival in primary sclerosing cholangitis, independent of dominant stenosis. Aliment Pharmacol Ther 2014;40:1292‐1301. [DOI] [PubMed] [Google Scholar]
- 40. de Vries EM, Wang J, Leeflang MM, Boonstra K, Weersma RK, Beuers UH, et al. Alkaline phosphatase at diagnosis of primary sclerosing cholangitis and one year later: evaluation of prognostic value. Liver Int 2016;36:1867‐1875. [DOI] [PubMed] [Google Scholar]
- 41. FDA‐NIH Biomarker Working Group . Reasonably likely surrogate endpoint; in BEST (Biomarkers, EndpointS, and other Tools) resource. https://www.ncbi.nlm.nih.gov/books/NBK453485/. Accessed August 31, 2018.
- 42. U.S. Food and Drug Administration . FDA facts: biomarkers and surrogate endpoints. https://www.fda.gov/AboutFDA/Innovation/ucm512503.htm. 2017.