

Abstract
The mammalian cell nucleus contains numerous discrete suborganelles named nuclear bodies. While recruitment of specific genomic regions into these large ribonucleoprotein (RNP) complexes critically contributes to higher-order functional chromatin organization, such regions remain ill-defined. We have developed the high-salt–recovered sequences-sequencing (HRS-seq) method, a straightforward genome-wide approach whereby we isolated and sequenced genomic regions associated with large high-salt insoluble RNP complexes. By using mouse embryonic stem cells (ESCs), we showed that these regions essentially correspond to the most highly expressed genes, and to cis-regulatory sequences like super-enhancers, that belong to the active A chromosomal compartment. They include both cell-type–specific genes, such as pluripotency genes in ESCs, and housekeeping genes associated with nuclear bodies, such as histone and snRNA genes that are central components of Histone Locus Bodies and Cajal bodies. We conclude that HRSs are associated with the active chromosomal compartment and with large RNP complexes including nuclear bodies. Association of such chromosomal regions with nuclear bodies is in agreement with the recently proposed phase separation model for transcription control and might thus play a central role in organizing the active chromosomal compartment in mammals.
The interphasic nucleus of mammalian cells is a highly compartmentalized organelle. Chromosome conformation capture (3C)–derived technologies (Lieberman-Aiden et al. 2009) and molecular imaging methods (Wang et al. 2016b) have revealed several layers of chromosome organization. At the megabase scale, chromosomes are segregated into active (A) and inactive (B) compartments (median size ∼3 Mb), while at the sub-megabase scale, they are partitioned into discrete “topologically associating domains” (TADs; median size ∼880 kb) (Dixon et al. 2012; Nora et al. 2012). However, the molecular determinants and organization principles that control these two layers of organization remain enigmatic. In contrast to TADs, chromosomal compartments are cell type specific, even if only a subset of genes is affected by A/B compartment changes during cell differentiation (Dixon et al. 2012; Bonev et al. 2017). While cohesin and the CCCTC-binding factor (CTCF) are required for TAD organization, the A/B chromosomal compartments remain intact upon depletion of these factors, indicating that compartmentalization of mammalian chromosomes emerges independently of proper insulation of TADs (Nora et al. 2017; Schwarzer et al. 2017). It has been proposed that genome partitioning into chromosomal compartments may arise from contacts with specific nuclear bodies or other important architectural components of the nucleus, such as the nucleolus and the nuclear lamina for the B compartment or transcription factories for the A compartment (Gibcus and Dekker 2013; Ea et al. 2015a).
Nuclear bodies are composed of large ribonucleoprotein (RNP) complexes self-assembled onto specific chromatin regions, and recruitment of some genomic loci into nuclear bodies is known to be crucial for proper gene expression (Mao et al. 2011). One emblematic example is the U7 snRNA gene that is recruited, together with histone genes (for which it is maturating the pre-mRNAs) into “Histone Locus Bodies” (Frey and Matera 1995; Nizami et al. 2010). Impairment of nuclear body assembly has been evidenced in several pathologies, including spinal muscular atrophy (Sleeman and Trinkle-Mulcahy 2014). Despite their importance for nuclear functions, the genomic sequences associated with nuclear bodies remain largely unknown. Indeed, genomic profiling of such sequences is challenging because purification of nuclear bodies is laborious and complex.
Results
The HRS-seq method
We previously showed that high-salt treatment of nuclei preparations allows the mapping of active regulatory elements at mammalian imprinted genes (Weber et al. 2003; Braem et al. 2008; Court et al. 2011). More recently, extensive proteomic analyses have shown that high-salt treatments enable the recovery of known protein components of nuclear bodies, such as the nucleolus, the Cajal bodies, or the nuclear lamina (Engelke et al. 2014). We adopted this approach to develop a high-throughput method aiming at profiling nuclear body–associated genomic sequences. The method, which avoids formaldehyde crosslinking used in many currently available techniques (Dobson et al. 2017), involves three experimental steps.
First, the high-salt–recovered sequences (HRS) assay makes large RNP complexes, including nuclear bodies, insoluble through high-salt treatments in order to trap, purify, and sequence the genomic DNA associated with them (Fig. 1A). A detailed protocol is given in the Supplemental Methods. Briefly, a suspension containing 105 purified nuclei is placed onto an ultrafiltration unit and is treated with a 2 M NaCl buffer. Each nucleus forms a so-called “nuclear halo” composed of a dense core containing insoluble complexes with which parts of the genomic DNA remain tightly associated, surrounded by a pale margin of DNA loops corresponding to the rest of the genome (Fig. 1A). We digested nuclear halos with the StyI restriction enzyme (for enzyme choice, see Supplemental Methods; Supplemental Fig. S3C) and washed through the DNA loops (Loop fraction), leaving on the filter the insoluble complex-associated fraction containing the HRSs (HRS-containing fraction). Genomic DNA from each fraction is purified by proteinase K digestion, phenol/chloroform extraction, and ethanol precipitation.
Figure 1.
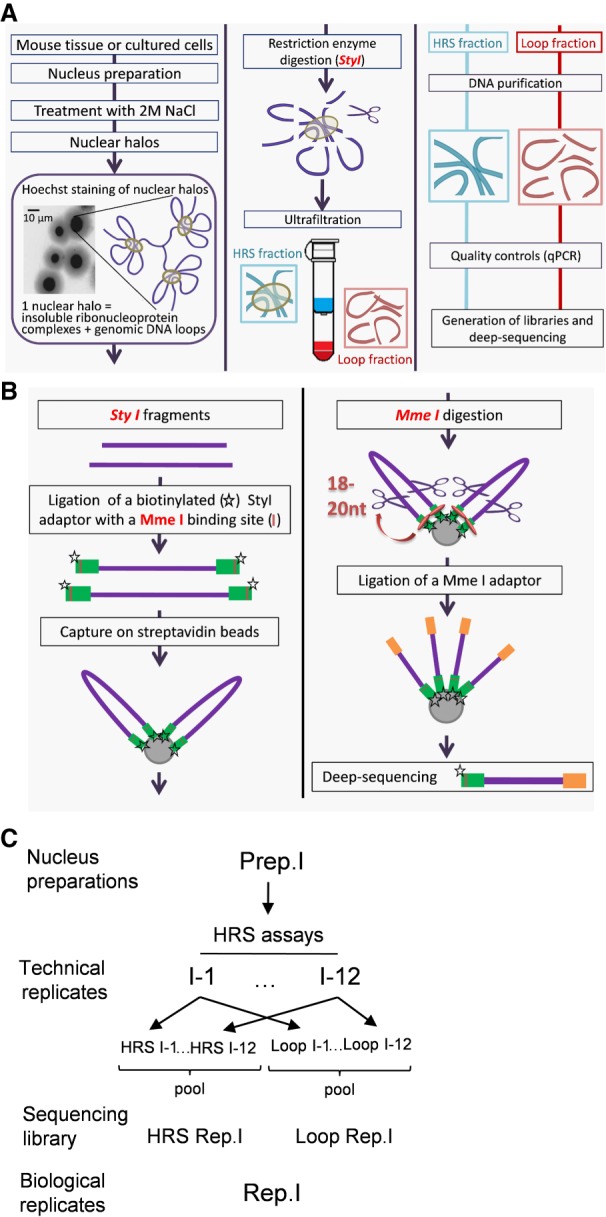
Flowchart of the HRS-seq method. The HRS-seq method consists of high-throughput sequencing of genomic DNA obtained from HRS assays. (A) HRS assay principle. Each HRS assay involves 105 nuclei that are treated with a 2 M NaCl buffer to obtain the so-called nuclear halos. Nuclear halos are digested with a restriction enzyme (here StyI), and the insolubilized fraction (HRS-containing fraction) is separated from the soluble Loop fraction by ultrafiltration. Genomic DNA is purified from each fraction, and controls are performed to ensure the quality of each assay. (B) Construction of HRS-seq libraries for deep-sequencing. For each HRS-seq experiment, two sequencing libraries are prepared: one from the HRS-containing fraction and one from the Loop fraction. A biotinylated StyI adaptor containing a MmeI binding site (green bars) is ligated to the StyI restriction fragments. Ligated fragments are captured on streptavidin beads and digested with MmeI to obtain StyI fragments having homogenous sizes (18 to 20 nt). The beads are washed several times, a MmeI adaptor (orange bars) is ligated, and these StyI/MmeI fragments are eluted from the beads. The StyI and MmeI adaptors are used for deep-sequencing. (C) Preparation of biological replicates. Each biological replicate (here Rep.1) is prepared from a different nuclei preparation (here Prep.1). A first sequencing library (here HRS Rep.1) is prepared from HRS fractions pooled from 12 HRS assays (technical replicates), and another one (Loop Rep.1) is prepared from Loop fractions pooled from the same 12 HRS assays. This procedure was applied to three distinct nuclei preparations to obtain six sequencing libraries representing three biological replicates.
Second, quality controls are performed in order to check the correct efficiency of each HRS assay. We used quantitative (q) PCR reactions targeting two positive controls corresponding to DNA sequences known to be constitutively enriched within the HRS-containing fraction in a wide range of experimental conditions (Weber et al. 2003; Court et al. 2011). The enrichment level of these controls (ratio of HRS to Loop fractions) was calculated for each HRS assay and normalized to the enrichment level of a negative control (Weber et al. 2003).
Third, the construction of DNA libraries for high-throughput sequencing is performed as detailed in the Methods section. Briefly, a first biotinylated StyI DNA adaptor, containing a binding site for the MmeI type IIS restriction enzyme, is ligated to both sides of the StyI fragments (Fig. 1B). Ligated products are captured onto streptavidin beads and digested with MmeI to homogenize the size of the StyI restriction fragments (∼18 to 20 nt). A second sequencing adaptor is ligated to MmeI-restricted sites, and DNA fragments are amplified on streptavidin beads using GEX PCR primers. The PCR reaction is purified on an acrylamide gel and used for high-throughput sequencing (50-nt single reads).
We applied our approach to the well-characterized e14Tg2a male mouse embryonic stem cells (ESCs) (Gaspard et al. 2008). We made three HRS-seq experiments, each performed on a distinct ESC nuclei preparation (biological replicates) (see Fig. 1C). In order to obtain enough material, for each of the three nuclei preparations, we selected 12 HRS assays displaying high enrichment levels of the positive controls (see Supplemental Fig. S1 and Methods). The HRS-containing fractions, on one side, or the Loop fractions, on the other side, of these 12 assays were then pooled, and 150 ng of genomic DNA from each pooled fractions was used for constructing sequencing libraries (see Methods). Therefore, for each of the three HRS-seq experiments, two sequencing libraries were prepared (HRS and Loop fractions). Each pair of libraries thus represents a biological replicate since it is made from only one of the three nuclei preparations (Fig. 1C).
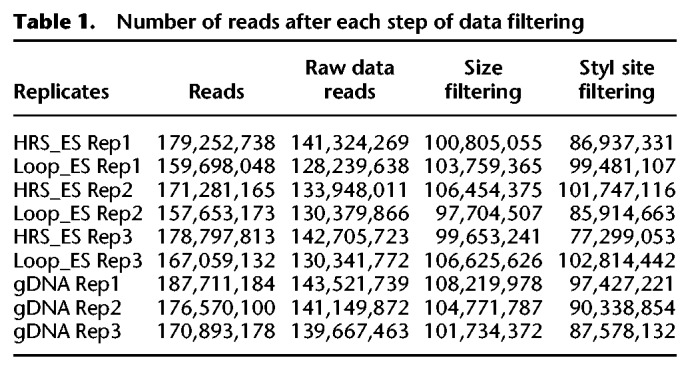
The reads obtained from each fraction in each replicate were mapped to the reference genome of e14Tg2a mouse ESCs (129P2 built from the mm9 assembly) (see Table 1 in Methods), and the number of reads mapping to each StyI fragment was counted. Among a total of 3,053,742 StyI fragments known in this reference mouse genome, 2,544,227 (83%) fragments were represented in the experiments performed on ESCs (509,515 StyI fragments were not sequenced and/or their corresponding reads did not map to a unique position on the mouse genome). Read counts of StyI fragments in both the Loop and the HRS-containing fractions were highly reproducible between biological replicates (R > 0.90) (Supplemental Fig. S2A,B; Supplemental Table S1) as well as in control libraries (gDNA control) constructed from StyI-digested genomic DNA (R > 0.90) (Supplemental Fig. S2D). In contrast, a poor correlation (R = 0.50) was found between read counts obtained from the HRS-containing and Loop fractions of each replicate, indicating that many StyI fragments were efficiently segregated into one of the two fractions (Supplemental Fig. S2C,D; Supplemental Table S1). By using the edgeR and DESeq R packages (Anders and Huber 2010; Robinson et al. 2010), we determined, for each informative StyI fragment, the significance of the overrepresentation of read counts in the HRS-containing fraction compared with the Loop fraction (see Supplemental Methods). The same approach was used to determine the overrepresentation of reads counts in the HRS-containing fraction compared with the gDNA control. As a result, 61,080 genomic regions overrepresented in the HRS-containing fraction relative to the gDNA control and/or to the Loop fraction have been identified in ESCs (Benjamini-Hochberg–corrected P-value <0.05) (Supplemental Table S2). They were termed HRSs. This ESC HRS set was used for subsequent bioinformatic analyses.
Table 1.
Number of reads after each step of data filtering
HRSs display chromosomal clustering
We first looked at the size distribution (Supplemental Fig. S3A) and nucleotide composition (Supplemental Fig. S3B) of the 61,080 ESC HRSs. We found that they are barely different from those obtained from 100 sets of 61,080 StyI fragments randomly selected in the mouse genome. We conclude that HRSs have sizes similar to those of regular StyI fragments and that their nucleotide composition is not globally biased toward A/T- or G/C-rich sequences, even if one can note that a small subset of HRSs is overrepresented in the range of 59%–73% of G/C (P-value <0.01) (Supplemental Fig. S3B). Globally, the G/C content of HRSs is distributed ∼43%, a value similar to the mean G/C content of the mouse genome that corresponds to the value expected for sequences located around regular StyI sites (Supplemental Fig. S3C). To demonstrate that the distribution of StyI sites in the mouse genome does not introduce biases for HRS identification, we performed a correlation study between StyI site density versus HRS density (i.e., the density of StyI sites associated with HRSs) in 100-kb bins. This analysis showed that HRS density does not correlate with the density of StyI sites in these bins (Spearman's correlation coefficient R = 0.129) (Supplemental Fig. S3D). Consistently, StyI density of HRS-containing bins is distributed around the mean StyI density in the mouse genome (117.33 StyI/100 kb) (vertical red line in Supplemental Fig. S3D). Overall, this demonstrates that HRSs are not specially found in bins with either high or low StyI density.
We then looked at the distribution of the 61,080 ESC HRSs along mouse chromosomes and found that they are spread over all chromosomes (Fig. 2A) with a mean genome-wide density of 23.47 HRSs per megabase. However, the mean density of HRSs was higher on Chromosomes 7, 11, 17, and 19 and lower on Chromosomes 12, 14, and 18 (Fig. 2A). Furthermore, HRSs seem to be not uniformly distributed along the chromosomes, but they appeared to cluster at specific loci. To demonstrate HRS clustering, we calculated the median distance between two consecutive HRSs (6 kb) and showed that it is much lower than the median distance obtained from 61,080 StyI fragments randomly selected in the mouse genome (32 kb) (Fig. 2B). This analysis demonstrated that HRSs are highly clustered in the genome of mouse ESCs.
Figure 2.

Chromosomal mapping of HRSs identified in mouse ESCs. (A) HRSs identified by HRS-seq performed in mouse ESCs have been mapped (brown bars) to mouse chromosomes. The mean densities of HRSs on each chromosome (HRS/Mb) are indicated on the figure. (B) The distance between consecutive HRSs (d) was determined. The graph shows the genome-wide distribution (1-kb bins) of non-null values for d corresponding to HRS (blue) and random (brown) StyI fragments. The median values of d for each distribution are indicated on the figure. The difference between the two distributions is highly significant, featuring a P-value lower than 10−100 (Wilcoxon rank-sum test). (C) The mean inter-chromosomal contact scores of 100-kb bins enriched in HRSs (red dots) were calculated from Hi-C data available for the same cell type (ESC) (Dixon et al. 2012) and compared to the mean contact scores obtained from 100 random sets of the same number of 100-kb bins (box plots). The box plot on the right represents the mean contact score and randomizations obtained when HRSs and random StyI fragments are taken only in the A compartment, while the box plot on the left represents the mean contact score and randomizations obtained from the whole genome. Bars, minimum and maximum values obtained in the 100 randomizations. The number of 100-kb bins (n) used for each randomizations is indicated on the figure. The P-value indicates the significance of the difference between the mean contact scores obtained for HRSs versus randomizations.
HRSs are associated with the active A chromosomal compartment
We then assessed whether HRSs also cluster in the three-dimensional (3D) space of the nucleus. By using published Hi-C data obtained from mouse ESCs (Dixon et al. 2012), we took the 100-kb bins most highly enriched in HRSs (hereafter called HRS bins) and calculated the mean score of inter-chromosomal contact frequency for all possible pairs of HRS bins in these cells (see Supplemental Methods). The score obtained was found to be significantly higher (P < 10−2) than the scores obtained from 100 sets of an equal number of 100-kb bins taken at random in the mouse genome (Fig. 2C, box plot on the left), thus demonstrating that HRSs located on distinct chromosomes are closer together in the 3D space of the nucleus. Among the 1125 HRS bins, 1102 (98%) were located in the active A chromosomal compartment. HRS bins located in this compartment also have a contact score higher than randomizations (Fig. 2C, box plot on the right), indicating that HRS bins found within the A compartment are also spatially clustered.
A global survey of ESC HRSs in a genome browser then suggested that HRSs are associated with gene-rich regions and with the active A chromosomal compartment (Fig. 3A; Lieberman-Aiden et al. 2009; Dixon et al. 2012). To assess this point, we determined the overlap score between this compartment and the HRSs (i.e., the number of base pairs located in HRSs and corresponding to the active A chromosomal compartment, divided by the total number of base pairs of the active compartment in the entire mouse genome) and found that, for each chromosome, the overlap score is systematically higher than the score obtained for a random set of StyI fragments. This demonstrates that HRSs are strongly associated with the active A compartment (Fig. 3B). In sharp contrast, HRSs are underrepresented in the inactive B compartment (Fig. 3C). We conclude that sequences identified by HRS-seq correspond to regions essentially associated with the mouse active A chromosomal compartment.
Figure 3.

HRSs are associated with the active chromosomal compartment. (A) Comparison between A and B compartments, StyI fragment, HRS, and gene densities along mouse Chromosome 1. (B) For each chromosome, the overlap score between ESC HRSs and the active A compartment has been calculated (red dot) and compared to the overlap scores obtained for 1000 randomizations (box plots). The overlap score represents the fraction of the genomic feature of interest (here A compartment) that is covered by HRS. The P-value (valid independently for each chromosome) assesses the difference between the overlap scores obtained for HRSs versus 1000 randomizations. (C) Analyses of overlap scores for the inactive B compartment were performed as described above.
HRSs are associated with highly expressed genes and super-enhancers
The preferential overlap of HRSs with the active A chromosomal compartment (Fig. 3B) and their weak overrepresentation in some G/C-rich sequences (Supplemental Fig. S3B) suggest that they might be associated with CpG islands and gene-rich regions. Indeed, 4817 HRSs (7.9%) are overlapping with CpG islands, which is significantly different from the mean count (623 ± 26) (1.1%) obtained from 1000 sets of 61,080 StyI fragments randomly selected in the mouse genome (Fig. 4A). Moreover, we found 3625 genes for which at least one TSS is located inside a HRS in ESCs, henceforth termed HRS-associated genes (listed in Supplemental Table S3). A randomization analysis showed that this number is much higher than expected by chance (632 ± 20) (Fig. 4B). Similar results were obtained separately for each individual chromosome (Supplemental Fig. S4). In contrast, overlap score analyses indicated that HRSs are underrepresented into lamina-associated domains (LADs) (Supplemental Fig. S5), which are associated with the inactive B compartment (Peric-Hupkes et al. 2010).
Figure 4.

HRSs are associated with active genes and super-enhancers. (A) The number of HRSs that overlap with CpG islands (UCSC, mm9 build) was counted (4817; left) and compared to the mean counts (623 ± 26, SD) obtained from “random permutation tests” with n = 1000 random resampling (1000 sets of equivalent number of random StyI fragments; right). The P-value indicates the significance of the difference between the counts obtained for HRSs versus 1000 randomizations. (B) The number of TSSs that map into the ESC HRS set was counted (brown dot). This number was compared to the counts obtained from “random permutation tests” with n = 1000 random resampling (1000 sets of 61,080 random StyI fragments; box plots, median value obtained from randomizations is indicated in purple). The P-value indicates the significance of the difference between the counts obtained for HRSs versus 1000 randomizations. (C) Based on RNA-seq data available from ESCs (Wamstad et al. 2012), mouse genes were classified into three sets. The first set corresponds to the 3000 genes having the highest expression levels, the second to 3000 moderately expressed genes, and the last to the 3000 genes with the weakest expression levels (mean of two replicates). For each set, the number of HRS-associated genes were counted and compared with the counts obtained for equivalent numbers of genes taken at random. The P-value indicates the significance of the difference between the counts obtained for HRSs versus 100 randomizations (box plots). It is valid independently for the differences observed in the highly and weakly expressed gene sets. (D) Identical analysis as described above in C was performed using ESC GRO-seq data (Min et al. 2011). (E) The numbers of super-enhancers (Khan and Zhang 2016) that overlap with the ESC HRSs (brown dots) were counted for all super-enhancers known in the mouse genome (left) and for those that are active in ESCs (middle) or in the cortex (right). These numbers were compared to “random permutation tests” (1000 random sets of 61,080 StyI fragments; box plots, median value indicated in purple). The P-value indicates the significance of the difference between the counts obtained for HRSs versus 1000 randomizations.
To assess whether HRS-associated genes belong to active or inactive genes, we used available RNA-seq data from ESCs (Wamstad et al. 2012) to design three sets of genes: The first set corresponds to the 3000 most highly expressed genes, the second set to 3000 genes that display moderate expression, and the third set to the 3000 genes that display the weakest expression levels. This analysis showed that HRS-associated genes are largely overrepresented in the first set of highly expressed genes. In contrast, the number of HRS-associated genes is comparable to those obtained from random sets in the moderately expressed gene set, while they are strongly underrepresented in the weakly expressed gene set (Fig. 4C). A similar result was obtained when GRO-seq data from ESCs (Min et al. 2011) were used instead of RNA-seq data (Fig. 4D). Furthermore, on each chromosome, HRSs are overrepresented in exon sequences (Supplemental Fig. S6A) and underrepresented in introns (Supplemental Fig. S6B).
By using the list of all super-enhancers known in the mouse genome (Khan and Zhang 2016), we found that super-enhancers are globally underrepresented in HRSs (5225 are overlapping with HRSs, while randomizations show that 5812 ± 78 should be expected) (Fig. 4E, left). However, among the 231 super-enhancers that possess active epigenetic marks in ESCs (Khan and Zhang 2016), 153 (66%) are found overlapping with HRSs, and this number is much higher than expected by chance (67 ± 6) (Fig. 4E, middle), while super-enhancers active in other cell types, like the cortex (Fig. 4E right), are not overrepresented in ESC HRS. Therefore, super-enhancers active in the ESCs are strongly associated with ESC HRS.
Finally, using data available in the literature (ENCODE Project), we showed that, in ESCs, HRSs are not correlated with trimethylation of lysine 9 on histone 3 (H3K9me3), which marks constitutive heterochromatin. In contrast, they overlap with trimethylation of lysine 36 on histone 3 (H3K36me3) (Fig. 5A,B), which marks transcriptionally active exon regions (Hon et al. 2009). This latter result was confirmed on each chromosome using appropriate randomizations (Supplemental Fig. S6C). We conclude that HRSs are associated with TSSs of highly expressed genes and active super-enhancers.
Figure 5.

HRSs are associated with active epigenetic marks. (A) Browser snapshot showing the HRS density at the Sox2 gene locus on mouse Chromosome 3 as determined by HRS-seq experiments performed in ESCs. Tracks displaying DNase I–sensitive sites, RNA PolII peaks as well as ChIP-seq data for the indicated epigenetic marks (ENCODE E14 ESC data) were plotted using the WashU EpiGenome Browser. (B) Heat map depicting the Pearson correlation coefficients obtained between ESC HRSs and sequences (10-kb bins) enriched in distinct epigenetic marks as indicated on the figure. (Black/red) High positive correlation coefficient; (white/blue) low null/negative correlation coefficient.
HRS-associated genes are housekeeping, as well as cell-type–specific, genes
By using DAVID functional annotation tool (Huang da et al. 2009), we carried out Gene Ontology (GO) analyses on genes with HRS-associated TSSs (Supplemental Table S4). Most of the ontology terms correspond to housekeeping genes often linked to known nuclear bodies, with terms such as “covalent chromatin modification” (P = 3.2 × 10−7), “intracellular RNP complexes” (P = 1.5 × 10−20), “spliceosome” (P = 3.9 × 10−7), “nucleolus” (P = 1.6 × 10−25), “cell cycle” (P = 9.3 × 10−17), and also “nuclear speckles” (P = 3.3 × 10−3) and “promyelocytic leukemia (PML) body” (P = 3.9 × 10−2) (Fig. 6). We also noted the term “stem cell population maintenance” (P = 2.6 × 10−4) (black arrow in Fig. 6), which reflects the presence in ESC HRSs of the TSSs of many pluripotency genes, e.g., Nanog, Tet1, or Sox2, which are very highly expressed in ESCs (Wamstad et al. 2012). HRS-seq data indicated the presence of HRSs at the Sox2, Klf4, Pou5f1, and Nanog loci (Fig. 5A; Supplemental Fig. S7). In contrast, DAVID ontology analysis of highly expressed genes that are not associated with HRSs (1656 genes among the 3000 genes used in Set1 of Fig. 4C) indicates that they essentially correspond to housekeeping genes involved in cell metabolism or cytoskeleton and membrane-associated processes, and no indication of cell-type–specific or nuclear body–associated genes could be evidenced (Supplemental Fig. S8A; Supplemental Table S5). By using the i-cisTarget tool (Herrmann et al. 2012), we then showed that the promoters of highly expressed genes associated with the HRSs preferentially bind cell-cycle regulators of the E2F family, while the promoters of highly expressed genes not associated with HRSs bind a whole series of factors belonging to the ETS family (ELF, ELK, GABPA…) (Supplemental Fig. S8B; Supplemental Table S6). This suggests that many HRS-associated genes are tightly regulated during cell cycle progression.
Figure 6.

HRS-associated genes are housekeeping, as well as cell-type–specific, genes. DAVID Gene Ontology analyses were performed on genes for which at least one TSS was mapping in the ESC HRSs. KEGG pathways (green) and GO terms related to biological processes (red), molecular functions (blue), and cellular components (yellow) are depicted by circles as a function of fold enrichments. For each indicated term, circle areas are proportional to gene counts. Only the most significant terms (P-value <0.05 and fold enrichment >1.80) are shown. P-values <5 × 10−6 are depicted by red squares (for exact values, see Supplemental Table S4).
Pluripotency genes are well known to be largely repressed when ESCs are differentiated into cortical neurons (see, e.g., Bonev et al. 2017). To further assess the functional significance of the association of pluripotency genes with HRSs, we differentiated ESCs into cortical neurons (Gaspard et al. 2009). In both cell types, we performed detailed analyses at the Sox2, Pou5f1, Nanog, and Klf4 loci using qPCR to determine the relative enrichment levels in the HRS-containing fraction of StyI fragments spread along these loci (HRS-qPCR experiments). At the Sox2 locus, we found that the enrichment levels within the gene body are 11 to 23 times higher than the mean local background in ESCs (Fig. 7A; for background definition, see Supplemental Methods), but they fall to five times the mean local background in cortical neurons (Fig. 7B). This region, which also maps with super-enhancers (Khan and Zhang 2016), is known to contain two Sox2 Regulatory Regions (SRR) (Zhou et al. 2014). As suggested by HRS-seq data (Fig. 5A), a second HRS region was found 107 kb downstream from the gene. It corresponds to a known super-enhancer (Khan and Zhang 2016), the Sox2 Regulatory Region 107 (SRR107), which is required to maintain a high expression level of this gene in ESCs (Zhou et al. 2014). Its enrichment level was three times lower in neurons (Fig. 7B) than in ESCs (Fig. 7A), confirming the previously described cell specificity of this region (Zhou et al. 2014). Similar results were obtained at the Pou5f1 and Nanog loci, where enrichment levels of the gene body and associated super-enhancers were high in ESCs but drastically reduced in neurons (Supplemental Fig. S9A,D). At the Klf4 locus, the gene body also displays much lower enrichment levels in neurons than in ESCs. However, the promoter as well as some part of the associated super-enhancer remained highly enriched in neurons (Supplemental Fig. S9E,F), suggesting that this locus may remain associated to a large RNP complex in cortical neurons even if transcription levels are largely reduced in this cell type.
Figure 7.

HRSs are associated with highly expressed cell-specific genes. The enrichment levels (HRS vs. Loop fractions) of StyI fragments at the Sox2 locus (Chr 3: 34,400,000–34,670,000, mm9 assembly) were determined by qPCR (HRS-qPCR) in ESCs (A; brown bars) or in neurons (B; blue bars). The red horizontal line corresponds to the mean local background level (value 1), and dashed lines depict the mean noise band as defined in the Supplemental Methods. The positions of StyI fragments identified by HRS-seq, as well as RefSeq genes, StyI sites, Super-enhancers (SE) (Khan and Zhang 2016) and Sox2 Regulatory Regions (SRR) (Zhou et al. 2014), are indicated below the histogram. The HRS-qPCR track indicates StyI fragments investigated in the experiment. Green bars represent HRSs (StyI fragments having enrichment levels above the noise band), red bars indicate StyI fragments that are not HRSs. n = 3 (technical replicates) for each experiment; error bars, SEM.
Overall, these results indicate that, while the vast majority of gene-associated HRSs correspond to housekeeping activities, some of them correspond to cell-type–specific genes (such as pluripotency genes in ESCs) and to their cis-regulatory sequences (such as super-enhancers).
HRSs include nuclear body–associated sequences
Former proteomic analyses indicated that high-salt treatments of nuclei preparations enable the recovery of protein components of nuclear bodies (Engelke et al. 2014). To check whether nuclear bodies are also recovered under our experimental conditions, we performed immunofluorescence microscopy on nuclear halos prepared as described above for the HRS assays (also see Methods). These experiments showed that Coilin, SMN (Survival of Motor Neuron), and PML foci are present within the insoluble material obtained after high-salt treatment of our nuclei preparations (Fig. 8A), indicating that PML bodies as well as nuclear bodies of the Cajal body family (Histone Locus Bodies, gems, or Cajal bodies) are indeed retained under these experimental conditions.
Figure 8.

HRSs include nuclear body–associated sequences. (A) Immunofluorescence (IF) microscopy experiments were performed on nuclear halos using the following antibodies: αSMN (Cajal Bodies and Gems) (top), αCoilin (Histone Locus Bodies and Cajal bodies) (middle), and αPML (PML bodies) (bottom). DAPI staining is shown on the left, IF in the center, and the merged picture on the right. (B) The enrichment levels (HRS vs. Loop fractions) of StyI fragments at the Hist1 locus were determined by qPCR (HRS-qPCR) in ESCs (brown bars). Tracks below the histogram are as described in Figure 7. tRNA genes are also indicated below the histogram. n = 3 (technical replicates); error bars, SEM. (C) The enrichment (given in percentage of total family members) in ESC HRSs of 1554 repeat families was calculated and compared to enrichments obtained from “random permutation tests” with n = 1000 random resampling (1000 random sets of equivalent numbers of StyI fragments). For each class of repeats, the percentage of repeat families that was found significantly overrepresented in HRSs (P-value <0.001) compared with randomizations was determined. (D) HRS-qPCR experiments were performed at the Malat1/Neat1 gene locus as indicated above (B). n = 3 (technical replicates); error bars, SEM. Tracks below the histogram are as described above (no tRNA gene is mapping to this locus) (B; Fig. 7).
Since nuclear bodies remain within the high-salt insoluble material of the HRS-containing fraction, we assessed whether known nuclear body–associated sequences are present among HRSs. From the list of the 3625 TSSs that map to HRSs in ESCs (Supplemental Table S3), GO analyses showed that 14 are associated with the term “nucleosome assembly” (P = 2.4 × 10−3) (Fig. 6). This term is linked to the presence of histone genes known to be an essential component of the Histone Locus Bodies, a class of nuclear bodies that share strong structural similarities with Cajal bodies (Nizami et al. 2010). Indeed, 35 U7-dependent histone genes (whose mRNA maturation involves the U7 snRNA) among 73, but only two TSSs of histone variant genes among 15 (including H2afx), are found into HRS. In the mouse, histone genes are clustered into three major histone loci: the Hist1 locus on Chromosome 13qA3.1, the Hist2 locus on Chromosome 3qF1-3qF2.1, and the Hist3 locus on Chromosome 11qB1.3. TSSs of genes belonging to all three clusters are found associated with HRSs. Therefore, in ESCs, histone genes are strongly associated with HRSs.
Given the strong association of the Hist1 locus genes with HRSs (Supplemental Fig. S10A), we performed a detailed analysis of this locus by HRS-qPCR. We found that the enrichment levels along the whole Hist1 locus, from Hist1h4h to Hist1h1e genes, are 1.5 to four times higher than the mean local background (Fig. 8B). While devoid from any coding genes, the upstream part of the Hist1 locus nevertheless displayed the highest enrichment levels, with some values rising to almost 100 times above the local background. These sequences, having high enrichment levels in the HRS-containing fraction, systematically contain tRNA genes. We conclude that, at the Hist1 locus, both histone genes and tRNA genes have high enrichment levels within the HRS-containing fraction.
Since, at the Hist1 locus, the tRNA genes appear highly enriched within the HRS-containing fraction, we assessed whether this is particular to this locus or whether a wider association of HRSs with tRNA genes exists in the mouse genome. Because tRNA genes are classified among repeat sequences, we analyzed the repeat content of the HRS. The enrichment levels of 1554 mouse repeat families (UCSC classification) in the HRSs were calculated and compared to the mean enrichment level obtained from 1000 random sets with the same number of StyI fragments. Repeats families that are significantly enriched (P-value <10−3) in HRSs belong to nine classes including SINE repeats, which are known to be overrepresented in gene-rich regions and in the A compartment (Cournac et al. 2016), as well as snRNA genes and tRNA genes (Fig. 8C). We conclude that HRSs are associated with snRNA and tRNA genes as well as SINE repeats, at the genome-wide level. This observation suggests that some repeat regions are associated with large RNP complexes in the mouse nucleus.
HRS-seq data indicated that one of the largest HRS clusters in ESCs maps to the metastasis associated lung adenocarcinoma transcript 1 (Malat1) / nuclear paraspeckle assembly transcript 1 (Neat1) locus (Supplemental Fig. S10B). Indeed, both genes are found in the HRS-associated genes (Supplemental Table S3). Given the importance of these genes and transcripts for the assembly of nuclear speckles and paraspeckles (Hutchinson et al. 2007; Clemson et al. 2009), we performed HRS-qPCR experiments and showed that both genes are strongly enriched within the HRS-containing fraction throughout their gene bodies and that enrichment levels for their TSS are particularly high (Fig. 8D). Therefore, nuclear speckles and paraspeckles might also be retained in HRS assays.
Overall, these results confirm that nuclear halos contain insoluble nuclear bodies, and show that many HRSs correspond to genomic regions known to be associated with nuclear bodies, including the Hist1 locus that is part of the Histone Locus Bodies (Nizami et al. 2010), as well as snRNA genes that are integral components of Cajal bodies, tRNA genes that are known to contact the perinucleolar compartment (Nemeth et al. 2010; Padeken and Heun 2014), and the Malat1/Neat1 genes that are required for the assembly of nuclear speckles and paraspeckles.
We conclude that the sequences identified by HRS-seq correspond to genomic regions associated with large high-salt insoluble RNP complexes, including nuclear bodies, that display preferential physical proximity and association with the mouse active A chromosomal compartment.
Discussion
The HRS-seq method provides an original genome-wide approach to identify genomic sequences physically associated in vivo with large RNP complexes, including several nuclear bodies. Our method is probing higher-order chromatin architecture at the supra-nucleosomal scale. It is therefore clearly different from previous methods such as FAIRE-seq (Giresi et al. 2007), ATAC-seq (Buenrostro et al. 2013), or MNase-seq (Henikoff et al. 2011; Valouev et al. 2011; Gaffney et al. 2012), which all aim at investigating accessibility of the chromatin nucleofilament (nucleosomal scale). Contrary to most genome-wide approaches developed so far to investigate higher-order chromatin architecture (e.g., DamID mapping or Hi-C) (Vogel et al. 2007; Lieberman-Aiden et al. 2009), the HRS-seq method is based on a simple and straightforward principle: high-salt treatments of nuclei preparations. Previous works in Drosophila (Henikoff et al. 2009), as well as proteomic analyses (Engelke et al. 2014) and immunofluorescence microscopy (Fig. 8A; Dobson et al. 2017), indicate that such treatments make large RNP complexes insoluble. Combined with restriction digestion (StyI) and ultrafiltration (Fig. 1), they allow an easy separation of DNA sequences that are trapped within such complexes (HRS-containing fraction) from sequences that are not interacting with them (Loop fraction). Our method avoids long purification procedures of nuclear bodies that may bias retention of genomic sequences. Such approaches proved to be efficient for the identification of the nucleolus-associated domains (NADs) (Nemeth et al. 2010), but they were so far mostly unsuccessful for other nuclear bodies. The HRS-seq method is also avoiding delicate chemical crosslinking procedures (Dobson et al. 2017) or the use of specific antibodies that may restrict retention of some genomic sequences. It should thus be very helpful to explore in further detail the impact of this level of chromatin organization on gene regulation and cell fate determination in a variety of physiological and pathological situations.
We have here performed the first global genomic profiling of HRSs and found that, in mouse ESCs, the major components of HRSs are (1) the histone genes and snRNA genes, which are known to contact the Histone Locus Bodies and the Cajal bodies, respectively; (2) tRNA genes, which are known to spatially cluster into the perinucleolar compartment (Thompson et al. 2003; Nemeth et al. 2010); and (3) many transcriptionally active genes contacting large RNP complexes that may correspond to RNA polymerase II foci. This latter result is in agreement with pioneering works in human HeLa cells (Linnemann et al. 2009) and Drosophila S2 cells (Henikoff et al. 2009), showing that salt-insoluble chromatin is enriched in actively transcribed regions. However, our method replaces nuclease treatments (Henikoff et al. 2009) and microarray-based profiling (Henikoff et al. 2009; Linnemann et al. 2009) used in these earlier works by restriction digestions and a high-throughput sequencing approach, which allow a more powerful high-resolution genome-wide profiling. Recent genome-wide identification of the so-called matrix attachment regions (MAR-seq) in human mammary epithelial cells showed that MARs are A/T-rich sequences that are overrepresented in the active A chromosomal compartment, even though no correlation was found between such MARs and active or inactive epigenetic marks (Dobson et al. 2017). While MAR-seq technique also involves high-salt treatments, it makes use of extensive crosslinking, and therefore, like 3C assays, it may also capture sequences involved in long-range chromatin contacts that are not necessarily associated with large RNP complexes. HRSs are associated not only with actively transcribed regions (Fig. 4C) but also with cell-type–specific super-enhancers (Fig. 4E) and, opposite to MARs (Dobson et al. 2017), with genomic regions that display active epigenetic marks like H3K36me3 (Fig. 5B). Finally, HRSs are not biased toward A/T-rich sequences (Supplemental Fig. S3B), and therefore, they appear to be clearly distinct from A/T-rich sequences like MARs or LADs. Indeed, although lamins are known to remain associated with the insoluble material upon high-salt extractions of nuclei preparations (Engelke et al. 2014), HRSs are underrepresented in LADs (Supplemental Fig. S5). Since it has been demonstrated that lamins are not required for LAD organization in mouse ESCs (Amendola and van Steensel 2015), it may be possible that LAD organization in these cells depends on a factor that is soluble upon high-salt treatments.
Our work shows that genomic sequences that are associated with large RNP complexes (HRS) are in close proximity in the 3D space of the nucleus and that such sequences are overrepresented in the active A chromosomal compartment. It has been suggested that this latter level of chromatin organization may be coordinated through contacts with some nuclear bodies (Gibcus and Dekker 2013; Ea et al. 2015a). It is indeed known that chromosomal compartments are established during the early G1 phase of the cell cycle, at the so-called “timing decision point” (TDP) (Dileep et al. 2015), when replication-timing programs are fixed and several major nuclear bodies, like Cajal bodies, are reassembled (Carmo-Fonseca et al. 1993). Recruitment into nuclear bodies may confine specific chromatin regions, thus limiting their diffusion into the nuclear space and favoring functional interactions required for genomic regulations during the interphase. This is particularly true for long-range inter-TAD chromatin interactions, since chromatin dynamics at this level displays extremely low contact frequencies while being essential for many genomic functions (Dixon et al. 2012; Nora et al. 2012; Rao et al. 2014; Ea et al. 2015b). For example, by use of 4C-seq, it was shown that Cajal body–associated regions are enriched in highly expressed histone genes and snRNA loci, thus forming intra- and inter-chromosomal clusters (Wang et al. 2016a). In the interphasic cell, chromosomal partitioning into the active or inactive compartments is cell type specific (Lieberman-Aiden et al. 2009). Hi-C experiments using an ESC differentiation model (Wamstad et al. 2012) have recently suggested that such hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation (Fraser et al. 2015; Bonev et al. 2017). Pluripotency genes (including Sox2, Pou5f1, Nanog, and Klf4) that are highly transcribed in ESCs (Wamstad et al. 2012) are found among HRSs in this cell type (Fig. 5A; Supplemental Fig. S7), but their association is drastically reduced in cortical neurons (Fig. 7; Supplemental Fig. S9). This observation indicates that HRS-associated genes correspond not only to highly expressed housekeeping genes but also to cell-type–specific genes that require high transcription levels. HRSs also include some cis-regulatory elements required for maintaining high expression levels, like cell-type–specific super-enhancers (Fig. 4E) and the SRR107 region found at the Sox2 locus (Fig. 7). This region, which is also described as a super-enhancer (Wei et al. 2016), is located within a major distal cluster of enhancers, named the Sox2 Control Region (SCR) (Zhou et al. 2014). It has been shown by 3C and Hi-C experiments that the SCR is required for maintaining high Sox2 expression levels in ESCs through long-range chromatin interactions with this gene (Zhou et al. 2014; Stadhouders et al. 2018). Furthermore, only the most highly expressed genes are found overrepresented in HRSs, while genes that are expressed at moderate or weak expression levels are not (Fig. 4C). These findings are in agreement with the recently proposed phase separation model for transcription control (Hnisz et al. 2017), suggesting the existence of a cell-type–specific transcriptional compartment where a subset of genes and their regulatory elements, including super-enhancers, are associated with large RNP complexes allowing high expression levels. Such complexes are likely to correspond to RNA polymerase II foci visualized by immunofluorescence microscopy (transcription factories/active chromatin hubs). However, given that sequences at the Malat1/Neat1 gene locus are found highly enriched within the HRS-containing fraction (Fig. 8D), one possibility could be that they correspond to RNA polymerase II complexes contacting nuclear speckles/paraspeckles, serving as “hubs” to link active transcription sites (Sutherland and Bickmore 2009; Cook 2010; Mao et al. 2011; Quinodoz et al. 2018).
Overall, our results provide a strong experimental support in favor of a model whereby nuclear bodies, and/or large RNP complexes associated with RNA polymerase II, play an important role in organizing the active chromosomal compartment through recruitment of highly expressed genes, including housekeeping and cell-type–specific genes with their cis-regulatory regions.
Methods
Cell culture
Cultures and in vitro corticogenesis of mouse ESCs (e14Tg2a strain, 129P2 genomic background) were performed as previously described (for details, see Supplemental Methods; Gaspard et al. 2009). Cells were tested for the absence of mycoplasma contamination, and their identity was confirmed by immunofluorescence microscopy (Supplemental Fig. S11; Supplemental Methods). All experimental designs and procedures are in agreement with the guidelines of the animal ethics committee of the French “Ministère de l'Agriculture” (European directive 2010/63/EU).
HRS assay
Nuclei preparations used for HRS assays were made from undifferentiated mouse ESCs or neurons as previously described for C2C12 myoblasts (Milligan et al. 2000). Such nuclei preparations are snap frozen into liquid nitrogen and can be stored at −80°C for several months. They were formerly used for nuclear run-on experiments to investigate transcriptional activity of mammalian genes (Milligan et al. 2000, 2002). The HRS assays were adapted from our previous publications (for details, see Supplemental Methods; Weber et al. 2003; Braem et al. 2008).
Real-time qPCR and quality check
The quality of each HRS assay was checked by real-time qPCR targeting StyI fragments that are known, from previously published works (Court et al. 2011), to be either highly enriched (positive control) or not enriched (negative control) in the HRS-containing fraction relative to the Loop fraction in diverse experimental conditions. Primer sequences used for HRS-qPCR analyses at Sox2, Pou5f1, Nanog, Klf4, Histone 1, and Malat1/Neat1 loci (Figs. 7, 8B,D; Supplemental Fig. S9) are given in Supplemental Tables S7 through S12, respectively.
The enrichment levels were calculated as the ratio of the amount of DNA target in the HRS-containing fraction versus the Loop fraction. They were normalized to the local background level according to an algorithm adapted from a previous work (for details, see Supplemental Methods; Braem et al. 2008).
HRS-seq library construction
HRS assays were performed from three distinct ESC nuclei preparations. For each nuclei preparation, DNA extracted from Loop fractions on one side and from HRS-containing fractions on the other side of 12 high-quality HRS assays were pooled (see Fig. 1C). These samples were used to make HRS-seq libraries (Supplemental Fig. S1B,C). Each biological replicate was thus composed of two HRS-seq libraries: one built from the DNA pooled from the Loop fractions and one built from that pooled from the HRS-containing fractions. Construction of HRS-seq libraries is done as follows: DNA samples are first redigested with StyI (Eco130I at 10 µ/µL, Fermentas ref ER0411) in order to ensure complete digestion. Thirty picomoles of biotynilated adaptor 1 with complementarity for StyI restriction sites (5′P-CWWGTCGGACTGTAGAACTCTGAACCTGTCCAAGGTGTGA-Biotin-3′ and 3′-AGCCTGACATCTTGAGACTTGGACA-5′) is ligated during 15 min at room temperature (RT) to 150 ng of StyI-digested genomic DNA (quick ligation kit, NEB ref. M2200S). One hundred micrograms of streptavidine beads (Dynabeads MyOne streptavidin C1 from Invitrogen, ref. 650.01) is resuspended into 50 µL of BW 2× buffer (10 mM Tris-HCl at pH 7.5, 1 mM EDTA, 2 M NaCl), and the adaptor 1 ligation reaction is added and incubated with the beads during 15 min at RT on a rotation wheel. Beads are washed three times by one volume of BW 1× buffer (10 mM Tris-HCl at pH 7.5, 1 mM EDTA, 2 M NaCl) and two times by one volume of TE buffer (10 mM Tris-HCl at pH7.5, 1 mM EDTA). They are resuspended into 10 µL of NEBuffer 4, and 10 µL of 10× SAM (made of 5 µL of 32 mM S-adenosylmethionin diluted in water to a final volume of 325 µL) are added, as well as 76 µL of water and 4 µL of MmeI restriction enzyme (NEB, ref. R0637S; final volume of 100 µL). This reaction is incubated 90 min at 37°C under agitation. The supernatant is then removed, and beads are washed three times with 50 µL of 1× BW buffer and two times with one volume of TE buffer. The following is then added to the beads: 5 µL of 10× T4 ligase buffer, 2 µL of 15 µM (30 pmol) of GEX adaptor 2 (5′-CAAGCAGAAGACGGCATACGANN-3′ and 3′-GTTCGTCTTCTGCCGTATGCT-P-5′), 1 µL of T4 DNA ligase (NEB M0202S), and 42 µL of water (final volume 50 µL). This reaction is incubated for 2 h at 20°C and agitated 15 sec each 2 min. Beads are then washed three times with 1× BW buffer and two times with one volume of TE buffer before being resuspended into 10 µL of distilled water. Two microliters of this reaction (DNA on beads) is then mixed with 10 µL of 5× HF Phusion buffer and 0.5 µL of Phusion DNA polymerase (Finnzymes, ref. F-530), 0.5 µL of dNTP mix (25 mM each), 0.5 µL of 25 µM GEX PCR primer 1 (5′-CAAGCAGAAGACGGCATACGA-3′), 0.5 µL of 25 µM GEX PCR primer 2 (5′-AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGA-3′), and 36 µL of water (final volume of 50 µL). This reaction is amplified in a thermocycler as follows: 30 sec at 98°C; followed by 15 cycles of 10 sec at 98°C, 30 sec at 60°C, and 15 sec at 72°C; and 10 min at 72°C. The PCR reaction is then run on a 6% 1× TBE acrylamide gel (Novex, Invitrogen) (Supplemental Fig. S1C), and the main DNA band (expected size 95–97 bp) is cut and purified (Spin-X-filter column from Sigma, and ethanol precipitation) before being resuspended into 10 µL of water. DNA concentration of the HRS-seq library is checked with an Agilent Bioanalyzer apparatus before being used for high-throughput sequencing (50-nt single reads) on a HiSeq 2000 apparatus (Illumina). The following primer was used to generate the clusters: 5′-CCACCGACAGGTTCAGAGTTCTACAGTCCGAC-3′. Five control sequencing libraries were also constructed exactly as described above by using 150 ng of mouse genomic DNA cut by StyI enzyme (gDNA libraries), and three of them were used for high-throughput sequencing (Supplemental Fig. S1B).
Raw data filtering
Sequencing tags were trimmed and aligned on the mouse reference genome of e14Tg2a mouse ESCs (129P2 built from mm9 assembly), and read positions were determined. Reads mapping to multiple positions and reads with more than two mismatches were removed. Two bioinformatic filters were then applied to exclude potentially aberrant reads. Indeed, according to our protocol, all relevant reads should have a size of 18 to 20 nt due to MmeI digestion (filter 1), and they should have one of their extremities next to a StyI site (filter 2). Table 1 summarizes the number of reads obtained at each step of data filtering. Note that tag alignments to the mm10 assembly would not be expected to improve raw data processing since 97% of StyI sites are identical between the two assemblies and that the missing 3% corresponds to additional sites that are essentially lying in telomeric regions of chromosomes and not in gene-rich regions where most HRSs are located.
For each fraction independently, we then calculated the total number of reads obtained for each StyI restriction fragments in the mouse genome (mm9 assembly) by including reads sequenced from both the 5′ and 3′ extremities. These processed data were then checked for technical reproducibility (Supplemental Fig. S2; Supplemental Table S1).
Statistical analyses
The processed data are discrete, consisting for each StyI fragment in read counts for three different biological replicates. The aim is to compare, for each experiment, the number of reads between the HRS-containing fraction and the Loop fraction or between the HRS-containing fraction and the gDNA control. Statistical significance of the overrepresentation of read counts for StyI fragments in the HRS-containing fraction compared with the Loop fraction (or gDNA control) has been assessed using the R packages DESeq (Anders and Huber 2010) and edgeR (see Supplemental Methods; Robinson et al. 2010). Only fragments being identified as differential between compared conditions (i.e., having a Benjamini-Hochberg–corrected P-value <5%) for both tests have been kept for further bioinformatic analyses (i.e., 61,080 HRSs for ESCs) (Supplemental Table S2).
Bioinformatic analyses
Mean inter-chromosomal contact scores were calculated from the Hi-C data obtained on mouse ESCs (SRR400251 to SRR400255, replicate 2 from Dixon et al. 2012). The analysis of 3D proximity of HRSs (Fig. 2C) was based on the comparison between mean contact scores of HRSs and randomizations (see Supplemental Methods).
The overlap score used in Figures 3 and 4 and Supplemental Figure S6 is the fraction of the genomic feature of interest that is covered by HRSs (or random sets with the same number of elements), i.e., the base-pair number of HRS regions corresponding to the genomic feature of interest divided by the total base-pair number of the genomic feature in the mouse genome. The null model used to generate the null hypothesis distribution was based on a random swapping procedure (see Supplemental Methods). For HRS distribution with respect to A/B compartments (Fig. 3B,C), all HRSs were uniformly randomized on the whole genome. To test the significance of the overlap between HRSs and H3K36me3 (ENCFF001KDY), exons or introns (UCSC mm9 assembly), the HRSs present in gene bodies were randomized only to gene body sequences. The distributions corresponding to 1000 random realizations were represented by their mean and the 95% confidence interval around this mean. A/B compartments were computed as described by Lieberman-Aiden et al. (2009) using Hi-C data sets from mouse ESCs (see Supplemental Methods; Dixon et al. 2012). All tracks were plotted with the WashU EpiGenome Browser (https://epigenomegateway.wustl.edu) (mm9 assembly). LAD analyses were performed on data available from DamID maps of lamin B1 in mouse ESCs (NimbleGen microarray probes) (Peric-Hupkes et al. 2010).
Gene content analyses (Fig. 4B; Supplemental Fig. S4) were performed using UCSC annotation data (reFlat.txt file, mm9 built). RNA-seq data from ESCs (used in Fig. 4C) were downloaded from the Gene Expression Omnibus repository (GSE47948) (Wamstad et al. 2012) and GRO-seq data from ESCs (used in Fig. 4D) from GSE27037 (Min et al. 2011). ChIP-seq data used in Figure 5B and Supplemental Figure S6C were downloaded from the ENCODE Project (ENCFF001KEV, ENCFF001KFB, ENCFF001KFH, ENCFF001KFN, ENCFF001KFT, ENCFF001ZHE, ENCFF001ZID; http://genome.cse.ucsc.edu/encode). Super-enhancer data (Fig. 4E) were downloaded from the dbSUPER database (Khan and Zhang 2016; http://asntech.org/dbsuper/). GO analyses (Fig. 6; Supplemental Fig. S8A) were performed using the functional annotation tool on the DAVID 6.8 ontology server (settings: GOTERM_BP_DIRECT, KEGG_PATHWAY, Fold Enrichment and Benjamini-Hochberg–corrected Fisher's exact test; all other settings were defaults) (Huang da et al. 2009; https://david.ncifcrf.gov/home.jsp). Comparative analysis of predicted transcription factor binding (Supplemental Fig. S8B) was performed using the i-cisTarget tool (Herrmann et al. 2012; https://gbiomed.kuleuven.be/apps/lcb/i-cisTarget/). All settings were defaults with a normalized enrichment score (NES) threshold of 0.3, corresponding to a P-value <0.01. For repeat analyses (Fig. 8C), we first identified repeat sequences mapping within ESC HRSs, and the number of repeats was then determined for each of the 1554 repeat families (UCSC, mm9 built). Several bioinformatic analyses performed on ESC HRSs were assessed statistically by randomization tests, with n = 100 or n = 1000 uniformly random resampling (see Supplemental Methods).
Immunofluorescence microscopy
Immunofluorescence microscopy on nuclear halos was performed on silanized coverslips, using antibodies targeting p80 coilin (polyclonal rabbit antibody, 1:400 dilution, gift from R. Bordonné) (Boulisfane et al. 2011), SMN (monoclonal antibody from BD Transduction Laboratories no. 610646, 1:1000 dilution), and PML (mouse monoclonal antibody, clone 36.1–104, Millipore no. MAB3738, 1:500 dilution, gift from P. Lomonte) (for further details, see Supplemental Methods).
Data access
Raw data and processed data from this study have been submitted to the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/)under accession number GSE106751. Fully processed data supporting the findings of this study are available within Supplemental Material files.
Supplementary Material
Acknowledgments
We thank Isabelle Degors, Sébastien This, Emeric Dubois, Françoise Carbonell, Marie-Noëlle Lelay-Taha, and Marjorie Drac (DNA combing platform) for technical assistance. We thank Florence Rage, Rémy Bordonné, and Patrick Lomonte for providing antibodies; Cyril Esnault and Jean-Christophe Andrau for help with bioinformatics; and Eric Soler and Mounia Lagha for critical reading of the manuscript. This work was supported by grants from the Institut National du Cancer (PLBIO 2012-129, INCa_5960 to T.F.), the AFM-Téléthon (no. 21024 to T.F.), the Ligue Contre le Cancer, comité de l'Hérault, the Agence Nationale de la Recherche (CHRODYT, ANR-16-CE15-0018-04), and the CNRS. M.-O.B. was supported by the University of Montpellier and Z.Y. by a fellowship from the IDEX Super 3DRNA.
Author contributions: M.-O.B. performed HRS assays, sequencing libraries, raw data filtering, and bioinformatic analyses, and edited the manuscript. A.C. performed bioinformatic analyses. F.C. designed the project and performed raw data filtering and bioinformatic analyses. M.S. conceived and optimized the sequencing library protocol. H.P. performed NGS sequencings. C. Reynes and R.S. analyzed genomic data and performed statistical analyses. T.B. provided the biological material and performed immunofluorescence microscopy (IF). Z.Y. performed bioinformatic analyses. M.T. and S.S. performed HRS-qPCR. C. Rebouissou performed HRS-qPCR and IF. G.C. designed the project and edited the manuscript. A.L. designed/assessed statistical analyses and edited the manuscript. J.M. performed bioinformatic analyses and edited the manuscript. L.J. designed the project, managed NGS sequencing, and edited the manuscript. T.F. conceived and designed the project, analyzed genomic data, performed bioinformatic analyses, and wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.237073.118.
References
- Amendola M, van Steensel B. 2015. Nuclear lamins are not required for lamina-associated domain organization in mouse embryonic stem cells. EMBO Rep 16: 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, et al. 2017. Multiscale 3D genome rewiring during mouse neural development. Cell 171: 557–572.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulisfane N, Choleza M, Rage F, Neel H, Soret J, Bordonne R. 2011. Impaired minor tri-snRNP assembly generates differential splicing defects of U12-type introns in lymphoblasts derived from a type I SMA patient. Hum Mol Genet 20: 641–648. [DOI] [PubMed] [Google Scholar]
- Braem C, Recolin B, Rancourt RC, Angiolini C, Barthes P, Branchu P, Court F, Cathala G, Ferguson-Smith AC, Forne T. 2008. Genomic matrix attachment region and chromosome conformation capture quantitative real time PCR assays identify novel putative regulatory elements at the imprinted Dlk1/Gtl2 locus. J Biol Chem 283: 18612–18620. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca M, Ferreira J, Lamond AI. 1993. Assembly of snRNP-containing coiled bodies is regulated in interphase and mitosis—evidence that the coiled body is a kinetic nuclear structure. J Cell Biol 120: 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A, Lawrence JB. 2009. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell 33: 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PR. 2010. A model for all genomes: the role of transcription factories. J Mol Biol 395: 1–10. [DOI] [PubMed] [Google Scholar]
- Cournac A, Koszul R, Mozziconacci J. 2016. The 3D folding of metazoan genomes correlates with the association of similar repetitive elements. Nucleic Acids Res 44: 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court F, Baniol M, Hagege H, Petit JS, Lelay-Taha MN, Carbonell F, Weber M, Cathala G, Forné T. 2011. Long-range chromatin interactions at the mouse Igf2/H19 locus reveal a novel paternally expressed long non-coding RNA. Nucleic Acids Res 39: 5893–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dileep V, Ay F, Sima J, Vera DL, Noble WS, Gilbert DM. 2015. Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res 25: 1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson JR, Hong D, Barutcu AR, Wu H, Imbalzano AN, Lian JB, Stein JL, van Wijnen AJ, Nickerson JA, Stein GS. 2017. Identifying nuclear matrix–attached DNA across the genome. J Cell Physiol 232: 1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea V, Baudement MO, Lesne A, Forné T. 2015a. Contribution of topological domains and loop formation to 3D chromatin organization. Genes (Basel) 6: 734–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea V, Sexton T, Gostan T, Herviou L, Baudement MO, Zhang Y, Berlivet S, Le Lay-Taha MN, Cathala G, Lesne A, et al. 2015b. Distinct polymer physics principles govern chromatin dynamics in mouse and Drosophila topological domains. BMC Genomics 16: 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelke R, Riede J, Hegermann J, Wuerch A, Eimer S, Dengjel J, Mittler G. 2014. The quantitative nuclear matrix proteome as a biochemical snapshot of nuclear organization. J Proteome Res 13: 3940–3956. [DOI] [PubMed] [Google Scholar]
- Fraser J, Ferrai C, Chiariello AM, Schueler M, Rito T, Laudanno G, Barbieri M, Moore BL, Kraemer DC, Aitken S, et al. 2015. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol Syst Biol 11: 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey MR, Matera AG. 1995. Coiled bodies contain U7 small nuclear RNA and associate with specific DNA sequences in interphase human cells. Proc Natl Acad Sci 92: 5915–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney DJ, McVicker G, Pai AA, Fondufe-Mittendorf YN, Lewellen N, Michelini K, Widom J, Gilad Y, et al. 2012. Controls of nucleosome positioning in the human genome. PLoS Genet 8: e1003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspard N, Bouschet T, Hourez R, Dimidschstein J, Naeije G, van den Ameele J, Espuny-Camacho I, Herpoel A, Passante L, Schiffmann SN, et al. 2008. An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature 455: 351–357. [DOI] [PubMed] [Google Scholar]
- Gaspard N, Bouschet T, Herpoel A, Naeije G, van den Ameele J, Vanderhaeghen P. 2009. Generation of cortical neurons from mouse embryonic stem cells. Nat Protoc 4: 1454–1463. [DOI] [PubMed] [Google Scholar]
- Gibcus JH, Dekker J. 2013. The hierarchy of the 3D genome. Mol Cell 49: 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. 2007. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17: 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG, Sakai A, Loeb GB, Ahmad K. 2009. Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res 19: 460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, Henikoff S. 2011. Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci 108: 18318–18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann C, Van de Sande B, Potier D, Aerts S. 2012. i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res 40: e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA. 2017. A phase separation model for transcriptional control. Cell 169: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon G, Wang W, Ren B. 2009. Discovery and annotation of functional chromatin signatures in the human genome. PLoS Comput Biol 5: e1000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A. 2007. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Zhang X. 2016. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res 44: D164–D171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnemann AK, Platts AE, Krawetz SA. 2009. Differential nuclear scaffold/matrix attachment marks expressed genes. Hum Mol Genet 18: 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao YS, Zhang B, Spector DL. 2011. Biogenesis and function of nuclear bodies. Trends Genet 27: 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan L, Antoine E, Bisbal C, Weber M, Brunel C, Forné T, Cathala G. 2000. H19 gene expression is up-regulated exclusively by stabilization of the RNA during muscle cell differentiation. Oncogene 19: 5810–5816. [DOI] [PubMed] [Google Scholar]
- Milligan L, Forné T, Antoine E, Weber M, Hemonnot B, Dandolo L, Brunel C, Cathala G. 2002. Turnover of primary transcripts is a major step in the regulation of mouse H19 gene expression. EMBO Rep 3: 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min IM, Waterfall JJ, Core LJ, Munroe RJ, Schimenti J, Lis JT. 2011. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev 25: 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth A, Conesa A, Santoyo-Lopez J, Medina I, Montaner D, Peterfia B, Solovei I, Cremer T, Dopazo J, Langst G. 2010. Initial genomics of the human nucleolus. PLoS Genet 6: e1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizami Z, Deryusheva S, Gall JG. 2010. The Cajal body and histone locus body. Cold Spring Harb Perspect Biol 2: a000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. 2017. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169: 930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padeken J, Heun P. 2014. Nucleolus and nuclear periphery: Velcro for heterochromatin. Curr Opin Cell Biol 28: 54–60. [DOI] [PubMed] [Google Scholar]
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. 2010. Molecular maps of the reorganization of genome–nuclear lamina interactions during differentiation. Mol Cell 38: 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. 2018. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174: 744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, Haering CH, Mirny L, et al. 2017. Two independent modes of chromatin organization revealed by cohesin removal. Nature 551: 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeman JE, Trinkle-Mulcahy L. 2014. Nuclear bodies: new insights into assembly/dynamics and disease relevance. Curr Opin Cell Biol 28: 76–83. [DOI] [PubMed] [Google Scholar]
- Stadhouders R, Vidal E, Serra F, Di Stefano B, Le Dily F, Quilez J, Gomez A, Collombet S, Berenguer C, Cuartero Y, et al. 2018. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet 50: 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland H, Bickmore WA. 2009. Transcription factories: gene expression in unions? Nat Rev Genet 10: 457–466. [DOI] [PubMed] [Google Scholar]
- Thompson M, Haeusler RA, Good PD, Engelke DR. 2003. Nucleolar clustering of dispersed tRNA genes. Science 302: 1399–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. 2011. Determinants of nucleosome organization in primary human cells. Nature 474: 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel MJ, Peric-Hupkes D, van Steensel B. 2007. Detection of in vivo protein–DNA interactions using DamID in mammalian cells. Nat Protoc 2: 1467–1478. [DOI] [PubMed] [Google Scholar]
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, et al. 2012. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 151: 206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Sawyer IA, Sung MH, Sturgill D, Shevtsov SP, Pegoraro G, Hakim O, Baek S, Hager GL, Dundr M. 2016a. Cajal bodies are linked to genome conformation. Nat Commun 7: 10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Su JH, Beliveau BJ, Bintu B, Moffitt JR, Wu CT, Zhuang X. 2016b. Spatial organization of chromatin domains and compartments in single chromosomes. Science 353: 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hagège H, Murrell A, Brunel C, Reik W, Cathala G, Forné T. 2003. Genomic imprinting controls matrix attachment regions in the Igf2 gene. Mol Cell Biol 23: 8953–8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Zhang S, Shang S, Zhang B, Li S, Wang X, Wang F, Su J, Wu Q, Liu H, et al. 2016. SEA: a super-enhancer archive. Nucleic Acids Res 44: D172–D179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Katsman Y, Dhaliwal NK, Davidson S, Macpherson NN, Sakthidevi M, Collura F, Mitchell JA. 2014. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev 28: 2699–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.