

Abstract
Fe-S clusters are ubiquitous cofactors of proteins involved in a variety of essential cellular processes. The biogenesis of Fe-S clusters in the cytosol and their insertion into proteins is accomplished through the cytosolic iron-sulphur protein assembly (CIA) machinery. The early- and middle-acting modules of the CIA pathway concerned with the assembly and trafficking of Fe-S clusters have been previously characterised in the parasitic protist Trypanosoma brucei. In this study, we applied proteomic and genetic approaches to gain insights into the network of protein-protein interactions of the late-acting CIA targeting complex in T. brucei. All components of the canonical CIA machinery are present in T. brucei including, as in humans, two distinct CIA2 homologues TbCIA2A and TbCIA2B. These two proteins are found interacting with TbCIA1, yet the interaction is mutually exclusive, as determined by mass spectrometry. Ablation of most of the components of the CIA targeting complex by RNAi led to impaired cell growth in vitro, with the exception of TbCIA2A in procyclic form (PCF) trypanosomes. Depletion of the CIA-targeting complex was accompanied by reduced levels of protein-bound cytosolic iron and decreased activity of an Fe-S dependent enzyme in PCF trypanosomes. We demonstrate that the C-terminal domain of TbMMS19 acts as a docking site for TbCIA2B and TbCIA1, forming a trimeric complex that also interacts with target Fe-S apo-proteins and the middle-acting CIA component TbNAR1.
Author summary
Cytosolic and nuclear proteins containing iron-sulphur clusters (Fe-S) are essential for the survival of every extant eukaryotic cell. The biogenesis of Fe-S clusters and their insertion into proteins is accomplished through the cytosolic iron-sulphur protein assembly (CIA) machinery. Recently, the CIA factors that generate cytosolic Fe-S clusters were characterised in T. brucei, a unicellular parasite that causes diseases in humans and animals. However, an outstanding question in this organism is the way by which the CIA machinery directs and inserts newly formed Fe-S clusters into proteins. We found that the T. brucei proteins TbCIA2B and TbCIA1 assemble at a region of the C-terminal domain of a third protein, TbMMS19, to form a complex labelled the CIA targeting complex (CTC). The CTC interacts with TbNAR1 and with Fe-S proteins, meaning that the complex assists in the transfer of Fe-S clusters from the upstream members of the pathway into target Fe-S proteins. T. brucei cells depleted of CTC had decreased levels of protein-bound cytosolic iron, and lower activities of cytosolic aconitase, an enzyme that depends upon Fe-S clusters to function.
Introduction
Iron-sulphur (Fe-S) clusters are simple and versatile cofactors involved in a plethora of cellular processes from bacteria to humans and theorised to have formed the ancient surfaces upon which prebiotic chemical reactions took place, laying the ground for the origin of life itself [1,2]. Biogenesis of Fe-S clusters and their subsequent incorporation into polypeptide chains are intricate processes involving dedicated compartmentalised pathways that comprise dozens of proteins [3,4]. At least three such pathways are conserved in eukaryotes, namely the cytosolic Fe-S protein assembly (CIA) machinery, the mitochondrial Fe-S cluster assembly (ISC) system and the plastidial sulphur mobilisation (SUF) system [4–6].
A cytosolic pathway for maturation of Fe-S proteins was first described in the early 2000’s when a genetic screen aimed at the reconstitution of the [4Fe-4S] cluster on human IRP1, also known as cytosolic aconitase, identified the cytosolic P-loop NTPase Cfd1 as essential for the maturation of IRP1 and other cytosolic, but not mitochondrial Fe-S proteins [7]. Since then, at least eight additional proteins (nine in yeast) have been associated with the CIA machinery, which has been implicated in the maturation of a growing list of cytosolic and nuclear Fe-S proteins [4].
The biogenesis of Fe-S proteins can be conveniently simplified in two discrete yet concerted steps: one for assembly of the clusters into a protein scaffold and another for their trafficking/insertion into client proteins. Functional studies have shown that the CIA machinery is highly conserved from yeast to man, and is organised into several sub-complexes that support different stages of the process [8], allowing the components of this pathway to be grouped in a modular fashion as follows: (i) an early-acting module encompassing proteins of the electron transfer chain Tah18 and Dre2 [9], and a heterotetrameric protein scaffold formed by Cfd1 and Nbp35, in which [4Fe-4S] clusters are initially assembled [10,11]; (ii) a middle-acting module, represented by Nar1 [11, 12] and concerned with the transfer and trafficking of the pre-formed Fe-S clusters to (iii) the late-acting or targeting module that facilitates the target-specific insertion of clusters into client proteins [13,14]. In yeast, the CIA targeting complex (CTC) is composed of Mms19, Cia1, and Cia2 [15], while human cells possess two isoforms of Cia2, labelled CIA2A and CIA2B, with the former displaying a notable specificity for the maturation of a subset of client proteins implicated in cellular iron homeostasis, while the latter is involved in canonical Fe-S cluster assembly.
Trypanosoma and Leishmania species are causative agents of human diseases that threaten hundreds of millions of people mostly in developing countries, as well as of major economically important veterinary diseases [16–19]. T. brucei is the best-studied member of the supergroup Excavata [20] serving as a model organism due to its genetic tractability [21–24]. The early- and middle-acting modules of the CIA pathway have been previously characterised in this parasite [25], however, the components of the late-acting part had yet to be studied. In addition to this, the Fe-S proteome of this divergent protist remains vastly unexplored, thus providing an excellent opportunity to study these two biological questions.
In this work, we demonstrate that the late-acting module of the CIA machinery is essential for the survival of this parasite in vitro, but not in vivo. TbCIA2B and TbCIA1 assemble at the C-terminal domain of TbMMS19 to form the canonical ternary targeting complex. Moreover, in both procyclic (PCF) and bloodstream stages (BSF) of T. brucei, binary configurations reminiscent of those observed in human cells were also present. Members of the CTC interacted with client Fe-S proteins and TbNAR1, while depletion of CTC components impaired cell growth and led to decreased protein-bound cytosolic iron levels and aconitase activity.
Results
Identification of CIA-targeting complex and subcellular localisation
Four proteins, termed TbCIA1 (Tb927.8.3860), TbCIA2A (Tb927.9.10360), TbCIA2B (Tb927.8.720) and TbMMS19 (Tb927.8.3920, Tb927.8.3500), were previously identified in T. brucei on the basis of their similarity to yeast and human CTC components [26,27]. Only TbCIA1 has been characterized to date [25]. T. brucei encodes two different MMS19 proteins, sharing 99.6% amino acid identity. As in humans, two genes encoding homologues of yeast Cia2 protein were found in T. brucei. The phylogenetic position of these proteins, designated TbCIA2A and TbCIA2B, has been analysed elsewhere [28].
We determined the subcellular localisation of TbCIA2A, TbCIA2B, and TbMMS19 by indirect immunofluorescence, crude digitonin fractionation and selective permeabilisation with digitonin. Cell lines expressing in situ C-terminally V5- or HA-tagged CIA proteins were produced (see Materials and Methods). Fixed parasites were probed with anti-V5 and anti-enolase antibodies (TbENO) [29] to detect the fusion proteins and the cytosolic marker, respectively. The co-localisation of all V5-tagged proteins with TbENO suggests their cytosolic localisation (Fig 1A). To further confirm this finding, the subcellular distribution of the CIA pathway components was analysed by a fractionation with digitonin. For this, we incubated the cells with a concentration of digitonin that liberates the cytosol, separating it from the mitochondrial fraction (Fig 1B). The signal for all of the CTC components co-localizes with that of the cytosolic marker (Cyt), TbENO. The mitochondrial marker, TbmtHSP70, is only present in the mitochondrial (M) fraction. The pellet (P) denotes the insoluble fraction after solubilizing the mitochondrial fraction, which exhibits proteins that are membrane-bound, such as part of TbmtHSP70. A parallel corroboration was performed by selective permeabilisation with digitonin. In this experiment, equal numbers of cells were incubated with increasing concentrations of the detergent, causing progressive cell membrane permeabilisation and sequential release of the cytosolic and organellar fractions. TbCIA2B-HA and TbMMS19-HA were co-released with the cytosolic control phospholipase A1 (TbPLA1) [30], while the trypanosome alternative oxidase (TbTAO), which served as a mitochondrial marker [31], was released only at higher detergent concentrations (Fig 1C). Taken together, immunofluorescence and detergent-based cell fractionation identified TbCIA1, TbCIA2A, TbCIA2B and TbMMS19 as cytosolic proteins.
Fig 1. The CIA targeting complex is localised in the cytosol of T. brucei.

(A) Confocal microscopy of PCF T. brucei cells expressing in-situ V5-tagged CIA components. Anti-V5 antibody (green) was used to detect the CIA proteins localized throughout the cell body. Enolase (red) was used as a cytosolic marker. DAPI (blue) stained DNA. Scalebar 1 μm. The merge displays co-localization of enolase with the V5-tagged proteins. (B) Isolation of mitochondrial fraction with digitonin. PCF trypanosomes were incubated with 0.4% (w/v) digitonin and fractions were separated by centrifugation. V5-tagged targets were visualized with anti-V5 monoclonal antibody. MtHSP70 and enolase were used as mitochondrial and cytosolic markers, respectively. P = pellet; M = mitochondrial fraction; Cyt = cytosolic fraction. All methods indicated that the proteins of the CIA targeting complex are present in the cytosol of PCF T. brucei.(C) Selective permeabilisation of whole PCF T. brucei cells with digitonin: supernatants of cells incubated with increasing amounts of digitonin were assessed by Western blot. Samples were probed against -HA (CIA components) and organelle markers: TbPLA1 (cytosolic marker); TbTAO (mitochondrial marker). The graphs represent densitometric quantifications of the Western blots for each experiment. Cells permeabilised with the highest concentration of digitonin were used as 100% release control.
Essentiality and functional analysis of the CIA targeting complex
Analysis of the function of the putative CTC members was carried out in uninduced and RNAi-induced PCF and BSF cell lines. The efficiency of the RNAi knockdowns was monitored for up to 8 days in the PCF and 6 days in the BSF and was further assessed by Western blot analysis (Fig 2A–2F and 2J–2O). While the growth rate of TbMMS19 RNAi in the BSF was mostly unaffected upon depletion, the same downregulation in the PCF exhibited considerable growth impairment (Fig 2A and 2J). On the other hand, the BSF TbCIA2A RNAi cell line showed a mild growth phenotype (Fig 2K), whereas in PCF this downregulation does not affect the growth rate (Fig 2B). Two days after the downregulation of TbCIA2B, a decrease in the growth of the PCF was observed (Fig 2C), but this effect was less pronounced in the BSF (Fig 2L). We have previously shown that depletion of the scaffold proteins TbCFD1 and TbNBP35 caused mild to severe growth impairment in PCF and BSF, but knocking down the expression of individual components upstream of the CTC did not affect the growth rate [25]. However, stringent pairwise knockdowns of the early-acting components of this pathway (e.g. TbTAH18 and TbDRE2) caused marked growth defects [25], suggesting an interaction of CIA factors, which only becomes critical upon simultaneous RNAi knockdown of more than one of them. We sought to employ this phenomenon by silencing the expression of two CTC members simultaneously. However, the observed phenotype of double knockdowns in PCF (TbCIA1-TbCIA2B and TbCIA2A-TbCIA2B) was no more pronounced than the phenotype observed following the depletion of TbCIA2B alone (S1 Fig).
Fig 2. The CIA targeting complex is essential for the cell growth of T. brucei and activity of cytosolic aconitase.

Growth curves of RNAi cell lines in PCF T. brucei of TbMMS19, TbCIA2A, and TbCIA2B (A-C), induced (Tet +) and uninduced (Tet -) with tetracycline (n = 3 ± SD). Western blots shown under each growth curve were probed with anti-HA, anti-V5 or specific antibodies and were used to assess protein expression before and after RNAi induction (D-F). Anti-tubulin or anti-enolase antibodies were used as loading controls. The activity of the Fe-S dependent enzyme TbACO was measured in cytosolic and mitochondrial fractions (G-I) of the above-mentioned cell lines (n = 3 ± SD). Growth curves for BSF RNAi cell lines of TbMMS19, TbCIA2A, and TbCIA2B (J-L, n = 3 ± SD). Western blots assessing downregulation of each BSF RNAi cell lines (M-O).
To assess the role of the CTC components on the pathogenicity of T. brucei, we infected mice with BSF RNAi cell lines of TbMMS19 and TbCIA2B. As shown in S2 Fig, these infection experiments suggest that neither protein is essential in BSF, in agreement with the mild in vitro growth phenotypes described above, as well as with the initial observation of the double-knockdown cell lines in this life stage, in which at least two components of the pathway had to be ablated in order to obtain a clearer growth phenotype (S1 Fig) [25]. We next asked whether depleting the cells of individual CTC members would impact the activity of known Fe-S proteins. Aconitase (TbACO), a Fe-S enzyme that catalyses the reversible isomerisation of citrate to isocitrate, is encoded by a single gene and has a dual subcellular localisation, being ca. 70% in the cytosol and 30% in the mitochondrion [32,33]. These features qualify it as a suitable surrogate for Fe-S cluster-dependent enzymatic activity in these two cellular compartments [27]. As shown in Fig 2G and 2I, cytosolic TbACO activity was reduced in 60% and 40%, when TbMMS19 and TbCIA2B were knocked down, respectively, whereas the mitochondrial activity remained unchanged. Furthermore, the depletion of TbCIA2A did not affect aconitase activity in these cellular compartments (Fig 2H). Hence, TbMMS19 and TbCIA2B seem to be required for the maturation of this Fe-S protein, providing a functional link between the CTC and the transfer of Fe-S clusters to target proteins.
Several lines of evidence have linked the pathways for Fe-S cluster biogenesis to DNA repair processes in humans, yeast, and plants [13,14,34–37]. Surprisingly, even efficient depletion of the CTC members did not affect the ability of T. brucei to cope with DNA damage caused by various genotoxic agents as determined by Alamar blue assays, and in some cases the EC50 was in fact higher for the CIA-depleted parasites (S1 Table).
Since TbMMS19 and TbCIA2B exhibited essentiality in T. brucei (Fig 2A and 2C), we addressed the influence of these CTC components on the iron metabolism of the parasite. For this purpose, we used deferoxamine (DFO), a siderophore that chelates Fe3+ but has no effect on iron bound to either haem or transferrin [38] and which starves the cells by sequestering the labile iron pool [39–41]. TbCIA2B RNAi cell lines were induced with tetracycline (Tet) for 24 hours and then challenged with different concentrations of DFO for 2 or 3 days (PCF and BSF, respectively), when cell proliferation was measured. When depleted of TbCIA2B, both life stages were significantly more susceptible to DFO compared to those with normal levels of this protein (Fig 3A and 3C), suggesting a decrease in the pool of available intracellular iron. A similar effect was observed upon TbMMS19 knock down in PCF cells (Fig 3E). Additionally, WT PCF cells grown in the presence of Tet and treated with DFO under the same conditions displayed identical EC50 values as those grown in the absence of the antibiotics (Fig 3I), confirming that this result was specifically due to TbCIA2B or TbMMS19 knockdown and not to the synergistic effects of DFO and Tet, which is also a chelator of polyvalent metal cations [42]. In agreement with this finding, no DFO toxicity was observed when TbCIA2B or TbMMS19 RNAi parasites were treated with drug pre-saturated with an excess of Fe3+ (Fig 3B, 3D, 3F and 3H), strongly indicating that the enhanced sensitivity can be specifically attributed to iron depletion and not to off-target effects of DFO. Furthermore, ferene assays suggested that the content of iron bound to proteins in the cytosolic lysates of TbCIA2B knockdowns was lower than that found in uninduced cells, whereas protein-bound iron levels did not change in the organellar fractions (Fig 3J).
Fig 3. Knockdown of CIA members affects iron levels and sensitivity to iron depletion.

Wild type (WT), TbCIA2B, and TbMMS19 RNAi cells were grown without (blue, Tet -) or with (red, Tet +) tetracycline for 24 hours and then treated with different concentrations of deferoxamine (DFO). After 2 or 3 days of incubation (PCF and BSF parasites, respectively), cell growth was measured by the Resazurin method for determination of EC50s. Representative DFO concentration-response curves are shown in (A) TbCIA2B PCF, (C) TbCIA2B BSF, (E) TbMMS19 PCF, (G) TbMMS19 BSF, or (I) WT PCF. Representative plots of DFO pre-incubated with an excess of iron before adding to (B) TbCIA2B PCF, (D) TbCIA2B BSF, (F) TbMMS19 PCF, (H) TbMMS19 BSF. The values shown in the inset of the curves are the mean DFO EC50s for induced or uninduced cultures. The bar charts on the right side of the curves are the mean ± SEM EC50s of 3 independent experiments performed in quadruplicate. ns = non-significant; * = p< 0.05; *** = p<0.001 (two tailed paired t test). (J) PCFTbCIA2B RNAi cells were grown for 4 days in the presence (red) or absence (blue) of tetracycline and the content of iron bound to proteins was measured in the cytosolic and organellar fractions of digitonin permeabilised parasites. The purity of the cellular fractions was validated by Western blot using anti-HA (TbCIA2B-HA), anti-TbPLA1 (cytosolic marker), or anti-TbTAO (mitochondrial marker).
Yeast complementation assay
For functional complementation assays, TbCIA2A, TbCIA2B and TbMMS19 were PCR-amplified from genomic DNA and cloned into yeast expression vectors under the control of the TDH3 or MET25 promoters of Saccharomyces cerevisiae [25,43]. Plasmids without insert or plasmids encoding endogenous yeast CIA genes were used as controls. Subsequently, these constructs were transformed into regulatable yeast Gal-CIA mutants, in which the expression of the cognate CIA gene is induced in the presence of galactose and repressed by the presence of glucose as described elsewhere [25]. The growth defect of Mms19-depleted cells on glucose-containing medium was not restored by TbMMS19 expression, even when TbMMS19 was co-expressed with either TbCIA2A or TbCIA2B (Fig 4A). Expression of TbCIA2B partially rescued the growth of Cia2-depleted cells, but TbCIA2A failed to do so (Fig 4B). For both TbCIA2 proteins, co-expression with TbMMS19 not only failed to enhance the rescue, but exhibited a dominant negative phenotype (Fig 4). Interestingly, when TbMMS19 is co-expressed with Cia2 from S. cerevisiae (Fig 4B), the same dominant negative-like effect is observed. These findings show that TbCIA2B can partially take over the role of its yeast counterpart, suggesting that it performs an orthologous function.
Fig 4. TbCIA2B, but not TbCIA2A, functionally replace yeast homologue ScCia2.

Plasmids p424, p426 or p416, empty (Ø), or with the indicated genes, under the control of the strong promoters MET25 or TDH3, and the natural promoter (NP) of S. cerevisiae CIA2 were transformed into W303 cells, strains GalL-MMS19 (A) and Gal-CIA2 (B). Cells were grown for 16 h in liquid minimal medium supplemented with glucose (2%). After washing, 10-fold serial dilutions were spotted onto agar plates containing minimal medium supplemented with galactose or glucose and incubated at 30°C for 2 days. The result was reproduced at least three times with independent transformations.
Protein-protein interactions of the CTC
Individual interactions of the CTC proteins had only been mapped in detail for a few representatives of the eukaryotic supergroup Opisthokonta [44]. Moreover, the progress made in the field of Fe-S biology in the past decade suggests Fe-S proteins are diverse and abundant in a typical eukaryotic cell, but remained overlooked due to the difficulties related to their instability under aerobic conditions. To the best of our knowledge, the dynamics of protein-protein interactions of the CTC had not been studied in any Excavata, with only a few examples of identification and functional studies of CTC components [45]; despite ~0.6% of the annotated proteins of T. brucei being predicted to contain Fe-S clusters [46–48], its Fe-S proteome remains largely unexplored.
One of the most valuable tools that contributed to expanding the list of mammalian Fe-S proteins was the use of mass spectrometry (MS) and affinity purifications to detect potential Fe-S proteins interacting with the human CIA targeting complex [13,14,49]. Therefore, aiming to gain insight into the composition of the T. brucei CTC and its interactions with cytosolic and nuclear Fe-S proteins, three complementary strategies for affinity purification/MS were devised: (i) in situ PTP-tagged CTC members in PCF were affinity purified by a two-step approach [50], (ii) V5-tagged CTC members in PCF and iii) V5-tagged CTC members in BSF were immunoaffinity purified using a technique suited for the detection of transient and/or weak interactions [51]. In all cases, MS detected proteins co-purifying with the tagged baits.
Tandem affinity purifications were performed using PTP-TbCIA1, PTP-TbCIA2B, or TbMMS19-PTP as baits, and mock purifications with wild type PCF parasites served as negative controls. Unfortunately, we were not able to purify PTP-TbCIA2A complexes by this method for reasons that remain unclear, but may be related to the PTP tag (~19 kDa) being larger than TbCIA2A, which is a protein of ~17 kDa. SYPRO Ruby stained SDS-PAGE gels of the final PTP elutions are shown in Fig 5A–5D. This exercise revealed that PTP-tagged CTC components can be reciprocally co-purified, proving the existence of the canonical ternary complex (TbCIA1-TbMMS19-TbCIA2B). In TbCIA2A-V5 pull-down assays (S3 and S4 Tables), TbCIA1, but not TbCIA2B or TbMMS19, was found interacting with the bait protein, in a configuration reminiscent of that described for the human CTC [13–15]. Abundant proteins such as tubulins and the eukaryotic elongation factor 1α (EF1α) were present in control PTP purifications, but no detectable levels of CIA proteins were seen in these samples (Fig 5A). A summary of the PTP/MS data for the co-purified CTC members is shown in S2 Table. In addition to this, affinity pull-downs with V5-tagged TbCIA1, TbCIA2A, TbCIA2B, and TbMMS19 in PCF and BSF confirmed the reciprocal nature of the interactions and the presence of the similar complex configurations in both life stages (S3 and S4 Tables), and also showed in PCF that TbCIA1, TbCIA2A and TbMMS19, but not TbCIA2B, co-immunoprecipitated with TbNAR1, the upstream CIA component that mediates the transfer of Fe-S clusters from the early-acting part of the pathway to the CTC (S3 Table). Moreover, co-IP performed with lysates of a double-tagged strain of PCF parasites co-expressing PTP-TbNAR1 and TbMMS19-HA further validated this interaction (Fig 5E).
Fig 5. Protein-protein interaction profile of the CIA targeting complex.

(A)-(D) Tandem affinity purification from WT parasites (mock) or PTP-tagged CIA components. Eluates were resolved in Bis-Tris gels and sections were analysed by mass spectrometry for protein identification. (E) Co-IP of PTP-TbNAR1 with TbMMS19-HA. Lysates of double tagged PCF parasites were incubated with IgG-Sepharose and the bound material subjected to SDS-PAGE and immunostaining with anti-HA and anti-protein A (PTP) antibodies. (F) Pull-down of TbFHc-HA from PCF T. brucei extracts by GST alone, rTbMMS19-NTD, rTbMMS19-CTD. (G) Pull-down of TbXPD-HA, or TbACO-HA from T. brucei PCF extracts by HIS-tagged rTbCIA2B. The bound material of the pull-downs was resolved by SDS-PAGE and immunostained with anti-HA, anti-Strep Tag II, or anti-HIS antibodies. (H) Schematic representation of the protein-protein interaction profile of the T. brucei CIA targeting complex. The shaded blue bubble represents the identified members of the canonical CIA targeting complex and the green bubble represents the binary complex formed by TbCIA2A-TbCIA1.
Next, in an attempt to identify potential target Fe-S proteins, we investigated other proteins co-eluting with members of the CTC. The combined TAP/MS and co-IP experiments identified over 200 such proteins, most of them in association with TbMMS19 and/or TbCIA1 (S5 Table). To inquire if these proteins were known or could be predicted to contain Fe-S clusters, the amino acid sequences of the hits were retrieved from the TriTryp database [46] and analysed with MetalPredator [48], a tool to predict Fe-S clusters in polypeptide chains based on the presence of known Fe-S domains and metal-binding motifs. This analysis returned three positive hits: the catalytic subunit of Pol δ (TbPOLD1, Tb927.2.1800), the class I cytosolic fumarate hydratase (TbFHc, Tb927.3.4500), and a putative radical SAM tRNA modification enzyme (Tb927.6.3510) (S5 Table).
In order to further examine the interactions of the CTC with Fe-S proteins, the amino- or carboxy-terminal domains of TbMMS19 (respectively, recombinant (r) TbMMS19-NTD and rTbMMS19-CTD) were expressed in E. coli as GST-Strep-Tag II fusion proteins. Equimolar amounts of purified recombinant proteins or glutathione S-transferase (GST), used as a negative control, were coupled to glutathione Sepharose 4B beads, incubated with soluble extracts of PCF parasites expressing HA-tagged TbFHc, and the interactions were assessed by Western blotting. This pull-down confirmed the interaction detected by TAP/MS and further showed that TbFHc was able to interact with rTbMMS19-NTD, but not rTbMMS19-CTD or GST alone (Fig 5F). Also, a relatively low number of proteins were detected in PCF TAP/MS or V5 co-IP/MS experiments with TbCIA2B (S5 Table). In order to verify possible protein-protein interactions of TbCIA2B that could not be detected by other methods, this protein was expressed in E. coli as a fusion with an N-terminal Strep-Tag II and a C-terminal hexahistidine tag (rTbCIA2B), then immobilised to Strep-Tactin Sepharose and incubated with extracts of parasites expressing either tagged aconitase (TbACO-HA) or the DNA helicase XPD (Xeroderma pigmentosum group D homologue,TbXPD-HA). As shown in Fig 5G, TbXPD interacts with rTbCIA2B. Moreover, TbACO also interacted with rTbCIA2B (Fig 5G). This is in accordance with the results depicted in Fig 2G and 2I, where the silencing of TbCIA2B led to decreased cytosolic activity of TbACO. These results validate the position of the CTC as the late-acting module of the CIA machinery at the interface between the upstream TbNAR1 and the client Fe-S proteins.
A summary of interactions detected by TAP/MS experiments and additionally confirmed by co-IPs is depicted in Fig 5H. The interaction with TbNAR1 was not observed in the V5-tagged co-immunoprecipitations performed in BSF trypanosomes (S4 Table). This difference may reflect stage-specific requirement of the CIA pathway. Importantly, mutual interactions of the CTC members are the same in the BSF and PCF cells, although the sets of their targets differ from one another and require further analysis to determine their capability to bear an Fe-S cluster.
Interestingly, several proteins captured by the PCF TAP/MS methodology show multiple clustered cysteine residues, including Cys-Pro dipeptide sequences (Tb927.3.4360, Tb927.7.4390, Tb927.2.5130 and Tb927.8.4890). We are currently pursuing the possibility that these proteins harbour an Fe-S cluster by heterologous expression and purification.
The CTC is assembled at the C-terminal domain of TbMMS19
Aiming to better understand the dynamics of the interactions amongst the CTC members, it was of interest to identify the site through which members of this complex interact. However, the amino acid sequence of TbMMS19 is poorly conserved when compared to its human or yeast homologues [52], and no crystal structures of this protein have been elucidated so far. Nevertheless, in silico homology modelling of the tertiary structure of TbMMS19 using the Phyre2 server [53,54] suggested the overall architecture of an Armadillo-like protein that contains α-helical HEAT repeat motifs throughout its sequence [55,56], as previously predicted for MMS19 in higher eukaryotes [57]. In human cells, CIA2B and CIAO1 interact with the tightly spaced HEAT repeats at a region of the MMS19 C-terminal domain [58], whereas in TbMMS19 these motifs seem to be more loosely distributed and are more numerous (S3 Fig). We hypothesised that the binding site for the CIA proteins would be different in trypanosomes, given the divergent amino acid composition and apparent different distribution of the repeats.
To clarify this question, TbCIA2B-HA cell lines of PCF parasites were transfected with constructs derived from the pLEW82 vector that integrates into the non-transcribed spacer of the rDNA locus and allows strong ectopic overexpression of proteins in the presence of Tet [21]. We engineered cell lines in which the PTP-tagged full-length TbMMS19 (PTP-TbMMS19), its N-terminal (PTP-TbMMS19-NTD) or C-terminal domain (PTP-TbMMS19-CTD) can be conditionally overexpressed. Both the overexpression of the control PTP tag alone and PTP-TbMMS19 did not impact the cell growth (Fig 6A and 6B, respectively). However, overexpression of PTP-TbMMS19-NTD caused a mild cell growth delay (Fig 6C), whereas a prominent dominant-negative phenotype developed when excess PTP-TbMMS19-CTD was produced, causing near-arrest of the cell growth after two days in culture (Fig 6D). Interestingly, overexpressing PTP-TbMMS19 or PTP-TbMMS19-CTD for two days resulted in ~2 and ~3-fold higher levels of TbCIA2B-HA, respectively, and this effect was sustained after 4 days of overexpression (Fig 6E, 6G and 6H). Conversely, in cells induced to overexpress PTP-TbMMS19-NTD, the levels of TbCIA2B-HA remained unaltered (Fig 6F and 6H).
Fig 6. Overexpression of the C-terminal domain of TbMMS19 is detrimental for cell growth and increases TbCIA2B levels.

Growth curves of PCF T. brucei carrying an in situ HA-tagged copy of TbCIA2B and overexpressing an ectopic inducible copy of (A) PTP tag, (B) PTP-MMS19, (C) PTP-MMS19-NTD, or (D) PTP-MMS19-CTD. Cell numbers were assessed in the presence (Tet +) or absence (Tet -) of tetracycline in the culture medium for the specified number of days. Data points represent the mean ± SD of 2 independent experiments. (E)-(G) Parasites were grown for 2 or 4 days in the presence or absence of tetracycline. Total cell lysates were probed by Western blot using anti-Protein A (PTP-tag), anti-HA and anti-tubulin antibodies. (H) Protein expression was calculated by densitometry and the HA/tubulin ratio in induced cells was normalised to the respective ratio of uninduced cultures (dashed line). Bars represent the mean ± SEM of two experiments.
To investigate if the up-regulation of TbCIA2B by TbMMS19 or its C-terminal domain was dependent on their interaction, we performed co-IPs with extracts of parasites induced overnight to overexpress PTP-tagged TbMMS19, TbMMS19-NTD or TbMMS19-CTD. Cells overexpressing only the PTP-tag or those not transfected with pLEW82 constructs were used as controls. Although lower levels of PTP-TbMMS19-NTD were observed when compared to those achieved for PTP-TbMMS19 or PTP-TbMMS19-CTD after induction, co-IP assays indicated that TbCIA2B-HA was able to bind PTP-TbMMS19 and PTP-TbMMS19-CTD, but not PTP-TbMMS19–NTD (Fig 7B). Additional TAP/MS assays revealed that TbCIA1 was only detected in eluates from PTP-TbMMS19 and PTP-TbMMS19-CTD, but not PTP-TbMMS19-NTD (S4 Fig), which implicates that TbMMS19-CTD acts as a docking site for the assembly of the ternary complex.
Fig 7. The C-terminal domain of TbMMS19 is the binding site of TbCIA2B.

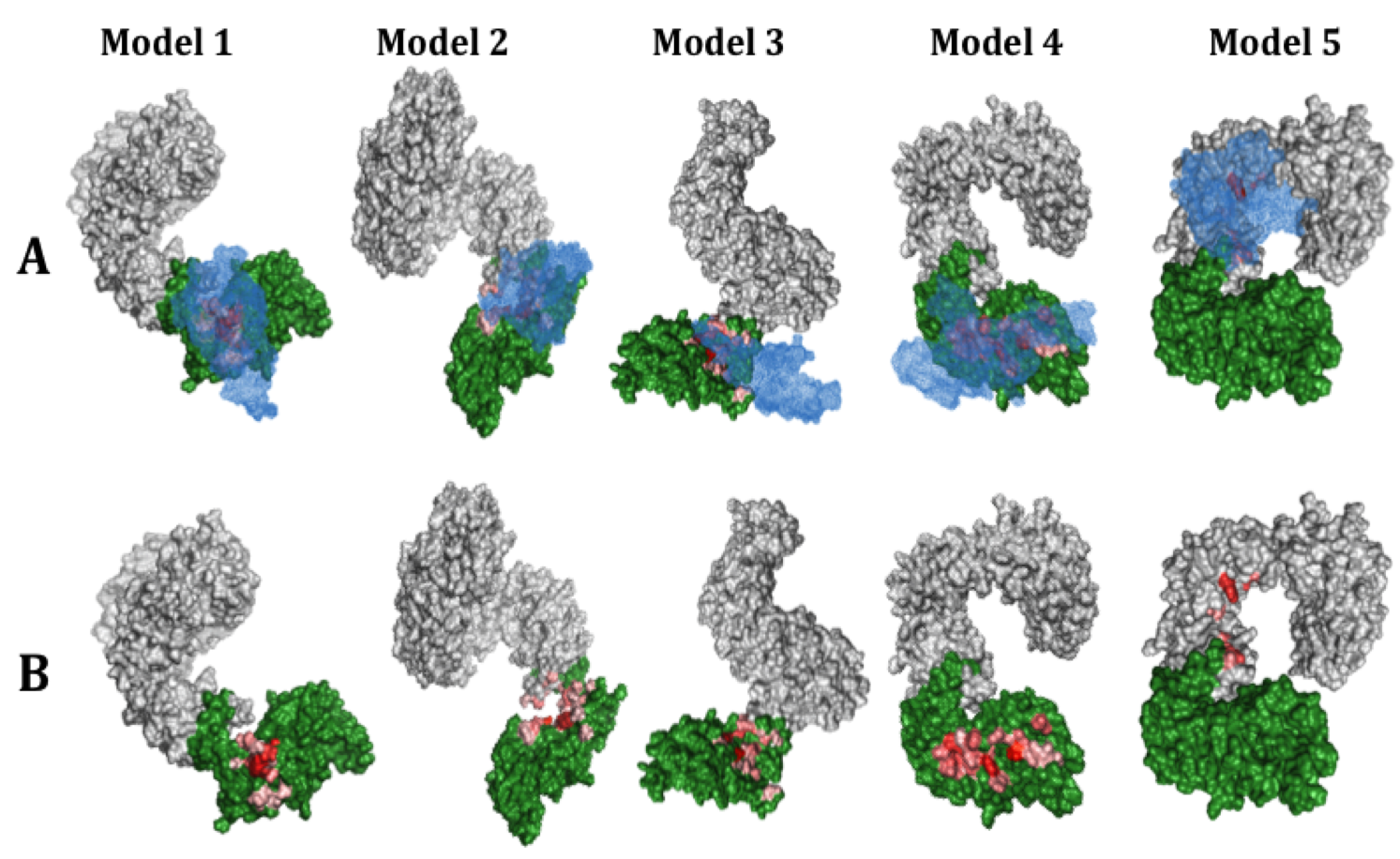
(A) Schematic representation of T. brucei MMS19. The N-terminal domain is depicted in blue and the C-terminal domain in red. Grey boxes show the position of the 11 HEAT repeats identified in TbMMS19. C1-C5 represent the position of GST/Strep-tagged recombinant fragments of TbMMS19-CTD used in pull-down experiments. (B) Co-IP of TbCIA2B with TbMMS19. Lysates of TbCIA2B-HA cells overexpressing the PTP tag (empty vector), PTP-TbMMS19, PTP-TbMMS19-NTD, or PTP-TbMMS19-CTD were incubated with IgG Sepharose. The bound proteins were subjected to Western blot and probed with Anti-HA and Anti-Protein A (PTP) antibodies. Identification of the TbCIA2B binding site at TbMMS19. Cell extracts of T. brucei expressing HA-tagged TbCIA2B (C), or rTbCIA2B (D) were incubated with GST/Strep-tagged fragments (C1-C5) immobilised in glutathione Sepharose beads. Bound material was resolved by SDS-PAGE and immunoblotted with anti-HA or anti-HIS tag antibodies as indicated. (E) The 3D structures of TbMMS19 and TbCIA2B were modelled by homology using the Phyre2 server. The grey and green surfaces represent respectively TbMMS19-NTD, and TbMMS19-CTD while the blue ribbon corresponds to TbCIA2B. The TbMMS19-TbCIA2B complex was modelled in silico with ClusPro and correctly predicted that TbCIA2B binds to TbMMS19-CTD. This theoretical complex was analysed with PredHS to identify TbMMS19 residues at the surface of interaction with TbCIA2B. The surface of interaction is represented in a red scale according to their PredHS SVM-Hot spot score. Labelled residues at the inset had the highest SVM-Hot spot scores. PyMol (Schrödinger, LLC) was used to visualise and generate the figures. The binding site of TbCIA2B was found to be in a region comprising ~185 amino acids at the C-terminal domain of TbMMS19.
To pinpoint the binding site of TbCIA2B within TbMMS19-CTD, we used recombinant fragments of the latter (named C1-C5, Fig 7A), expressed as GST-Strep-Tag II fusions. Equimolar amounts of C1-C5 or GST were bound to glutathione Sepharose 4B beads and incubated with cellular extracts of T. brucei expressing HA-tagged TbCIA2B (Fig 7C) or purified rTbCIA2B (Fig 7D). Fragments C1-C4 were able to bind TbCIA2B-HA from cell lysates, although fragment C2 appears to bind with higher affinity, while the C5 fragment, containing only 1 HEAT domain, has very weak affinity (Fig 7C). Moreover, fragments C1, C2, C4 and C5 captured purified rTbCIA2B and also in this case, C2 displayed the highest binding capacity (Fig 7D). Curiously, C1 was less capable of binding to TbCIA2B than the smaller C2 or C3 fragments. One possible explanation is that C1 adopted a fold that hinders the ability of TbCIA2B to reach the interaction surface which could, in turn, be more accessible in C2. We also observed that the C3 and C4 fragments have an enhanced ability to bind TbCIA2B in cell extracts in comparison to the purified recombinant protein, hinting at the presence of a factor that stabilises the complex. The C5 fragment interacted (albeit weakly) with recombinant and native TbCIA2B (Fig 7C and 7D), suggesting that this repeat is the minimal structural unit necessary to form the TbMMS19-TbCIA2B complex. This fragment contains 50 amino acids that roughly correspond to the most C-terminal HEAT repeat in TbMMS19, although such interaction likely spans a much larger contact surface, involving at least two HEAT repeats localized between the residues Val763 and Lys947 of TbMMS19.
Given the paucity of structural analyses for the CIA proteins, individually or in a complex, the 3D structures of TbMMS19 and TbCIA2B were modelled by homology using the Phyre2 server [53]. The predicted structures were subsequently used to generate models of protein-protein interaction by in silico docking with ClusPro [59]. Corroborating our experimental findings, the top scoring model for the TbMMS19-TbCIA2B complex correctly predicted that TbCIA2B should bind to TbMMS19–CTD (Fig 7E). In fact, most of the top scoring models also pointed to the binding site of the C-terminal domain of TbMMS19 (S5 Fig). Given this reassuring overlap between the experimental data and in silico predictions, we aimed to refine this analysis by examining the best complex model with PredHS, a tool that integrates analysis of structural and energetic properties to identify regions at the contact surface, which are more likely to be crucial for protein-protein interactions (i.e. hot spots or hot regions of interaction) [60,61]. This analysis suggested that although Thr808 seems to be important, a contiguous region of 12 amino acids in the TbMMS19-CTD (Phe762-Thr773) could be essential for the interaction with TbCIA2B. These residues are depicted in a scale of red in Fig 7E. This model fits satisfactorily our experimental data, which indicated that the C2 fragment (Val763-Lys947) binds tightly to rTbCIA2B, while the C3 fragment (Thr808-Lys947) interacted (very) weakly with it, although this association was stabilised when the native protein was present in cell lysates. Collectively, these results indicate that TbCIA2B binds directly and tightly to the C-terminal domain of TbMMS19, but this interaction is likely to require additional factors to stabilise the complex.
Discussion
Since the subcellular localisation of the CIA components seems to depend upon the organism under study [34,62–66], we aimed to clarify the cellular compartment in which the late-acting module of the CIA machinery was present in trypanosomes. For this aim, a combination of immunofluorescence and immunoblot analyses of detergent-permeabilised cell extracts localised all four studied proteins to the cytosol, in agreement with data from mammalian cells [13,15,67]. However, Mms19 in Schizosaccharomyces pombe, and Cia1 in S. cerevisiae are predominantly nuclear [34,68]. In the plant Arabidopsis thaliana, MMS19 is exclusively cytosolic, although other members of the CTC exist both in the nuclear and cytosolic compartments [66]. Moreover, Giardia intestinalis exhibits a dual localisation of Cia2, between the intermembrane space of the mitosome and the cytosol [45].
TbMMS19 and TbCIA2B were shown to be essential for the survival of PCF, but their depletion exhibited only marginal defects in BSF trypanosomes. In human cells, the levels of CIA2B are greatly reduced when MMS19 is ablated [10,12,49], yet MMS19 remains steady regardless of the absence of CIA2B, suggesting a tight regulation of CIA2B rather than reciprocal stabilisation between the interacting partners, since MMS19 prevents proteasomal degradation of CIA2B in a binding-dependent manner [49]. Interestingly, overexpressing the C-terminal domain of TbMMS19 produced a dominant-negative phenotype with severe defects on the cell growth and concomitant up-regulation of the TbCIA2B levels. One plausible explanation for this finding concerns the modes of interaction within the CTC, since the C-terminal domain of TbMMS19 appears to be the docking site of the targeting complex, as recently described also for human cells [58]. It is possible that high levels of this truncated protein can sequester TbCIA2B, TbCIA1, as well as client proteins into non-functional complexes, thus depleting the cell of at least two CTC members and mimicking the effect of a double knockdown. On the other hand, the depletion of TbCIA2A does not affect PCF, and only has a mild effect in BSF. Though the MS data suggests non-redundant functions, such as the formation of different subcomplexes among various components of the CTC, the growth phenotype in the RNAi cell lines, as well as the capability of infection of BSF RNAi cell lines, hint at the possibility of function overlapping. However, residual proteins escaping RNAi knockdown may be sufficient to maintain the functionality of the CIA machinery.
The effect of RNAi-mediated depletion of the late-acting CIA factors was monitored through the activity of TbACO. The CIA2A protein aids the maturation of iron regulatory protein 1 (IRP1), the human homologue of TbACO, and stabilizes IRP2 by Fe-S independent mechanisms, whereas CIA2B has a role in the maturation of numerous cytosolic and nuclear Fe-S proteins [15]. Conversely, the CIA proteins do not exert a direct impact on iron regulation in S. cerevisiae, and an IRP1-like mechanism has not been implicated in T. brucei iron regulation [32,69] Regardless, TbMMS19 and TbCIA2B were found to be essential for the activity of the cytosolic but not the mitochondrial fraction of this enzyme. This is in line with previous studies, which demonstrated that the mitochondrial pool of this enzyme is matured by the ISC pathway, the mitochondrial machinery for Fe-S biogenesis [70–72], while the cytosolic fraction requires both the ISC and CIA machineries to obtain its cluster [25,73]. Furthermore, TbACO was shown to interact with TbCIA2B in dedicated pull-down assays. The growth complementation of Cia2-depleted yeast cells by TbCIA2B presents independent evidence for the functional conservation of this protein in the CTC. Taken together, the functional and physical interactions of the CTC with TbACO provide an example of a maturation mechanism of cytosolic Fe-S proteins in T. brucei.
We observed that upon silencing of TbCIA2B or TbMMS19, PCF cells displayed an enhanced sensitivity to the iron chelator deferoxamine, with EC50 values about 1.5 times lower than in uninduced controls. The specificity of this effect was confirmed by incubating trypanosomes with deferoxamine pre-saturated with iron, which abolished its toxicity. Furthermore, BSF parasites depleted of TbCIA2B also displayed equally lower EC50 values. This effect was consistent, although not as pronounced as in conditional null mutants of the cation channel mucolipin 1 that delivers iron to the cytosol of BSF flagellates [74]. Although IRP1-like mechanisms implicating the CIA machinery in iron sensing and regulation, such as those described in human cells [15], seem unlikely to exist in T. brucei [32,69], a role for unknown Fe-S cluster-containing factors in iron regulation cannot be completely ruled out. Deferoxamine acts by scavenging the cellular labile iron pool (LIP), thus preventing incorporation of this element into the newly synthesised apo-proteins [75]. The precise composition of LIP is uncertain, but free iron is seldom present in the intracellular milieu, given its capacity to generate reactive oxygen species via the Fenton reaction [26,76]. The source of iron for the assembly of Fe-S clusters in the cytosol remains unknown, although one line of thought speculates that the scaffold proteins for Fe-S cluster assembly can bind LIP directly [77]. If this was the case, LIP depletion by deferoxamine would magnify an already impaired CIA function in cells depleted of TbCIA2B or TbMMS19, thus explaining the increased sensitivity. The LIP is expected to account for 0.2 to 3% of total cellular iron, with its bulk bound to the cytosolic and/or mitochondrial proteins [78]. Lower levels of protein-bound iron were observed in the cytosol of PCF flagellates depleted of TbCIA2B but remained unchanged in organellar fractions, indicating that Fe-S proteins may comprise a considerable portion of the cytosolic iron content in T. brucei.
Collectively, these data demonstrate that the CTC is essential for the survival of T. brucei in vitro but does not seem to have an influence on the pathogenicity of the parasite in in vivo mouse experiments. The CTC further functions in both the iron metabolism and the maturation of target Fe-S proteins. However, the processes of DNA damage repair appear to be more resilient to the depletion of the CTC in this excavate protist when compared to other eukaryotic systems, where they are strongly linked to the functionality of Fe-S assembly pathways [13,14,35–37]. This observation can be partially attributed to the unique mechanisms of nucleotide excision repair (NER) utilised by this parasite. In yeast and humans, XPD is part of the transcription factor complex TFIIH [79]. Along with XPB, XPD forms the core of this complex, acting together in transcription initiation and DNA repair [79]. In T. brucei, TbXPD and TbXPB are not part of the same complex [80], nor do they respond to DNA damage in the same fashion [80,81]. Moreover, XPB exhibits two orthologues in this flagellate, known as TbXPB and TbXPB-R (or TbXPBz), of which only the latter seems to be involved in NER independently of TFIIH [80,81]. Yet, contrary to yeast and humans, TbXPD knock-downs in PCF and BSF exhibit different growth phenotypes, the protein does not influence NER proficiency and seems to be mostly involved in transcription initiation [80,81]. The genetic, functional and physical interactions of XPD (Rad3 in yeast) with the late-acting members of the CIA machinery have been well described in various organisms, and the ternary CTC is necessary for efficient maturation of this protein [12,35,36,82]. Interestingly, TbXPD was undetectable in our TAP/MS and V5 co-IP/MS assays, which could suggest a lower affinity of this transient association with the CTC in T. brucei than that observed for its human and yeast counterparts, although an interaction with TbCIA2B was seen in a dedicated pull-down assay. It is also plausible that down-regulation of the CIA machinery can trigger compensatory mechanisms of DNA repair, which are Fe-S independent. An alternative explanation is that residual levels of the CTC components upon RNAi knockdown may be sufficient to maintain adequate levels of maturation of Fe-S proteins involved in DNA repair.
We used a combination of TAP/MS, co-IP/MS and dedicated pull-downs to detect potential client Fe-S proteins of the CTC. This approach validated the interactions amongst the late-acting members of the CIA machinery. A relatively large number of proteins was found in (transient) association with the CTC, with only a few of them predicted to contain Fe-S clusters. These analyses revealed that the three core components of the canonical ternary CTC could indeed be reciprocally co-purified showing that the CTC is conserved to both life stages of T. brucei. Interestingly, in both PCF and BSF cells, TbCIA2A was only observed in complexes purified from PTP-TbCIA1 or TbCIA1-V5, while TbMMS19 and TbCIA2B were not detected in co-IPs with V5-tagged TbCIA2A (S2, S3 and S4 Tables). Consistent with our findings, CIA2A was not reported as a core CTC member in the seminal studies that established the role of the ternary complex CIAO1-MMS19-CIA2B in the maturation of Fe-S proteins [13,14]. However, in HeLa cells CIAO1 was shown to associate with both CIA2A and CIA2B in a mutually exclusive fashion, with these complexes interacting selectively with distinct subsets of target proteins [15]. The lack of interaction, between TbCIA2A and either TbCIA2B or TbMMS19 indicates the existence of a binary complex comprised of TbCIA1 and TbCIA2A in a configuration that is reminiscent of that described in mammalian cells [15], although the biological purpose of TbCIA2A or the complex it forms with TbCIA1 remains elusive at this time. It is worth mentioning here that a very weak interaction between TbCIA2A and TbMMS19 was detected in the BSF cells.
We demonstrate that TbCIA2B interacts with the C-terminal domain of TbMMS19. A schematic representation of the proposed model for the ternary T. brucei CTC is depicted in Fig 8. The C-terminal domain of TbMMS19 (TbMMS19-CTD) acts as a docking site for the other two members of the trimeric complex, namely TbCIA2B and TbCIA1. We can also conclude from our pull-down assays that TbCIA2B independently interacts with TbMMS19-CTD. In humans, the interaction between CIA2B and MMS19 has been shown to be vital not only for the stability of the CTC itself, but also for the association with client Fe-S proteins [58,83]. Interestingly, van Wietmarschen and colleagues [67] reported that in vitro translated murine CIA2B and MMS19 were not able to bind directly to each other, although both could interact with CIAO1. However, in support of our observations, Odermatt and Gari [58] showed that CIA2B binds to the C-terminal HEAT repeats of MMS19 in HeLa cells, and similar results were observed in pull-down assays with purified human proteins [83]. The status of TbCIA1 in the CTC of T. brucei is less clear, since from our results we cannot distinguish whether its interaction with TbMMS19 depends on the presence of TbCIA2B. However, recombinant fragments of the C-terminal domain of TbMMS19 had an enhanced ability to bind TbCIA2B in cell extracts if compared to the purified recombinant protein. In agreement with these observations, human CIAO1 was reported to stabilise the interaction between CIA2B and the HEAT repeats at the C-terminal domain of MMS19, forming a trimeric complex [58]. Thus, we favour the interpretation that TbCIA2B independently interacts with the C-terminal domain of TbMMS19, yet this interaction may be further strengthened by other proteins, with TbCIA1 being a prime candidate. Considering that both TbCIA1 and TbCIA2B are involved in the maturation of Fe-S proteins, it is possible that the assembly of the clusters into apo-proteins takes place at this C-terminal docking site [58].
Fig 8. Functional model of the CIA targeting complex of T. brucei.

TbNAR1 receives a Fe-S cluster from the early-acting CIA machinery and interacts with members of the CIA targeting complex (CTC). TbCIA2B binds tightly to the C-terminal domain of TbMMS19 in and interaction possibly stabilised by TbCIA1 (as indicated by the dotted lines), forming the canonical ternary CTC, although binary complexes also exist. The function of the complex formed by TbCIA1 and TbCIA2A is unknown. The targeting complex directly interacts with cytosolic and nuclear Fe-S proteins. TbPOLD1 was found in association with TbCIA1 and TbMMS19, and TbXPD with TbCIA2B. TbFHc interacts with TbMMS19 at its N-terminal domain and also co-purifies with TbCIA1. TbACO interacts with TbCIA2B but also requires TbCIA1 for maturation. Other potential Fe-S proteins also interact with the complex, but their status as bona fide Fe-S proteins is unknown.
The binding site of TbCIA2B, as supported by in silico modelling of theTbCIA2B-TbMMS19 complex, was narrowed down to a region between the residues Val763-Lys947 of the C-terminal domain of TbMMS19. The remarkable complementarity of the experimental observations with the in silico predictions allowed us to model the interface between the two proteins and identify residues likely involved in their interaction. However, bearing in mind that our data also strongly suggested the CTC exists in both binary and ternary versions, these simulations may not exactly reflect the whole scenario taking place at a cellular level. Since a reliable structural model for TbCIA1 could not be generated, we did not attempt to dock a ternary TbCIA2B-TbCIA1-TbMMS19 complex, or predict binary interactions of that protein. It is also important to recognise the caveats associated with this method, as homology-based structural models may not accurately reflect the minutia of biologically relevant conformations of proteins or complexes. Nevertheless, a similar approach has been successfully used to study the specificity of binding of the trans-acting acyltransferase to acyl-carrier proteins [84] and to design inhibitors of the human tumour necrosis factor [85]. Altogether, we believe our model provides a valuable snapshot of the TbMMS19-TbCIA2B interaction. The comprehensive analysis of protein-protein interactions for the CTC presented herein sheds light on the flexibility, as well as on the level of conservation of this ubiquitous eukaryotic pathway.
Materials and methods
Parasite cultivation and transfection
T. brucei PCF 29–13 [21], and SmOxP927 [86] cell lines co-expressing T7 RNA polymerase (T7RNAP) and the Tet repressor (TetR) are referred to as wild-type in this study. The conditions for cultivation have been described elsewhere [87,88]. BSF cells used throughout were the single marker strain that constitutively expresses T7RNAP and TetR [21], and were grown in Hirumi modified Iscove’s medium 11 (HMI-11) [89] supplemented with G418 (2.5 μg mL-1). BSF were grown at 37°C with 5% (v/v) CO2 in humidified atmosphere and kept at cell densities of 1 x 105 to 2 x 106 cells mL-1 and diluted with fresh HMI-11 media as required.
For transfections, 10 μg of linearised constructs (see below) were electroporated into 1 x 107 to 2 x 107 cells using an Amaxa Nucleofector 2b device or BTX electroporator, as previously described [87,88]. Stable transformants were selected by clonal dilution in media containing the appropriate selection drugs.
RNAi constructs
The sequences for all primers used in this study can be found in online supplementary material. RNAi constructs were prepared by amplifying fragments of TbCIA2A, TbCIA2B, and TbMMS19 flanked by BamHI and XhoI restriction sites and cloning into the p2T7-177 RNAi vectors [90]. TbMMS19 and TbCIA2A RNAi in PCF were obtained by Gibson assembly using the pTrypSon vector [91]. Double RNAi constructs were generated by ligating a second gene fragment in previously generated single RNAi constructs upon digestion with BamHI and SpeI as described before [25]. Constructs were linearized with NotI to allow integration into the silent 177 repeats of the T. brucei minichromosome prior to transfection into PCF 29–13, SmOx or BSF single marker cells. Selection was carried out with 1.25 to 5 μg mL-1 phleomycin, or 4 μg mL-1 hygromycin B (for the BSF TbMMS19 RNAi cell line).
Constructs for epitope tagging
C-terminal in situ V5 tagging of the TbCIA proteins was performed as described [92]. PCR tagging was performed using a modified version of the pPOTv4 vector in which eYFP was replaced by the sequence of a triple V5 tag. PCR products were electroporated into PCF SmOxP927 cell line and selection was performed with 50 μg mL-1 hygromycin B. For C-terminal HA- or PTP-tagging, ~400–1,000 bp upstream of the termination codon of the genes of interest were inserted into the vectors pC-HA-BLA [93] or pC-PTP-PURO [94] using the KpnI and AflII restriction sites. N-terminal PTP-tagging constructs were generated by ligating pN-PTP-PURO [94] with ~400–1,200 bp downstream of the start codon of the respective CIA gene using NotI and KpnI restriction sites. The resulting plasmids were linearised at restriction sites within the inserts and transfected into PCF 29–13 or BSF single marker cells then selected with 20 μg mL-1 blasticidin or 2 μg mL-1 puromycin. The vector for conditional ectopic overexpression of N-terminally tagged proteins was constructed by amplifying the PTP-tag sequence from pN-PTP-PURO and ligating into to the plasmid pLEW82v4 [21] using 5’ PacI and 3’ HindIII restriction sites and adding a KpnI recognition sequence downstream of PacI to allow the introduction of the TbMMS19 ORF or sequences corresponding to its N- or C-terminal domain in frame with the PTP tag. pLEW82-PTP constructs were linearised with NotI for integration at the rRNA locus, transfected into TbCIA2B-HA PCF cells and selected with 5 μg mL-1 puromycin.
Western blot analyses
Proteins were resolved by SDS-PAGE, transferred to PVDF or nitrocellulose membranes and blocked in phosphate-buffered saline (PBS) with 5% milk for 1 hr at room temperature (RT). Blots were incubated with primary antibodies (see below) overnight at 4 oC, washed three times in PBS-T (PBS supplemented with 0.1% Tween 20), and incubated with the corresponding secondary antibodies for 1 hr at RT before further washes in PBS-T. Fluorescent signals were captured using the Odyssey CLx digital Imaging system (Li-Cor Biosciences) or chemiluminescent signals developed using the Clarity ECL substrate (BioRad). Data for semi-quantitative Western blots were obtained by densitometry using the FIJI package for ImageJ [95]. The following primary antibodies were used in this study: mouse monoclonal α-V5 (1:1.000; Invitrogen), α-tubulin (1:10000), α-TAO (1:100) [31], and α-Strep-Tag (1:500; IBA Life Sciences), mouse polyclonal α-TbPLA1 (1:1000) [30], rabbit polyclonal α-TbENO (1:2000; a gift from Paul A.M. Michels), α-mtHSP70 (1:1000), α-protein A (PAP antibody; 1:500; Sigma-Aldrich), and rat monoclonal 3F10 α-HA (1:1000; Sigma-Aldrich). In some experiments TbCIA2A and TbCIA2B were detected with specific antibodies (both at 1:200) raised in rabbit using protocols described elsewhere [70–73]. Fluorescent secondary antibodies were goat IgG α-rabbit, α-mouse, or α-rat conjugated to IRDye680 or IRDye800 (Li-Cor Biosciences). HRP-conjugated reagents were from Sigma-Aldrich.
Confocal imaging
Preparation of C-terminal in situ-tagged TbCIA1-V5, TbCIA2A-V5, TcCIA2B-V5 and TbMMS19-V5 for confocal imaging was performed as described elsewhere, with minor modifications [88]. Cells were fixed with 4% (w/v) paraformaldehyde in phosphate buffered saline (PBS), permeabilised with 0.2% (v/v) Triton X-100 in PBS on microscopy slides and then probed with primary antibodies in PBS/gelatin. Monoclonal α-V5 (Life Technologies) and polyclonal anti-TbENO antibodies were used at 1:1000 and 1:2000 dilution, respectively. As secondary antibodies, Alexa Fluor 488 anti-mouse and Alexa Fluor 555 anti-rabbit (Life Technologies) were used. DNA was visualized using ProLong Gold antifade reagent with DAPI (Life Technologies). Confocal microscopy was performed using an inverted IX81 motorized FluoView FV1000 confocal (Olympus) microscope and detection was carried out with FV1000 software (Olympus). Image analysis was performed using Magic Montage plugin for ImageJ [96] and FIJI [95].
Homology modelling of protein structures
For generation of structural homology models, amino acid sequences of proteins were submitted to Protein Homology/analogy Recognition Engine v. 2.0 (Phyre2) [53], available at http://www.sbg.bio.ic.ac.uk/phyre2/, using either the normal or intensive modelling modes. The resulting PDB files with the 3D structure of proteins were visualised with MacPyMOL (Schrodinger).
In silico protein-protein docking and interaction hot-spots
PDB files with the 3D structures of proteins were used as input to the ClusPro docking server [59,97], available at https://cluspro.bu.edu/home.php. TbMMS19 was defined as the receptor and TbCIA2B as ligand and all settings were kept as default. Output PBD files containing the top highest scoring models according to the balanced method were downloaded and visualised with MacPyMOL (Schrodinger). PDB files with protein complexes were uploaded to the PredHS server [61], available at http://www.predhs.org/. The predicted interaction hot-spots on the surface of proteins were identified by the SVM algorithm and superimposed on the 3D structure of the complex using MacPyMOL (Schrödinger).
Recombinant protein expression and purification
PCR amplified sequences corresponding to the N- or C-terminal domains of TbMMS19, or fragments of the latter were cloned into using pGEX-6P-1 (GE Healthcare), using the BamHI and NotI restriction sites. The sequence for a Strep-Tag II was included in the antisense primers to generate a C-terminal Strep-Tag II fusion in addition to the N-terminal GST tag encoded in the expression vector. TbCIA2B was cloned into pASK-IBA7plus (IBA Life Sciences) using EcoRI and EcoRV restriction sites and the sequence for a hexahistidine tag was included in the antisense primer to generate a C-terminal 6XHIS fusion in addition to the N-terminal Strep-Tag II present in the vector. Recombinant proteins were expressed in C43 (DE3) pLysS E. coli [98] carrying the pRARE plasmid for rare codons, grown in terrific broth. rTbCIA2B was purified by immobilised metal affinity chromatography with Ni-NTA agarose (Qiagen) and eluted in EB1 (50 mM Tris.HCl, pH 9, 250 mM NaCl, 0.1% Triton X-100, 1 mM EDTA, 1 mM DTT, 400 mM imidazole, 10% glycerol). Fragments and domains of TbMMS19 were batch purified with Glutathione Sepharose 4B beads (GE Healthcare), followed by Strep-Tactin (IBA Life Sciences) affinity purification and eluted in EB2 (50 mM Tris.HCl, pH 9, 250 mM NaCl, 1% Triton X-100, 0.5% sarkosyl, 1 mM EDTA, 1 mM DTT, 5 mM desthiobiotin, and 10% [v/v] glycerol).
TAP/MS
Tandem affinity purifications were performed following a standard protocol as described [99], with minor modifications. Briefly, 2.5 litres of PCF expressing PTP tagged proteins were grown to late log phase, centrifuged and washed in ice-cold PBS. Cell pellets were suspended in TLB buffer (20 mM Hepes KOH pH 7.7, 150 mM potassium glutamate, 150 mM sucrose, 3 mM MgCl2, 2 mM DTT, 1% [v/v] Triton X-100, Roche cOmplete EDTA-free protease inhibitor cocktail) and lysed on ice with a Dounce homogenizer. Lysates were cleared by centrifugation, filtered into a 10 mL Poly-Prep column (Bio-Rad) and incubated with pre-equilibrated IgG Sepharose 6 Fast Flow resin (GE Healthcare). The resin was washed with PA-150, equilibrated with TEV buffer and incubated overnight with 400U of AcTEV protease (Invitrogen). TEV eluates were collected, added to buffer PC-150 supplemented with 1 mM CaCl2 and protease inhibitors, then bound to a pre-equilibrated Anti-Protein C affinity matrix (Sigma-Aldrich) in another Poly-Prep. After extensive washes, proteins were eluted in 1.8 mL of EDTA/EGTA buffer and concentrated with StrataClean resin (Agilent). The resin was pelleted, resuspended in NuPAGE LDS sample buffer (Invitrogen), boiled at 95°C for 10 minutes, and the proteins were resolved in NuPage 4–12% Bis-Tris gels (Invitrogen) before staining with SYPRO Ruby (Molecular Probes). Images were captured in a Typhoon FLA 7000 laser scanner (GE Healthcare). Trypsin digests of excised gel sections were analysed by LC/MS in an ABSciex TripleTOF 5600+ mass spectrometer and the spectra were searched against a T. brucei protein database [46] using MASCOT. Proteins hits with less than 2 unique peptides were disregarded.
Co-immunoprecipitation
PCF parasites were washed with ice cold PBS, resuspended in TLB buffer and quickly disrupted with glass beads in a FastPrep machine (MP Biomedicals). Lysates were cleared by centrifugation (16,000 g, 30 minutes, 4°C) and transferred to 1.5 mL tubes containing 25 μL of pre-equilibrated IgG Sepharose 6 Fast Flow resin (GE Healthcare) (for assays with double-tagged cell lines) or 200 pmoles of TbMMS19 GST-fusion proteins immobilised to 25 μL of Glutathione Sepharose 4B, and incubated with rotation for two hours at 4°C. Alternatively, immobilised proteins were incubated with 400 pmoles of purified rTbCIA2B. The resins were washed 4 times with 1 mL of TLB, resuspended in 25 μL of 2 X SDS-PAGE sample buffer and subsequently boiled at 95°C for 10 min. For Strep Tag pull-downs, 400 pmoles of rTbCIA2B were immobilised in 25 μL of Strep-Tactin resin and everything else performed as described above. Interactions were analysed by Western blot after SDS-PAGE.
For V5 co-IP/MS, pellets of 3 x 109 PCF or BSF cells were suspended in PBS, snap-frozen in liquid nitrogen and grinded using a CryoGrinder (OPS Diagnostic) [51]. The cell powder was suspended in 500 μL of lysis buffer (20 mM HEPES, pH 7.4, 150 mM Na-Citrate, 1 mM MgCl2, 0.2 mM CaCl2, 0.1% [v/v] Triton X-100, and Roche cOmplete EDTA-free protease inhibitor cocktail). Cleared lysates were added to 12 μL of DynaBeads pre-cross-linked with anti-V5 antibody and incubated for two hours at 4°C. Beads were further washed with lysis buffer and proteins were eluted in 100 μL of elution buffer (25 mM Tris.HCl, pH 7.5, 2% [v/v] SDS) at 72°C for 10 minutes. Proteins were precipitated with ethanol, resolved by SDS-PAGE, visualised by silver staining and analysed by Western blot or mass spectrometry.
Selective permeabilization with digitonin
Cell fractionation using a digitonin gradient was performed as described elsewhere [100]. For co-localisation, 1 x 107 cells were suspended in 100 μL of FB (20 mM Tris-HCl, 0.6 M sorbitol, 1 mM DTT, Roche cOmplete protease inhibitor cocktail; pH 7.5) containing concentrations of 0.01 to 1 mg mL-1 of digitonin. Cells were incubated on ice for 5 min and centrifuged at 16,000 g for 5 min at 4°C. The supernatant was transferred to a clean 1.5 mL tube and evaporated in a SpeedVac until almost dry. The pellet was suspended in 20 μL of 2 X SDS-PAGE sample buffer, boiled for 10 min at 95°C and resolved by SDS-PAGE. Protein release in each fraction was detected by a semi-quantitative Western blot. Cytosolic and organellar fractions for other assays were prepared by suspending 1 x 108 cells in 1 mL FB containing 0.15 mg mL-1 digitonin. The soluble supernatant was considered the cytosolic fraction. The pellet was washed once with 1 mL of FB, incubated for 10 min on ice with 1 mL FB with 0.5% (v/v) Triton X-100 and centrifuged at 16,000 g for 5 min at 4°C. The resulting supernatant was considered the organellar fraction.
Crude cell fractionation analysis
The cytosol was separated from the organellar fraction as described elsewhere [101]. Mid-log cells expressing the V5-tagged proteins were harvested at 1000 g for 10 min at 4°C, washed with ice cold SHE buffer (25 mM HEPES, pH 7.4, 250 mM sucrose, 1 mM EDTA), resuspended in fresh SHE buffer to a final concentration of 5 x 109 cells mL-1, and the protein concentration was determined according to Bradford. One milligram protein aliquots were suspended in Hanks’ balanced salt solution (HBSS) (1.26 mM CaCl2, 5.33 mM KCl, 0.44 mM KH2PO4, 0.81 mM MgSO4, 138 mM NaCl, 4 mM NaHCO3, 0.3 mM Na2HPO4, 5.6 mM glucose, pH 7.3) and digitonin was added to the final concentration of 0.4 μg μL-1. After vortexing, the suspension was incubated at RT for 5 min, and followed by centrifugation at 14000 g at RT for 2 min. The supernatant represented the cytosolic fraction, while the pellet was washed once with HBSS and then resuspended in HBSS containing 0.1% (v/v) Triton X-100 and incubated on ice for 5 min. After centrifugation, the supernatant was collected as the organellar fraction. The pellet was washed once more with HBSS and then resuspended in a volume equal to the previous two fractions and analyzed by Western blotting. This final pellet fraction contains proteins that are insoluble or strongly associated to membranes.
Aconitase activity measurement
Aconitase activity was measured as previously described by monitoring the increase of the absorbance at 240 nm due to the conversion of isocitrate into cis-aconitate [102]. Two hundred microliters of lysates were added to 1.3 mL of aconitase buffer (50 mM Tris.HCl, 1 mM DTT, 20 mM DL-isocitric acid or sodium citrate; pH 7.4) and incubated at 25°C. The rate of increase of the absorbance at 240 nm per min (ΔA240 nm/min) was monitored for 30 min in a Varian Cary 50 UV/Vis spectrophotometer. A blank reaction without cell lysate was run in parallel. Specific activity was obtained by dividing the measured aconitase activity (mU mL-1) by protein concentration in the sample. Uninduced controls were considered as 100% of activity.
Measurement of protein-bound iron
Cellular fractions from digitonin fractionation were concentrated in a SpeedVac (Thermo) and the iron content measured by the Ferene method, as described by [103],. The pellets were thoroughly suspended in 100 μL of milliQ water, mixed with 100 μL of 1% HCl, incubated for 10 min at 100°C, quickly cooled down on ice and centrifuged (12,000 g, 5 min). Subsequently, 500 μL of 7.5% ammonium acetate, 100 μL 4% ascorbic acid and 100 μL of 2.5% SDS were added to the samples and vortexed. The samples were centrifuged again (12,000 g, 10 min) and 855 μL of the supernatant was transferred to a semi-micro cuvette to which 95 μL of 6.2 mM Ferene (Sigma) were added. The absorbance of the ferrous-ferene complex at 593 nm was corrected for turbidity by subtraction of the absorbance at 800 nm and measured in a Varian Cary 50 UV/Vis spectrophotometer. Iron content was estimated by interpolation from a standard curve of ferrous sulphate (2,000–12.5 ng) using least squares linear regression.
Resazurin cell viability assay
The Alamar Blue assay was used to assess viability of cells exposed to DNA damaging agents or DFO. In this assay, the resazurin salt is reduced to resorufin, which emits a fluorescent signal proportional to the number of viable cells [104]. Cell densities of exponentially growing cells were adjusted to 1 x 106 or 5 x 104 cells mL-1 for PCF and BSF trypanosomes, respectively, to generate a 2x working cell suspension. One hundred microliters of cell suspension were added in quadruplicate to 96-well plates containing 100 μL per well of 2-fold serial dilutions of drugs. Wells without drugs or without cells served as maximum growth control and blank, respectively. PCF cells were grown for 48 hrs at 28°C, while BSF were incubated for 72 hrs at 37°C, after which 10 μL of a 1.1 mg mL-1 solution of resazurin (Sigma) were dispensed to each well and the plates were incubated for another 6 hrs. The fluorescent signal was measured in a FLx800TM Microplate reader (BioTek) with excitation wavelength set at λ530 and emission at λ590. All EC50s (concentration of a compound that reduces cell growth by 50%) were calculated by nonlinear regression using the software Prism 7.0 (GraphPad Inc.). DFX, methyl methane sulfonate (MMS), 4-nitroquinoline 1-oxide (4NQO), hydroxyurea, and camptothecin were purchased from Sigma-Aldrich, and phleomycin (Zeocin) was purchased from Thermo Fisher.
Yeast complementation
Complementation experiments were carried out in Saccharomyces cerevisiae strain W303-1A as WT (MATa, ura3-1, ade2-1, trp1-1, his3-11,15, leu2-3,112). The galactose-regulatable mutants used were GalL-MMS19 and Gal-CIA2 [14,43]. The latter mutant strain was constructed by homologous recombination in which the upstream promoter region of CIA2 was replaced by a PCR product containing the NatNT2 resistance marker gene and the GAL promoter. PCR analysis of chromosomal DNA confirmed correct insertion of the promoter. Yeast cells were grown in minimal (SC) media, containing galactose or glucose at a concentration of 2% (m/v) [105]. The yeast MMS19-encoding gene was cloned into the SmaI and XhoI sites of the pRS424-TDH3 vector [106]. For control of the rescue by yeast Cia2, the Cia2-encoding sequence with 500 bp natural promoter (NP) sequence was amplified from yeast DNA and cloned into the SacI and XhoI sites of pRS416-MET25. TbCIA2A and TbCIA2B genes were amplified from T. brucei DNA and cloned into the BamHI and SalI sites of pRS416-MET25. TbMMS19 (Tb927.8.3920) was cloned into pRS424-TDH3 in two steps. SpeI-BamHI and BamHI-ClaI fragments were consecutively PCR-amplified and cloned. The BamHI site, which is lacking in the TbMMS19 gene, introduces silent mutations at amino acids 514–515 (Gly-Ser). After transformation of plasmids into GalL-MMS19 or Gal-CIA2 cells, growth in liquid minimal media supplemented with 2% galactose was carried out for 16 h. Then cells were shifted to the same medium, but with 2% glucose for 16 h (Gal-CIA2) or 16 and 24 h (GalL-MMS19). Cell suspensions were diluted to an optical density of 0.5 at 600 nm and 5 μl aliquots, including four consecutive 10-fold serial dilutions, were spotted on agar plates. Plates containing minimal media supplemented with 2% galactose or glucose were incubated at 30°C for 48 h and photographed.
In vivo infectivity
Mice had food and fresh water ad libitum. The experiment was approved by our institution’s Animal Ethics Committee. To determine the infectivity of trypanosomes depleted for TbCIA2B or TbMMS19, six groups of BALB/C mice (uninduced and RNAi-induced TbCIA2B, uninduced and RNAi-induced TbMMS19, wild type single marker [WT SM] cells with and without doxycycline). Each group consisted of 5 females (8 to 9 weeks old) which were infected intraperitoneally with 10,000 BSF cells. In their drinking water, the induced groups received 1 mg/ml doxycycline sweetened with 50 mg/ml sucrose, starting 2 days before the infection. The survival was recorded twice a day. Survival data was plotted using Prism 7.
Supporting information
Growth curves of double RNAi cells lines for TbCIA1-TbCIA2B and TbCIA2A-TbCIA2B in PCF (A and B) and BSF (C and D) cells were grown in presence (Tet+) and absence (Tet-) of tetracycline for 10 and 8 days, respectively.
(TIF)
Survival of mice infected with BSF TbMMS19 (A) and TbCIA2B (B) RNAi cell lines, uninduced (-) and induced (+) with doxycycline. Wild type (SM) was used as controls, also in the absence (-) and presence (+) of doxycycline. Five mice per group were used. Induced (+) cell lines are nudged in the graph for easier visualisation of the overlapping curves.
(TIF)
(A) Primary sequence of TbMMS19 with the centres of the HEAT repeats highlighted in a scale of red according to the probability of the respective residue to be in the centre of a HEAT repeat unit, as calculated by Ard2 [55].(B) Predicted 3D structure of TbMMS19 with HEAT repeats highlighted in red. The homology model for TbMMS19 was created using Phyre2 [53].(C) The N-and C-terminal domains of TbMMS19 are represented in blue and red, respectively. The boundaries of the domains were defined by alignment with human MMS19. Letters A-C represent functional domains of the human protein. Grey boxes correspond to HEAT repeats annotated in the Uniprot database for MMS19 (accession number Q9T76), or identified as described in (B) for TbMMS19.
(PNG)
{kind=link}
N- and C-termini of TbMMS19 interact with TbCIA1. Tandem affinity purifications of PTP-TbMMS19 (3), PTP-TbMMS19-NTD (4), and PTP-TbMMS19-CTD (5), mock (1) and (2) empty plasmid, observed in a SYPRO Ruby-stained SDS-PAGE gel.
(PNG)
{kind=link}
Three-dimensional models for the proteins were created using Phyre2 [53] and the best predictions were used for docking with ClusPro [59]. The five highest-scoring complexes (balanced score), were analysed with PredHS [61] to identify the key residues for interaction with TbCIA2B at the binding surface of TbMMS19. TbCIA2B is depicted as blue mesh in (A) and the N and C-terminal domains of TbMMS19 are shown in (A) and (B) as grey and green surfaces, respectively. TbCIA2B was omitted in (B) to uncover the residues at the contact surface, which are shown in a scale of red according to their associated SVM hot-spot score. Except in model 5, the residues more likely to be hot-spots of interaction are predicted to be in the C-terminal domain of TbMMS19. MacPyMOL (Schrödinger, LLC) was used to generate the figures based on the output of PredHS, ClusPro and Phyre2.
(PNG)
{kind=link}
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(XLSX)
Acknowledgments
We are grateful to the University of St Andrews mass spectrometry facility for collecting and processing MS data. We thank Paul A.M. Michels (University of Edinburgh, UK) and Minu Chaudhuri (Meharry Medical College, USA) for kindly providing antibodies, and Paula Castañeda (University of South Bohemia, Czech Republic) for technical help.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Support from the Czech Grant Agency (16-18699S to JL) and partial funding by CAPES/Science without Borders (BEX1333/13-5 to MLT) is kindly acknowledged. We are grateful to the University of St. Andrews mass spectrometry facility for collecting and processing MS data and to other members of the TKS and SM groups for their assistance with this project. Work of RL and AJP was supported by the Deutsche Forschungsgemeinschaft (Koselleck grant (to RL) and SPP 1927) and a Coordenação de aperfeiçoamento de pessoal de nivel superior (CAPES - 1333/2013-05) for the financial support to this project. We acknowledge networking support from the COST Action FeSBioNet (Contract CA15133). JL was supported by ERD Funds, The Czech Ministry of Education, project OPVVV 16_019/0000759. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Wächtershäuser G. On the chemistry and evolution of the pioneer organism. Chem Biodivers. 2007; 4:584–602. 10.1002/cbdv.200790052 [DOI] [PubMed] [Google Scholar]
- 2.Beinert H. Iron-sulfur proteins: ancient structures, still full of surprises. J Biol Inorg Chem. 2000; 5:2–15. 10.1007/s007750050002 [DOI] [PubMed] [Google Scholar]
- 3.Lill R. and Function biogenesis of iron–sulphur proteins. Nature. 2009; 460:831–838. 10.1038/nature08301 [DOI] [PubMed] [Google Scholar]
- 4.Paul VD, Lill R. Biogenesis of cytosolic and nuclear iron–sulfur proteins and their role in genome stability. BBA-Molecular Cell Research. 2015; 1853:1528–1539. 10.1016/j.bbamcr.2014.12.018 [DOI] [PubMed] [Google Scholar]
- 5.Braymer JJ, Lill R. Iron–sulfur cluster biogenesis and trafficking in mitochondria. J Biol Chem. 2017; 292:12754–12763. 10.1074/jbc.R117.787101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Couturier J, Touraine B, Briat J-F, Gaymard F, Rouhier N. The iron-sulfur cluster assembly machineries in plants: current knowledge and open questions. Front Plant Sci. Frontiers; 2013; 4:1–22. 10.3389/fpls.2013.00259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roy A. A novel eukaryotic factor for cytosolic Fe-S cluster assembly. EMBO J. 2003; 22:4826–4835. 10.1093/emboj/cdg455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lill R, Dutkiewicz R, Freibert SA, Heidenreich T, Mascarenhas J, Netz DJ, et al. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur J Cell Biol. 2015; 94:280–291. 10.1016/j.ejcb.2015.05.002 [DOI] [PubMed] [Google Scholar]
- 9.Netz DJA, Stümpfig M, Doré C, Mühlenhoff U, Pierik AJ, Lill R. Tah18 transfers electrons to Dre2 in cytosolic iron-sulfur protein biogenesis. Nat Chem Biol. 2010; 6:758–765. 10.1038/nchembio.432 [DOI] [PubMed] [Google Scholar]
- 10.Netz DJA, Pierik AJ, Stümpfig M, Mühlenhoff U, Lill R. The Cfd1-Nbp35 complex acts as a scaffold for iron-sulfur protein assembly in the yeast cytosol. Nat Chem Biol. 2007; 3:278–286. 10.1038/nchembio872 [DOI] [PubMed] [Google Scholar]
- 11.Netz DJA, Pierik AJ, Stumpfig M, Bill E, Sharma AK, Pallesen LJ, et al. A bridging [4Fe-4S] cluster and nucleotide binding are essential for function of the Cfd1-Nbp35 complex as a scaffold in iron-sulfur protein maturation. J Biol Chem. 2012; 287:12365–12378. 10.1074/jbc.M111.328914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balk J, Pierik AJ, Netz DJA, Mühlenhoff U, Lill R. The hydrogenase-like Nar1p is essential for maturation of cytosolic and nuclear iron-sulphur proteins. EMBO J. 2004; 23:2105–2115. 10.1038/sj.emboj.7600216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gari K, Ortiz AML, Borel V, Flynn H, Skehel JM, Boulton SJ. MMS19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science. 2012; 337:243–245. 10.1126/science.1219664 [DOI] [PubMed] [Google Scholar]
- 14.Stehling O, Vashisht AA, Mascarenhas J, Jonsson ZO, Sharma T, Netz DJA, et al. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science. 2012; 337:195–199. 10.1126/science.1219723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stehling O, Mascarenhas J, Vashisht AA, Sheftel AD, Niggemeyer B, Rösser R, et al. Human CIA2A-FAM96A and CIA2B-FAM96B integrate iron homeostasis and maturation of different subsets of cytosolic-nuclear iron-sulfur proteins. Cell Metab. 2013; 18:187–198. 10.1016/j.cmet.2013.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.WHO | Epidemiological situation. WHO. World Health Organization. Available: http://www.who.int/trypanosomiasis_african/country/en/
- 17.Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis worldwide and global estimates of its incidence. Kirk M, editor. PLoS ONE. 2012; 7:e35671 10.1371/journal.pone.0035671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.WHO | Chagas disease (American trypanosomiasis). WHO. World Health Organization. Available: http://www.who.int/mediacentre/factsheets/fs340/en/
- 19.Auty H, Torr SJ, Michoel T, Jayaraman S, Morrison LJ. Cattle trypanosomosis: the diversity of trypanosomes and implications for disease epidemiology and control. Rev—Off Int Epizoot. 2015; 34:587–598. doi: 10.20506/rst.34.2.2382 [DOI] [PubMed] [Google Scholar]
- 20.Adl SM, Simpson AGB, Lane CE, Lukeš J, Bass D, Bowser SS, et al. The revised classification of eukaryotes. J Eukaryot Microbiol. 2012; 59:429–514. 10.1111/j.1550-7408.2012.00644.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wirtz E, Leal S, Ochatt C, Cross GA. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999; 99:89–101. 10.1016/S0166-6851(99)00002-X [DOI] [PubMed] [Google Scholar]
- 22.Clayton CE. Genetic manipulation of kinetoplastida. Parasitol Today. 1999; 15:372–378. 10.1016/S0169-4758(99)01498-2 [DOI] [PubMed] [Google Scholar]
- 23.LaCount DJ, Bruse S, Hill KL, Donelson JE. Double-stranded RNA interference in Trypanosoma brucei using head-to-head promoters. Mol Biochem Parasitol. 2000; 111:67–76. 10.1016/S0166-6851(00)00300-5 [DOI] [PubMed] [Google Scholar]
- 24.Alibu VP, Storm L, Haile S, Clayton C, Horn D. A doubly inducible system for RNA interference and rapid RNAi plasmid construction in Trypanosoma brucei. Mol Biochem Parasitol. 2005; 139:75–82. 10.1016/j.molbiopara.2004.10.002 [DOI] [PubMed] [Google Scholar]
- 25.Basu S, Netz DJ, Haindrich AC, Herlerth N, Lagny TJ, Pierik AJ, et al. Cytosolic iron-sulphur protein assembly is functionally conserved and essential in procyclic and bloodstream Trypanosoma brucei. Mol Microbiol. 2014; 93:897–910. 10.1111/mmi.12706 [DOI] [PubMed] [Google Scholar]
- 26.Basu S, Horáková E, Lukeš J. Iron-associated biology of Trypanosoma brucei. BBA—General Subjects. 2016; 1860:363–370. 10.1016/j.bbagen.2015.10.027 [DOI] [PubMed] [Google Scholar]
- 27.Lukeš J, Basu S. Fe/S protein biogenesis in trypanosomes—A review. Biochim Biophys Acta. 2015; 1853:1481–1492. 10.1016/j.bbamcr.2014.08.015 [DOI] [PubMed] [Google Scholar]
- 28.Tsaousis AD, Gentekaki E, Eme L, Gaston D, Roger AJ. Evolution of the cytosolic iron-sulfur cluster assembly machinery in Blastocystis species and other microbial eukaryotes. Eukaryot Cell. 2014; 13:143–153. 10.1128/EC.00158-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hannaert V, Albert MA, Rigden DJ, Giotto M, Thiemann O, Garratt RC, et al. Kinetic characterization, structure modelling studies and crystallization of Trypanosoma brucei enolase. Eur J Biochem. 2003; 270:3205–3213. 10.1046/j.1432-1033.2003.03692.x [DOI] [PubMed] [Google Scholar]
- 30.Richmond GS, Smith TK. A novel phospholipase from Trypanosoma brucei. Mol Microbiol. 2007; 63:1078–1095. 10.1111/j.1365-2958.2006.05582.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaudhuri M. and Biochemical molecular properties of the Trypanosoma brucei alternative oxidase. Mol Biochem Parasitol. 1998; 95:53–68. 10.1016/S0166-6851(98)00091-7 [DOI] [PubMed] [Google Scholar]
- 32.Saas J, Ziegelbauer K, Haeseler von A, Fast B, Boshart M. A developmentally regulated aconitase related to iron-regulatory protein-1 is localized in the cytoplasm and in the mitochondrion of Trypanosoma brucei. J Biol Chem. 2000; 275:2745–2755. 10.1074/jbc.275.4.2745 [DOI] [PubMed] [Google Scholar]
- 33.van Weelden SWH, Fast B, Vogt A, van der Meer P, Saas J, van Hellemond JJ, et al. Procyclic Trypanosoma brucei do not use Krebs cycle activity for energy generation. J Biol Chem. 2003; 278:12854–12863. 10.1074/jbc.M213190200 [DOI] [PubMed] [Google Scholar]
- 34.Balk J, Pilon M. Ancient and essential: the assembly of iron-sulfur clusters in plants. Trends Plant Sci. 2011;16: 218–226. 10.1016/j.tplants.2010.12.006 [DOI] [PubMed] [Google Scholar]
- 35.Netz DJA, Stith CM, Stümpfig M, Köpf G, Vogel D, Genau HM, et al. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol. 2012; 8:125–132. 10.1038/nchembio.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo D, Bernard DG, Balk J, Hai H, Cui X. The DUF59 family gene AE7 acts in the cytosolic iron-sulfur cluster assembly pathway to maintain nuclear genome integrity in Arabidopsis. Plant Cell. 2012; 24:4135–4148. 10.1105/tpc.112.102608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuss JO, Tsai C-L, Ishida JP, Tainer JA. Emerging critical roles of Fe-S clusters in DNA replication and repair. Biochim Biophys Acta. 2015; 1853:1253–1271. 10.1016/j.bbamcr.2015.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lentz DJ, Henderson GH, Eyring EM. Kinetics of aqueous iron (III) complexation by desferrioxamine B. Mol Pharmacol. 1973; 9:514–519. [PubMed] [Google Scholar]
- 39.Tenopoulou M, Doulias P-T, Barbouti A, Brunk U, Galaris D. Role of compartmentalized redox-active iron in hydrogen peroxide-induced DNA damage and apoptosis. Biochem J. 2005; 387:703–710. 10.1042/BJ20041650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurz T, Gustafsson B, Brunk UT. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J. 2006; 273:3106–3117. 10.1111/j.1742-4658.2006.05321.x [DOI] [PubMed] [Google Scholar]
- 41.Macdonald RL, Weir B. Hematology. In: Macdonald RL, Weir B, editors. Cerebral Vasospasm. 2001.
- 42.Neuvonen PJ. Interactions with the absorption of tetracyclines. Drugs. 1976; 11:45–54. 10.2165/00003495-197611010-00004 [DOI] [PubMed] [Google Scholar]
- 43.Pierik AJ, Netz DJA, Lill R. Analysis of iron–sulfur protein maturation in eukaryotes. Nat Protoc. 2009; 4:753–766. 10.1038/nprot.2009.39 [DOI] [PubMed] [Google Scholar]
- 44.Rouault TA, Maio N. Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J Biol Chem. 2017; 292:12744–12753. 10.1074/jbc.R117.789537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pyrih J, Pyrihová E, Kolísko M, Stojanovová D, Basu S, Harant K, et al. Minimal cytosolic iron-sulfur cluster assembly machinery of Giardia intestinalis is partially associated with mitosomes. Mol Microbiol. 2016; 102:701–714. 10.1111/mmi.13487 [DOI] [PubMed] [Google Scholar]
- 46.Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010; 38:D457–62. 10.1093/nar/gkp851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005; 309:416–422. 10.1126/science.1112642 [DOI] [PubMed] [Google Scholar]
- 48.Valasatava Y, Rosato A, Banci L, Andreini C. MetalPredator: a web server to predict iron–sulfur cluster binding proteomes. Bioinformatics. 2016; 32:2850–2852. 10.1093/bioinformatics/btw238 [DOI] [PubMed] [Google Scholar]
- 49.Rouault TA. Iron-sulfur proteins hiding in plain sight. Nat Chem Biol. 2015; 11:442–445. 10.1038/nchembio.1843 [DOI] [PubMed] [Google Scholar]
- 50.Schimanski B, Nguyen TN, Günzl A. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot Cell. 2005; 4:1942–1950. 10.1128/EC.4.11.1942-1950.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Field MC, Adung'a V, Obado S, Chait BT, Rout MP. Proteomics on the rims: insights into the biology of the nuclear envelope and flagellar pocket of trypanosomes. Parasitology. 2012; 139:1158–1167. 10.1017/S0031182011002125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsaousis AD, Nývltová E, Sutak R, Hrdy I, Tachezy J. A nonmitochondrial hydrogen production in Naegleria gruberi. Genome Biol Evol. 2014; 6:792–799. 10.1093/gbe/evu065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015; 10:845–858. 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kelley LA, Sternberg MJE. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4: 363–371. 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 55.Fournier D, Palidwor GA, Shcherbinin S, Szengel A, Schaefer MH, Perez-Iratxeta C, et al. Functional and genomic analyses of alpha-solenoid proteins. PLoS ONE. 2013; 8:e79894 10.1371/journal.pone.0079894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Groves MR, Barford D. Topological characteristics of helical repeat protein. Curr Opin Struct Biol. 1999; 9:383–389. 10.1016/S0959-440X(99)80052-9 [DOI] [PubMed] [Google Scholar]
- 57.Queimado L. Cloning the human and mouse MMS19 genes and functional complementation of a yeast mms19 deletion mutant. Nucleic Acids Res. 2001; 29:1884–1891. 10.1093/nar/29.9.1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Odermatt DC, Gari K. The CIA targeting complex is highly regulated and provides two distinct binding sites for client iron-sulfur proteins. Cell Rep. 2017; 18:1434–1443. 10.1016/j.celrep.2017.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. The ClusPro web server for protein–protein docking. Nat Protoc. 2017; 12:255–278. 10.1038/nprot.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng L, Guan J, Wei X, Yi Y, Zhang QC, Zhou S. Boosting prediction performance of protein–protein interaction hot spots by using structural neighborhood properties. J Comput Biol 2013;20: 878–891. 10.1089/cmb.2013.0083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng L, Zhang QC, Chen Z, Meng Y, Guan J, Zhou S. PredHS: a web server for predicting protein–protein interaction hot spots by using structural neighborhood properties. Nucleic Acids Res. 2014; 42:W290–W295. 10.1093/nar/gku437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ali V, Nozaki T. Iron-sulphur clusters, their biosynthesis, and biological functions in protozoan parasites. Advances in Parasitology. 2013. pp. 1–92. 10.1016/B978-0-12-407705-8.00001-X [DOI] [PubMed] [Google Scholar]
- 63.Anwar S, Dikhit MR, Singh KP, Kar RK, Zaidi A, Sahoo GC, et al. Interaction between Nbp35 and Cfd1 proteins of cytosolic fe-s cluster assembly reveals a stable complex formation in Entamoeba histolytica. PLoS ONE. 2014; 9:e108971–13. 10.1371/journal.pone.0108971 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Duan C-G, Wang X, Tang K, Zhang H, Mangrauthia SK, Lei M, et al. MET18 connects the cytosolic iron-sulfur cluster assembly pathway to active dna demethylation in Arabidopsis. PLoS Genet. 2015; 11:e1005559 10.1371/journal.pgen.1005559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Freibert SA, Goldberg AV, Hacker C, Molik S, Dean P, Williams TA, et al. Evolutionary conservation and in vitro reconstitution of microsporidian iron-sulfur cluster biosynthesis. Nat Commun. 2016; 8:1–12. 10.1038/ncomms13932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han Y-F, Huang H-W, Li L, Cai T, Chen S, He X-J. The cytosolic iron-sulfur cluster assembly protein MMS19 regulates transcriptional gene silencing, DNA repair, and flowering time in Arabidopsis. PLoS ONE. 2015; 10:e0129137 10.1371/journal.pone.0129137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Wietmarschen N, Moradian A, Morin GB, Lansdorp PM, Uringa EJ. The mammalian proteins MMS19, MIP18, and ANT2 are involved in cytoplasmic iron-sulfur cluster protein assembly. J Biol Chem. 2012; 287:43351–43358. 10.1074/jbc.M112.431270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li F, Martienssen R, Cande WZ. Coordination of DNA replication and histone modification by the Rik1–Dos2 complex. Nature. 2011; 475:244–248. 10.1038/nature10161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fast B, Kremp K, Boshart M, Steverding D. Iron-dependent regulation of transferrin receptor expression in Trypanosoma brucei. Biochem J. 1999; 342:691–696. 10.1042/bj3420691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Long S, Changmai P, Tsaousis AD, Skalický T, Verner Z, Wen Y-Z, et al. Stage-specific requirement for Isa1 and Isa2 proteins in the mitochondrion of Trypanosoma brucei and heterologous rescue by human and Blastocystis orthologues. Mol Microbiol. 2011; 81:1403–1418. 10.1111/j.1365-2958.2011.07769.x [DOI] [PubMed] [Google Scholar]
- 71.Long S, Jirků M, Ayala FJ, Lukeš J. Mitochondrial localization of human frataxin is necessary but processing is not for rescuing frataxin deficiency in Trypanosoma brucei. Proc Natl Acad Sci USA. 2008; 105:13468–13473. 10.1073/pnas.0806762105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Changmai P, Horáková E, Long S, Černotíková-Stříbrná E, McDonald LM, Bontempi EJ, et al. Both human ferredoxins equally efficiently rescue ferredoxin deficiency in Trypanosoma brucei. Mol Microbiol. 2013; 89:135–151. 10.1111/mmi.12264 [DOI] [PubMed] [Google Scholar]
- 73.Horáková E, Changmai P, Paris Z, Salmon D, Lukeš J. Simultaneous depletion of Atm and Mdl rebalances cytosolic Fe-S cluster assembly but not heme import into the mitochondrion of Trypanosoma brucei. FEBS J. 2015; 282:4157–4175. 10.1111/febs.13411 [DOI] [PubMed] [Google Scholar]
- 74.Taylor MC, McLatchie AP, Kelly JM. Evidence that transport of iron from the lysosome to the cytosol in African trypanosomes is mediated by a mucolipin orthologue. Mol Microbiol. 2013; 89:420–432. 10.1111/mmi.12285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stijlemans B, Beschin A, Magez S, Van Ginderachter JA, De Baetselier P. Iron Homeostasis and Trypanosoma brucei associated immunopathogenicity development: a battle/quest for iron. BioMed Res Int. 2015;2015: 819389 10.1155/2015/819389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taylor MC, Kelly JM. Iron metabolism in trypanosomatids, and its crucial role in infection. Parasitology. 2010; 137:899–917. 10.1017/S0031182009991880 [DOI] [PubMed] [Google Scholar]
- 77.Outten FW. A stress-responsive Fe-S cluster biogenesis system in bacteria–the suf operon of Gammaproteobacteria. In: De Gruyter (ed.) Iron-sulfur clusters in chemistry and biology. 10.1515/9783110308426.297 [DOI] [Google Scholar]
- 78.Dlouhy AC, Outten CE. The iron metallome in eukaryotic organisms Metallomics and the Cell. Springer; Netherlands; 2012. pp. 241–278. 10.1007/978-94-007-5561-1_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Compe E, Egly J-M. Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu Rev Biochem. 2016; 85:265–290. 10.1146/annurev-biochem-060815-014857 [DOI] [PubMed] [Google Scholar]
- 80.Badjatia N, Nguyen TN, Lee JH, Günzl A. Trypanosoma brucei harbours a divergent XPB helicase paralogue that is specialized in nucleotide excision repair and conserved among kinetoplastid organisms. Mol Microbiol. 2013; 90:1293–1308. 10.1111/mmi.12435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Machado CR, Vieira-da-Rocha JP, Mendes IC, Rajão MA, Marcello L, Bitar M, et al. Nucleotide excision repair in Trypanosoma brucei: specialization of transcription-coupled repair due to multigenic transcription. Mol Microbiol. 2014; 92:756–776. 10.1111/mmi.12589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vashisht AA, Yu CC, Sharma T, Ro K, Wohlschlegel JA. The association of the Xeroderma pigmentosum group D DNA helicase (XPD) with transcription factor IIH is regulated by the cytosolic iron-sulfur cluster assembly pathway. J Biol Chem. 2015; 290:14218–14225. 10.1074/jbc.M115.650762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Seki M, Takeda Y, Iwai K, Tanaka K. IOP1 protein is an external component of the human cytosolic iron-sulfur cluster assembly (CIA) machinery and functions in the MMS19 protein-dependent CIA pathway. J Biol Chem. 2013; 288:16680–16689. 10.1074/jbc.M112.416602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ye Z, Musiol EM, Weber T, Williams GJ. Reprogramming acyl carrier protein interactions of an Acyl-CoA promiscuous trans-acyltransferase. Chem Biol. 2014; 21:636–646. 10.1016/j.chembiol.2014.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Steeland S, Puimège L, Vandenbroucke RE, Van Hauwermeiren F, Haustraete J, Devoogdt N, et al. Generation and characterization of small single domain antibodies inhibiting human tumor necrosis factor receptor 1. J Biol Chem. 2015; 290:4022–4037. 10.1074/jbc.M114.617787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Poon SK, Peacock L, Gibson W, Gull K, Kelly S. A modular and optimized single marker system for generating Trypanosoma brucei cell lines expressing T7 RNA polymerase and the tetracycline repressor. Open Biol. 2012; 2:110037 10.1098/rsob.110037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tulloch LB, Menzies SK, Fraser AL, Gould ER, King EF, Zacharova MK, et al. Photo-affinity labelling and biochemical analyses identify the target of trypanocidal simplified natural product analogues. PLoS Negl Trop Dis. 2017; 11:e0005886 10.1371/journal.pntd.0005886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peña-Diaz P, Vancová M, Resl C, Field MC, Lukeš J. A leucine aminopeptidase is involved in kinetoplast DNA segregation in Trypanosoma brucei. PLoS Pathog. 2017; 13:e1006310 10.1371/journal.ppat.1006310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol. 1989; 75:985–989. 10.2307/3282883 [DOI] [PubMed] [Google Scholar]
- 90.Wickstead B, Ersfeld K, Gull K. Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol Biochem Parasitol. 2002; 125:211–216. 10.1016/S0166-6851(02)00238-4 [DOI] [PubMed] [Google Scholar]
- 91.McAllaster MR, Sinclair-Davis AN, Hilton NA, de Graffenried CL. A unified approach towards Trypanosoma brucei functional genomics using Gibson assembly. Mol Biochem Parasitol. 2016; 210:13–21. 10.1016/j.molbiopara.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dean S, Sunter J, Wheeler RJ, Hodkinson I, Gluenz E, Gull K. A toolkit enabling efficient, scalable and reproducible gene tagging in trypanosomatids. Open Biol. 2015; 5:140197–140197. 10.1098/rsob.140197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee JH, Nguyen TN, Schimanski B, Günzl A. Spliced leader RNA gene transcription in Trypanosoma brucei requires transcription factor TFIIH. Eukaryot Cell. 2007; 6:641–649. 10.1128/EC.00411-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schimanski B, Nguyen TN, Günzl A. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot Cell. 2005; 4:1942–1950. 10.1128/EC.4.11.1942-1950.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012; 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012; 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2003; 20:45–50. 10.1093/bioinformatics/btg371 [DOI] [PubMed] [Google Scholar]
- 98.Dumon-Seignovert L, Cariot G, Vuillard L. The toxicity of recombinant proteins in Escherichia coli: a comparison of overexpression in BL21(DE3), C41(DE3), and C43(DE3). Protein Expr Purif. 2004; 37:203–206. 10.1016/j.pep.2004.04.025 [DOI] [PubMed] [Google Scholar]
- 99.Günzl A, Schimanski B. Tandem Affinity Purification of Proteins. Current Protocols in Protein Science. 2009. pp. 19.19.1–19.19.16. 10.1002/0471140864.ps1919s55 [DOI] [PubMed] [Google Scholar]
- 100.Schneider A, Charrière F, Pusnik M, Horn EK. Isolation of mitochondria from procyclic Trypanosoma brucei. In: Structural Genomics and Drug Discovery. 2007. pp. 67–80. 10.1007/978-1-59745-365-3_5 [DOI] [PubMed] [Google Scholar]
- 101.Smíd O, Horáková E, Vilímová V, Hrdy I, Cammack R, Horváth A, et al. Knock-downs of iron-sulfur cluster assembly proteins IscS and IscU down-regulate the active mitochondrion of procyclic Trypanosoma brucei. J Biol Chem. 2006; 281:28679–28686. 10.1074/jbc.M513781200 [DOI] [PubMed] [Google Scholar]
- 102.Miller FJ Jr., Griendling KK. Functional evaluation of nonphagocytic NAD(P)H oxidases. Redox Cell Biology and Genetics Part B. 2002. pp. 220–233. 10.1016/S0076-6879(02)53050-0 [DOI] [PubMed] [Google Scholar]
- 103.Molik S, Lill R, Mühlenhoff U. Methods for studying iron metabolism in yeast mitochondria. Mitochondria. 2007. pp. 261–280. 10.1016/S0091-679X(06)80013-0 [DOI] [PubMed] [Google Scholar]
- 104.Gould MK, Vu XL, Seebeck T, de Koning HP. Propidium iodide-based methods for monitoring drug action in the kinetoplastidae: Comparison with the Alamar Blue assay. Anal Biochem. 2008; 382:87–93. 10.1016/j.ab.2008.07.036 [DOI] [PubMed] [Google Scholar]
- 105.Sherman F. Getting started with yeast. Guide to Yeast Genetics and Molecular and Cell Biology—Part B. 2002. pp. 3–41. 10.1016/S0076-6879(02)50954-X [DOI] [Google Scholar]
- 106.Mumberg D, Müller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995; 156:119–122. 10.1016/0378-1119(95)00037-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Growth curves of double RNAi cells lines for TbCIA1-TbCIA2B and TbCIA2A-TbCIA2B in PCF (A and B) and BSF (C and D) cells were grown in presence (Tet+) and absence (Tet-) of tetracycline for 10 and 8 days, respectively.
(TIF)
Survival of mice infected with BSF TbMMS19 (A) and TbCIA2B (B) RNAi cell lines, uninduced (-) and induced (+) with doxycycline. Wild type (SM) was used as controls, also in the absence (-) and presence (+) of doxycycline. Five mice per group were used. Induced (+) cell lines are nudged in the graph for easier visualisation of the overlapping curves.
(TIF)
(A) Primary sequence of TbMMS19 with the centres of the HEAT repeats highlighted in a scale of red according to the probability of the respective residue to be in the centre of a HEAT repeat unit, as calculated by Ard2 [55].(B) Predicted 3D structure of TbMMS19 with HEAT repeats highlighted in red. The homology model for TbMMS19 was created using Phyre2 [53].(C) The N-and C-terminal domains of TbMMS19 are represented in blue and red, respectively. The boundaries of the domains were defined by alignment with human MMS19. Letters A-C represent functional domains of the human protein. Grey boxes correspond to HEAT repeats annotated in the Uniprot database for MMS19 (accession number Q9T76), or identified as described in (B) for TbMMS19.
(PNG)
N- and C-termini of TbMMS19 interact with TbCIA1. Tandem affinity purifications of PTP-TbMMS19 (3), PTP-TbMMS19-NTD (4), and PTP-TbMMS19-CTD (5), mock (1) and (2) empty plasmid, observed in a SYPRO Ruby-stained SDS-PAGE gel.
(PNG)
Three-dimensional models for the proteins were created using Phyre2 [53] and the best predictions were used for docking with ClusPro [59]. The five highest-scoring complexes (balanced score), were analysed with PredHS [61] to identify the key residues for interaction with TbCIA2B at the binding surface of TbMMS19. TbCIA2B is depicted as blue mesh in (A) and the N and C-terminal domains of TbMMS19 are shown in (A) and (B) as grey and green surfaces, respectively. TbCIA2B was omitted in (B) to uncover the residues at the contact surface, which are shown in a scale of red according to their associated SVM hot-spot score. Except in model 5, the residues more likely to be hot-spots of interaction are predicted to be in the C-terminal domain of TbMMS19. MacPyMOL (Schrödinger, LLC) was used to generate the figures based on the output of PredHS, ClusPro and Phyre2.
(PNG)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.