

Abstract
Traumatic brain injury (TBI)1 is a major public health problem estimated to affect nearly 1.7 million people in the United States annually. Due to the often debilitating effects of TBI, novel preventative agents are highly desirable for at risk populations. Here, we tested a whey protein supplement, Immunocal®, for its potential to enhance resilience to TBI. Immunocal® is a non-denatured whey protein preparation which has been shown to act as a cysteine delivery system to increase levels of the essential antioxidant glutathione (GSH). Twice daily oral supplementation of CD1 mice with Immunocal® for 28 days prior to receiving a moderate TBI prevented an ~25% reduction in brain GSH/GSSG observed in untreated TBI mice. Immunocal® had no significant effect on the primary mechanical injury induced by TBI, as assessed by MRI, changes in Tau phosphorylation, and righting reflex time or apnea. However, pre-injury supplementation with Immunocal® resulted in statistically significant improvements in motor function (beam walk and rotarod) and cognitive function (Barnes maze). We also observed a significant preservation of corpus callosum width (axonal myelination), a significant decrease in degenerating neurons, a reduction in Iba1 (microglial marker), decreased lipid peroxidation, and preservation of brainderived neurotrophic factor (BDNF) in the brains of Immunocal®-pretreated mice compared to untreated TBI mice. Taken together, these data indicate that pre-injury supplementation with Immunocal® significantly enhances the resilience to TBI induced by a moderate closed head injury in mice. We conclude that Immunocal® may hold significant promise as a preventative agent for TBI, particularly in certain high risk populations such as athletes and military personnel.
Keywords: Traumatic brain injury, Glutathione, Motor function, Cognitive function, Neuroprotection
Graphical Abstract
Introduction
Approximately 1.7 million incidences of TBI occur in the United States each year (Faul et al., 2010). Of these cases, nearly 75% are categorized as forms of mild TBI including concussions. Chronic traumatic encephalopathy and other forms of dementia have been linked to repetitive mild TBI caused by sports related concussive and subconcussive head trauma in football, hockey, soccer, and wrestling (Guskiewicz et al., 2005; McKee et al., 2009; Broglio et al., 2011; Gavett et al., 2011). In a similar manner, blastrelated TBI which is estimated to affect 10–20% of veterans returning from the wars in Iraq and Afghanistan, is also associated with an increased risk of chronic traumatic encephalopathy and other types of dementia, as well as posttraumatic stress disorder (Elder and Cristian, 2009; Goldstein et al., 2012). Regardless of the cause or severity of TBI, even mild TBI appears to be a significant risk factor for development of dementia including Alzheimer’s disease (Gavett et al., 2010; Johnson et al., 2010; Shively et al., 2012; Lee et al., 2013; Gardner and Yaffe, 2015). Thus, identification of new strategies to enhance resilience against TBI is of particular importance to people participating in “high risk” occupations, such as athletes or military personnel.
Oxidative and nitrosative stress are key elements of the secondary injury processes following TBI (Bains and Hall, 2012; Abdul-Muneer et al., 2015). GSH is an essential antioxidant that works in concert with GSH peroxidases, GSH transferases, and peroxiredoxins to detoxify hydroperoxides and other electrophilic species produced during periods of oxidative and nitrosative stress. Several studies suggest that endogenous GSH plays an important protective role against TBI. Brain GSH levels are significantly reduced following TBI induced by controlled cortical impact in rats (Tyurin et al., 2000). Genetic variations in the activity of glutathione-S-transferase-4, a GSH-dependent enzyme that reduces 4hydroxynonenal, is a determining factor in the extent of neurodegeneration after TBI (Al Nimer et al., 2013). Moreover, mice homozygous for deletion of glutathione peroxidase-1 display enhanced susceptibility to brain mitochondrial dysfunction induced by TBI (Xiong et al., 2004). These findings suggest that a strategy aimed at sustaining or enhancing brain GSH levels may be a viable approach to mitigate secondary injury and the subsequent long term cognitive, physical, and emotional deficiencies induced by TBI.
Along these lines, several studies have shown that administration of the GSH precursor, N-acetylcysteine, just prior to or immediately after TBI, significantly preserved brain tissue and mitochondrial GSH levels, reduced oxidative damage, and preserved neuronal survival (Xiong et al., 1999; Hicdonmez et al., 2006). Similarly, treatment with gamma-glutamylcysteine ethyl ester reduced indices of oxidative and nitrosative stress and preserved blood-brain-barrier (BBB) function when given immediately post-TBI (Reed et al., 2009; Lok et al., 2011). Finally, the GSH analog, S-nitrosoglutathione, decreased BBB disruption, minimized neuronal loss, and increased the expression of neurotrophic factors when administered post-TBI to rats subjected to controlled cortical impact (Khan et al., 2009, 2011) Each of these results indicates the potential of enhancing GSH as a therapeutic approach for TBI. Unfortunately, few of these previous studies evaluated the effects of GSH precursor supplementation on cognitive or motor deficits induced by TBI and as a result, it is presently unclear what therapeutic benefit this strategy might realistically hold for patients suffering from TBI.
The nutritional supplement, Immunocal®, is a non-denatured whey protein designed to augment the available intracellular GSH pool. Cellular GSH concentrations are highly dependent on the availability of cysteine, which is the limiting precursor in GSH synthesis (Tateishi et al., 1974; Meister, 1984). The cysteine precursor, cystine, occurs at high levels in Immunocal®, as does the direct GSH precursor, glutamylcysteine (Baruchel and Viau, 1996; Baruchel et al., 1998). Immunocal® has been shown to significantly increase blood or lymphocyte GSH levels in HIV-seropositive or cystic fibrosis patients, respectively (Bounous et al., 1993; Grey et al., 2003). Immunocal® is one of only a handful of nutritional supplements that are included in the Physician’s Desk Reference and is comprised of natural food protein placing it in the FDA category of generally recognized as safe (Physicians’ Desk Reference, 2016). We have recently found that Immunocal® preserves blood and spinal cord GSH levels and delays disease onset in a transgenic mouse model of amyotrophic lateral sclerosis (Ross et al., 2014). Similarly, Immunocal® restored GSH homeostasis in the CNS and ameliorated behavioral deficits in a mouse model of schizophrenia (Song et al., 2017). Based on the above findings, we hypothesized that supplementation with Immunocal® prior to TBI in mice would provide enhanced resilience against oxidative damage, neuronal cell death, and cognitive and motor impairments induced by a closed head impact injury.
Materials and Methods
Animal care and treatment
All animal work was conducted under a protocol approved by the University of Denver Institutional Animal Care and Use Committee. Male CD1 Elite mice (35 days-old) were purchased from Charles River Laboratories (Hollister, CA). Mice received a numbered ear tag upon arrival for identification purposes, and then were allowed one week to acclimate to the animal facility at the University of Denver before beginning the study. Mice were then randomly assigned and evenly distributed among one of three treatment groups: Sham, TBI, or TBI+Immunocal®. Mice in the TBI+Immunocal® group were dosed twice daily by oral gavage with 0.25mL of a 3.3% solution of Immunocal® in sterile drinking water. Dosing was performed 5 days a week over a period of 28 days prior to TBI. This dosing regimen has previously been shown to yield positive therapeutic effects in a mouse model of amyotrophic lateral sclerosis (Ross et al., 2014). The day on which TBI was induced was considered “day 0”. Following TBI, mice were monitored closely each day for signs of infection, bleeding, and general distress until the main study concluded on day 18. The magnetic resonance imaging (MRI) session (described below) concluded on day 3.
Traumatic brain injury
Following the 28-day Immunocal® dosing regimen, TBI was induced by controlled cortical impact using the Leica Impact One system (Leica Biosystems, Buffalo Grove, IL). Briefly, mice were anesthetized using an isoflurane vaporizer (VetEquip, Inc., Livermore, CA) and monitored throughout the procedure for the depth of anesthesia by toe pinch reflex. While anesthetized, temperature was maintained at approximately 37±1˚C by placement on a thermal pad. A midline incision approximately 1cm in length was made along the head and the skin was pulled aside using small bulldog clamps. Bupivacaine (0.25% solution in sterile water) was applied generously to the open incision. With the skull exposed, a dental scraper was used to partially remove the fascia in order to better visualize anatomical markers. Bregma was located, and a concave 22-gauge stainless steel disk, 5mm in diameter, was affixed to the skull using tissue adhesive just caudal to this point. Animals were then placed into a stereotaxic frame (Braintree Scientific Inc., Braintree, MA) and the head was secured to prevent movement during impact. The arm of the impactor was then positioned such that the impactor probe (5mm diameter) was directly centered over the metal disk. The probe was then set to the desired impact depth of 2.75mm and a velocity of 5.875 (±0.125) m/s (dwell time 100msec) to induce a moderate injury as described by Lloyd et al. (2008). Mice in the TBI and TBI+Immunocal® groups were subjected to injury at this time, after which animals were monitored for signs of TBI-induced apnea. Once apnea was overcome, animals were removed from the stereotaxic frame. Sham animals were not subjected to impact and were simply removed from the stereotaxic frame following identification. Mice were returned to the thermal pad, the metal disk was removed from the skull, and the incision was then closed using tissue adhesive. The mice were allowed to recover on the thermal pad during which time their righting reflex times were measured. Righting reflex was defined as the point at which the animal was able to return to and maintain a sternal position after being placed on its side during recovery from anesthesia. Mice were returned to their home cage once they became fully ambulatory.
Behavioral assays of cognitive and motor function
Challenging beam walk task
The challenging beam walk task for motor function and coordination was performed as previously described by Fleming et al. (2013). The apparatus for the challenging beam walk was composed of four segments supplied by Starks Plastics (Forest Park, OH), each of which was 25cm in length. The first segment had a width of 3.5cm, with each subsequent section decreasing by one centimeter in width to a final measurement of 0.5cm. These segments were secured together and suspended at a height of approximately 14.5cm above a level surface. Mice were allowed a two-day training period prior to TBI on days −2 and −1. On the first training day, mice were placed at the wide end of the beam. The investigator then held an empty cage containing clean bedding on its side a few centimeters in front of the mouse as incentive for the animal to navigate the beam. As the mouse moved toward the cage, the investigator pulled the cage away from the mouse such that the animal was forced to traverse the beam and the mouse was only allowed to enter the cage once it had successfully reached the end of the beam. This procedure was repeated until the mouse could traverse the beam without the need for prompting or correction from the investigator. On the following day, a cage with clean bedding was placed on its side at the narrow end of the beam in a fixed position. Mice were then placed on the wide end of the beam and allowed to traverse the full length of the beam to reach the empty cage. This phase of training was repeated until the mouse could consistently traverse the entire beam without prompting or correction from the investigator. Following TBI induction (or Sham surgery) on day 0, mice entered the testing phase of the challenging beam walk task on day 1. For this phase, wire grids with openings measuring 1cm2 were placed securely over each beam segment creating a space between the top of the grid and the surface of the beam. This was done to increase the difficulty of the task and to enhance visual scoring of foot faults. As before, an empty cage was placed at the narrow end of the beam and served as the goal for successful completion of the beam walk. Mice were placed at the wide end of the beam on top of the grid and allowed to traverse the entire length of the beam a total of three times, with each traversal of the beam recorded using a video camera. The number of foot faults for the right hind foot was quantified for each animal on each segment of the beam and averaged across the three attempts. Foot faults were defined as any point at which the mouse stepped through the metal grid or gripped the plastic beam instead of the wire grid. The time it took the mouse to traverse the full length of the beam was also recorded for each of the three attempts on the beam, and the percentage change in time taken to traverse the beam between trial 1 and trial 3 for each mouse was calculated.
Accelerating Rotarod
Rotarod testing for motor coordination and function was performed on days 9 and 16 following TBI (or Sham surgery). Mice were placed on a rod, 30 mm in diameter, rotating at 4 rpm. Animals were placed in individual lanes to prevent interference between mice while the test was being conducted. When the mice had acclimated to the slow speed, the rod was accelerated from 4 rpm to 40 rpm over the course of 5 minutes. Mice were given three attempts on the apparatus before testing ended. The duration of time that the mouse spent on the rod was recorded by depression of a lever triggered upon the mouse falling and the recorded values were averaged across the three attempts.
Barnes maze
Barnes maze (ANY-maze, Wood Dale, IL) testing was performed on days 10–16 post-TBI, as described by Mouzon et al. (2012). The first 6 days of testing comprised the acquisition phase, followed by a single probe/test day. The circular maze was divided into quadrants with an arrow on the wall used as a visual cue to identify the location of the escape pod. During the acquisition phase, mice were placed in each quadrant and allowed 90sec to find the escape pod. If the mice were unable to find the pod after the allotted time, they were directed to it and remained in the pod for 30sec. If they found the pod and entered on their own, the pod was then covered and they remained there for 30sec. Videos were reviewed and latency times to find the escape pod were recorded. On the probe day, the pod was blocked so that mice could not enter. Mice were placed in the middle of the maze and allowed to search the maze for 60sec. Videos were reviewed and latency times to the escape pod zone (encompassing the escape pod and either pod directly adjacent to it) were recorded.
Reagents
Primary antibodies to beta actin, S100beta and Iba-1 were purchased from Abcam (Cambridge, MA). The primary antibodies to Tau phosphorylated on Ser396, Thr231, and Ser404, as well as total Tau, were purchased from Invitrogen (Carlsbad, CA). Primary antibody to BDNF was from Alomone Labs (Jerusalem, Israel). Primary antibody to GFAP was purchased from Abcam (Cambridge, MA). Purified oxidized (GSSG) and reduced (GSH) glutathione were purchased from Sigma Aldrich Co. LLC (St. Louis, MO). Cy3-conjugated secondary antibody was purchased from Jackson Immunoresearch (Westgrove, PA). Fluoro-Jade C staining kit was purchased from Biosensis (Temecula, CA). Luxol fast blue staining kit was purchased from American Mastertech (Lodi, CA). Malondialdehyde (MDA) lipid peroxidation assay kit was obtained from Abcam (Cambridge, MA) and the assay was conducted essentially as described by the manufacturer.
Fluoro-Jade C staining for degenerating neurons
Tissue processing
Frozen whole brains, excluding cerebellum, were cryosectioned either by the Histology Core at the University of Colorado Anschutz medical campus or AML Laboratories Inc. (St. Augustine, FL). Briefly, 12μm coronal sections were created starting at bregma and proceeding towards the posterior of the brain. Tissue sections were mounted on adhesive microscope slides discarding three to four tissue sections between each mounting. Following mounting, tissue was fixed in 4% paraformaldehyde for one hour.
Slide staining
Fluoro-Jade C staining was performed as specified by the manufacturer. Briefly, coronal brain sections were immersed in a 1:9 (v/v) solution of 1% sodium hydroxide and 70% ethanol for five minutes, followed by a two-minute wash in 70% ethanol. Next, tissue sections were immersed in a 1:9 (v/v) solution of 0.06% potassium permanganate and distilled water for ten minutes and then washed with distilled water for two minutes. Tissue was then incubated in a 1:2:8 (v/v/v) solution of DAPI, 0.0004% Fluoro-Jade C and distilled water for ten minutes, taking precaution to protect the solution from light. Sections were then washed three times in distilled water and dried at 50–60ºC for ten minutes. Sections were imaged under 40x magnification on a Zeiss Axiovert-200M fluorescence microscope using a FITC filter and in a blinded fashion, to identify fluorescent foci of degenerating neurons. The total number of Fluoro-Jade C-positive foci were then quantified for at least two tissue sections per mouse.
Luxol fast blue staining
Tissue processing was done as described above for Fluoro-Jade C staining. Brain sections were incubated in Luxol fast blue stain solution at 60°C overnight, followed by washing with distilled water. Sections of gray and white matter were differentiated by dipping brain tissue into 0.05% lithium carbonate and 70% ethanol. Slides were then immersed in cresyl violet stain for ten minutes followed by further differentiation in 70% ethanol. Following the staining process, tissue sections were imaged at 20x magnification to visualize the corpus callosum. Images of the mid-body of the corpus callosum were captured for at least two tissue sections per animal. The health of the corpus callosum was assessed by measuring the maximum width of the mid-body.
Magnetic resonance imaging (MRI)
All MRI studies were performed in the Colorado Animal Imaging Shared Resources (University of Colorado Anschutz Medical Campus, Aurora, CO). All animals underwent an MRI session 72 hours after TBI (or Sham surgery), using pre- and post-gadolinium-enhanced (0.2mmol/kg Omniscan® IV) T1-weighted sequences (Frey et al., 2014). The mice were anesthetized with 2.5% isoflurane, placed into an animal holder and inserted into a 4.7 Tesla Bruker PharmaScan. A quadrature birdcage coil (inner diameter 38mm) tuned to the 1H frequency of 200.27 MHz, was used for RF transmission and reception. T1-weighted MR images (for BBBD assessment) were acquired using a multi-slice multi-echo (MSME, Bruker manufacturer label for a spin echo sequence, in this case with one echo) sequence, before and 5 minutes after administration of 0.2mmol/kg Omniscan® via tail vein. The following acquisition parameters were used: FOV = 36mm; TR/TE = 900/11msec; slice thickness =1mm with no gaps applied; number of slices = 16; number of averages = 2; matrix size = 128×256; total acquisition time = 3min 50sec. All images were acquired in the axial plane. All images analysis was performed using Bruker ParaVision v4.1 software.
Western blotting
Whole half brains, excluding cerebellum, were thawed from liquid nitrogen. A 1mL aliquot of lysis buffer was added, with 1μL of leupeptin (5mg/mL) and 1 μL of aprotinin (5mg/mL), per half brain. The brains were then homogenized using a Dounce glass/glass homogenizer by 20 strokes with the loose pestle followed by 20 strokes with the tight pestle. Samples were centrifuged for 5min at 10,000rpm, and the supernatant was isolated. The samples were diluted 1:100 for a BCA protein assay. Western immunoblotting was done to immunochemically detect proteins immobilized on polyvinylidene difluoride (PVDF) membranes. Protein samples (80μg/lane) were resolved by SDS-PAGE and proteins were then transferred to PVDF membranes. Non-specific binding sites were blocked using 1% BSA in phosphate-buffered saline (pH 7.4) containing 0.1% Tween-20 (PBS-T) for 1h at 25°C. The blocking buffer was drained and the membrane was allowed to incubate in primary antibody diluted in blocking buffer overnight at 4°C. The membrane was washed 3x for 15min in PBS-T and was then incubated with the secondary antibody for 1.5h at 25°C. The secondary was then removed and the membrane was washed again in PBS-T, 3x for 15min. Immunoreactive proteins were detected using enhanced chemiluminescence (GE Healthcare; Pittsburgh, PA) and films were developed using a CP 1000 developer (AGFA; Mortsel, Belgium). Re-probing of blots was performed by stripping in 0.1M Tris-HCl (pH 8.0), 2% SDS, and 100 mM β-mercaptoethanol for 30min at 52°C. The blots were rinsed twice in PBS-T and processed as above with a different primary antibody. In general, blots shown are representative of duplicate gels ran on at least three sets of animals with each set consisting of a Sham, TBI, and TBI+ICAL mouse.
Immunofluorescence microscopy
Sections of cortex were stained for the astrocyte marker, glial fibrillary acidic protein (GFAP), and nuclei were stained with DAPI, using a standard immunohistochemistry protocol. GFAP-positive astrocytes were detected using a Cy3-conjugated secondary antibody. Images shown are representative of triplicate slides ran on at least three sets of animals with each set consisting of a Sham, TBI, and TBI+ICAL mouse.
High performance liquid chromatography with electrochemical detection (HPLC-ECD)
Tissue processing
Full half brains, excluding cerebellum, were obtained from mice 72h post-TBI (or Sham surgery) and were immediately frozen in liquid nitrogen. For HPLC-ECD analysis, we utilized a previously published procedure (Ross et al., 2014). Briefly, 2.5M perchloric acid was added to each half brain and the brains were roughly chopped using pointed surgical scissors. Samples were then sonicated 3 times for 15s intervals. Samples were then centrifuged for 5min at 13,000rpm and the supernatant was removed. A 2μL aliquot of the supernatant was used for a BCA protein assay. The remainder of each solution was neutralized with 500μL of 4M KOH and vortexed thoroughly. Samples were then centrifuged for 15min at 13,000rpm, and stored at −80°C until separation and analysis by HPLC-ECD.
HPLC-ECD
GSH and GSSG in samples and known standards were separated by reversed-phase HPLC on a C18 bonded silica column at 35°C (5μm, 4.6 × 250mm) from Dionex, Inc. (Sunnyvale, CA). Analytes were detected using a CoulArray® detector (model 5600, ESA) on three coulometric array cells in series; electrochemical detectors were set between 0 and 900mV at increments of 75mV. Concentrations were determined with a standard curve of each identified analyte. Mobile phase consisted of 50mM lithium acetate and 1% acetonitrile in water, pH 3.8. The flow rate was set to 0.4mL/min for all samples. CoulArray® software was used for baseline correction and peak analysis.
Experimental design and statistical analysis
Data presented are shown as the mean±SEM for the number (n) of independent experiments performed. An independent set of mice consisted of a single mouse from each group (Sham, TBI, TBI+ Immunocal®). Statistical differences between groups were evaluated using either one-way ANOVA with a post-hoc Tukey’s test or unpaired Student’s t-tests. Effect sizes and corresponding 95% confidence intervals are also shown within the Figure Legends. Data analysis of behavioral tests was performed by observers blinded to the group assignments of the mice. Similarly, microscopic analysis of Fluorojade-C-and luxol fast blue-stained slides was performed by observers blinded to the group assignments of the mice. Finally, MRI analysis of BBB permeability was also performed and quantified by an observer blinded to the group assignments of the mice.
Results
Pre-injury supplementation with Immunocal® does not affect the primary mechanical injury induced by a moderate TBI
Throughout the study, mice were equally divided into the following three groups: Group 1, Sham surgery controls; Group 2, untreated TBI mice; and Group 3, mice pretreated with Immunocal® for 28 days prior to TBI. The extent of brain injury was initially assessed at 72h post-TBI by MRI analysis of BBB disruption. In mice subjected to TBI, T1 weighted images taken with gadolinium contrast showed areas of hyper-intensity which indicate BBB disruption (Figures 1A and 1B, see asterisk in the TBI image which marks an area of BBB disruption). In general, areas of BBB disruption were exclusively observed in mice subjected to TBI but not Sham controls. In addition, areas of BBB disruption appeared primarily in the outer layers of the cortex and were most often seen in the region caudal to bregma (i.e., the region of impact). However, brain injury was not confined solely to the midline of the brain, but also extended to either side of the midline. This latter observation is characteristic of this TBI model where the impactor probe hits a metallic disk affixed to the closed skull, causing a diffusion of the injury throughout the cortex. Quantification of the volume of BBB disruption indicated by the T1 weighted, gadoliniumenhanced images revealed no significant differences between the untreated TBI group and the Immunocal®-pretreated TBI group (Figure 1C). In a scatter plot of these data, it is evident that 3 out of 5 untreated TBI mice and 4 out of 5 Immunocal®-pretreated TBI mice displayed measurable BBB disruption (Figure 1D). These data indicate that the overall magnitude of brain injury induced by the TBI procedure, measured as BBB disruption, was comparable for each group of mice and moreover, preinjury supplementation with Immunocal® had no discernible protective effect against the primary mechanical injury induced by a moderate TBI.
Figure 1. MRI analysis of BBB disruption reveals that Immunocal® does not protect mouse brain from the primary mechanical injury induced by TBI.

A) Representative gadolinium-enhanced MSME T1-weighted MR images of BBB disruption at 72h post-TBI. The white asterisk indicates a hyper-intense area of BBB disruption in a mouse that received a moderate TBI (left) compared to a Sham mouse (right). B) The areas demarcated by the boxes in (A) are magnified 300% to emphasize the area of hyper-intensity in the TBI mouse. C) The graph shows quantification of the volume of BBB disruption in mm3. Results are shown as mean±SEM, n=5 mice per group; oneway ANOVA, p=0.230. D) The graph shows a scatter plot of the data shown in (C). Abbreviations used: BBB, blood-brain-barrier; ICAL, Immunocal®.
Following MRI analysis, mice were euthanized and brains removed at 72h post-TBI. One-half of each brain was flash frozen in liquid nitrogen for subsequent HPLC analysis of GSH (discussed below). 32The other half of the brain was dounce homogenized in lysis buffer and protein samples were resolved by SDS-PAGE and western blotted to assess the phosphorylation status and expression level of the microtubule bundling protein Tau. Several TBI-induced changes in Tau phosphorylation were observed by western blot including an enhanced electrophoretic mobility (downward shift) of Tau phosphorylated on Ser396, Thr231, and Ser404 (Figure 2A). In addition, TBI caused a marked reduction in the amount of total Tau observed in brain lysates, while no difference in actin was apparent in the same samples (Figure 2A). Finally, quantification of Tau phosphorylated at specific residues and normalized to total Tau revealed no statistically significant increases in Tau phosphorylation at 72h post-TBI; however, there was a trend towards increased Tau phosphorylation on Ser396 in untreated TBI mice compared to Sham and this trend persisted for TBI mice which had been pretreated with Immunocal® (Figure 2B). All of these TBI-induced changes in the electrophoretic mobility of various phospho-Tau forms and the expression of total Tau were observed regardless of whether the samples were obtained from untreated TBI mice or mice pretreated with Immunocal® (Figure 2). It is unclear precisely what the changes in electrophoretic mobility represent for these various forms of phospho-Tau, but most likely they reflect changes in phosphorylation at other sites on the molecule. Nonetheless, there are detectable changes in Tau phosphorylation and expression induced in this TBI model and they are completely unaffected by preinjury supplementation with Immunocal®, suggesting that they may represent biochemical changes caused by, or in response to, the primary mechanical injury induced by TBI.
Figure 2. Western blotting for Tau phosphorylation and expression in brain tissue of mice subjected to TBI.

At 72h post-TBI, one half of the brain (excluding cerebellum) was dissected and homogenized in lysis buffer. Whole brain tissue lysates were resolved by SDS-PAGE and proteins transferred to PVDF membranes. A) Blots were sequentially stripped and re-probed with antibodies against Tau phosphorylated on Ser396, Thr231, and Ser404, total Tau, and actin (as a loading control). Asterisks indicate prominent phospho-Tau bands and MW markers are shown for estimation of size. The blots shown are from two independent sets of mice (Sham, TBI, TBI+ICAL) which displayed similar results. B) Densitometric analysis of each form of phospho-Tau was performed on three independent sets of mice. Phospho-Tau was normalized to total Tau and this value was set to 1.00 for each Sham mouse; the ratio of phospho-Tau to total Tau was then expressed relative to the Sham control for each set of mice. No statistically significant differences were observed; however, there was a trend towards increased Tau phosphorylation on Ser396 in untreated TBI mice compared to Sham and this trend persisted for TBI mice which had been pretreated with Immunocal® (one-way ANOVA, p=0.096). Abbreviations used: ICAL, Immunocal®; pTau, phospho-Tau.
Finally, in addition to the MRI and Tau analyses described above, several clinical indicators also demonstrate that pre-injury supplementation with Immunocal® did not significantly affect the magnitude of the primary mechanical injury induced by a moderate TBI in mice. Immunocal®-pretreated mice had a significantly lower body weight (~10% decrease) than either Sham control mice or untreated TBI mice when weight was assessed just prior to TBI (Figure 3A). This decrease in body weight may reflect some mild stress due to the 28-day chronic oral dosing procedure or it could represent the animals becoming leaner due to the whey protein administration. TBI induced a statistically significant, nearly three-fold increase in the righting reflex time in comparison to Sham control mice, and this effect was comparable in untreated TBI mice and those pretreated with Immunocal® (Figure 3B). In a similar manner, both untreated TBI mice and Immunocal®-pretreated TBI mice displayed substantial apnea times which were comparable to one another, while Sham control mice did not show any signs of apnea (Figure 3C). These clinical measures further support the conclusion that pre-injury supplementation with Immunocal® had no significant effect on the magnitude of the primary brain injury that the mice experienced in response to a moderate TBI.
Figure 3. Clinical measures following TBI or Sham surgery.

A) Body weights (mean±SEM) were assessed on day 0, just prior to TBI or Sham surgery. Mice pretreated for 28 days with Immunocal® displayed a statistically significant (p<0.01, n=40 mice per group) reduction in body weight compared to both Sham mice (**) and untreated TBI mice (##); one-way ANOVA, p=0.0005. B) Righting reflex times (mean±SEM) were measured immediately post-TBI or Sham surgery. Both untreated and Immunocal®-pretreated mice subjected to TBI displayed statistically significant (p<0.001, n=40 mice per group) increases in righting reflex times when compared to Sham mice (***); one-way ANOVA, p=0.0001. C) Apnea times (mean±SEM) were documented immediately post-TBI or Sham surgery. All of the mice subjected to TBI, including those pretreated with Immunocal®, displayed substantial periods of apnea following impact, with no significant difference observed between groups (n=20 mice per group; unpaired t-test, p=0.995). By comparison, no mice subjected to Sham surgery displayed any apnea. Abbreviations used: ICAL, Immunocal®; s, seconds.
Pre-injury supplementation with Immunocal® significantly improves motor and cognitive deficits induced by a moderate TBI
Animals were tested for TBI-induced deficits in motor function using the challenging beam walk task and performance on an accelerating rotarod. In the challenging beam walk task, mice were trained prior to TBI to traverse a beam with progressively narrower width segments. On the day of testing (24h post-TBI), a wire grid was placed over the beam to create a more challenging motor paradigm for the mice. In general, animals in all three groups performed very well on the beam walk with approximately 10% or fewer right hind foot faults observed on the first three beam segments (Figures 4A-C). However, on the narrowest width segment of the beam, both Sham control mice and untreated TBI mice had significant difficulty traversing the beam and each group displayed greater than 40% right hind foot faults (Figure 4D). No significant difference was observed between the Sham control and untreated TBI groups, demonstrating that this effect was not related to TBI but instead reflected the overt difficulty of the task. Notably, Immunocal®-pretreated mice that were subjected to TBI performed consistently better than either Sham control mice or untreated TBI mice on the narrowest width segment of the beam, though the difference observed was only statistically significant when compared to untreated TBI mice (Figure 4D).
Figure 4. Performance on the challenging beam walk task of mice subjected to TBI.

A-D) Percentage of right hind foot faults quantified in narrowing beam segments at 24h post-TBI. Segments 1–4 represent sections of the beam with progressively narrower widths. All groups showed increasing foot faults as the beam became progressively narrower. In the narrowest section of the beam (section 4), the percentage of right hind foot faults for Immunocal®-pretreated mice that were subjected to TBI was statistically significantly lower than the corresponding value for untreated TBI mice (#p=0.045, n=8 mice per group; unpaired t-test; effect size [95% confidence intervals] = −1.10 [−2.09 to 0.00]). E) The percentage change in the time taken to traverse the entire beam between trial #1 and trial #3 is shown for each group. Untreated TBI mice showed significantly less improvement than Sham mice (*p<0.05; effect size [95% confidence intervals] = 1.53 [0.15 to 2.68]) and Immunocal®-pretreated mice that were subjected to TBI showed significantly more improvement than untreated TBI mice (#p<0.05, n=6 mice per group; one-way ANOVA, p=0.012; effect size [95% confidence intervals] = −1.42 [−2.55 to −0.06]). Abbreviations used: ICAL, Immunocal®.
In addition to assessing right hind foot faults on the beam walk, we also utilized this motor function test as a type of learning paradigm. Because the mice were trained on the beam without the wire grid, addition of this grid on the day of testing represented a new challenge for the mice (i.e., they had to learn to traverse the beam by walking on the wire grid). To assess their capacity to learn this new task, the amount of time taken to traverse the beam on the first of three trials was compared to that on the final of the three trials on the day of testing. Sham control mice clearly learned to traverse the wire grid as evidenced by a nearly 70% reduction in the time taken to traverse the beam between the first and last trial (Figure 4E). In contrast, untreated TBI mice only improved their time to traverse the beam on average by approximately 20% from the first to the last trial, which was statistically significantly different than the Sham control group. Pre-injury supplementation with Immunocal® corrected this deficit after TBI and these mice displayed an improvement in time to traverse the beam which was indistinguishable from that of the Sham control group (Figure 4E).
Next, we evaluated motor performance on an accelerating rotarod at 9 days and 16 days post-TBI. On day 9 post-TBI, Sham control mice spent an average of approximately 20s on the accelerating rotarod before falling off of the apparatus. Untreated TBI mice only remained on the rotating rod for approximately half this time, a statistically significant decrease compared to the Sham control group (Figure 5A). However, mice pretreated with Immunocal® prior to TBI showed a latency time to fall which was significantly greater than untreated TBI mice and not statistically different from the Sham control group (Figure 5A). By day 16 post-TBI, all three groups had increased their performance on the accelerating rotarod and displayed greater latency times to fall than they showed at 9 days post-TBI. In addition, the untreated TBI group appeared to recover their motor function on this task and no longer displayed a significant difference from the Sham control group (Figure 5B).
Figure 5. Pre-injury supplementation with Immunocal® significantly improves rotarod performance post-TBI.

Rotarod performance was assessed on day 9 (A) and day 16 (B) post-TBI. On day 9 post-TBI, time spent on the accelerating rotarod apparatus significantly decreased in untreated TBI mice compared to Sham control mice (*p<0.05; effect size [95% confidence intervals] = −1.40 [−2.46 to −0.15]). This motor deficit was completely prevented by pre-injury supplementation with Immunocal® (#p<0.05 versus untreated TBI mice, n=7 mice per group; one-way ANOVA, p=0.026; effect size [95% confidence intervals] = 1.70 [0.38 to 2.78]). On day 16 postTBI, no significant differences in latency time were observed between groups (n=6 mice per group; one-way ANOVA, p=0.686). Abbreviations used: ICAL, Immunocal®; s, seconds.
We also evaluated the effects of TBI on cognitive function using the Barnes maze to assess spatial learning and memory on days 10–16 post-TBI. During the six-day acquisition phase of the Barnes maze test, Sham control mice progressively learned to find the escape pod as evidenced by a shortening of the average delay time from approximately 85s on day 1 compared to less than 40s on day 6 (Figure 6A). Over this same time frame, untreated TBI mice appeared to learn less quickly than Sham control mice to find the escape pod and demonstrated a plateau in average delay time of approximately 75s. Immunocal®-pretreated mice displayed average delay times that were intermediate between the Sham control group and untreated TBI mice (Figure 6A). Statistical analysis of the acquisition phase data revealed a statistically significant difference between the delay times for the untreated TBI mice and the Sham control group on day 6 (Figure 6B). Furthermore, on day 6 of the acquisition phase, Immunocal®pretreated mice that were subjected to TBI showed a statistically significant improvement in delay time to find the escape pod in comparison to untreated TBI mice (Figure 6B). Finally, in the probe phase of the Barnes maze test, Sham control mice took on average approximately 6s to find the escape pod zone. Untreated TBI mice took greater than 10s on average to find the escape pod zone, an effect which was not statistically significantly different from the Sham control group, probably due to the relatively high error in the untreated TBI group (Figure 6C). However, mice which had received Immunocal® treatment prior to TBI displayed an average delay time of less than 5s to find the escape pod zone, an effect which was statistically significantly different than untreated TBI mice (Figure 6C).
Figure 6. Pre-injury supplementation with Immunocal® significantly improves various aspects of Barnes maze performance post-TBI.

A) The mean values calculated for the delay time taken to find the escape pod are shown for each group of mice (Sham, TBI, TBI+ICAL) during the six-day acquisition phase of the Barnes maze test (days 10–15 post-TBI). Error bars are not shown in (A) for clarity. B) Data for day 6 of the acquisition phase of the Barnes maze test are shown as the mean±SEM for the delay times observed. On day 6, untreated TBI mice displayed a statistically significant increase in delay time to find the escape pod when compared to Sham control mice (**p<0.01, n=7–8 mice per group; one-way ANOVA, p=0.003; effect size [95% confidence intervals] = 2.15 [0.92 to 3.20]) and Immunocal®-pretreated mice that were subjected to TBI showed a statistically significant decrease in delay time in comparison to untreated TBI mice (#p<0.05, n=7–8 mice per group; one-way ANOVA, p=0.003; effect size [95% confidence intervals] = −1.52 [−2.56 to −0.29]). C) Delay times to find the escape pod zone for the probe phase of the Barnes maze test (day 16 post-TBI) are shown as the mean±SEM for each treatment group. The difference between the Sham control group and the untreated TBI group did not reach statistical significance due to the high error in the TBI group. Immunocal®-pretreated mice that were subjected to TBI showed a statistically significant decrease in delay time in comparison to untreated TBI mice (#p<0.05, n=7–8 mice per group; one-way ANOVA, p=0.049; effect size [95% confidence intervals] = −1.15 [−2.17 to 0.00]). Abbreviations used: ICAL, Immunocal®; s, seconds.
Pre-injury supplementation with Immunocal® preserves brain GSH/GSSG ratio and ameliorates biochemical and histopathological indices of oxidative damage and neuronal injury induced by a moderate TBI
Several biochemical and histopathological indices of neuronal injury were evaluated in mice subjected to TBI. First, we measured brain levels of GSH and the ratio of reduced GSH to oxidized GSSG at 72h post-TBI using HPLC with electrochemical detection. The concentrations of GSH and GSSG measured in mouse brain are shown in Figures 7A and 7B, respectively. No significant differences were observed between groups with respect to total GSH or total GSSG, although there was a trend towards enhanced GSSG levels in the untreated TBI group (Figure 7B, p=0.09). The ratio of GSH to GSSG was on average, approximately 250 to 1 in the brains of Sham control mice. Untreated TBI mice displayed an approximately 25% reduction in the brain GSH to GSSG ratio in comparison to Sham control mice (Figure 7C). Pre-injury supplementation with Immunocal® completely preserved the brain GSH to GSSG ratio measured at 72h post-TBI at a level significantly higher than that measured in untreated TBI mice and similar to that of the Sham control group (Figure 7C). Finally, we calculated the amount of GSSG as a percentage of total GSH equivalents (GSH +2 GSSG). The %GSSG trended towards an increase in untreated TBI mice in comparison to Sham control mice, although this change did not reach statistical significance (Figure 7D, p=0.09). Immunocal®-pretreated TBI mice displayed a %GSSG in brain which was statistically significantly less than that observed in untreated TBI mice (Figure 7D).
Figure 7. Pre-injury supplementation with Immunocal® preserves the brain GSH/GSSG ratio following TBI in mice.

At 72h post-TBI, one half of the brain (excluding cerebellum) was dissected and extracted for analysis of reduced GSH (A) and oxidized GSSG (B) by HPLC with electrochemical detection, as described in the Materials and Methods. No significant differences were observed between groups with respect to total GSH or GSSG, although there was a trend towards enhanced GSSG levels in the untreated TBI group (p=0.09; one-way ANOVA). C) Immunocal®-pretreated mice that were subjected to TBI showed a statistically significant increase in the GSH/GSSG ratio in comparison to untreated TBI mice (#p<0.05, n=4–6 mice per group; one-way ANOVA, p=0.017; effect size [95% confidence intervals] = 2.13 [0.51 to 3.39]). D) Immunocal®-pretreated mice that were subjected to TBI showed a statistically significant decrease in the %GSSG in comparison to untreated TBI mice (#p<0.05, n=4–5 mice per group; one-way ANOVA, p=0.025; effect size [95% confidence intervals] = −2.17 [−3.46 to −0.45]). Abbreviations used: ICAL, Immunocal®.
Next, we assessed the effects of Immunocal® pretreatment on lipid peroxidation and expression of brain-derived neurotrophic factor (BDNF) measured at 72h post-TBI. Untreated TBI mice displayed a statistically significant, nearly two-fold increase in brain MDA content when compared to Sham control mice and this effect was essentially reversed by pretreatment with Immunocal® (Figure 8A). Analysis of brain BDNF expression revealed an approximately 35% decrease in untreated TBI mice compared to Sham control mice at 72h post-TBI. Pretreatment with Immunocal® significantly preserved brain BDNF expression at this time point (Figures 8B and 8C).
Figure 8. Pre-injury supplementation with Immunocal® reduces brain lipid peroxidation and preserves BDNF expression following TBI in mice.

A) At 72h post-TBI, one half of the brain (excluding cerebellum) was dissected and homogenized in lysis buffer. Whole brain tissue lysates were assayed for lipid peroxidation using detection of malondialdehyde (MDA) as a marker of oxidative damage. MDA absorbance was normalized to total protein. Untreated TBI mice displayed a statistically significant, approximately two-fold increase in brain MDA content compared to Sham control mice (*p<0.05, n=3–4 mice per group; one-way ANOVA, p=0.012) and Immunocal®-pretreated mice that were subjected to TBI showed a statistically significant decrease in brain MDA content in comparison to untreated TBI mice (#p<0.05, n=3–4 mice per group; one-way ANOVA, p=0.012). B) Whole brain tissue lysates were resolved by SDS-PAGE and proteins transferred to PVDF membranes. Blots were sequentially stripped and reprobed with antibodies against pro-BDNF/BDNF and Actin. The blots shown are representative of data obtained from three independent sets of mice (Sham, TBI, TBI+ICAL) which displayed similar results. C) Densitometric analysis of pro-BDNF/BDNF expression was performed on three independent sets of mice. Total BDNF (pro-BDNF+BDNF) was normalized to actin and this value was set to 1.00 for each Sham mouse; the ratio of total BDNF to actin was then expressed relative to the Sham control for each set of mice. Untreated TBI mice displayed an approximately 35% reduction in brain BDNF compared to Sham control mice (p<0.05, one-way ANOVA with a post-hoc Dunnett’s test). Immunocal®-pretreated mice that were subjected to TBI showed a statistically significant increase in brain BDNF expression in comparison to untreated TBI mice (##p<0.01, n=3 mice per group; unpaired t-test).
We also evaluated neuroinflammation by western blotting whole brain lysates harvested at 72h post-TBI for the microglial marker, Iba1, and the astrocyte marker, S100beta. In paired sets of mice (i.e., mice which had been subjected to TBI or Sham surgery on the same day), Iba1 immunoreactivity was increased in the brains of untreated TBI mice in comparison to both Sham control mice and Immunocal®pretreated mice subjected to TBI (Figure 9A). In contrast, no apparent change in immunoreactivity for S100beta was observed in these brain lysates. To further assess reactive astrocytes, we stained for the astrocyte marker, GFAP, in brain sections taken from mice at 18 days post-TBI. No significant differences in the number or morphology of GFAP-positive astrocytes were observed between any of the treatment groups (Figure 9B).
Figure 9. Assessment of neuroinflammation in mice subjected to TBI.

A) At 72h post-TBI, one half of the brain (excluding cerebellum) was dissected and homogenized in lysis buffer. Whole brain tissue lysates were resolved by SDS-PAGE and proteins transferred to PVDF membranes. Blots were sequentially stripped and reprobed with antibodies against Iba1 (a microglial/macrophage marker), Actin (as a loading control), and S100beta (an astrocyte marker). The blots shown are from two independent sets of mice (Sham, TBI, TBI+ICAL) which displayed similar results. Iba1 levels trended higher in untreated TBI mouse brains than in either the corresponding Sham controls or Immunocal®-pretreated TBI mouse brains. S100beta levels did not appear to differ substantially between groups. B) Sections of cerebral cortex taken from near the midline and just caudal to bregma were stained for the astrocyte marker GFAP (shown in red) and nuclei were stained with DAPI (shown in blue). Images of 40x fields shown are representative of results observed in multiple sets of mice at 18 days post-TBI. Abbreviations used: ICAL, Immunocal®.
Axonal myelination was assessed by staining brain tissue harvested at 18 days post-TBI with Luxol fast blue and measuring the maximal width of the mid-body of the corpus callosum. Sham control mice displayed intact corpus callosum mid-bodies with deep Luxol fast blue staining indicative of extensive axonal myelination (Figure 10A). Untreated TBI mice displayed either much narrower midbodies or corpus callosum with large regions devoid of staining, while Immunocal®-pretreated mice subjected to TBI showed mostly intact mid-bodies with continuous staining (Figure 10A). Quantification of the maximum width of the mid-body of the corpus callosum, which stained positively with Luxol fast blue, demonstrated that untreated TBI mice had a statistically significant decrement of approximately 50% in myelinated axons compared to Sham control mice. Pre-injury supplementation with Immunocal® significantly but only partially, rescued axonal myelination of the corpus callosum following a moderate TBI (Figure 10B).
Figure 10. Pre-injury supplementation with Immunocal® improves axonal myelination of the corpus callosum in mice subjected to TBI.

A) Panels show Luxol fast blue-stained imaging of the mid-body of the corpus callosum at 20x magnification taken at 18 days post-TBI. Red lines indicate maximum width of mid-body and white arrows indicate area of demyelination observed in an untreated TBI mouse. B) Quantification of corpus callosum mid-body measurements. Untreated TBI mice displayed a statistically significant decrease in the maximum width of the corpus callosum mid-body when compared to Sham control mice (***p<0.001, n=5–7 mice per group; one-way ANOVA, p=0.001; effect size [95% confidence intervals] = −3.40 [−4.84 to −1.44]. Immunocal®-pretreated mice that were subjected to TBI showed a statistically significant increase in the maximum width of the corpus callosum midbody in comparison to untreated TBI mice (#p<0.05, n=5–6 mice per group; one-way ANOVA, p=0.001; effect size [95% confidence intervals] = 1.35 [−0.06 to 2.53]). Abbreviations used: ICAL, Immunocal®.
Finally, we evaluated brain tissue harvested at 18 days post-TBI for areas of degenerating neurons using Fluoro-Jade C staining. Most Fluoro-Jade C-positive foci were observed in the outer layers of the cerebral cortex, although some isolated regions of staining were also observed in subcortical structures (Figures 11A and 11B). Fluoro-Jade C-positive foci were scored across entire coronal sections of brain and the number of 40x fields containing either single or multiple foci were quantified relative to the Sham control group. Untreated TBI mice displayed statistically significant, approximately 4-fold and 2.5-fold increases in the number of fields with single and multiple Fluoro-Jade C-positive foci, respectively, in comparison to the Sham control group (Figures 11C and 11D). Pre-injury supplementation with Immunocal® markedly attenuated neuronal degeneration induced by a moderate TBI, resulting in statistically significant decreases in the numbers of fields with single or multiple FluoroJade C-positive foci in comparison to the untreated TBI group (Figures 11C and 11D).
Figure 11. Pre-injury supplementation with Immunocal® markedly reduces neuronal degeneration in the brains of mice subjected to TBI.

Sections of cerebral cortex taken from near the midline and just caudal to bregma were stained with Fluoro-Jade C to label degenerating neurons at 18 days post-TBI. A) Panel shows an example of two Fluoro-Jade C-positive foci (indicated by the white arrows) in an untreated TBI mouse brain. Foci are shown imposed onto a bright field image of the tissue viewed at 40x magnification. B) An example of a diffuse Fluoro-Jade C-positive foci in an untreated TBI mouse brain. Image magnification increased to show finer detail. C, D) Quantification of the number of 40x fields with a single Fluoro-Jade C-positive foci (or multiple foci) measured as the fold change compared to Sham control mice. Untreated TBI mice displayed statistically significant increases in the number of fields with single (C) and multiple (D) foci when compared to Sham control mice (**p<0.01, n=3–4 mice per group; effect size for single foci [95% confidence intervals] = 3.01 [0.73 to 4.50]. Immunocal®-pretreated mice that were subjected to TBI showed statistically significant decreases in the number of fields with single (C) and multiple (D) foci in comparison to untreated TBI mice (##p<0.01, n=3–4 mice per group; effect size for single foci [95% confidence intervals] = −3.18 [−4.70 to −0.83]; one-way ANOVA for (C), p=0.007; one-way ANOVA for (D), p=0.004). Abbreviations used: ICAL, Immunocal®.
Discussion
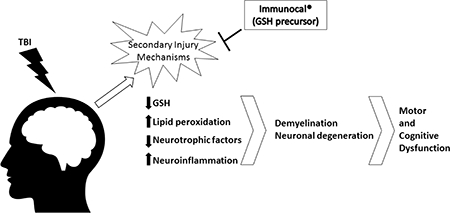
The pathophysiological processes underlying the short and long term injury sequelae associated with TBI are complex. The primary injury is mechanical, resulting from an external force, and leads to tissue deformation, tearing of blood vessels and neuronal axons, necrotic cell death, and initiation of secondary injury processes. Secondary injury mechanisms may include intracranial hemorrhage, excitotoxicity, ionic disturbances, decreased cerebral blood flow, edema, inflammation, mitochondrial dysfunction, oxidative stress, nitrosative stress, and (neuronal and glial) cell death by apoptosis. Although many patients might in theory, be able to significantly recover from the primary mechanical injury of TBI given appropriate acute surgical interventions and supportive care, the detrimental consequences of secondary injury often lead to long term physical, cognitive, and emotional impairments that markedly reduce quality of life. Given the multi-factorial nature of secondary injury, many different therapeutic approaches have been investigated in an attempt to mitigate the post-acute neuronal damage caused by TBI including antioxidants, neurorestorative therapies, neuroprotective pharmacological agents, and drugs that modulate neuroinflammation (Hall et al., 2010; Xiong et al., 2010; Lulic et al., 2011; Kumar and Loane, 2012; McConeghy et al., 2012; Gruenbaum et al., 2016; Simon et al., 2017). Yet, despite some compelling results with specific agents in pre-clinical animal models of TBI and Phase I/II trials in patients, there are currently no FDA approved drugs for TBI which have shown significant therapeutic efficacy in large, randomized Phase III clinical trials. Therefore, novel therapeutic approaches for TBI are critically needed.
In the present study, we evaluated the potential of a cysteine-rich, whey protein supplement, Immunocal®, to enhance resilience to a moderate TBI induced by controlled cortical impact in mice. Untreated CD1 mice subjected to TBI displayed ample evidence of a primary mechanical injury, including regions of BBB disruption detected by MRI, alterations in Tau phosphorylation and expression, and substantial increases in righting reflex times and apnea, in comparison to Sham control mice. None of these indices of primary injury were significantly altered by pre-injury supplementation with Immunocal®. On the other hand, Immunocal®-pretreated mice subjected to TBI performed significantly better than untreated TBI mice on several aspects of the challenging beam walk task, rotarod performance, and the Barnes maze test, demonstrating marked improvements in these motor and cognitive tasks. Moreover, pre-injury supplementation with Immunocal® completely preserved the brain GSH to GSSG ratio in mice subjected to TBI, whereas untreated TBI mice showed a nearly 25% reduction in this ratio, which is indicative of oxidative damage. Notably, pre-injury supplementation with Immunocal® also significantly attenuated lipid peroxidation and preserved BDNF expression in the brain following TBI. Finally, Immunocal®-pretreated mice subjected to TBI displayed significantly less demyelination of the corpus callosum and reduced numbers of foci of degenerating neurons, when compared to untreated TBI mice. Taken collectively, these results demonstrate that pre-injury supplementation with Immunocal® significantly increases resilience to a moderate TBI induced by a closed head injury in mice. Thus, Immunocal® may have significant utility as a preventative and restorative agent for TBI, particularly in populations at high risk of brain trauma.
As mentioned previously, several studies have reported that brain GSH levels are reduced following TBI and genetic variations in GSH-dependent, peroxide/electrophile-detoxifying enzymes, such as glutathione-S-tranferase-4 and glutathione peroxidase-1, can sensitize mice and rats to brain injury induced by TBI (Tyurin et al., 2000; Xiong et al., 2004; Al Nimer et al., 2013). In a similar manner, genetic deletion of the excitatory amino acid carrier type 1 (EAAC1), a glutamate transporter which also participates in the neuronal uptake of cysteine for GSH synthesis, significantly sensitized mice to TBI induced by controlled cortical impact, resulting in enhanced neuronal death and increased microglial activation (Aoyama et al., 2006; Choi et al., 2016). These findings suggest that a strategy aimed at sustaining or enhancing brain GSH may be a viable approach to mitigate secondary injury processes induced by TBI. In this context, a prior study using a novel closed skull injury model in mice demonstrated that transcranial administration of GSH ameliorated brain injury and neuroinflammation (Roth et al., 2014). Moreover, multiple studies have shown that administration of various GSH precursors, including N-acetylcysteine and gamma-glutamylcysteine ethyl ester, as well as the GSH analog, S-nitrosoglutathione, provide antioxidant and neuroprotective effects in mouse and rat models of TBI (Xiong et al., 1999; Hicdonmez et al., 2006; Reed et al., 2009; Lok et al., 2011; Khan et al., 2009, 2011; Henderson et al., 2016). Although these studies are quite supportive of a therapeutic role for GSH in TBI, most are somewhat limited in scope in that they only evaluated neuronal degeneration and various indices of oxidative or nitrosative stress while neglecting to assess cognitive or motor deficits induced by TBI. As a result, these prior studies do not clearly delineate the specific therapeutic benefit that this strategy might realistically hold for patients suffering from TBI.
In the present study, we observed that Immunocal® not only preserved the brain GSH to GSSG ratio and ameliorated neuronal injury, but it also significantly improved motor and cognitive function in mice tested post-TBI. One previous study using N-acetylcysteine amide did show a beneficial effect on cognitive function in rats treated post-TBI, as assessed by a modified Morris water maze test (Pandya et al., 2014). However, motor function was not evaluated in this prior study. One may argue that our findings are less relevant to TBI in patients because we administered Immunocal® orally to mice for 28 days prior to inducing TBI. Certainly, there are a large number of TBI cases which cannot be predicted and therefore, pre-injury administration of a protective therapy is not possible. However, for those individuals in occupations with a high risk of experiencing a TBI, such as military personnel and athletes in contact sports, our data may be highly relevant. Indeed, the identification of novel agents that significantly increase resilience to TBI would provide valuable preventative options to limit the brain injury caused by this type of trauma. It should be noted that historically, it is quite challenging to ensure nutritional regimen adherence even in a population at high risk such as athletes in contact sports or military personnel, so the feasibility of using a nutritional supplement pre-injury can be challenging. However, given the relatively new recognition that TBI early in life may lead to devastating consequences such as chronic traumatic encephalopathy, it should be possible to convince athletes and military personnel of the importance of limiting TBI-induced brain damage by adhering to a pre-injury nutritional supplement regimen (Ban et al., 2016).
One aspect of the TBI model that we employed which was unexpected was the relative lack of a large neuroinflammatory response. In particular, we did not observe increases in the reactive astrocyte marker, S100beta, in whole brain lysates assessed by western blotting at 72h post-TBI. Nor did we observe any notable increases in GFAP immunoreactivity in brain tissue of untreated TBI mice when assayed at 18 days post-TBI. This apparent lack of a global reactive astrogliosis response was not anticipated based on prior studies using a controlled cortical impact paradigm to induce TBI. Although it is interesting to note that Lloyd et al (2008) only showed significant increases in brain S100beta immunoreactivity using this model of moderate TBI induced by controlled cortical impact at 28 days post-TBI. Therefore, it is possible that we simply missed the peak time point of reactive astrogliosis in our assessments at 72h and 18 days post-TBI. Another possibility is that there were discrete regions of reactive astrogliosis throughout the brains of our untreated TBI mice that we did not identify. During the immunofluorescence imaging of GFAP-stained brain sections, entire coronal sections were evaluated for reactive astrocytes. Although some untreated TBI mice clearly had fields with large numbers of astrocytes, this was also the case for Sham controls and Immunocal®-pretreated mice subjected to TBI. In fact, quantitative analysis did not reveal any significant differences between groups in the numbers of GFAP-positive cells per mouse brain section, even when different morphological variants were specifically counted (e.g., ramified versus amoeboid; data not shown). In contrast to the lack of reactive astrocytes, we did observe an increase in Iba1 immunoreactivity in whole brain lysates assessed by western blotting at 72h post-TBI, an effect which was mitigated by Immunocal® pretreatment. Interestingly, we have found that Immunocal® suppresses lipopolysaccharide-induced nitric oxide production in cultured BV2 mouse microglial cells, suggesting that this whey protein may possess some anti-neuroinflammatory properties (Khatter and Linseman, unpublished data). Thus, there does seem to be a component of microgliosis in the TBI model that we employed, which was significantly influenced by pretreatment with Immunocal®.
How might pre-injury administration of Immunocal® enhance resilience to TBI at the molecular level? Our major hypothesis in this regard is based on the observation that pre-injury administration of Immunocal® significantly preserved the brain GSH to GSSG ratio in mice subjected to TBI. By preserving brain GSH, Immunocal® could mitigate several of the secondary injury mechanisms that are activated by TBI. In this context, we recently demonstrated that Immunocal® is broadly neuroprotective in vitro and rescues primary cerebellar neurons and various neuronal cell lines from a number of stressors including oxidative damage, nitric oxide, and excitotoxicity (Winter et al., 2017). All of these stressors are believed to contribute to the secondary injury processes post-TBI. Importantly, the neuroprotective effects of Immunocal® observed in cell culture are dependent on the de novo synthesis of GSH (Winter et al., 2017). Thus, it seems probable that at least the neuroprotective actions of Immunocal® observed in this mouse model of TBI are due largely to its capacity to act as a cysteine delivery system and thereby, a precursor pool for GSH synthesis. Downstream of preserving GSH, pre-injury supplementation with Immunocal® significantly attenuated lipid peroxidation and preserved BDNF expression in the brain following TBI. Similar effects on BDNF expression have previously been observed in rat models of controlled cortical impact injury with procyanidin antioxidants and S-nitrosoglutathione (Khan et al., 2011; Mao et al., 2015). Collectively, these findings suggest that the neuroprotective mechanism of action of Immunocal® in TBI likely stems from its capacity to preserve GSH and in turn, to limit oxidative damage and maintain neurotrophic factors. It is presently unclear if Immunocal® has any effect on nuclear factor erythroid 2-related factor 2 expression or function as a regulator of antioxidant and phase II detoxification genes and in particular, enzymes required for GSH synthesis. This possibility will be explored in future studies. Lastly, the potential anti-neuroinflammatory actions of Immunocal® and whether they might contribute to its protective capacity against TBI, will also require further investigation.
In summary, pre-injury oral administration of the cysteine-rich, whey protein supplement, Immunocal®, significantly enhanced resilience to TBI induced by controlled cortical impact in mice. Although Immunocal® did not protect mice from the primary mechanical injury induced by a moderate TBI, it did preserve the brain GSH to GSSG ratio, reduce lipid peroxidation, sustain BDNF expression, and attenuate demyelination and neuronal degeneration. Perhaps most significantly, the therapeutic actions of Immunocal® pretreatment were evidenced by significant improvements in motor and cognitive deficits induced by TBI. We conclude that Immunocal® holds significant promise as a preventative agent for TBI-induced damage, particularly in those individuals at high risk including military personnel and athletes in contact sports.
Highlights.
Prior studies have shown neuroprotective effects of antioxidants in models of TBI.
Very few have demonstrated significant recovery of motor or cognitive function.
Immunocal® significantly enhanced resilience to TBI induced by closed head injury in mice.
Immunocal® preserved brain GSH/GSSG and attenuated demyelination and neuronal degeneration.
Most significantly, Immunocal® improved motor and cognitive deficits induced by TBI.
Immunocal® holds significant promise as a preventative agent for TBI.
Acknowledgments
Funding
This study was supported in large part by funding from Immunotec Inc. (Quebec, Canada), which is the manufacturer of Immunocal®. Funding was also provided by a pilot grant from the Knoebel Institute for Healthy Aging at the University of Denver. The University of Colorado Animal Imaging Shared Resources are supported by the University of Colorado Cancer Center (NCI P30 CA046934) and the Colorado Clinical and Translational Sciences Institute (NIH/NCATS UL1 TR001082).
Abbreviations used
- BBB
blood brain barrier
- BBBD
blood brain barrier disruption
- EAAC1
excitatory amino acid carrier type 1
- GFAP
glial fibrillary acidic protein
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HPLC-ECD
high performance liquid chromatography with electrochemical detection
- Iba1
ionized calcium binding adaptor molecule 1
- ICAL
Immunocal®
- MRI
magnetic resonance imaging
- PBS-T
phosphate-buffered saline with 0.1% Triton X-100
- PHF
paired helical filament
- PVDF
polyvinylidene difluoride
- SDS-PAGE
sodium dodecylsulfate polyacrylamide gel electrophoresis
- TBI
traumatic brain injury
Footnotes
Conflicts of Interest
The authors have received funding from Immunotec Inc. (Quebec, Canada) to support research on the therapeutic effects of Immunocal® in pre-clinical mouse models of TBI.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul-Muneer PM, Chandra N, Haorah J (2015) Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol 51:966–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Nimer F, Strom M, Lindblom R, Aeinehband S, Bellander BM, Nyengaard JR, Lidman O, Piehl F (2013) Naturally occurring variation in the glutathione-s-transferase 4 gene determines neurodegeneration after traumatic brain injury. Antioxid Redox Signal 18:784–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA (2006) Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci 9:119–126. [DOI] [PubMed] [Google Scholar]
- Bains M, Hall ED (2012) Antioxidant therapies in traumatic brain and spinal cord injury. Biochem Biophys Acta 1822:675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban VS, Madden CJ, Bailes JE, Batjer HH, Lonser RR (2016) The science and questions surrounding chronic traumatic encephalopathy. Neurosurg Focus 40:E15. [DOI] [PubMed] [Google Scholar]
- Baruchel S, Viau G (1996) In vitro selective modulation of cellular glutathione by a humanized native milk protein isolate in normal cells and rat mammary carcinoma model. Anticancer Res 16:1095–1100. [PubMed] [Google Scholar]
- Barucehl S, Viau G, Olivier R, Bounous G, Wainberg MA (1998) Nutraceutical modulation of glutathione with a humanized native milk serum protein isolate, Immunocal®: application in AIDS and cancer,” in Oxidative Stress in Cancer, AIDS, and Neurodegenerative Diseases, Montagnier L, Olivier R and Pasquier C, Eds., vol. 1, Chapter 42, pp. 447–462, Marcel Dekker, Inc., New York, NY, USA. [Google Scholar]
- Bounous G, Baruchel S, Falutz J, Gold P (1993) Whey proteins as a food supplement in HIVseropositive individuals. Clin Invest Med 16:204–209. [PubMed] [Google Scholar]
- Broglio SP, Eckner JT, Martini D, Sosnoff JJ, Kutcher JS, Randolph C (2011) Cumulative head impact burden in high school football. J Neurotrauma 28:2069–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BY, Kim IY, Lee BH, Lee SH, Kho AR, Jung HJ, Sohn M, Song HK, Suh SW (2016) Decreased cysteine uptake by EAAC1 gene deletion exacerbates neuronal oxidative stress and neuronal death after traumatic brain injury. Amino Acids 48:1619–1629. [DOI] [PubMed] [Google Scholar]
- Elder GA, Cristian A (2009) Blast-related mild traumatic brain injury: mechanisms of injury and impact on clinical care. Mt Sinai J Med 76:111–118. [DOI] [PubMed] [Google Scholar]
- Faul M, Xu L, Wald MM, Coronado VG (2010) Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Atlanta (GA): Center for Disease Control and Prevention, National Center for Injury Prevention and Control. [Google Scholar]
- Fleming SM, Ekhator OR, Ghisays V (2013) Assessment of sensorimotor function in mouse models of Parkinson’s disease. J Vis Exp 76:e50303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey L, Lepkin A, Schickedanz A, Huber K, Brown MS, Serkova N (2014) ADC mapping and T1weighted signal changes on post-injury MRI predict seizure susceptibility after experimental traumatic brain injury. Neurol Res 36:26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Yaffe K (2015) Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci 66:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavett BE, Stern RA, Cantu RC, Nowinski CJ, McKee AC (2010) Mild traumatic brain injury: a risk factor for neurodegeneration. Alzheimers Res Ther 2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavett BE, Stern RA, McKee AC (2011) Chronic traumatic encephalopathy: a potential late effect of sportrelated concussive and subconcussive head trauma. Clin Sports Med 30:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LE et al. (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4:134–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey V, Mohammed SR, Smountas AA, Bahlool R, Lands LC (2003) Improved glutathione status in young adult patients with cystic fibrosis supplemented with whey protein. J Cyst Fibros 2:195–198. [DOI] [PubMed] [Google Scholar]
- Gruenbaum SE, Zlotnik A, Gruenbaum BF, Hersey D, Bilotta F (2016) Pharmacologic neuroprotection for functional outcomes after traumatic brain injury: a systematic review of the clinical literature. CNS Drugs 30:791–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guskiewicz KM, Marshall SW, Bailes J, McCrea M, Cantu RC, Randolph C, Jordan BD (2005) Association between recurrent concussion and late-life cognitive impairment in retired professional football players. Neurosurgery 57:719–726. [DOI] [PubMed] [Google Scholar]
- Hall ED, Vaishnav RA, Mustafa AG (2010) Antioxidant therapies for traumatic brain injury. Neurotherapeutics 7:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson M, Rice B, Sebastian A, Sullivan PG, King C, Robinson RA, Reed TT (2016) Neuroproteomic study of nitrated proteins in moderate traumatic brain injured rates treated with gamma glutamyl cysteine ethyl ester administration post injury: insight into the role of glutathione elevation in nitrosative stress. Proteomics Clin Appl 10:1218–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicdonmez T, Kanter M, Tiryaki M, Parsak T, Cobanoglu S (2006) Neuroprotective effects of Nacetylcysteine on experimental closed head trauma in rats. Neurochem Res 31:473–481. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH (2010) Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci 11:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Im YB, Shunmugavel A, Gilg AG, Dhindsa RK, Singh AK, Singh I (2009) Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation 6:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Sakakima H, Dhammu TS, Shunmugavel A, Im YB, Gilg AG, Singh AK, Singh I (2011) Snitrosoglutathione reduces oxidative injury and promotes mechanisms of neurorepair following traumatic brain injury in rats. J Neuroinflammation 8:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Loane DJ (2012) Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 26:1191–1201. [DOI] [PubMed] [Google Scholar]
- Lee YK, Hou SW, Lee CC, Hsu CY, Huang YS, Su YC (2013) Increased risk of dementia in patients with mild traumatic brain injury: a nationwide cohort study. PLoS One 8:e62422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd E, Somera-Molina K, Van Eldik LJ, Watterson DM, Wainwright MS (2008) Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J Neuroinflammation 5:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok J, Leung W, Zhao S, Pallast S, van Leyen K, Guo S, Wang X, Yalcin A, Lo EH (2011) Gammaglutamylcysteine ethyl ester protects cerebral endothelial cells during injury and decreases blood-brain barrier permeability after experimental brain trauma. J Neurochem 118:248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lulic D, Burns J, Bae EC, van Loveren H, Borlongan CV (2011) A review of laboratory and clinical data supporting the safety and efficacy of cyclosporin A in traumatic brain injury. Neurosurgery 68:1172–1185. [DOI] [PubMed] [Google Scholar]
- Mao X, Hao S, Zhu Z, Zhang H, Wu W, Xu F, Liu B (2015) Procyanidins protects against oxidative damage and cognitive deficits after traumatic brain injury. Brain Inj 29:86–92. [DOI] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68:709–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConeghy KW, Hatton J, Hughes L, Cook AM (2012) A review of neuroprotection pharmacology and therapies in patients with acute traumatic brain injury. CNS Drugs 26:613–636. [DOI] [PubMed] [Google Scholar]
- Meister A (1984). New aspects of glutathione biochemistry and transport - selective alteration of glutathione metabolism. Nutr Rev 42:397–410. [DOI] [PubMed] [Google Scholar]
- Mouzon B, Chaytow H, Crynen G, Bachmeier C, Stewart J, Mullan M, Stewart W, Crawford F (2012) Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotraum 29:2761–2773. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Readnower RD, Patel SP, Yonutas HM, Pauly JR, Goldstein GA, Rabchevsky AG, Sullivan PG (2014) N-acetylcysteine amide confers neuroprotection, improves bioenergetics and behavioral outcome following TBI. Exp Neurol 257:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Physicians’ Desk Reference (PDR). (2016). PDR Network (Montvale, NJ: ); 70th 2016. ed. [Google Scholar]
- Reed TT, Owen J, Pierce WM, Sebastian A, Sullivan PG, Butterfield DA (2009) Proteomic identification of nitrated brain proteins in traumatic brain-injured rats treated postinjury with gammaglutamylcysteine ethyl ester: insights into the role of elevation of glutathione as a potential therapeutic strategy for traumatic brain injury. J Neurosci Res 87:408–417. [DOI] [PubMed] [Google Scholar]
- Ross EK, Winter AN, Wilkins HM, Sumner WA, Duval N, Patterson D, Linseman DA (2014) A cystine-rich whey supplement (Immunocal®) delays disease onset and prevents spinal cord glutathione depletion in the hSOD1 (G93A) mouse model of amyotrophic lateral sclerosis. Antioxidants 3:843–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB (2014) Transcranial amelioration of inflammation and cell death after brain injury. Nature 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shively S, Scher AI, Perl DP, Diaz-Arrastia R (2012). Dementia resulting from traumatic brain injury: what is the pathology? Arch Neurol 69:1245–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM (2017) The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol 13:171–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Tavitian A, Cressatti M, Galindez C, Liberman A, Schipper HM (2017) Cysteine-rich whey protein isolate (Immunocal(®)) ameliorates deficits in the GFAP.HMOX1 mouse model of schizophrenia. Free Radic Biol Med 110:162–175. [DOI] [PubMed] [Google Scholar]
- Tateishi N, Higashi T, Shinya S, Naruse A, Sakamoto Y (1974) Studies on the regulation of glutathione level in rat liver. J Biochem 75:93–103. [DOI] [PubMed] [Google Scholar]
- Tyurin VA, Tyurina YY, Borisenko GG, Sokolova TV, Ritov VB, Quinn PJ, Rose M, Kochanek P, Graham SH, Kagan VE (2000) Oxidative stress following traumatic brain injury in rats: Quantitation of biomarkers and detection of free radical intermediates. J Neurochem 75:2178–2189. [DOI] [PubMed] [Google Scholar]
- Winter AN, Ross EK, Daliparthi V, Sumner WA, Kirchhof DM, Manning E, Wilkins HM, Linseman DA (2017) A cystine-rich whey supplement (Immunocal(®)) provides neuroprotection from diverse oxidative stress-inducing agents in vitro by preserving cellular glutathione. Oxid Med Cell Longev 2017:3103272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Peterson PL, Lee CP (1999) Effect of N-acetylcysteine on mitochondrial function following traumatic brain injury in rats. J Neurotrauma 16:1067–1082. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Shie FS, Zhang J, Lee CP, Ho YS (2004) The protective role of cellular glutathione peroxidase against trauma-induced mitochondrial dysfunction in the mouse brain. J Stroke Cerebrovasc Dis 13:129–137. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M (2010) Neurorestorative treatments for traumatic brain injury. Discov Med 10:434–442. [PMC free article] [PubMed] [Google Scholar]