

Abstract
Background
Glioblastoma multiforme, the most prevalent and aggressive brain tumour, has a poor prognosis. The molecular mechanisms underlying gliomagenesis remain poorly understood. Therefore, molecular research, including various markers, is necessary to understand the occurrence and development of glioma.
Method
Weighted gene co-expression network analysis (WGCNA) was performed to construct a gene co-expression network in TCGA glioblastoma samples. Gene ontology (GO) and pathway-enrichment analysis were used to identify significance of gene modules. Cox proportional hazards regression model was used to predict outcome of glioblastoma patients.
Results
We performed weighted gene co-expression network analysis (WGCNA) and identified a gene module (yellow module) related to the survival time of TCGA glioblastoma samples. Then, 228 hub genes were calculated based on gene significance (GS) and module significance (MS). Four genes (OSMR + SOX21 + MED10 + PTPRN) were selected to construct a Cox proportional hazards regression model with high accuracy (AUC = 0.905). The prognostic value of the Cox proportional hazards regression model was also confirmed in GSE16011 dataset (GBM: n = 156).
Conclusion
We developed a promising mRNA signature for estimating overall survival in glioblastoma patients.
Electronic supplementary material
The online version of this article (10.1186/s12920-018-0407-1) contains supplementary material, which is available to authorized users.
Keywords: GBM, WGCNA, TCGA, Cox proportional hazards regression module
Background
Glioblastoma multiforme (GBM), the most prevalent and aggressive primary intracranial tumour, displays heterogeneity, rapid proliferation and extensive invasion, with a median survival of approximately 15 months [1, 2]. Therefore, developing appropriate and effective biomarkers to predict prognosis is crucial for glioblastoma patients. Previous genomic analyses of glioblastoma have identified some molecular markers, including epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor alpha (PDGFRA), vascular endothelial growth factor (VEGF), insulin-like growth factor 1 (IGF-1), P53 and isocitrate dehydrogenase 1 (IDH1), and X-linked alpha thalassemia mental retardation syndrome gene (ATRX) [3, 4]. In addition, methylation levels of the promoter of O6methylguanineDNA methyltransferase (MGMT) are related to sensitivity of temozolomide therapy and the prognosis of patients. The 1p/19q loss is another prognosis marker and indicates a better prognosis [5].
With the development of high-throughput sequencing and bioinformatics, abundant sequencing data provide a remarkable opportunity to detect glioblastoma-associated key genes, networks and pathways. However, identification of these features remains challenging. Weighted gene co-expression network analysis (WGCNA) is a system biology method used for describing the correlation patterns among genes and finding highly correlated modules. In this study, we performed WGCNA for RNASeq data derived from The Cancer Genome Atlas (TCGA) and reconstructed gene co-expression networks. Then, we identified gene modules related to survival and recurrence time. Using Kaplan-Meier survival analysis and multivariate Cox regression analysis, we identified an independent prognostic model. This finding provides new insights into the molecular mechanism of GBM.
Methods
Data download and pre-processing
RNA sequencing data (RNA-Seq2 level 3 data) from human glioblastoma samples were obtained from the TCGA data portal (https://portal.gdc.cancer.gov), which contains 152 glioblastoma tissues [3]. These data were updated to January 28, 2016. According to the instructions for WGCNA, fragments per kilobase per million (FPKM) is recommended as the data type for subsequent analysis. As some genes without significant changes in expression between samples will be highly correlated in WGCNA, the 5000 most differentially expressed genes were used in the following WGCNA studies. The clinical metadata of 152 samples were also downloaded and filtered for useful information. Because the clinical data of TCGA database was constantly updated, the survival time of the death patients was more accurate. The age, gender, survival time and recurrence time data of 75 deceased patients were selected and divided into two groups according to the median (Table 1). Subtype data of 152 samples was downloaded from GlioVis database (http: //gliovis.bioinfo.cnio.es/) [6]. The GSE36245, GSE16011 and GSE50161 datasets were included in the study, and both originated from an Affymetrix Human Genome U133 Plus 2.0 Array [7–9]. GSE36245 dataset only contained 46 glioblastoma samples, so it was used to validate whether the modules which are obtained from TCGA database were reproducible. GSE16011 dataset (GBM: n = 159) was used to validate whether the Cox proportional hazards regression model was reproducible. GSE50161 dataset (GBM: n = 34; Normal control: n = 13) contained glioblastoma and normal brain samples and was used to perform difference analysis.
Table 1.
Information for 75 glioblastoma patients
| TCGA Datasets | |
|---|---|
| Variables | Case number(N = 75) |
| Age | |
| < 59 | 38 |
| > =59 | 37 |
| Gender | |
| Female | 55 |
| Male | 20 |
| Survival time | |
| < 448 | 39 |
| > =448 | 36 |
| Recurrence time | |
| < 178 | 38 |
| > =178 | 37 |
Weighted gene co-expression network analysis and module preservation
WGCNA was performed using the R package WGCNA [10]. First, RNASeq data were filtered to reduce outliers. A co-expression similarity matrix was composed of the absolute value of the correlation between the expression levels of transcripts. For an unsigned network, the correlation coefficient between gene i and j was defined as Sij: Sij = |cor(i,j)|. The co-expression similarity matrix was then transformed into the adjacency matrix by choosing 7 as a soft threshold (Fig. 1a). A topological matrix was created using the topological overlap measure (TOM) [11, 12]. Finally, we chose the dynamic hybrid cut method, a bottom-up algorithm, to identify co-expression gene modules [13]. To identify the significance of each module, we calculated gene significance (GS) to measure the correlation between genes and sample traits. Module significance (MS) was defined as the average GS within modules and was calculated to measure the correlation between modules and sample traits (age, gender, survival time and recurrence time) [14, 15]. Statistical significance was determined using the correlation P value. The module Preservation function in the WGCNA R package was used to calculate the Z summary to evaluate whether a module was conserved [16].
Fig. 1.

Weighted gene co-expression network of glioblastoma. a Identification of the soft threshold according to the standard of the scale-free network. b Dendrogram of consensus module eigengenes. The red line represented merging threshold. Modules with a correlation coefficient greater than 0.75 were merged. c Identification of a co-expression module in glioblastoma. The branches of the cluster dendrogram correspond to the 19 different gene modules. Each piece of the leaves on the cluster dendrogram corresponds to a gene. d Correlation between the gene module and clinical traits. The clinical traits include age, gender, survival time and recurrence time. The correlation coefficient in each cell represented the correlation between the gene module and the clinical traits, which decreased in size from red to blue. The corresponding P value is also annotated
Hub gene identification and module visualisation
Hub genes were identified using “network screening” within the R package WGCNA [10]. This method identifies genes that have high GS and MS. We selected the q. weighted < 0.01 as a cutoff to obtain the hub genes [17]. The targeted module visualisation was performed using Cytoscape3.5.1. Cytoscape is an open source software for visualising molecular interaction network (http://www.cytoscape.org/index.html) [18].
Functional enrichment analysis
Gene ontology (GO) and pathway-enrichment analysis (Kyoto Encyclopedia of Genes and Genomes (KEGG)) were performed using the R package clusterProfiler (https://guangchuangyu.github.io/clusterProfiler) [19–21]. Enriched ontological terms and pathways with P < 0.05 were selected.
Cox proportional hazards regression model
The prognostic value of each hub gene was first assessed by univariate Cox proportional hazards regression. Then, statistically significant genes were used to construct the multivariate Cox regression model as follows: Risk score = (0.2844* expression level of oncostatin M receptor (OSMR)) + (− 0.1682* expression level of SRY-Box 21 (SOX21)) + (1.3462* expression level of mediator complex subunit 10 (MED10)) + (0.3776* expression level of protein tyrosine phosphatase, receptor type N (PTPRN)). Glioblastoma samples were divided into high-score and low-score groups based on the median of the risk score. K-M survival curves were generated to assess the prognostic value of the model using the R package “survival” (https://CRAN.R-project.org/package=survival). The receiver operating characteristic curve (ROC) was generated to assess the accuracy of the model with the R package “survivalROC” (https: //CRAN.R-project.org/package = survivalROC) [22].
Statistical analysis
The Pairwise t tests and Tukey’s Honest Significant Difference test were used to perform differentail analysis. All statistical tests and graphing were performed using RStudio (www.rstudio.com) and GraphPad Prism 7.0. P values < 0.05 were considered statistically significant [23]. Statistical significance was indicated in the figures as follows: *P < 0.05, **P < 0.01 and ***P < 0.001.
Results
Pre-processing of TCGA RNA sequencing and clinical data
Glioblastoma RNASeq data were downloaded from TCGA and constructed into a matrix RNASeq with gene symbols as the rows and patient barcodes as the column names. Furthermore, expression estimates in less than 20% of cases were removed, and the top 5000 most differentially expressed genes were used in WGCNA studies. Simultaneously, the corresponding clinical data were also downloaded to relate co-expression modules to clinical phenotypes. After outliers were removed, we selected data from 75 deceased patients among the 152 samples, including 5000 genes (Table 1).
Gene co-expression network analysis
WGCNA was performed to construct a gene co-expression network to identify biologically meaningful gene modules and better understand the molecular mechanism of glioblastoma. WGCNA defined gene modules as a set of genes with topological overlaps. The specific approach was to establish a hierarchical clustering tree based on dynamic hybrid cut. Each piece of the leaves on this tree corresponded to a gene, and the different gene modules were the branches of the tree. Identification of co-expression modules could facilitate identification of hub genes that drive and maintain important functions. Ultimately, 19 gene modules were identified. The grey module includes genes that were not assigned to any gene modules (Fig. 1b, c).
Calculation of module-trait correlations in GBMs
To analyse the relationship between gene modules and sample clinical information, we used the module eigengene (ME) as the overall gene expression level of corresponding modules and calculated correlations with clinical phenotypes, such as age, gender, survival time and recurrence time. The yellow and dark green modules were significantly associated with survival time (Fig. 1d and Additional file 1: Table S6).
Module preservation statistics
To validate whether the modules were reproducible (or preserved), we selected 4644 genes which from GSE36245 (GBM: n = 46) to construct a weighted gene co-expression network. Then, the Z summary score was calculated to determine module preservation. Modules with a Z summary score > 10 were regarded as preserved [24]. That is, the modules of the TCGA dataset also existed in the network of the Gene Expression Omnibus (GEO) dataset. The 10 modules were highly conserved, including the yellow module, while the dark green module was poorly conserved (Fig. 2a). Thus, we focused on analysis of the yellow module in the follow-up study.
Fig. 2.

Module preservation and visualisation. a Module preservation statistics of TCGA modules in GEO modules (y - axis) vs module size (x - axis). b Visualisation of the gene co-expression network of the yellow module was generated using Cytoscape. Based on weight, not all genes corresponding to each module were represented
Identification of the hub gene
The function of the WGCNA R package, called network Screening, was used to search for the hub gene in the yellow module. We used the q. weighted < 0.01 and obtained 228 survival-related genes. These intramodular hub genes were centrally located in their respective modules and may thus be critical components within the modules [25].
Module visualisation of network connections
To further depict the expression network of module genes related to survival time, we exported the co-expression network of the yellow module into Cytoscape. The nodes were defined as individual genes in the network, and edges were defined as the interactions between genes. As shown in Figures, the yellow module included 311 nodes and 21,557 edges. The hub genes of the modules were marked as orange nodes (Fig. 2b).
GO and pathway-enrichment analysis of hub genes
To explore the cellular component (CC), molecular function (MF) and biological process (BP), we performed GO enrichment analysis. A total of 228 hub genes were significantly enriched in the following subclasses of GO classification (Fig. 3): focal junction (GO: 0005925, P = 3.17E - 15), cell adhesion molecule binding (GO: 0050839, P = 1.07E - 15), collagen binding (GO: 0005518, P = 1.56E - 11), extracellular matrix organisation (GO: 0030198, P = 2.67E - 20), and extracellular structure organisation (GO: 0043062, P = 1.10E - 21). KEGG pathway analysis showed that the top enriched terms were focal adhesion (hsa04510, P = 1.53E - 10) and ECM-receptor interaction (hsa04512, P = 1.39E - 07) based on P value. These results suggest that these genes were closely related to the cell adhesion function (Fig. 3a–d and Additional files 2, 3, 4, 5: Table S2–5).
Fig. 3.

Enrichment analysis of hub genes. a Enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for 228 hub genes related to survival time. Molecular Function b, Biological Process c and Cellular Component d of Gene Ontology (GO) enrichment analysis were shown separately
Construction of the cox proportional hazards regression model based on hub genes and Kaplan-Meier analysis
We further narrowed down and selected the top 20 genes significantly related to survival time by univariate Cox analysis of 228 hub genes (Additional file 6: Table S1). Then, we used the 20 genes to perform multivariate Cox analysis and construct a Cox proportional hazards regression model from 152 glioblastoma patients. The risk score for predicting survival time was calculated with a formula based on the above mentioned four genes: risk score = (0.2844 * expression level of OSMR) + (− 0.1682 * expression level of SOX21) + (1.3462 * expression level of MED10) + (0.3776 * expression level of PTPRN) (Fig. 4a–c). We divided 152 patients into high-risk (N = 76) and low-risk (N = 76) groups according to the median of the risk score. The five-year survival rate of the high-risk group was significantly poorer than that of the low-risk group (Fig. 5a). The model was reproducible in GSE16011 dataset (Additional file 7: Figure S1 and Additional file 8: Table S7). Elevated expression of OSMR, MED10 and PTPRN was associated with an increased risk score, but SOX21 produced the opposite effect. The area under the ROC curve was 0.905 (Fig. 5c), indicating a higher predictive value. Moreover, K-M curves confirmed that the four genes could function as an independent predictive indicator for the survival of glioblastoma patients (Fig. 5b).
Fig. 4.
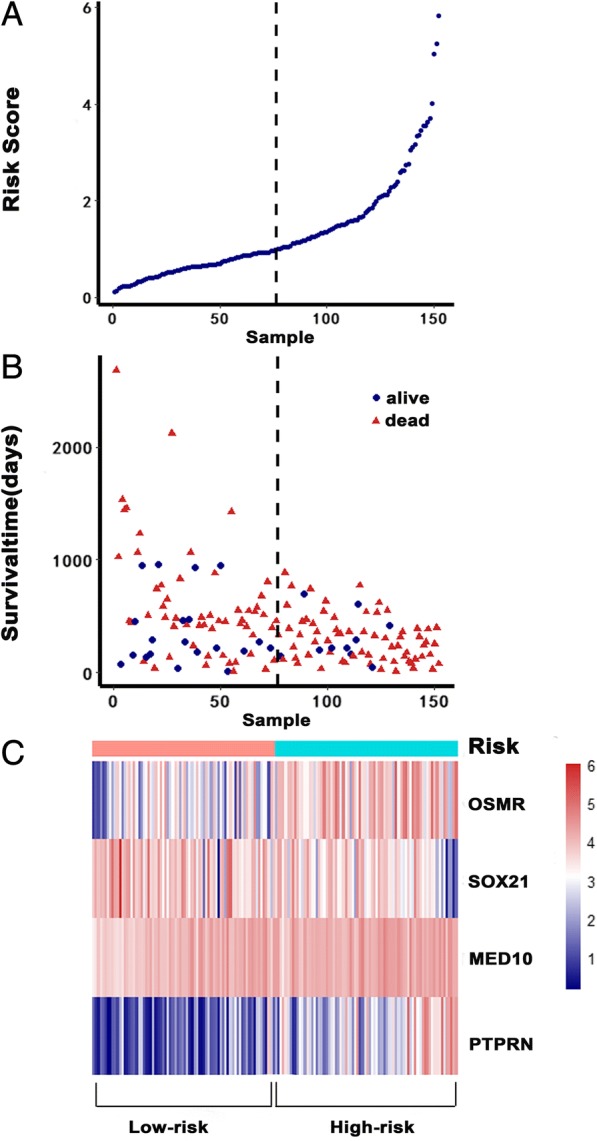
Cox proportional hazards regression model. a The risk score of the low-risk and high-risk groups. b Survival status and time of 152 glioblastomas. c Heatmap of the model genes
Fig. 5.

Kaplan-Meier curves and receiver operating characteristic (ROC). a Kaplan-Meier curves show that the high-risk group had greater mortality than does the low-risk group (P = 0). b Kaplan-Meier curves of the four different genes. c Time-dependent ROC curves indicated higher predictive value. The area under the ROC curve (AUC) was 0.905
Difference analysis of the four genes
To assess the expression level of the four genes between normal and glioblastoma tissues, we chose the GSE50161 datasets (normal brain tissue = 13, glioblastoma tissue = 34) to perform difference analysis. Interestingly, OSMR (P = 0.0011) and PTPRN (P < 0.0001) were differentially expressed (Fig. 6a, d), while MED10 (P = 0.5332) and SOX21 (P = 0.2831) were not (Fig. 6b, c). Subsequently, we assessed the mRNA expression levels of the four genes within each subtype (Classical, Mesenchymal and Proneural) [26]. The results showed that mRNA expression levels of the four genes in proneural subtype were significantly different from the other two subtypes (Fig. 7a). Meanwhile, the four genes had a better prognosis in proneural subtype (Fig. 7b).
Fig. 6.

Expression of the four genes between normal and glioblastoma tissue in GSE50161. a OSMR; b SOX21; c MED10; d PTPRN; **P < 0.01, ****P < 0.0001
Fig. 7.

Expression of the four genes in different molecular subtypes of glioblastoma. a mRNA expression levels of OSMR, SOX2, MED10 and PTPRN in three molecular subtypes of glioblastoma. Statistical significance was indicated in the figures, **P < 0.01, ***P < 0.001. b Survival curves for the four genes set in different subtypes
Discussion
Due to the diffusely infiltrative nature of glioblastoma, completely removing tumours is difficult, and these tumours also resist radiation therapy and chemotherapy. Thus, molecular studies, including various markers, are necessary to understand gliomagenesis and development. In addition, some molecular markers are important for determining molecular subtypes, identifying individualised treatments and judging clinical prognosis. For instance, overexpression or amplification of EGFR, mutations in IDH1 and IDH2 and phosphatase and tensin homologue (PTEN) mutations contribute to the pathogenesis of glioblastoma [27]. With the rapid development of high-throughput sequencing and bioinformatics methods, exploiting the great potential of RNASeq data requires new analytic approaches that move beyond gene difference analysis.
Instead of relating thousands of genes to a clinical trait, we used a recently developed methodology to construct a weighted gene co-expression network in 75 glioblastoma samples from TCGA, revealing survival time-specific modules (yellow, p < 0.01). As the most important gene in the module, the hub gene is the main feature of the gene module and closely related to the corresponding clinical information. Thus, we identified 228 intramodular hub genes based on GS and MS. The enrichment analysis of GO and KEGG showed that adhesion function and adhesion molecules accounted for the highest proportion of hub genes. These results can partly explain why glioblastoma tumours exhibit high invasiveness, and adhesion molecules can play an important role in gliomagenesis. By constructing the Cox proportional hazards regression model, we selected an optimal four-gene model (OSMR + SOX21 + MED10 + PTPRN) for prognosis prediction. Among the genes in this model, OSMR and SOX21 have been previously reported in glioblastoma studies [28–31]. OSMR encodes a member of the type I cytokine receptor family. OSMR forms a complex with EGFRvIII, the most common EGFR mutation that occurs in glioblastoma, and regulates glioblastoma tumour growth. Overexpression of OSMR and low methylation level was reported to have a poor survival time in GBM [28]. According to our research and previous reports [29], expression level of OSMR was higher in mesenchymal and classical subtypes than proneural subtype. SOX21, the counteracting partner of SOX2, functions as a tumour suppressor during gliomagenesis by negatively regulating SOX2 [30, 31]. Currently, MED10 is known only as a component of the coactivator for DNA-binding factors that activate transcription via RNA polymerase II. The protein encoded by PTPRN is a member of the protein tyrosine phosphatase family and may be involved in cancer initiation and progression [32]. However, MED10 and PTPRN have not been previously reported in glioblastoma-related studies. Each gene was confirmed to have independent prognostic significance. The difference analyses were performed in the GSE50161 datasets. Although MED10 (P = 0.5332) and SOX21 (P = 0.2831) exhibited no differential expression in glioblastoma and normal tissues, they may exhibit differential expression between glioblastoma and low-grade glioma. Thus, further studies are needed.
WGCNA used a statistical method to make the gene network consistent with the scale-free distribution; the resulting gene modules are more in line with biological phenomena and can be more finely divided. To date, there are a few similar studies on glioblastoma. Aoki K used the Cox proportional hazards regression model to investigate the effects of genetic alterations in 308 diffuse lower-grade gliomas (LGGs) and verified the results using the dataset from TCGA. The authors reported that IDH mutation, 1p19q deletion, Notch homologue 1 (NOTCH1) mutations and phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1) mutations were significantly associated with poor prognosis in LGGs [33]. However, glioblastomas were not examined. Horvath S adopted WGCNA to detect oncogenic modules and confirm abnormal spindle-like microcephaly-associated protein (ASPM) as a potential molecular target in glioblastoma [34]. Yu X used protein expression data of development process of macaque rhesus brain and RNA-seq data of GBM to identify several prognostic genes [35]. Similarly, Xiang Y applied WGCNA and K-means algorithm in gene expression data of GBM obtained from the TCGA database and found some prognosis sub-networks [36]. But, compared to the K-mean clustering method, WGCNA can construct a gene co-expression network to identify the hub genes associated with trait-related modules directly. Whether the two methods are used simultaneously was reasonable needed to further research. In addition, similar studies using WGCNA to predict prognostic molecules are rare. These results indicate that further analysis of this module may provide more clues to understand the occurrence and development of glioma. However, this study has some limitations. First, we did not validate the prognostic value of the four-gene model due to the lack of survival data in the GEO datasets. Thus, prediction of prognosis using the four-gene model needs further verification. Second, we selected only 5000 genes for analysis in WGCNA. These transcript changes can not represent all the genetic changes in glioblastomas. By increasing the number of genes in the study, we can find more molecular targets and key pathways. Third, these results were only detected using bioinformatics analysis and needed further experimental verification. Overall, our study provide a new perspective to identify the potential molecules and therapeutic targets for glioblastoma.
Conclusions
In conclusion, in this study we performed a WGCNA approach with GBM RNA-seq data from TCGA database to reveal a survival time-specific module. We also constructed a Cox proportional hazards regression model and identified four independent prognostic factors (OSMR, SOX21, MED10 and PTPRN). Although the specific mechanism remained to be studied, these genes could be considered as risk factors for GBM patients and novel therapeutics.
Additional files
Table S6. Specific data in Fig. 1d. (XLSX 10 kb)
Table S2. Biological process of hub genes. (CSV 60 kb)
Table S3. Cellular component of hub genes. (CSV 8 kb)
Table S4. Molecular function of hub genes. (CSV 5 kb)
Table S5. KEGG-enrichment of hub genes. (CSV 2 kb)
Table S1. Top ten prognostic genes identified from Cox regression analysis. (DOCX 21 kb)
Figure S1. Kaplan-Meier curves and receiver operating characteristic (ROC) in GSE16011 dataset. (TIF 2529 kb)
Table S7. Follow-up information of GSE16011 dataset. (XLSX 22 kb)
Acknowledgements
We gratefully acknowledge the TCGA project organizers as well as all study participants for making data and results available. Meanwhile, we would like to thank Sturm D and Donson AM.
Funding
The present study was supported by grants from the National Science Foundation of China (nos. 81572489).
Availability of data and materials
The RNA sequencing data of human glioblastoma samples were obtained from the TCGA data portal (https://portal.gdc.cancer.gov). GeneChip data were retrieved from the GEO data repository (http://www.ncbi.nlm.nih.gov/geo/) with the accession numbers GSE36245, GSE16011 and GSE50161. GlioVis database can be accessed with the http: //gliovis.bioinfo.cnio.es.
Abbreviations
- ATRX
X - linked alpha thalassemia mental retardation syndrome gene
- AUC
Area under the curve
- BP
Biological Process
- CC
Cellular Component
- EGFR
Epidermal growth factor receptor
- FPKM
Fragments per kilobase per million
- GBM
Glioblastoma multiforme
- GEO
Gene Expression Omnibus
- GO
Gene Ontology
- GS
Gene significance
- IDH1
Isocitrate dehydrogenase 1
- IDH2
Isocitrate dehydrogenase 2
- IGF-1
Insulin-like growth factor 1
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- MED10
Mediator complex subunit 10
- MF
Molecular Function
- MGMT
O6methylguanine DNA methyltransferase
- MS
Module significance
- OSMR
Oncostatin M receptor
- PDGFRA
Platelet-derived growth factor receptor alpha
- PTPRN
Protein tyrosine phosphatase, receptor type N
- ROC
Receiver operating characteristic curve
- SOX21
SRY (sex determining region Y)-box 21
- TCGA
The Cancer Genome Atlas
- TOM
Topological overlap measure
- VEGF
Vascular endothelial growth factor precursor
- WGCNA
Weighted Gene Co-expression Network Analysis
Authors’ contributions
P-FX designed the study, analysed the data, performed computational coding. J-AY, J-HL, XY, J-ML collected the data. P-FX, F-EY, Y X, B-HL, and Q-XC involved in drafting the manuscript and revising it critically for important intellectual content. All authors agreed to be accountable for all aspects of the work and approved the final manuscript.
Ethics approval and consent to participate
Ethical approval was waived since we used only publicly available data and materials in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Pengfei Xu, Email: 340241663@qq.com.
Jian Yang, Email: yjazwd@126.com.
Junhui Liu, Email: liusijia.zu@163.com.
Xue Yang, Email: snow2010666@163.com.
Jianming Liao, Email: ljm1992@whu.edu.cn.
Fanen Yuan, Email: 871460253@qq.com.
Yang Xu, Email: 920546330@qq.com.
Baohui Liu, Email: bliu666@163.com.
Qianxue Chen, Email: chenqx666@whu.edu.cn.
References
- 1.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 2.Alifieris C, Trafalis DT. Glioblastoma multiforme: pathogenesis and treatment. Pharmacol Ther. 2015;152:63–82. doi: 10.1016/j.pharmthera.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Network TC. Corrigendum: comprehensive genomic characterization defines human glioblastoma genes and core pathways. NATURE. 2013;494(7438):506. doi: 10.1038/nature11903. [DOI] [PubMed] [Google Scholar]
- 5.Westphal M, Lamszus K. Circulating biomarkers for gliomas. NAT REV NEUROL. 2015;11(10):556–566. doi: 10.1038/nrneurol.2015.171. [DOI] [PubMed] [Google Scholar]
- 6.Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncology. 2017;19(1):139–141. doi: 10.1093/neuonc/now247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 8.Griesinger AM, Birks DK, Donson AM, Amani V, Hoffman LM, Waziri A, Wang M, Handler MH, Foreman NK. Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol. 2013;191(9):4880–4888. doi: 10.4049/jimmunol.1301966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gravendeel LA, Kouwenhoven MC, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten LB, Kloosterhof NK, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69(23):9065–9072. doi: 10.1158/0008-5472.CAN-09-2307. [DOI] [PubMed] [Google Scholar]
- 10.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC BIOINFORMATICS. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li A, Horvath S. Network neighborhood analysis with the multi-node topological overlap measure. BIOINFORMATICS. 2007;23(2):222–231. doi: 10.1093/bioinformatics/btl581. [DOI] [PubMed] [Google Scholar]
- 12.Yip AM, Horvath S. Gene network interconnectedness and the generalized topological overlap measure. BMC BIOINFORMATICS. 2007;8:22. doi: 10.1186/1471-2105-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the dynamic tree cut package for R. BIOINFORMATICS. 2008;24(5):719–720. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 14.Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, Horvath S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm Genome. 2007;18(6–7):463–472. doi: 10.1007/s00335-007-9043-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, Castellanos R, Brozell A, Schadt EE, Drake TA, Lusis AJ, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2(8):e130. doi: 10.1371/journal.pgen.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7(1):e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu G, Wang LG, Yan GR, He QY. DOSE: an R/bioconductor package for disease ontology semantic and enrichment analysis. BIOINFORMATICS. 2015;31(4):608–609. doi: 10.1093/bioinformatics/btu684. [DOI] [PubMed] [Google Scholar]
- 20.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium NAT GENET. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu G, Wang LG, Han Y. He QY: clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang RX, Chen WJ, He RQ, Zeng JH, Liang L, Li SK, Ma J, Luo DZ, Chen G. Identification of a RNA-Seq based prognostic signature with five lncRNAs for lung squamous cell carcinoma. ONCOTARGET. 2017;8(31):50761–50773. doi: 10.18632/oncotarget.17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loraine AE, Blakley IC, Jagadeesan S, Harper J, Miller G, Firon N. Analysis and visualization of RNA-Seq expression data using RStudio, bioconductor, and integrated genome browser. Methods Mol Biol. 2015;1284:481–501. doi: 10.1007/978-1-4939-2444-8_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He D, Liu ZP, Chen L. Identification of dysfunctional modules and disease genes in congenital heart disease by a network-based approach. BMC Genomics. 2011;12:592. doi: 10.1186/1471-2164-12-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104(21):8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, DeCarvalho AC, Lyu S, Li P, Li Y, et al. Tumor evolution of Glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32(1):42–56. doi: 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garrett-Bakelman FE, Melnick AM. Differentiation therapy for IDH1/2 mutant malignancies. Cell Res. 2013;23(8):975–977. doi: 10.1038/cr.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang W, Zhao Z, Wu F, Wang H, Wang J, Lan Q, Zhao J. Bioinformatic analysis of gene expression and methylation regulation in glioblastoma. J Neuro-Oncol. 2018;136(3):495–503. doi: 10.1007/s11060-017-2688-1. [DOI] [PubMed] [Google Scholar]
- 29.Natesh K, Bhosale D, Desai A, Chandrika G, Pujari R, Jagtap J, Chugh A, Ranade D, Shastry P. Oncostatin-M differentially regulates mesenchymal and proneural signature genes in gliomas via STAT3 signaling. NEOPLASIA. 2015;17(2):225–237. doi: 10.1016/j.neo.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caglayan D, Lundin E, Kastemar M, Westermark B, Ferletta M. Sox21 inhibits glioma progression in vivo by forming complexes with Sox2 and stimulating aberrant differentiation. Int J Cancer. 2013;133(6):1345–1356. doi: 10.1002/ijc.28147. [DOI] [PubMed] [Google Scholar]
- 31.Ferletta M, Caglayan D, Mokvist L, Jiang Y, Kastemar M, Uhrbom L, Westermark B. Forced expression of Sox21 inhibits Sox2 and induces apoptosis in human glioma cells. Int J Cancer. 2011;129(1):45–60. doi: 10.1002/ijc.25647. [DOI] [PubMed] [Google Scholar]
- 32.Frankson R, Yu ZH, Bai Y, Li Q, Zhang RY, Zhang ZY. Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res. 2017;77(21):5701–5705. doi: 10.1158/0008-5472.CAN-17-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aoki K, Nakamura H, Suzuki H, Matsuo K, Kataoka K, Shimamura T, Motomura K, Ohka F, Shiina S, Yamamoto T, et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro-Oncology. 2018;20(1):66–77. doi: 10.1093/neuonc/nox132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A. 2006;103(46):17402–17407. doi: 10.1073/pnas.0608396103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu X, Feng L, Liu D, Zhang L, Wu B, Jiang W, Han Z, Cheng S. Quantitative proteomics reveals the novel co-expression signatures in early brain development for prognosis of glioblastoma multiforme. ONCOTARGET. 2016;7(12):14161–14171. doi: 10.18632/oncotarget.7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiang Y, Zhang CQ, Huang K: Predicting glioblastoma prognosis networks using weighted gene co-expression network analysis on TCGA data. BMC BIOINFORMATICS 2012, 13 Suppl 2: S12. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S6. Specific data in Fig. 1d. (XLSX 10 kb)
Table S2. Biological process of hub genes. (CSV 60 kb)
Table S3. Cellular component of hub genes. (CSV 8 kb)
Table S4. Molecular function of hub genes. (CSV 5 kb)
Table S5. KEGG-enrichment of hub genes. (CSV 2 kb)
Table S1. Top ten prognostic genes identified from Cox regression analysis. (DOCX 21 kb)
Figure S1. Kaplan-Meier curves and receiver operating characteristic (ROC) in GSE16011 dataset. (TIF 2529 kb)
Table S7. Follow-up information of GSE16011 dataset. (XLSX 22 kb)
Data Availability Statement
The RNA sequencing data of human glioblastoma samples were obtained from the TCGA data portal (https://portal.gdc.cancer.gov). GeneChip data were retrieved from the GEO data repository (http://www.ncbi.nlm.nih.gov/geo/) with the accession numbers GSE36245, GSE16011 and GSE50161. GlioVis database can be accessed with the http: //gliovis.bioinfo.cnio.es.