

Graphical Abstract
Keywords: Conformational change, Double electron-electron resonance, Double quantum coherence, Pulsed electron paramagnetic resonance, Maltose binding protein, Maximum entropy method, Methanethiosulfonate, Molten globule state, Molecular dynamics simulation, Site-directed mutagenesis, Tikhonov regularization
Abstract
An intensively investigated intermediate state of protein folding is the molten globule state (MG), which contains secondary but hardly any tertiary structure. In previous work we have determined the distances between interacting spins within maltose binding protein (MBP) in its native state using continuous wave and double electron-electron resonance (DEER) EPR spectroscopy. Seven double mutants had been employed to investigate the structure within the two domains of MBP. DEER data nicely corroborated the previously available X-ray data.
Even in its MG state, MBP is known to still bind its ligand maltose. We therefore hypothesized that there must be a defined structure around the binding pocket of MBP already in the absence of tertiary structure.
Here we have investigated the functional and structural difference between native and MG state in the open and closed form with a new set of MBP mutants. In these the spin label positions were placed near the active site. Binding of its ligands leads to a conformational change from open to closed state, where the two domains are more closely together.
The complete set of MBP mutants was analyzed at pH 3.2 (MG) and pH 7.4 (native state) using double quantum coherence (DQC) EPR. The values were compared with theoretical predictions of distances between the labels in biradicals constructed by molecular modeling from the crystal structures of MBP in open and closed form and were found to be in excellent agreement.
Measurements show a defined structure around the binding pocket of MBP in MG, which explains maltose binding. A new and important finding is that in both states ligand-free MBP can be found in open and closed form, while ligand-bound MBP only appears in closed form due to maltose binding.
INTRODUCTION:
The complex mechanisms of protein folding are still not well understood, although they play an important role in protein function and biological processes [1]. Most models of protein folding introduce so called “folding pathways”, where protein folding is a process of multidimensional routes and intermediate conformations strongly influenced by their environment instead of a single route [2].
The molten globule state (MG) is an intermediate in this process and is therefore extensively examined as a key in understanding protein folding [3]. It contains a native-like secondary structure but fluctuating tertiary structures [4]. MG states contain exposed hydrophobic pockets that allow for reaction with the dye 8-anilinonaphthalene-1-sulfonic acid (ANS). Circular dichroism, fluorescence spectroscopy and NMR have been employed to examine the MG [5].
Relative to the above methods, EPR together with site-directed spin labeling, yields complementary information about the distance between the spin labels at selected positions within the biological molecule [6, 7]. In combination with molecular dynamics (MD) simulations measured distances of spin labels can be related to specific protein models and conformations [8]. The reason to use DQC EPR is that in most cases a considerably better Tikhonov reconstruction of P(r) could be achieved (see in the Supplement) [6,9,10,11,12]. It works very well over the broad distances range, which includes the short distance range down to ~1 nm, yielding very good sensitivity [13, 14, 15,16]. Low-background dipolar traces in DQC simplify the reconstruction by typically used L-curve regularization method [11, 17]. Maltose binding protein (MBP) is a 370 amino acid, periplasmic protein of E. coli involved in chemotaxis for the uptake of a range of maltose sugars. MBP is a single-chain monomeric two-domain protein devoid of any cofactors [18]. Binding of its ligands leads to a conformational change from open (Figure 1) to closed state, where the two domains are more closely together. It has been employed extensively for monitoring folding dynamics and to determine the corresponding thermodynamic parameters. Unlike other large proteins, MBP undergoes reversible unfolding and the MG state can be stabilized at pH 3.2 [4]. ITC measurements show that even in its MG state MBP can still bind its ligand maltose [19, 20].
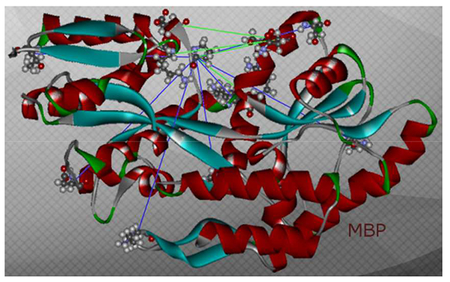
Figure 1:
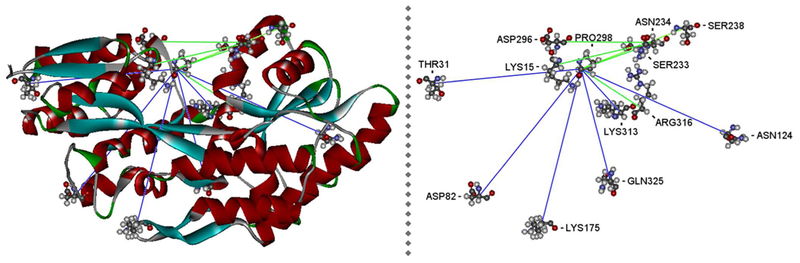
Overview of the complete set of MBP mutants in open state MBP (PDB: 1OMP). Blue lines indicate C(alpha)-amino-acid-distances of mutants MBP 01 – 06. Green lines indicate C(alpha)-amino-acid-distances of mutants MBP 07 – 11.
MATERIALS AND METHODS
Materials
All reagents were from commercial sources and of analytical grade. (1-Oxyl-2,2,5,5-tetramethyl-pyrroline-3-methyl)-methanethiosulfonate (MTS) was a kind gift from Kálmán Hideg (University of Pécs, Pécs, Hungary).
Mutants and Plasmids
The plasmids of the double mutants MBP 09 (P298C_R316C), 10 (N234C_D296C) and 11 (N234C_K015C) were constructed by site-directed mutagenesis employing standard techniques [21]. The mutants MBP 01 (P298C_T031C), 02 (P298C_D082C), 03 (P298C_N124C), 04 (P298C_K175C), 05 (P298C_K313C), 06 (P298C_Q325C) and 07 (P298C_S238C) are described in [22]. An overview of the complete set of mutants and their positions is given in Figure 1.
The complete MBP sequences of all mutants were determined by Sanger sequencing at Seq-it, Kaiserslautern, Germany
Protein purification, spin labeling and sample preparation
Wild type (WT) (UniProtKB - P0AEX9) and mutant proteins were expressed in the E. coli strains DH5α [4]. Cells were grown in TB medium containing 100 μg/ml ampicillin at 37 °C. The culture was induced with 2 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at an OD600 of 2. After 12 hours of incubation, cells were pelleted at 6,000 rpm, 4 °C, and washed by resuspending in 20 mM Tris-NaC1 buffer, pH 7.4. Cell lysis was achieved through processing in a French press. The lysate was subjected to centrifugation at 13,000 rpm at 4 °C before extracting the proteins by affinity chromatography on an amylose column at 4 °C [23].
To remove maltose, MBP was dialyzed at room temperature in Tris-NaCl buffer containing 6 M urea to completely release maltose from MBP. MBP was slowly refolded by drop dilution in Tris-NaCl buffer and concentrated on an Q-sepharose column. Protein concentration was estimated by the method of Lowry with bicinchoninic acid (BCA) and purity of the proteins was assayed by SDS-PAGE and MALDI-TOF-MS. The removal of maltose was proven by a coupled maltose assay (Sigma Aldrich) where maltose is degraded to glucose (alpha-glucosidase), and further phosphorylated (hexokinase) and dehydrogenated (glucose-6-phosphate dehydrogenase) to 6-phosphogluconate [20].
Spin Labeling was performed as described in [22] with the exception that desalting and buffer exchange were achieved through dialysis (under dark conditions) instead of a PD-10 column and a 3-fold molar excess of MTS was used. The protein samples for MG measurements were slowly adjusted to pH 3.2 in dialyses (under dark conditions).
The EPR samples were prepared in CGH5 (citrate, glycine, and HEPES, 5 mM each) buffer with 50–100 μM protein, 20% (v/v) glycerol and 20 mM maltose if required.
All of the samples were shock frozen and stored at −80 °C until used.
EPR experiment
The DQC experiments were performed at ACERT, Cornell University, Ithaca, NY, in the laboratory of Jack Freed by Peter Borbat.
Pulse dipolar EPR spectroscopy (PDS) experiments on MBP samples were performed at 60 K and 17.3 GHz frequency using a home-built Ku-band PDS spectrometer at ACERT [24, 25]. All MBP samples were measured by DQC using the 6-pulse sequence π/2-tp- π -tp- π /2-td- π -td- π /2-(tm-tp)- π -(tm-tp)- echo for all conditions [10, 11]. In addition, DEER measurements were carried out on selected samples, which all had distances well in the “DEER range” for all four conditions used [26, 27]. DQC in most cases used π/2 and π pulses of 3 and 6 ns, respectively, and the sequence was applied at the center of the spectrum. DEER used 16 and 32 ns pulses for detection and 16 ns pump pulse. The setup for DEER was standard with the detection sequences applied at the low-field edge of the spectrum [28].
The examples of DQC and (some) DEER data are shown in the Supplement. The rest of the data that is not shown was of the same quality. The data were collected usually in 1–4 hours, but ~12 hours were used in several cases to yield high SNR. The time-domain DEER data were always in very good agreement with the respective DQC data and the distance distributions, P(r)’s, were thus close to that obtained from DQC, agreeing well, for example, in the bimodal characters and other details. This also indicated that these details were unlikely to originate from (usually weak) orientation effects.
The L-curve Tikhonov regularization followed by the refinement with the maximum-entropy method was used for distance reconstruction [17, 29]. Since the DQC data had only very small background the backgrounds were approximated accurately by linear function and subtracted out. The stability of distance reconstruction was high in DQC, and the results have shown only very minor contribution of spurious peaks, which is characteristically for DQC and is clear from the examples in the Supplement.
MD simulation
For the MD simulation of MBP the software Accelrys Discovery Studio 2.5.5 and the X-ray structures 1ANF and 1OMP from the rcsb protein database were used [30]. The structures were typed by CHARMm-forcefield and minimized. For all simulations the implicit water model Generalized Born with a simple Switching (Dielectric Constant: 1, Implicit Solvent Dielectric Constant: 80, Non-polar Surface Area: True, Salt Concentration: 0, Van-der-Waals-radii, Molecular Surface: True), Dynamic Integrator: Leapfrog Verlet, Non-bond List Radius: 14, Electrostatics: Spherical Cut-off and SHAKE constraint: False were used. For energy minimization a minimization cascade of Steepest Descent followed by Conjugated Gradient and Adopted Basis Newton-Raphson algorithm was used. For the simulation of 1OMP the ligand maltose was removed. Water molecules were removed from both structures.
All mutants were simulated through mutation of the pair of respective cysteines, addition of MTS fragments to the sulfide groups and re-typing by CHARMm-forcefield. To find an appropriate starting position for dynamic simulation the complete system was at first minimized with fixed constraints over the complete protein area including ligand without cysteines and MTS. The minimization was continued in harmonic restraints with force constant 8 over the same area, and again in harmonic restraints with force constant 4 over all hydrophob amino acids including the ligand without cysteines (Hydrophob-Cys). Then an explicit water environment was placed around the MTS binding site using Explicit Spherical Boundary with Harmonic Restraint in a sphere radius of 1.5 nm to obtain a realistic environment for the spin label [31]. For the dynamic simulation of the complete system harmonic restraints with force constant 0.2 for all water molecules and the restraints Hydrophob-Cys were used. The MTS distance area was determined in the 40 ps production step of a dynamic cascade simulation (Minimization, Heating & Cooling, Equilibration, Production).
RESULTS & DISCUSSION
Open and closed form can be detected for ligand-free MBP
The EPR measurements show in most cases more rotamers of MBP and a different proportion of distances in the ligand-free MBP than in the MBP samples with maltose excess.
The MTS distances in open and closed state MBP are calculated through MD simulation and fit very well in comparison to DQC data. Due to this comparison these rotamers can be assigned to open and closed state MBP. The loss of signal for the rotamers assigned to open state MBP in maltose-bound DQC measurements confirm these assignments (Figure 3).
Figure 3:
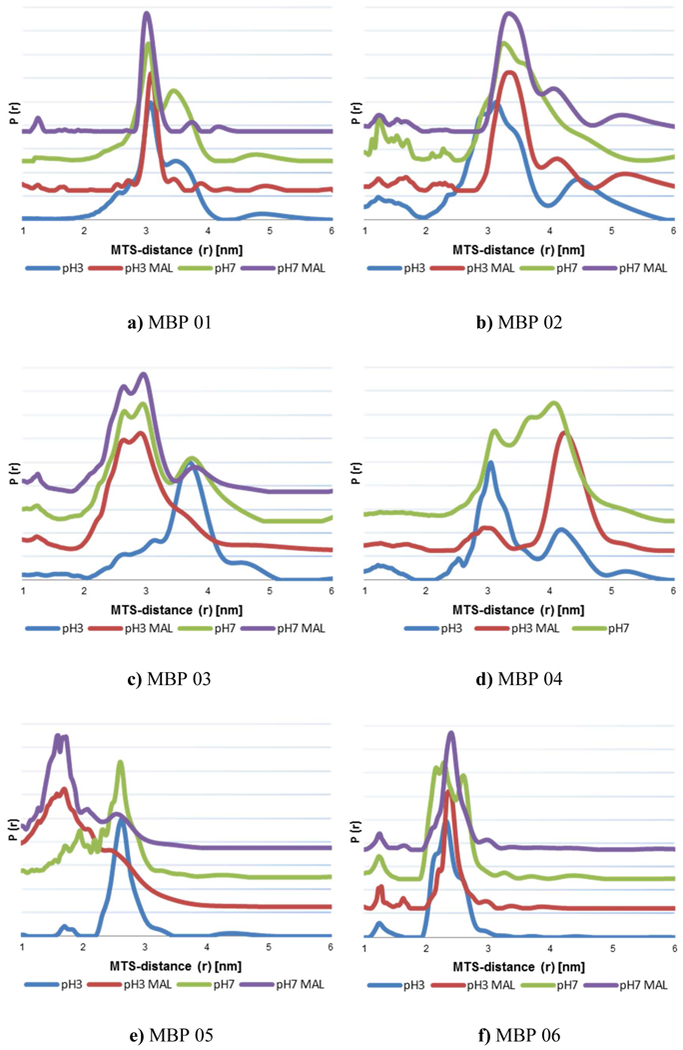
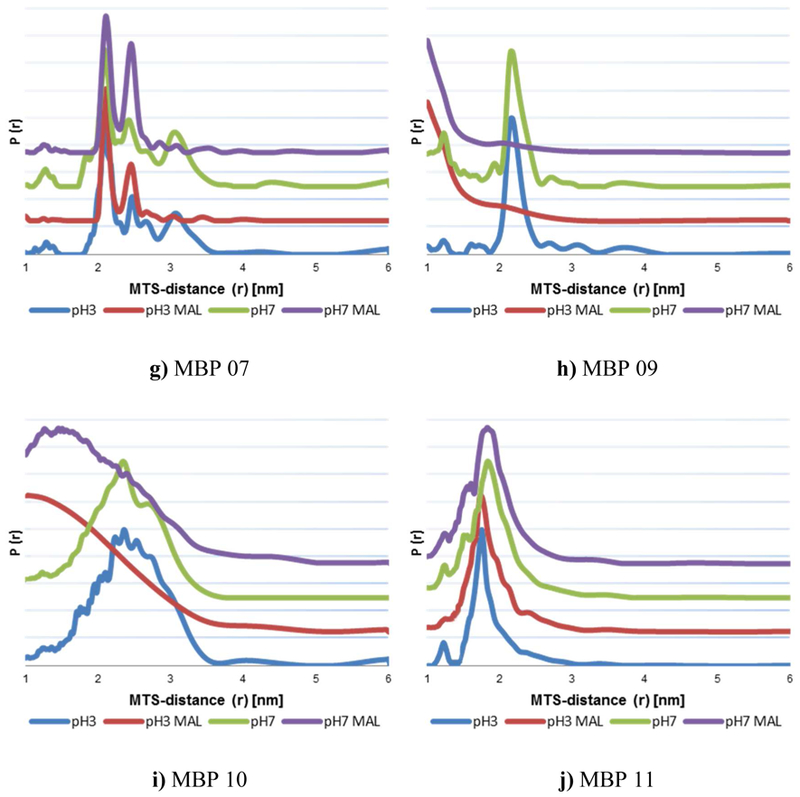
Distance distribution of MTS distances in mutants MBP 01 – 11 in native (pH 7) and MG (pH 3.2) state with and without maltose. More rotamers can be seen in measurements without maltose (blue & green) indicating open state distances.
As an example the distance distribution of MBP 07 pH 7 without maltose is shown in Figure 2 and the rotamers of closed and open state MBP are marked. The measurement of the same sample with maltose only shows the MTS distances of the rotamers of closed state MBP in native and MG, as shown in Figure 3g and Supplement.
Figure 2:
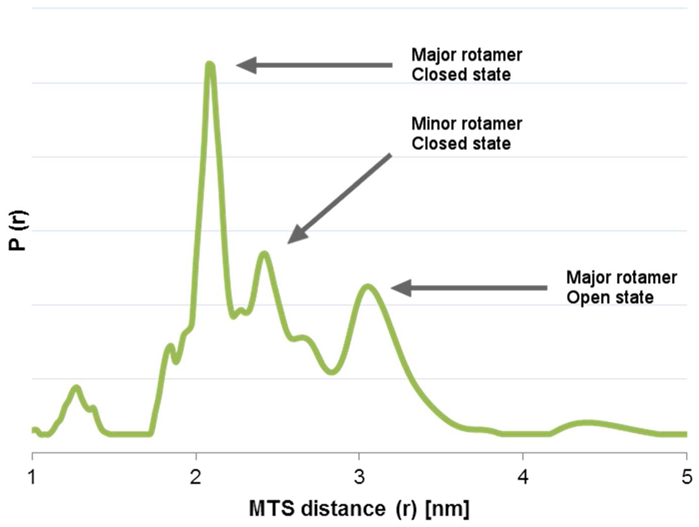
Distance distribution of MTS distances for MBP 07 (P298C S238C) pH 7 without maltose. The rotamers of closed and open state MBP are marked.
The comparison between simulated distances and DQC data for open and closed state MBP for all mutants is shown in Table 1. DQC distance distributions can be seen in Figure 3.
Table 1:
Comparison between simulated spin label distances and DQC data for open and closed state MBP. (Specifically, the distance between the nitroxide groups of MTS The simulated distances fit very well for measured DQC data).
| Mutant | State | Simulated MTS distance [nm] | DQC MTS distance [nm] | |
|---|---|---|---|---|
| (area) | Peak | rmsd | ||
| MBP01 | Open | 3.5–3.6 | 3.6 | 0.3 |
| Closed | 3.1–3.2 | 3 | 0.2 | |
| MBP02 | Open | 3.3–3.5 | 3.3 & 3.5 | 0.3 & 0.3 |
| Closed | 3.4–3.6 | 3.4 | 0.2 | |
| MBP03 | Open | 3.5–3.6 | 3.7 | 0.3 |
| Closed | 2.6–2.8 | 2.6 & 3.0 | 0.2 & 0.2 | |
| MBP04 | Open | 3.0–3.1 | 3.1 | 0.2 |
| Closed | 4.1–4.3 | 3.6 & 4.0 | 0.2 & 0.2 | |
| MBP05 | Open | 2.5–2.6 | 2.6 | 0.2 |
| Closed | 1.7–1.8 | 1.6 & 1.7 | 0.1 &0.1 | |
| MBP06 | Open | 2.6–2.7 | 2.3 & 2.6 | 0.2 & 0.1 |
| Closed | 2.3–2.4 | 2.4 | 0.1 | |
| MBP07 | Open | 2.6–2.8 | 3 | 0.2 |
| Closed | 2.0–2.2 | 2.1 & 2.4 | 0.1 &0.1 | |
| MBP09 | Open | 1.9–2.1 | 2.2 | 0.1 |
| Closed | 0.9–1.1 | X | X | |
| MBP 10 | Open | 2.2–2.4 | 2.3 | 0.5 |
| Closed | 0.5–0.7 | X | X | |
| MBP 11 | Open | 1.1 – 1.3 | 1.2 | 0.1 |
| Closed | 1.7–1.8 | 1.8 | 0.3 | |
For the mutants MBP 09 and MBP 10 the closed state could not be measured due to the distance smaller than 1 nm. In case of MBP 04 pH7 with maltose no DQC distance could be obtained and due to the already high agreement of spectra between maltose-bound native and MG structure the measurement was not repeated.
Table 1 shows high agreement of MD simulated data and DQC measurements for all mutants. Mutants MBP 01 – 07 represent distances covering a broad range of the structure of the complete protein as described in [22]. Mutants 09 – 11 provide further insight in the structure near the active binding site. In combination, a good insight about the overall protein conformation is obtained.
The supplement provides typical examples and their detailed interpretation of the DQC spectra for mutants 01 (Figure S5), 07 (Figure S6) and 09 (Figure S7).
In most cases the distances in open state MBP are larger than in closed state, but as shown in simulated data and accompanying loss of signal at maltose excess measurements, mutants MBP 04 and MBP 02 are an exception. In those the MTS distance in closed state is larger than in open state which can be seen in the MD simulation (Table 1) and explained by the cysteine position and adjustment of spin label. The measurements of MBP 02 show only a small difference, while MBP 04 presents a large difference about 1 nm in the MTS distance between both states.
For ligand-bound mutant MBP 02 the major closed state rotamer can be found at 3.4 nm in addition to a minor rotamer at 4.1 nm, which is not seen in the absence of maltose, when the open form dominates. As open and closed forms overlap at 3.3 and 3.6 nm, individual rotamers are not clearly visible.
Mutant MBP 03 exhibits a higher proportion of rotamers in the open state, both at pH 7 and pH 3, as compared to spectra in the presence of maltose. Mutant MBP 05 shows a relatively broad distribution of rotamers in the closed state indicating a higher mobility of the labels as well as of the protein backbone in this region. This shows especially well in the broader distribution of pH 3 Mal as compared to pH 7 Mal. The difference between open and closed forms of MBP is best seen in Fig. 3e.
Hence, the measurements allow for detection of the open and closed conformation of MBP. Even without ligand MBP is found in the open and in the closed conformation.
Even in the MG state the structure of MBP resembles the structure of the native state
As known by ANS binding and CD measurements MBP is in a stabilized molten globule state at pH 3.2 [19]. We verified that all MBP double mutants also show the loss of tertiary structure by ANS binding measurements at pH 3.2 as shown for example in [22].
DQC measurements between native and MG state only show a small broadening of peaks in case of pH 3.2 as can be seen in Figure 3. All rotamers of open and closed state MBP can be found in native and MG measurements. So the simulated MTS distances in Table 1 can be applied to determine open and closed state of MBP in the MG state. Earlier findings of DEER measurements for MBP 03 at pH3.3 published in [22] pointing to individual helices pointing in all possible directions for MBP in MG can now be newly interpreted. Those earlier data showed two broad peaks at 2.67 nm and 3.85 nm which can be now connected to open and closed state MBP through loss of maltose through sample preparation and pH adjustment, exactly explaining the newly formed peak in the pH 3.3 measurement.
In the measurements of label positions further away from the maltose binding pocket (MBP 01 – 06) a stronger tendency to the open state in case of ligand-free MBP can be seen for MG state. In the presence of maltose excess, only the closed state of MBP can be detected even in the MG state. In the vicinity of the active site (MBP 07 – 11) the structures hardly differ between the MG and native state as shown by EPR spectroscopy. These corroborate previous findings that MBP can still bind maltose in the MG state and confirm the assumption that a defined structure of MBP exists near the binding pocket even in the MG state [4, 33].
CONCLUSION
In combination with MD simulation the analysis of MTS-distances in Mutants MBP 01 – 11 reveal the changes between open and closed state and ligand binding. Even in absence of its ligand MBP can be found in open and closed state. This corroborates previous findings of Reif and co-workers that MalF interacts with MBP independently of maltose [20]. Through DQC-measurements it was proven that MBP keeps a native-like structure, especially around the binding pocket, in the molten globule state, thus explaining the ability of MBP to bind its ligands in MG. Open and closed forms of MBP exist in the absence of maltose, both in the native as well as in the MG form.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported in part by NIH grant P41GM103521.
ABBREVIATIONS
- ANS
8-anilinonaphthalene-1-sulfonic acid
- CW
continuous wave
- DEER
double electron-electron resonance
- DQC
double quantum coherence
- EPR
electron paramagnetic resonance
- ITC
isothermal titration calorimetry
- MBP
maltose binding protein
- MD
molecular dynamic
- MG
molten globule state
- MTS
(1-Oxyl-2,2,5,5-tetramethyl-pyrroline-3-methyl)-methanethiosulfonate
Footnotes
Publisher's Disclaimer: “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
SUPPORTING INFORMATION
1. An illustration to PDS data, mostly DQC and some DEER used in this work (Figures S1–S3)
2. Example DQC data and according distance distributions (Figures S4–S7)
REFERENCES
- 1.Alberts B, Johnson A, Lewis J, Morgan D, Raff M, Roberts K, Walter P (2002) Molecular Biology of the Cell. 4th Edition, Garland Science, New York [Google Scholar]
- 2.Wolynes P, Onuchic J, and Thirumalai D (1995) Navigating the folding routes, Science 267, 1619–1620 [DOI] [PubMed] [Google Scholar]
- 3.Kuwajima K (1989) The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure, Proteins 6(2):87–103 [DOI] [PubMed] [Google Scholar]
- 4.Prajapati RS, Indu S, Varadarajan R (2007) Identification and Thermodynamic Characterization of Molten Globule States of Periplasmic Binding Proteins, Biochemistry, 46, 10339–10352 [DOI] [PubMed] [Google Scholar]
- 5.Ohgushi M, Wada A (1983) ‘Molten-globule state’: a compact form of globular proteins with mobile side-chains, FEBS Lett, 164 (1): 21– 24 [DOI] [PubMed] [Google Scholar]
- 6.Borbat PP, Freed JH (1999) Multiple-quantum ESR and distance measurements, Chem. Phys. Letters 313, 145–154, Elsevier [Google Scholar]
- 7.Jeschke G, Polyhach Y (2007) Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance, Phys. Chem. Chem. Phys, 9, 1895–1910 [DOI] [PubMed] [Google Scholar]
- 8.Steinhoff HJ, Radzwill N, Thevis W, Lenz V, Brandenburg D, Antson A, Dodson G, Wollmer A (1997) Determination of Interspin Distances between Spin-Labels Attached to Insulin: Comparison of EPR Data with the X-ray analysis Structure, Biophysical Journal 73, 3287–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borbat PP, Costa-Filho AJ, Earle KA, Moscicki J, Freed JH. (2001) Electron spin resonance in studies of membranes and proteins, Science (New York, N.Y.) 291(5502): 266–269 [DOI] [PubMed] [Google Scholar]
- 10.Borbat PP, McHaourab HS, Freed JH (2002) Protein structure determination using long-distance constraints from double-quantum coherence ESR: Study of T4 Lysozyme, Biophysical Journal 82(1): 360A-360A [DOI] [PubMed] [Google Scholar]
- 11.Borbat PP, Freed JH (2017) Dipolar Spectroscopy - Single-resonance Methods, Emagres 6(4): 465–493 [Google Scholar]
- 12.Borbat PP and Freed JH (2013) Pulse Dipolar Electron Spin Resonance: Distance Measurements, Springer-Verlag Berlin Heidelberg, DOI: 10.1007/430_2012_82 [Google Scholar]
- 13.Gaffney BJ, Bradshaw MD, Frausto S., Wu F, Freed JH, Borbat PP (2012) Locating a Lipid at the Portal to the Lipoxygenase Active Site, Biophysical Journal 103(10): 2134–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merz GE, Borbat PP, Pratt AJ, Getzoff ED, Freed JH, Crane BR (2014) Copper-Based Pulsed Dipolar ESR Spectroscopy as a Probe of Protein Conformation Linked to Disease States, Biophysical Journal 107(7): 1669–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fafarman AT, Borbat PP, Freed JH, Kirshenbaum K (2007) Characterizing the structure and dynamics of folded oligomers: Pulsed ESR studies of peptoid helices, Chem. Commun(4): 377–379 [DOI] [PubMed] [Google Scholar]
- 16.Orlando BJ, Borbat PP, Georgieva ER, Freed JH, Malkowski MG (2015) Pulsed Dipolar Spectroscopy Reveals That Tyrosyl Radicals Are Generated in Both Monomers of the Cyclooxygenase-2 Dimer, Biochemistry 54(50): 7309–7312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiang YW, Borbat PP, Freed JH (2005) The determination of pair distance distributions by pulsed ESR using Tikhonov regularization, JMR 172(2): 279–295 [DOI] [PubMed] [Google Scholar]
- 18.Spurlino JC, Quiocho FA, Lu GY (1991) The 2.3-Å resolution structure of the maltose- or maltodextrin- binding protein, a primary receptor of bacterial active transport and chemotaxis, J. Biol. Chem 266(8):5202–5219 [DOI] [PubMed] [Google Scholar]
- 19.Sheshadri S, Lingaraju GM, Varadarajan R (1999) Denaturant mediated unfolding of both native and molten globule states of maltose binding protein are accompanied by large ∆Cp’s, Protein Science, 8, 1689–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nickolaus C (2017) The Molten Globule State of Maltose-Binding Protein: Structural Characterization by Electron Paramagnetic Resonance Spectroscopy, TU Kaiserslautern, Dissertation [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stratagene (2004) QuikChange® II XL Site-Directed Mutagenesis Kit - Instruction Manual, Revision #064001c, 2004
- 22.Reichenwallner J, Chakour M, Indu S, Varadarajan R, Trommer WE (2013) Maltose Binding Protein Is Partially Structured in Its Molten Globule State, Appl. Magn. Res Volume 44, Issue 8, pp 981–995 [Google Scholar]
- 23.New England Biolabs Inc. (2007) pMAL™ Protein Fusion and Purification System, Instruction Manual, manualE8200 [Google Scholar]
- 24.Borbat PP, Crepeau RH, Freed JH (1997) Multifrequency two-dimensional Fourier transform ESR: an X/Ku-band spectrometer, JMR 127(2): 155–167 [DOI] [PubMed] [Google Scholar]
- 25.Borbat PP, Georgieva ER, Freed JH (2013) Improved Sensitivity for Long-Distance Measurements in Biomolecules: Five-Pulse Double Electron-Electron Resonance, J Phys Chem Lett 4(1): 170–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banham JE, Baker CM, Ceola S, Day IJ, Grant GH, Groenen EJJ, Rodgers CT, Jeschke G, Timmel CR (2008) Distance measurements in the borderline region of applicability of CW EPR and DEER: A model study on a homologous series of spin-labelled peptides, JMR 191(2): 202–218 [DOI] [PubMed] [Google Scholar]
- 27.Borbat PP, Freed JH (2007) Measuring distances by pulsed dipolar ESR spectroscopy: Spin-labeled histidine kinases Two-Component Signaling Systems, Pt Simon BMI, Crane BR and Crane A. San Diego, Elsevier Academic Press Inc. 423: 52–116 [DOI] [PubMed] [Google Scholar]
- 28.Georgieva ER, Borbat PP, Norman HD, Freed JH (2015) Mechanism of influenza A M2 transmembrane domain assembly in lipid membranes, Sci Rep 5: 11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiang YW, Borbat PP, Freed JH (2005) Maximum entropy: A complement to Tikhonov regularization for determination of pair distance distributions by pulsed ESR, JMR 177(2): 184–196 [DOI] [PubMed] [Google Scholar]
- 30.Quiocho FA, Spurlino JC, Rodseth LE (1997) Extensive features of tight oligosaccharide binding revealed in high-resolution structures of the maltodextrin transport/chemosensory receptor, Structure, Aug 15;5(8):997–1015 [DOI] [PubMed] [Google Scholar]
- 31.White RP, Meirovitch H (2006) Minimalist explicit solvation models for surface loops in proteins, J. Chem. Theory Comput, 2(4): 1135–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacso T, Grote M, Daus ML, Schmieder P, Keller S, Schneider E, Reif B (2009) Periplasmic Loop P2 of the MalF Subunit of the Maltose ATP Binding Cassette Transporter Is Sufficient To Bind the Maltose Binding Protein MalE, Biochemistry, 48, 2216–2225 [DOI] [PubMed] [Google Scholar]
- 33.Selmke B, Chen N, Borbat P, Freed JH, and Trommer WE (2017) The molten globule state of maltose binding protein: structural characterization by EPR spectroscopy, Biophys. J, 112, 485A–486A [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.