

Abstract
The gut microbiome is sensitive to diet and environmental exposures and is involved in the regulation of host metabolism. Additionally, gut inflammation is an independent risk factor for the development of metabolic diseases, specifically atherosclerosis and diabetes. Exposures to dioxin-like pollutants occur primarily via ingestion of contaminated foods and are linked to increased risk of developing cardiometabolic diseases. We aimed to elucidate the detrimental impacts of dioxin-like pollutant exposure on gut microbiota and host gut health and metabolism in a mouse model of cardiometablolic disease. We utilized 16S rRNA sequencing, metabolomics, and regression modeling to examine the impact of PCB 126 on the microbiome and host metabolism and gut health. 16S rRNA sequencing showed that gut microbiota populations shifted at the phylum and genus levels in ways that mimic observations seen in chronic inflammatory diseases. PCB 126 reduced cecum alpha diversity (0.60 fold change; p=0.001) and significantly increased the Firmicutes to Bacteroidetes ratio (1.63 fold change; p=0.044). Toxicant exposed mice exhibited quantifiable concentrations of PCB 126 in the colon, upregulation of Cyp1a1 gene expression, and increased markers of intestinal inflammation. Also, a significant correlation between circulating Glucagon-like peptide-1 (GLP-1) and Bifidobacterium was evident and dependent on toxicant exposure. PCB 126 exposure disrupted the gut microbiota and host metabolism and increased intestinal and systemic inflammation. These data imply that the deleterious effects of dioxin-like pollutants may be initiated in the gut, and the modulation of gut microbiota may be a sensitive marker of pollutant exposures.
Keywords: dioxin, gut microbiota, PCB 126, inflammation, diabetes
Capsule
Dioxin-like PCB 126 increases gut inflammation, disrupts gut microbiota health, and modulates microbe/host metabolite and hormone formation in a mouse model of cardiometabolic disease.
Graphical abstract
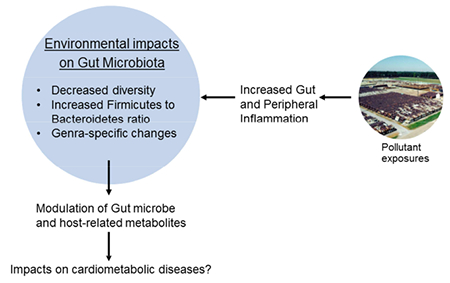
Introduction
Pollutant exposures are associated with numerous chronic inflammatory cardiometabolic diseases including cardiovascular diseases (CVD), obesity, and diabetes (Crinnion 2011; Dirinck et al. 2011; Kuehn 2011). Dioxin-like pollutants are a class of highly toxic lipophilic compounds known to accumulate in the adipose tissue of living animals and thus biomagnify up the food chain (Crinnion 2011). Using preclinical animal models including Ldlr and ApoE deficient mice as a tool to study cardiometabolic disorders, multiple groups have shown that dioxin-like pollutants can impact multiple pathologies through mechanisms that include induction of chronic inflammation in various organ systems (Perkins et al. 2016; Shan et al. 2014; Wu et al. 2011). The most common route of human exposure to these pollutants comes from consumption of contaminated food sources and because of this, the intestinal environment is a target organ that deserves further study.
The gut is home to trillions of bacteria which are sensitive to many factors including diet and environmental exposures (Rooks and Garrett 2016). Additionally, the gut microbiome plays an essential role in the maintenance of energy metabolism, immune function, neurological function, and overall host health and well-being (Rooks and Garrett 2016). Commensal bacteria exist in a mutualistic relationship with the host, relying on host energy and nutrient intake, and in turn, the gut microbiota protect against pathogen invasion, influence immune function, maintain gut barrier integrity, and produce metabolites that exert various effects on host physiology (Rooks and Garrett 2016). Therefore, alterations in gut microbial composition can have deleterious effects on host health. Such alterations are termed “dysbiosis” and have been associated with increased risk of cardiometabolic diseases (Koh and Kim 2017; Serino et al. 2014). Dysbiosis is commonly associated with a decrease in gut microbial diversity, an aspect that has been consistently observed in mice fed atherogenic diets, and human pathological conditions such as atherosclerosis and diabetes (Murphy et al. 2015). Also, chronic gut and systemic inflammation accompanies the observed disruption of gut flora homeostasis in these pathologies.
Chronic inflammation is an underlying aspect in many non-communicable diseases and has been strongly linked to contributing to the development of cardiovascular diseases (e.g., atherosclerosis) (Galkina and Ley 2009). Atherosclerosis is influenced by inflammation through a variety of mechanisms including cytokine production and release, production of reactive oxygen species, and certain immune responses (Galkina and Ley 2009). Additionally, it is well documented that low-grade chronic inflammation precedes and is predictive of the development of cardiometabolic diseases in adults (Luft et al. 2013). Emerging evidence now implicates a critical role of gut microbiota in systemic and intestinal inflammation, which may impact on other peripheral organ systems (e.g., vascular tissue) (Koh and Kim 2017). The initiation of these inflammatory processes that exert effects on the cardiovascular and metabolic systems, as well as the gut microbiota can come from behavioral factors such as diet as well as less-modifiable aspects including genetics and environmental pollutant exposure.
Although there is a large body of evidence linking dietary behaviors to modulation of gut microbiota, only a few studies have examined the interplay between persistent organic pollutants (POPs) and the gut microbiota. Zhang et al, found that exposure to 2,3,7,8 tetrochlorodibenzofuran in mice resulted in alterations in gut microbiota, bile acids, and short chain fatty acid (SCFA) metabolism (Zhang et al. 2015). Additionally, exposure to benzo[a]pyrene induces intestinal inflammation, ileal lesions, and shifts gut microbiota populations (Ribiere et al. 2016). When examining the effects of polychlorinated biphenyls (PCBs), Choi et al. observed disruptions of intestinal barrier function through dysregulation of tight junction proteins (Choi et al. 2010). In a later study, Choi et. al found that exposure to a mixture of PCBs (PCB 153, PCB 138, PCB 180) acutely altered the gut microbiota and that exercise was able to blunt these effects (Choi et al. 2013). Although pollutant exposures have been shown to modulate gut microbiota in wild-type mice, it is not well established if dioxin-like pollutants can modulate gut microbiota in mice genetically predisposed to cardiometabolic disease. In humans, diabetes and related pathologies regularly accompany or exacerbate atherosclerosis, thus, utilizing Ldlr deficient mice is a useful tool to study multiple diseases related to metabolic dysfunction (Getz and Reardon 2012). Therefore, the specific objective of this study was to elucidate effects of the model dioxin-like pollutant PCB 126 on gut health, gut microbiota, and metabolism in a well established mouse model of cardiometabolic disease.
Materials and Methods
Animals, diet, and study design
The diet and dosing schedule utilized for this study was shown previously to be an effective model of PCB 126-accelerated atherosclerosis (Petriello et al. 2017). The results described herein were collected from a subset of mice from our previous study (Petriello et al. 2017). Briefly, seven week-old male Ldlr −/− mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and were randomly divided into 2 groups (n = 10 per group) with one group receiving 1 μmol/kg of PCB 126 (AccuStandard, CT, USA) and the other group receiving safflower oil vehicle (Dyets, Bethlehem, PA, USA) via oral gavage at weeks 2 and 4. This dose produces plasma PCB 126 levels that mimic human exposures of dioxin-like pollutants (Petriello et al. 2017). All mice were fed a lowfat high cholesterol Clinton/Cybulsky diet ad libitum (Research Diets, New Brunswick, NJ, USA; Product # D01061401C). Mice were housed at 22°C with 50% humidity, exposed to a 12-h light/ 12-h dark cycle, and given water ad libitum. Fecal samples were collected at 72h, 4 weeks, and at sacrifice (12 weeks) after first PCB 126 exposure. Prior to euthanasia, mice were fasted overnight, anesthetized, and blood was collected using retro-orbital bleed. At the conclusion of the study, intestinal samples, cecum, and cecum contents were also collected and immediately snap frozen in liquid nitrogen and stored at −80°C until analysis. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Kentucky.
DNA Extraction and 16S rRNA amplicon library preparation and sequencing
DNA extraction and 16S sequencing was conducted by the Environmental Sample Preparation and Sequencing Facility (ESPSF) at Argonne National Laboratory and analyzed by the program Quantitative Insights Into Microbial Ecology (QIIME). DNA was extracted using PowerSoil 96-well DNA Isolation Kit (MoBio, Carlsbad, CA, USA), according to the manufacturer’s protocol with the addition of a 65°C heating step after the addition of solution C1. PCR amplicon libraries targeting the 16S rRNA encoding gene present in metagenomic DNA were produced using a barcoded primer set adapted for the Illumina HiSeq2000 and MiSeq, and DNA sequence data was generated using Illumina paired-end sequencing (Caporaso et al. 2012). The V4 region of the 16S rRNA gene (515F-806R) was PCR amplified with region-specific primers that include sequencer adapter sequences used in the Illumina flowcell. The conditions for PCR were as follows: 94°C for 3 minutes to denature the DNA, with 35 cycles at 94°C for 45 s, 50°C for 60 s, and 72°C for 90 s; with a final extension of 10 min at 72°C to ensure complete amplification. Amplicons were quantified using PicoGreen (Invitrogen, Carlsbad, CA) and a plate reader (Infinite 200 PRO, Tecan, Mannedorf, Switzerland) and then pooled into a single tube so that each amplicon is represented in equimolar amounts. This pool was then cleaned up using AMPure XP Beads (Beckman Coulter, Brea, CA), and quantified using a fluorometer (Qubit, Invitrogen, Carlsbad, CA). After quantification, the molarity of the pool was determined and diluted to 2 nM, denatured, and diluted to a final concentration of 6.75 pM with a 10% PhiX spike for sequencing on the Illumina MiSeq. Amplicons were sequenced on a 151 bp x 12bp x 151 bp MiSeq run using customized sequencing primers and procedures. Operational taxonomic units (OTUs) were selected using open reference OTU picking against the Greengenes database and picked at 97% sequence identity. OTUs were then filtered based on the default parameters provided by QIIME and sequences were rarified to a sampling depth of 10000 reads per sample. Before statistical analyses, OTUs present in less than 50% of samples were filtered from the OTU table.
RNA Extraction and qPCR
For mRNA extraction, intestinal samples from jejunum and colon were homogenized in TRIzol (Invitrogen, Carlsbad, CA) and isolated according to manufacturer’s protocols. mRNA quality and concentrations were determined using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA). Complementary DNA was generated using the AMV reverse transcription system (Promega, Madison, MI) following manufacturer’s protocols. Expression of inflammatory, metabolic, and gut health markers were determined via quantitative qPCR utilizing Taqman fast reagents (Thermo Scientific, Waltham, MA) in an CFX96 Real-Time PCR system (Bio-Rad, Hercules, CA). β-actin was used as the housekeeping gene for jejunum and 18S was used as the housekeeping gene for colon. Differences in gene expression were calculated using ΔΔCt relative quantification compared to control animals.
Analyses of circulating cytokines and proteins related to metabolic function
To measure plasma metabolic hormones and cytokines the Milliplex Map Mouse Metabolic Hormone Magnetic Bead Panel- Metabolism Multiplex Assay and the Milliplex Map Mouse Cytokine/Chemokine Magnetic Bead Panel 25-plex (Millipore Corp, Billerica, MA, USA) were used, respectively, following manufacturer’s protocols. Panels were measured on the Luminex Xmap MAGPIX system (Luminex Corp, Austin, TX, USA) following manufactures instructions. For statistical analyses, values below the standard curve were represented as zero.
Metabolomics Analysis
At the conclusion of the study, livers were snap frozen and shipped to Metabolon (Durham, NC) for targeted metabolomics analysis as detailed previously (Beattie et al. 2017). Briefly, Samples were prepared using the automated MicroLab STAR (Hamilton Company, Reno, NV) and analysed by Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectroscopy (UPLC-MS/MS). All methods utilized a Waters ACQUITY ultra-performance liquid chromatography (UPLC) and a Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer interfaced with a heated electrospray ionization (HESI-II) source and Orbitrap mass analyzer. A series of proprietary standards of known concentration was added to each sample extract. 4 separate analytical methodologies were utitlized which were opitimized depending on the characteristics of compounds of interest (e.g., hydrophilic compounds vs. hydrophobic compounds). The MS analysis alternated between MS and data-dependent MSn scans using dynamic exclusion. For biochemical identification, three criteria were utilized: retention time, accurate mass match to the library, and the MS/MS compared against authentic standards. Peaks were quantified using area-under-the-curve.
Quantitation of atherosclerotic lesions and gluocse sensitivity
To quantify atherosclerosis, aortic roots were frozen in OCT, sectioned at 10 pm per section, and stained with Oil Red O as described before (Paigen et al. 1987; Petriello et al. 2017). Briefly, serial sections were collected as close as possible to the emergence of the three valves, and sections were placed on microscope slides (Probe-on Plus; Fisher Scientific, Pittsburgh, PA) until the aortic valves disappeared. Frozen sections were lipid stained with Oil Red O, and images were taken using a Nikon Eclipse 55iUpright microscope attached to a 12 MP color camera. For the results shown in Suppl. Fig. 3, on average a mean of 6 serial sections per mouse were used for quantification. To quantify glucose sensitivity a gluocse tolerance test was performed at 5 and 12 weeks post initial PCB exposure. Mice were fasted for 6h (7am-1pm), and fasting blood glucose levels were measured with a hand-held glucometer (Accu-check Avivia, Roche,Basel, Switzerland) using 1-2μL of blood collected through the tail vein. Glucose was given via IP injection (2mg/g body weight, sterile saline) and blood glucose levels were measured at 15, 30, 60, 90, and 120 minutes post injection.
PCB 126 Quantitation
PCB 126 from colons, livers, and plasma was extracted using a modified dispersive solid phase extraction method as before (Petriello et al. 2017). Briefly, tissue/plasma, internal standard (13C12-PCB 126; Cambridge Isotopes, Tewksbury, MA), deionized water, and acetonitrile containing 1% acetic acid were added to each tube and homogenized. The upper layer was transferred to an Agilent Bond Elut QuEChERS fatty sample dispersive 2 ml SPE column. PCB 126 was analyzed using an Agilent GC-triple quadrupole MS (GC-MS/MS) 7000C system equipped with a multimode inlet and a HP-5MS UI column (30m, 0.25mm, 0.25μm) in multiple reaction monitoring (MRM) mode. Ion transitions monitored were 325.9/255.9 for PCB 126 and 337.9/267.9 for 13C12-PCB 126 internal standard. Relative quantitation was done by comparing peak area of the sample to peak area of an internal standard sample of known concentration.
Statistical analyses
qPCR and MAGPIX results were analyzed using a Student’s t-test to compute comparisons of means between treatment groups. Analyses were conducted using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). Metabolomics results were analyzed using a Welch’s two-sample t-test (n=6 per group). These analyses were performed using ArrayStudio (Qiagen, Hilden, Germany) on log transformed data. For statistical analyses of changes in fecal bacterial genera over time, linear mixed-effects model was utilized with unstructured correlation matrix accounting for dependence among observations over time and within an animal. Tukey’s pair-wise multiple comparison procedure followed to assess the significance of change in bacterial genera at each time point. Gut hormones and metabolic markers were correlated with bacterial species using one of the three hypothesized linear models: the full model that accounted for a possible interaction between PCB 126 exposure and a biomarker on the gut hormones level; the reduced model that had an additive effect of PCB 126 exposure and a metabolic biomarker only; and the simple model that did not account for the PCB 126 exposure. The most appropriate model was determined based on the largest value of the adjusted-R2, and its results were Bonferroni-adjusted to account for multiple comparisons. Results were considered statistically significant with an observed p-value <0.05.
Results:
PCB exposure induces shifts in bacterial populations over time
It is hypothesized that the gut microbiota of Ldlr −/− mice will shift as cardiometabolic disease progresses, but it is unknown if pollutant exposures will exacerbate or modify these changes. Using 16S rRNA sequencing and the QIIME analysis platform, we quantified the bacterial populations in fecal samples collected 72 h, 4 weeks, and 12 weeks after first PCB or vehicle gavage (depicted as phlya level changes in Figure 1 and genra level changes in Figure 2). At the phyla level, PCB 126 decreased relative Bacteroidetes (0.75 fold change, p=0.011) and increased relative Verrucomicrobia (1.41 fold change, p=0.008) and species in the feces at 4 weeks post gavage, but not at 12 weeks. Using these relative abundances we then analyzed the Firmicutes to Bacteriodetes ratio over the four timepoints (Figure 1B). We found that in general this ratio increased throughout the study for all mice (which was expected as mice were fed the atherogenic diet), but interestingly, PCB treated mice had a significantly higher ratio at the conclusion of the study in both feces and cecum contents (feces: 3.29 fold change, p=0.0332; cecum: 1.63 fold change, p=0.044).
Figure 1.
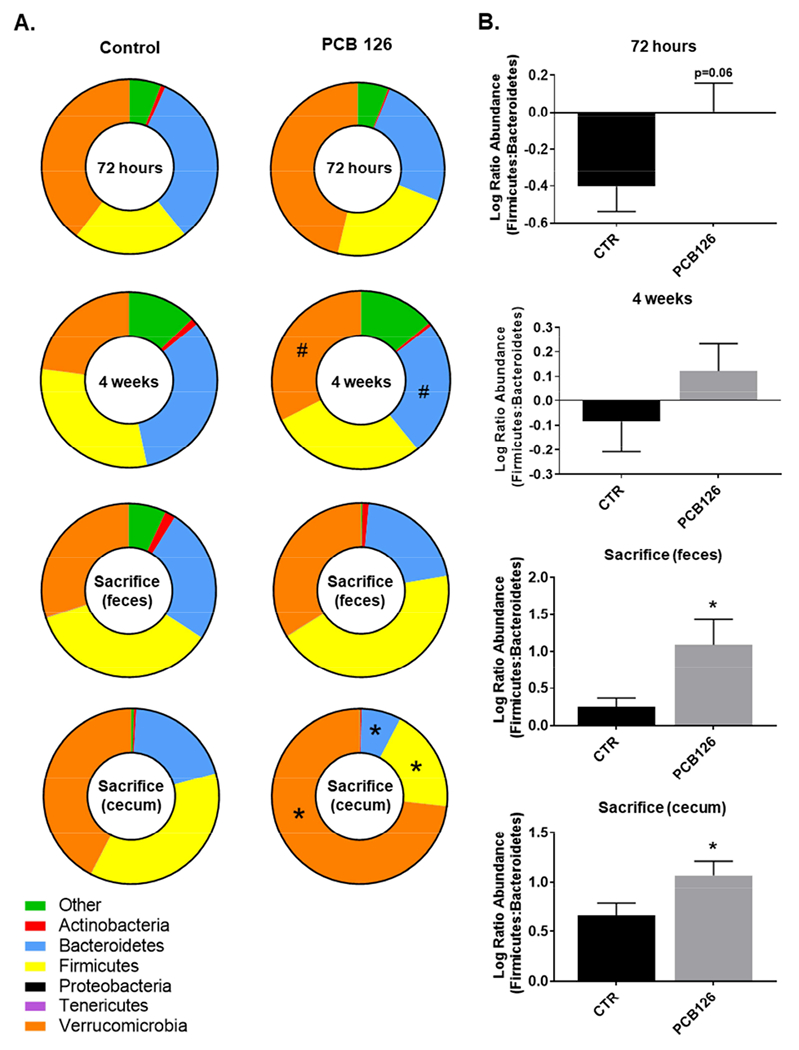
Exposure to PCB 126 drives phyla level alterations in bacterial populations over time. Male Ldlr−/− mice were fed an atherogenic diet for 14 weeks and exposed to PCB 126 (1μmol/kg) at weeks 2 and 4. Fecal samples were collected at the start of the study, 72 hours, 4 weeks, and at sacrifice (12 weeks) post first PCB 126 exposure. Cecum contents were also collected at the conclusion of the study. 16S rRNA sequencing was conducted and data was analyzed using QIIME. A. Changes in the taxonomic composition of the gut microbiota over time at the phylum level. Significant differences were observed in the cecum contents at sacrifice. Cecum contents displayed a significant increase in Verrucomicrobia and decreases in Firmicutes and Bacteriodetes. Data are presented as relative abundances. B. Firmicutes / Bacteroidetes ratio changes over time. PCB 126 increased the ratio of Firmicutes / Bacteriodetes in both the feces and cecum contents at sacrifice. Data are presented as mean ± S.E.M (n=10 per group; Student’s t-test). Statistical significance is denoted by * (p<0.05). # represents (p<0.1).
Figure 2.
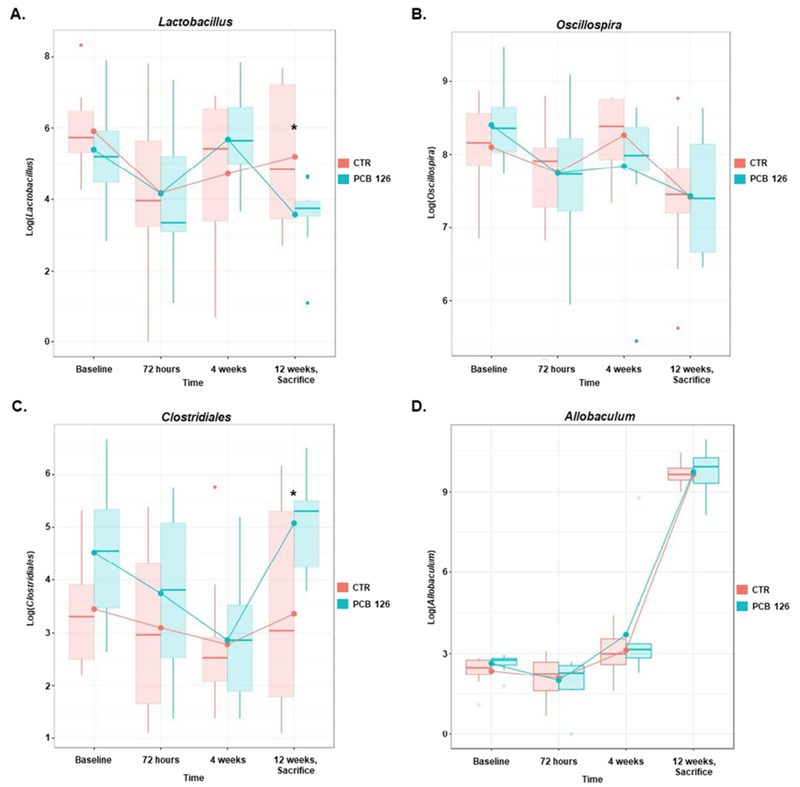
PCB 126 modulates gut microbiota populations at the genra level over time. Male Ldlr−/− mice were fed an atherogenic diet for 14 weeks and exposed to PCB 126 (1 μmol/kg) at weeks 2 and 4. Fecal samples were collected at the start of the study, 72 hours, 4 weeks, and at sacrifice (12 weeks) post first PCB 126 exposure. A representiative example of each of the 4 observed trends is depicted. For all genras, please see Suppl. Table 1. A linear mixed-effects model was utilized with Tukey’s pairwise multiple comparison procedure to assess the significance of change in bacterial genera at each time point. Statistical significance is denoted by * (p<0.05). (n=10 per group; Student’s t-test).
Next, using mixed-effects modeling, we increased the resolution of our QIIME analyses to determine which genra differed between PCB and vehicle-treated mice over time. In general, we determined four major trends with which generas changed over time. The first pattern was one in which PCB-treated mice displayed decreased bacterial genera abundance, while the abundance of vehicle-treated mice was stable across the course of the study. For instance, by the conclusion of the study, Lactobacillus genera in PCB-treated mice decreased by 84% relative to the baseline values (Tukey-adjusted pTA = 0.0021) and was 80% lower than that of the vehicle-treated mice (pTA = 0.0339), among which the abundance of Lactobacillus genera did not appreciably change from baseline measurements (Figure 2A). The second pattern was one in which neither vehicle nor PCB-treated mice displayed a statistically significant deviation from each other or from the baseline genera values. For example, the abundance of Oscillospira genera was not affected by PCB exposure, nor did it change or over time (Figure 2B). The third pattern was one in which PCB-treated mice displayed increased abundance over time, while the abundance among vehicle-treat mice did not change. For example, by the conclusion of the study, the abundance of Clostridiales genera was 420% higher among PCB-exposed mice than among vehicle-treated mice (pTA = 0.0248; Figure 2C). Finally, the last pattern was one in which both vehicle and PCB-treated mice displayed increased abundance over time but this difference was equivalent between the two groups. For example, relative to the baseline levels, the abundance of Allobaculum genera increased over tenfold by the conclusion of the study in both groups (week 14) (both pTA < 0.0001; Figure 2D). A summary of all temporal results can be found in Supplemental Table 1.
PCB exposure alters cecal microbial diversity and bacterial genera
The cecum contains the greatest abundance of microbiota and is a primary site of fermentation and bacterial metabolism. Using 16S rRNA sequencing and QIIME analysis software, we also examined the bacterial populations in cecum samples collected at the end of the study (week 14). Similarly to the fecal results, PCB 126-treated mice exhibited significantly reduced relative Bacteroidetes (0.38 fold change, p=0.002) and Firmicutes (0.54 fold change, p=0.004) and greater relative Verrucomicrobia (1.71 fold change, p=0.021) abundances compared to vehicle control (Figure 1A). Alpha diversity is a common measurement of microbial diversity within a sample and takes into account the richness (count of different microbes) and evenness (distribution of different microbes) of a sample. PCB 126 decreased the alpha diversity (Shannon diversity index) in cecal samples compared to vehicle control (0.60 fold change, p=0.001) (Figure 3A). We also examined beta-diversity in these samples and determined that PCB-exposed mice exhibited a significant difference in microbial community structures compared to vehicle treated mice (R=0.2924, p=0.010) (Supplemental Figure 1). We next examined genra level changes in the cecum contents and determined this source of bacteria to be more sensitive to pollutant-induced changes compared with the fecal samples from the same timepoint (i.e., conclusion of study). The abundance of S24.7 and Clostridiales as well as the genera Bifidobacterium, Lactobacillus, Ruminococcus, and Oscillospira were all significantly decreased in PCB exposed mice by similar magnitudes compared to vehicle control (0.39–0.90 fold change). Furthermore, PCB 126 increased the abundance of Akkermansia (1.71 fold change, p=0.021) in the cecal samples compared to vehicle control (Figure 3B).
Figure 3.

Exposure to PCB 126 alters cecal bacterial genera and alpha diversity. Male Ldlr−/− mice were fed an atherogenic diet for 14 weeks and exposed to PCB 126 (1 μmol/kg) at weeks 2 and 4. Cecum contents were collected at the conclusion of the study. (A) Alpha diversity of cecum contents quantified using the Shannon Diversity Index. PCB 126 exposure decreased alpha diversity in cecum contents. (B) Order and genera level changes in cecal gut microbiota. PCB 126 decreased abundance of S24.7, Clostridiales, Bifidobacterium, Lactobacillus, Ruminococcus, and Oscillospira and increased Akkermansia abundance. Data are presented as mean ± S.E.M (n=10 per group; Student’s t-test). Statistical significance is denoted by * (p<0.05).
PCB exposure increases systemic inflammation and alters cardiometabolic disease parameters
Systemic inflammation may be a marker of altered gut health, and has been shown to be increased by PCB 126 (Liu et al. 2015; Petriello et al. 2014; Petriello et al. 2017). Using MAGPIX multiplex technology, we quantified the levels of circulating cytokines in plasma at the conclusion of the study and found that PCB 126 significantly increased interferon gamma-induced protein (IP-10; Figure 4A). Additionally, macrophage inflammatory protein-1 β (Mip-1 β), regulated on activation, normal T cell expressed and secreted (Rantes), and interleukin 12 (IL-12 p70) both increased in PCB 126 exposed mice, however this increase did not quite attain the statistical significance threshold (0.05 < p < 0.1). Since gut health is also directly related to endogenous metabolism, we also quantified levels of circulating mediators of gluocse/insulin signaling and determined that PCB 126 significantly increased levels of insulin (2.74 fold change, p=0.019), c-peptide (1.63 fold change, p=0.015), and glucose-dependent insulinotropic peptide (GIP) (1.41 fold change, p=0.046) as well as significantly decreased levels of glucagon-like peptide 1 (GLP-1; Figure 4B) (0.31 fold change, p=0,048). At 5 weeks post initial PCB gavage and again at the conclusion of the study, we completed a glucose tolerance test and observed no significant differences in glucose sensitivity during either of the challenges. However, fasting blood glucose of PCB-exposed mice trended higher at both timepoints (p= 0.07, p= 0.16) compared to vehicle treated controls (Supplemental Figure 2). Finally, we quantified Oil Red O-staining within aortic roots to examine the extent of atherosclerosis in these mice. We previously showed acceleration of atherosclerosis due to PCB 126 exposure using this same model in studies of shorter duration (e.g., 8 or 10 weeks post first PCB gavage) (Petriello et al. 2017). We determined in this current study that PCB-exposed mice displayed similar trends as seen before, but the differences in lipid accumulation between the two groups was not as evident as in the shorter studies (Mean control; 2.05×105 ± 2.53×104 μm2, Mean PCB; 2.60 ×105 ± 3.42×104 μm2, p=0.2; Supplemental Figure 3).
Figure 4.

Analysis of circulating cytokines and biomarkers of energy metabolism revealed that PCB 126 increases inflammation and alters insulin and related markers. Male Ldlr−/− mice were fed an atherogenic diet for 14 weeks and exposed to PCB 126 (1 μmol/kg) at weeks 2 and 4. Fecal samples were collected at the start of the study, 72 hours, 4 weeks, and at sacrifice (12 weeks) post first PCB 126 exposure. (A) Quantification of circulating cytokines (MAGPIX technology). PCB 126 increased Interferon gamma-induced protein (IP-10) plasma levels. (B) Quantification of circulating markers of energy metabolism (MAGPIX technology). PCB 126 increased plasma levels of insulin, c-peptide, and glucose-dependent insulinotropic peptide (GIP). PCB 126 reduced plasma levels of glucagon-like peptide 1 (GLP-1) Data are presented as mean ± S.E.M (n=9–10 per group; Student’s t-test). Statistical significance is denoted by * (p<0.05).
The intestine is a target of PCB 126 toxicity
There is currently limited evidence that PCB 126 can elicit an inflammatory response in intestinal tissue (Chen et al. 2018; Choi et al. 2010). Thus, we utilized qPCR to examine gene expression of markers related to inflammation, gut health, and metabolism in the jejunum (small intestine) and the colon (large intestine). In general, PCB 126 elicited similar responses in both the jejenum and colon (Supplemental Figure 4A and 4B). PCB 126 significantly increased expression of Cyp1a1, a marker of aryl hydrocarbon receptor (AhR) activation, in the jejunum and the colon (11.23 fold change, p<0.0001; 23.00 fold change, p<0.0001). Additionally, PCB 126 increased expression of the inflammatory markers hepcidin (Hamp) and tumor necrosis factor alpha (Tnfα) in the colon and interleukin 6 (II-6) and interleukin 18 (II-18) in the jejunum. Expression of the tight junction proteins occludin (Ocel) and claudin (Cldn3) were significantly increased only in the colon. Furthermore, mucin 2 (Muc2) only trended towards increased expression by PCB 126 in the colon (0.05 < p < 0.1). Toll like receptor 4 (Tlr4), a transmembrane receptor that functions in pathogen recognition and activation of innate immunity, was significantly decreased in the jejunum and colon of PCB 126 exposed mice. Interestingly, peroxisome proliferator-activated receptor delta (Pparδ), an important regulator of intestinal cell differentiation and potential inhibitor of inflammatory bowel disease, was significantly decreased in the colon of PCB treated mice (0.46 fold change, p=0.008). Gene expression of glucagon (Gcg), the gene responsible for the production of glucagon like peptide 1 (GLP-1), was significantly increased in both the jejunum and colon of PCB treated mice. Finally, to better elucidate the presence of PCB in gut tissue, and possible causitive mediator of gut inflammation, we followed up these gene expression observations by quantitating PCB 126 in colon tissue via GC-MS and determined that the average concentration was 0.767 ± 0.608 (Mean ± S.E.M.) pmoles/mg tissue. In comparison, hepatic levels averaged 6.388 ± 0.688 (Mean ± S.E.M.) pmoles/mg and plasma levels were undetectable in most mice analyzed.
PCB 126 alters hepatic metabolism in ways that mirror metabolic diseases
With the close proximity of the gut and liver, hepatic metabolism and gut health are tightly intertwined, exerting continual influence on each other. It is known that changes in hepatic metabolism occur as cardiometabolic disease progresses, but it is unknown if dioxin-like pollutant exposure can modify global metabolomic pathways in mice genetically predisposed to cardiometabolic disease. Thus, using targeted metabolomics we next examined the impact of PCB 126 on multiple metabolites related to glycolysis, lipogenesis, and gut microbiota (Table 1). PCB 126 exposure significantly decreased glycolytic intermediates including glucose 6-phosphate, 2-phosphoglycerate, and 3-phosphoglycerate and increased glycerol, glycerol 3-phosphate, and 3-hydroxybutyrate. Furthermore we observed PCB induced alterations in metabolites that can be influenced by interactions between gut microbiota and host systems including reductions in N-acetylphenylalanine, hippurate, and 5-hydroxyindoleacetate, and elevations in dimethylglycine, N-oleoyltaurine, and O-methyltyrosine.
Table 1.
Hepatic Metabolomics Analysisa
| Metabolite Category | Fold Change PCB vs. Vehicle | P-value | Pathway |
|---|---|---|---|
|
| |||
| Carbohydrate Metabolism | |||
|
| |||
| Glucose 6-phosphate | 0.70 | 0.014 | glycolysis, gluconeogenesis, and pyruvate metabolism |
| 2-phosphoglycerate | 0.50 | <0.010 | |
| 3-phosphoglycerate | 0.66 | 0.011 | |
|
| |||
| Lipid Metabolism | |||
|
| |||
| Glycerol | 2.05 | <0.001 | glycerolipid metabolism |
| Glycerol 3-Phosphate | 2.38 | <0.001 | glycerolipid metabolism |
| 3-hydroxybutryate | 1.49 | 0.0298 | Ketone bodies |
|
| |||
| Gut Microbiota | |||
| Influenced Metabolites | |||
|
| |||
| 5-hydroxyindoleacetate | 0.55 | <0.01 | Tryptophan metabolism |
| Retinol | 0.58 | 0.015 | Vitamin A metabolism |
| Retinal | 0.52 | <0.01 | Vitamin A metabolism |
| Hypotaurine | 0.47 | <0.01 | Methionine, cystine, SAM |
| 1-methylhistamine | 0.33 | 0.081 | Histidine metabolism |
| N-acetylarginine | 0.64 | <0.010 | Urea cycle |
| hypoxanthine | 0.79 | 0.012 | Purine metabolism |
| N-acetylphenylalanine | 0.35 | <0.010 | Phenylalanine metabolism |
| Hippurate | 0.52 | 0.067 | Benzoate metabolism |
| Creatinine | 0.52 | 0.086 | Creatine metabolism |
| Argininate | 0.72 | 0.014 | Urea cycle |
| N-oleoyltaurine | 1.74 | 0.049 | Endocannabinoid |
| O-methyltyrosine | 1.54 | 0.050 | Tyrosine metabolism |
| Urate | 2.13 | <0.01 | Purine metabolism |
| dimethylglycine | 1.41 | 0.022 | Glycine, serine, and threonine metabolism |
| glutarate | 1.79 | 0.097 | Fatty acid, dicarboxylate |
n=6 per group, statistically significant differences determined by Welch’s two-sample t-tests.
PCB-induced bacterial alterations associated with metabolic markers
Finally, little is still known about how alterations in gut microbiota are related to overall host health, therefore, using regression analysis we examined the relationship between quantitative markers of inflammation and metabolism with cecal bacterial genera counts. In general, of the correlations examined, there were very few statistically significant associations between cecal genra counts and circulating mediators of metabolism and inflammation. For example, no significant association was observed between any of the cecal genras and Il-6, c-peptide, GIP, or insulin. However, we did observe a statistically significant association between absolute counts of Bifidobacterium and GLP-1 (Supplemental Figure 5A). Specifically, there was a positive association between Bifidobacterium counts and GLP-1 among PCB-treated mice (Bonferroni-adjusted p-value for the number of genras considered pBA = 0.044), while among vehicle-treated mice GLP-1 levels were decreasing with an increase in Bifidobacterium counts (pBA = 0.031). Furthermore, we also observed an inverse association between Akkermansia and fasting blood glucose in both PCB 126 exposed mice and vehicle treated mice (Supplemental Figure 5B). Moreover, at the conclusion of the study, overall fasting glucose levels were 23 points higher (pBA = 0.0342) among PCB-treated mice than among unexposed mice (Supplemental Figure 5B).
Discussion
Currently, a major exposure source of dioxin-like pollutants such as PCB 126 is through consumption of contaminated foods (Carpenter 2006). Thus, the gastrointestinal system is susceptible to a relatively high concentration of pollutants, but is a comparably under studied target organ. Importantly, the gut can have drastic impacts systemically because of its critical role in maintenance of overall health (Janssen and Kersten 2015). We previously showed that PCB 126 could accelerate atherosclerosis and increase systemic inflammation in mice genetically predisposed to cardiometabolic disease, but any impacts of PCB 126 on gut microbiota in this model were yet to be examined (Petriello et al. 2017). Therefore in this study we employed the same Ldlr −/− mouse model fed a high cholesterol diet to examine the effect of PCB 126 on gut microbiota populations, gut health, and the interplay of these effects with inflammation and metabolic complications. We observed that PCB 126 dramatically altered gut microbial populations, primarily in the cecum, and consistently increased intestinal and systemic inflammation.
The gut microbiota are very sensitive to environmental factors including diet and pollutant exposure. Dysbiosis, or the abnormal distribution of bacterial populations, is associated with numerous health conditions from inflammatory bowel diseases to obesity and cardiometabolic disease (Janssen and Kersten 2015). In these conditions, an increase in the ratio of Firmicutes/Bacteriodetes has been consistently observed and has been linked to increased susceptibility to inflammation, infection, oxidative stress, and insulin resistance (Hakansson and Molin 2011; Mafra et al. 2014). In Figure 1B we showed that this ratio was increased due to PCB 126 exposure by the end of the study in both feces and cecum. Furthermore, individuals with type 2 diabetes, have been demonstrated to have a unique microbial signature coprised of a reduction in the number of Clostriadiales, including Roseburia spp, and Faecalibacerium prausnitzii (Karlsson et al. 2013; Qin et al. 2012). In these patients with diabetes or insulin resistance, it has been observed that these specific alterations in gut microbial composition persist without alterations in carbohydrate metabolism (Karlsson et al. 2013; Qin et al. 2012). Research examining the relationship between cardiometabolic disease and gut microbiota has been of great interest in the field lately. Studies have identified a strong link between a gut microbial dependent metabolite, trimethylamine N-oxide (TMAO), and the incidence of cardiometabolic disease (Tang et al. 2015; Warrier et al. 2015; Zhu et al. 2016). In our metabolomics work described herein we did observe an increase in dimethylglycine, a possible TMAO precursor, but hepatic TMAO levels were not significantly different between groups (plasma metabolomics was not completed). Because the microbiota seem to have a strong ability to sense and respond to systemic inflammation, examining known causes of inflammation, such as pollutant exposure, on gut microbiota populations and functionality is greatly needed.
Using 16S rRNA sequencing of fecal samples we examined the change in bacterial populations over time (Figure 2). At the conclusion of the study, we determined that PCB treated mice displayed a significant drop in S24.7 (30%) and Lactobacillus (80%), as well as a significant increase in Clostridales (80.9%). The repeated collection of feces samples also revealed changes in gut microbiota as cardiometabolic disease progressed in the Ldlr −/− mice. For example, irrespective of treatment group, mice showed a significant ~70% decrease in Lachnospiraceae (family) counts, a near ablation of Anaerostipes counts, and a drastic increase (>1100 times) in Allobaculum from baseline to study conclusion (Supplemental Table 1). Surprisingly, few studies in Ldlr or ApoE −/− mice exist describing genra level changes throughout the progression of cardiometabolic disease. There is some work looking at progressive changes due to ageing and glucose intolerance/diabetes, but more work focused on vascular diseases is needed (Odamaki et al. 2016; Zhang et al. 2013). In addition to studying gut microbiota changes over time using the collection of feces, we also examined gut microbiota differences due to PCB 126 at the conclusion of the study in cecum contents. This medium was more sensitive to changes due to PCBs with significant alterations in S24.7, Clostridiales, Akkermansia, Bifidobacterium, Lactobacillus, Ruminococcus, and Oscillospira (Figure 3B).
Interestingly, all mice, regardless of treatment, exhibited high levels of Verrucomicrobia (phylum level) and subsequently Akkermansia (genus level) throughout the entirety of the study compared to what is typically observed in the murine gut microbiota (Lagkouvardos et al. 2016). We hypothesize that the high cholesterol diet all mice received caused an increase in Akkermansia. Although Akkermansia is usually thought of as a healthful bacterial species, we are not the first to observe increased abundance during high cholesterol feeding. Hamilton et. al reported that a obesogenic diet may increase the level of this bacteria similarly to what we observed (Hamilton et al. 2015). Furthermore Carmody et al. observed a high proportion of Verrucomicrobia and thus Akkermansia with high fat, high sucrose feeding (Carmody et al. 2015). Although the authors of both articles attributed this to the fat content, these diets were also high in cholesterol which may be driving the increase in this bacteria observed in both studies as well as ours. Further research needs to be done to examine this effect.
We did observe that PCB 126 exposed mice exhibited higher cecum levels of the phyla Verrucomicrobia and Akkermansia, a genus falling within this phyla, at the end of the study, compared to vehicle treated mice. In support of this, a smaller but still significant increase in Verrucomicrobia following PCB treatment was observed by Choi et al (Choi et al. 2013). The Akkermansia genus contains mucin degrading bacteria, specifically A. muciniphila, and there are reports that an abnormally high abundance of Akkermansia may actually be detrimental due to the excessive degradation of the mucus layer lining the intestine in such a way that the mucin producing goblet cells cannot compensate (Ganesh et al. 2013; Hamilton et al. 2015). In support of this, we observed trends toward an increase in mucin (Muc2) gene expression in the colon of mice exposed to PCB 126. Although we did collect and histologicaly stain Ileum tissues (H&E) in the current study, pathology analyses did not reveal any conclusive differences between the two groups (necrosis was observed in 50% of PCB treated mice and 33.3% of vehicle treated mice; data not shown).
Overall, cecum bacterial populations were impacted to a greater degree by PCB 126 exposure compared to feces. The cecum is characterized by a greater abundance of bacterial species and is often more representative of actual gut microbial populations than fecal samples (Gu et al. 2013). We observed that PCB 126 exposed mice had reduced levels of the genera Oscillospira, Bifidobacterium, Lactobacillus, and Ruminoccoccaea. Oscillospira has recently been observed to be positively associated with leanness and health while decreases in this genera are observed in inflammatory diseases (Konikoff and Gophna 2016). In our previous study, utilizing the same mice, we showed that PCB 126 exposed mice had higher body fat and lower lean mass percentages (Petriello et al. 2017). Bifidobacterium and Lactobacillus, two of the most commonly recognized genera and prebiotics in the food system, are both associated with improved gut health, glucose tolerance and reduced risk of disease (Gomes et al. 2014). Specifically, lower levels of Bifidobacteria has been observed in the fecal microbiota of patients with irritable bowel syndrome, a condition characterized by high levels of inflammation, while Lactobacillus spp. supplementation has been demonstrated to aid in the prevention of intestinal inflammation (Kerckhoffs et al. 2009; Madsen et al. 1999). Importantly, both Bifidobacterium spp. and Lactobacillus spp. have been demonstrated to be increased by prebiotic supplementation, which in turn was associated with improved glucose tolerance and metabolic health (Cani et al. 2007; Kootte et al. 2012). Thus, the reductions in these bacterial genera in PCB treated mice may be associated with the observed alterations in metabolic health.
To link the gut microbiota alterations to systemic inflammation and responses to PCB 126 exposure, we conducted regression analysis examining the relationship between markers of inflammation and metabolism with cecal bacterial genera. Using this approach, we observed an inverse association between Akkermansia and fasting blood glucose, regardless of treatment group. This observation has been noted by other groups previously, demonstrating that decreased levels of Akkermansia were associated with glucose intolerance and supplementation of Akkermansia muciniphila to high fat fed mice improved glucose tolerance (Egshatyan et al. 2016). Interestingly we observed a toxicant-dependent association between Bifidobacterium and GLP-1, where increased levels of Bifidobacterium were associated with increased levels of GLP-1, only in the PCB 126 treated group. This is important due to GLP-1’s involvement in managing insulin levels. As such, it has been demonstrated that Bifidobacterium adolescentis supplementation improves insulin sensitivity in an animal model of metabolic syndrome (Chen et al. 2012).
This current study is significant because we were able to examine PCB 126-induced changes in gut health over time. Importantly, we observed that even 10 weeks following a final dose of PCB 126, gene expression of Cyp1a1 remained highly elevated in the jejunum and colon. Cyp1a1 is a marker of AhR activation, observed with exposure to PCB 126 and other dioxin-like pollutants. Our observations of quantifiable levels of PCB 126 in the colons of exposed mice at the end of the study highlight that PCBs have a long lasting impact on the gut. Other investigators have discussed the circulation of these pollutants through enterohepatic circulation, which involves the cycling of drugs and other compounds from circulation into the intestine where reuptake and transport to the liver occurs (Jandacek and Tso 2007). Because we observed such an elevation in Cyp1a1 gene expression, we hypothesize that these compounds are continually being cycled through enterohepatic circulation, thus exerting a continual impact on the gut microbiota and contributing to chronic increases in gut inflammation.
Intestinal inflammation poses a risk for systemic inflammatory and metabolic conditions including diabetes and cardiovascular disease. For example, patients with inflammatory bowel disease have increased endothelial dysfunction and atherosclerotic lipid profiles (Roifman et al. 2009). This association with inflammatory diseases and risk of cardiometabolic disease is well founded, for example in patients with rheumatoid arthritis and psoriasis (Han et al. 2006). It is important to note that these associations are independent of traditional cardiometabolic disease risk factors. Importantly, we observed increases in intestinal markers of inflammation including Tnf-α, Il-6, Il-18. It has been demonstrated that intestinal upregulation of Il-18 results in production of chemoattractants and cytokines that can reach circulation and initiate inflammatory responses systemically (Siegmund 2010). Interestingly, it has been shown that the gut microbiota and antigen-presenting cells in the intestine interact and stimulate the release of cytokines including TNF-α and IL-6 and that this results in the exacerbation of inflammatory bowel disease as well as whole body inflammation (Mudter and Neurath 2007). Although we did see similar trends in our previous work showing PCB 126 accelerated atherosclerosis at earlier time points (Petriello et al. 2017), this was no longer significantly different in mice sacrificed at 12 weeks post first PCB gavage. However, if we combined data points from our previously published work and this current study to increase statistical power, the acceleration of atherosclerosis due to PCB 126 becomes highly significant (p=0.004; data not shown). It is not unexpected that as cardiometabolic disease progresses in all of the Ldlr deficient mice, the impact of PCB 126 on atherosclerosis is blunted at later time points. To investigate the impact of dioxin-like pollutants in future longer term studies, additional toxicant exposures (i.e., additional gavages) may be useful. In fact, circulating levels in plasma of PCB 126 were undetectable in many of the mice examined at the end of this current study. In our previous study (Petriello et al. 2017) circulating levels of PCB 126 at 8 weeks post first PCB exposure were detected in all mice analyzed with an average of 0.104 picomoles PCB 126/μL plasma.
This is the first study to link PCB 126-induced disruption of the gut to changes in systemic metabolism. We observed that PCB treated mice had increased circulating levels of insulin, c-peptide, and glucose-dependent insulinotropic peptide (GIP) and reduced levels of glucagon-like peptide 1 (GLP-1). While we did not observe differences in glucose sensitivity during a glucose tolerance test (Supplemental Figure 3), the increased levels of insulin and c-peptide in PCB exposed mice may indicate reduced insulin sensitivity (i.e., increased level of insulin required to stimulate cellular glucose uptake). Furthermore, GLP-1 and GIP are both insulinotropic incretins, but there is evidence that hyperinsulinemia and insulin resistance can cause impairments in GLP-1 secretion from intestinal L cells (Lim et al. 2009). Because it is well understood that systemic inflammation can lead to insulin resistance, we believe that the high level of inflammation observed in PCB 126 exposed mice is responsible for the dysregulation of insulin signaling and subsequently further disruption of GLP-1 secretion from the intestine.
This disruption in metabolism was also observed in our hepatic metabolomic analyses where we found that mice exposed to PCB 126 displayed disruptions of glycolytic metabolism, evidenced by decreased levels of glucose 6-phosphate, 2-phosphoglycerate, 3-phosphoglycerate, and phosphoenolpyruvate. Furthermore, PCB 126 treated mice exhibited increases in glycerol, glycerol-3 phosphate, and the ketone body 3-hydroxybutryate, indicating a shift towards a dominance of fatty acid metabolism for energy resulting in ketone body formation. This shift away from carbohydrate metabolism to fatty acid metabolism could be due to the high circulating levels of insulin indicating a requirement for more insulin to stimulate glucose uptake, thus causing fatty acid utilization and ketone body formation to be upregulated to meet energy demands more efficiently.
Conclusion
Our current study demonstrates that exposure to PCB 126 in a compromised model of cardiometabolic disease disrupts gut microbial populations, contributes to intestinal inflammation, systemic inflammation, and metabolic disruption. We hypothesize that the effects on systemic inflammation, intestinal inflammation, and gut dysbiosis with PCB 126 exposure occur simultaneously, and exert a feed-forward effect on each other. More studies are needed to determine if toxicant-initiated changes in gut microbiota are a causitive mechanism linking exposures to dioxin-like pollutants to increased risk of pro-inflammatory diseases in humans.
Supplementary Material
Highlights:
PCB126 exposure reduces gut microbial diversity and alters critical taxa.
Toxicant-exposed mice exhibit modulations in gut microbial influenced metabolites.
Pollutant exposure results in drastic increases in circulating insulin levels.
Interplay of microbiota and pollutants increase risk of cardiometabolic disease
Acknowledgments
Funding: This work was supported by the National Institute of Environmental Health Sciences at the National Institutes of Health [P42ES007380] and the University of Kentucky Agricultural Experiment Station. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that there are no competing financial interests.
References
- Beattie SR, Mark KMK, Thammahong A, Ries LNA, Dhingra S, Caffrey-Carr AK, et al. 2017. Filamentous fungal carbon catabolite repression supports metabolic plasticity and stress responses essential for disease progression. PLoS Pathog 13:e1006340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, et al. 2007. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50:2374–2383. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. 2012. Ultra-high-throughput microbial community analysis on the illumina hiseq and miseq platforms. ISME J 6:1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody RN, Gerber GK, Luevano JM Jr., Gatti DM, Somes L, Svenson KL, et al. 2015. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter DO. 2006. Polychlorinated biphenyls (pcbs): Routes of exposure and effects on human health. Rev Environ Health 21:1–23. [DOI] [PubMed] [Google Scholar]
- Chen J, Wang R, Li XF, Wang RL. 2012. Bifidobacterium adolescentis supplementation ameliorates visceral fat accumulation and insulin sensitivity in an experimental model of the metabolic syndrome. Br J Nutr 107:1429–1434. [DOI] [PubMed] [Google Scholar]
- Chen L, Zhang W, Hua J, Hu C, Lai NL, Qian PY, et al. 2018. Dysregulation of intestinal health by environmental pollutants: Involvement of the estrogen receptor and aryl hydrocarbon receptor. Environ Sci Technol. [DOI] [PubMed] [Google Scholar]
- Choi JJ, Eum SY, Rampersaud E, Daunert S, Abreu MT, Toborek M. 2013. Exercise attenuates pcb-induced changes in the mouse gut microbiome. Environ Health Perspect 121:725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Seelbach MJ, Pu H, Eum SY, Chen L, Zhang B, et al. 2010. Polychlorinated biphenyls disrupt intestinal integrity via nadph oxidase-induced alterations of tight junction protein expression. Environ Health Perspect 118:976–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crinnion WJ. 2011. The role of persistent organic pollutants in the worldwide epidemic of type 2 diabetes mellitus and the possible connection to farmed atlantic salmon (salmo salar). Altern Med Rev 16:301–313. [PubMed] [Google Scholar]
- Dirinck E, Jorens PG, Covaci A, Geens T, Roosens L, Neels H, et al. 2011. Obesity and persistent organic pollutants: Possible obesogenic effect of organochlorine pesticides and polychlorinated biphenyls. Obesity (Silver Spring) 19:709–714. [DOI] [PubMed] [Google Scholar]
- Egshatyan L, Kashtanova D, Popenko A, Tkacheva O, Tyakht A, Alexeev D, et al. 2016. Gut microbiota and diet in patients with different glucose tolerance. Endocr Connect 5:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkina E, Ley K. 2009. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol 27:165–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh BP, Klopfleisch R, Loh G, Blaut M. 2013. Commensal akkermansia muciniphila exacerbates gut inflammation in salmonella typhimurium-infected gnotobiotic mice. PLoS One 8:e74963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz GS, Reardon CA. 2012. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol 32:1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AC, Bueno AA, de Souza RG, Mota JF. 2014. Gut microbiota, probiotics and diabetes. Nutr J 13:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Chen D, Zhang JN, Lv X, Wang K, Duan LP, et al. 2013. Bacterial community mapping of the mouse gastrointestinal tract. PLoS One 8:e74957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakansson A, Molin G. 2011. Gut microbiota and inflammation. Nutrients 3:637–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton MK, Boudry G, Lemay DG, Raybould HE. 2015. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am J Physiol Gastrointest Liver Physiol 308:G840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Robinson DW Jr., Hackett MV, Paramore LC, Fraeman KH, Bala MV. 2006. Cardiovascular disease and risk factors in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. J Rheumatol 33:2167–2172. [PubMed] [Google Scholar]
- Jandacek RJ, Tso P. 2007. Enterohepatic circulation of organochlorine compounds: A site for nutritional intervention. J Nutr Biochem 18:163–167. [DOI] [PubMed] [Google Scholar]
- Janssen AW, Kersten S. 2015. The role of the gut microbiota in metabolic health. FASEB J 29:3111–3123. [DOI] [PubMed] [Google Scholar]
- Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, et al. 2013. Gut metagenome in european women with normal, impaired and diabetic glucose control. Nature 498:99–103. [DOI] [PubMed] [Google Scholar]
- Kerckhoffs AP, Samsom M, van der Rest ME, de Vogel J, Knol J, Ben-Amor K, et al. 2009. Lower bifidobacteria counts in both duodenal mucosa-associated and fecal microbiota in irritable bowel syndrome patients. World J Gastroenterol 15:2887–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JH, Kim WU. 2017. Dysregulation of gut microbiota and chronic inflammatory disease: From epithelial defense to host immunity. Exp Mol Med 49:e337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konikoff T, Gophna U. 2016. Oscillospira: A central, enigmatic component of the human gut microbiota. Trends Microbiol 24:523–524. [DOI] [PubMed] [Google Scholar]
- Kootte RS, Vrieze A, Holleman F, Dallinga-Thie GM, Zoetendal EG, de Vos WM, et al. 2012. The therapeutic potential of manipulating gut microbiota in obesity and type 2 diabetes mellitus. Diabetes Obes Metab 14:112–120. [DOI] [PubMed] [Google Scholar]
- Kuehn BM. 2011. Environmental pollutants tied to atherosclerosis. JAMA 306:2081. [DOI] [PubMed] [Google Scholar]
- Lagkouvardos I, Pukall R, Abt B, Foesel BU, Meier-Kolthoff JP, Kumar N, et al. 2016. The mouse intestinal bacterial collection (mibc) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat Microbiol 1:16131. [DOI] [PubMed] [Google Scholar]
- Lim GE, Huang GJ, Flora N, LeRoith D, Rhodes CJ, Brubaker PL. 2009. Insulin regulates glucagon-like peptide-1 secretion from the enteroendocrine l cell. Endocrinology 150:580–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Perkins JT, Petriello MC, Hennig B. 2015. Exposure to coplanar pcbs induces endothelial cell inflammation through epigenetic regulation of nf-kappab subunit p65. Toxicol Appl Pharmacol 289:457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luft VC, Schmidt MI, Pankow JS, Couper D, Ballantyne CM, Young JH, et al. 2013. Chronic inflammation role in the obesity-diabetes association: A case-cohort study. Diabetol Metab Syndr 5:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen KL, Doyle JS, Jewell LD, Tavernini MM, Fedorak RN. 1999. Lactobacillus species prevents colitis in interleukin 10 gene-deficient mice. Gastroenterology 116:1107–1114. [DOI] [PubMed] [Google Scholar]
- Mafra D, Lobo JC, Barros AF, Koppe L, Vaziri ND, Fouque D. 2014. Role of altered intestinal microbiota in systemic inflammation and cardiovascular disease in chronic kidney disease. Future Microbiol 9:399–410. [DOI] [PubMed] [Google Scholar]
- Mudter J, Neurath MF. 2007. Il-6 signaling in inflammatory bowel disease: Pathophysiological role and clinical relevance. Inflamm Bowel Dis 13:1016–1023. [DOI] [PubMed] [Google Scholar]
- Murphy EA, Velazquez KT, Herbert KM. 2015. Influence of high-fat diet on gut microbiota: A driving force for chronic disease risk. Curr Opin Clin Nutr Metab Care 18:515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odamaki T, Kato K, Sugahara H, Hashikura N, Takahashi S, Xiao JZ, et al. 2016. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol 16:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. 1987. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 68:231–240. [DOI] [PubMed] [Google Scholar]
- Perkins JT, Petriello MC, Newsome BJ, Hennig B. 2016. Polychlorinated biphenyls and links to cardiovascular disease. Environ Sci Pollut Res Int 23:2160–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petriello MC, Han SG, Newsome BJ, Hennig B. 2014. Pcb 126 toxicity is modulated by cross-talk between caveolae and nrf2 signaling. Toxicol Appl Pharmacol 277:192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petriello MC, Brandon JA, Hoffman J, Wang C, Tripathi H, Abdel-Latif A, et al. 2017. Dioxin-like pcb 126 increases systemic inflammation and accelerates atherosclerosis in lean ldl receptor deficient mice. Toxicol Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. 2012. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490:55–60. [DOI] [PubMed] [Google Scholar]
- Ribiere C, Peyret P, Parisot N, Darcha C, Dechelotte PJ, Barnich N, et al. 2016. Oral exposure to environmental pollutant benzo[a]pyrene impacts the intestinal epithelium and induces gut microbial shifts in murine model. Sci Rep 6:31027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roifman I, Sun YC, Fedwick JP, Panaccione R, Buret AG, Liu H, et al. 2009. Evidence of endothelial dysfunction in patients with inflammatory bowel disease. Clin Gastroenterol Hepatol 7:175–182. [DOI] [PubMed] [Google Scholar]
- Rooks MG, Garrett WS. 2016. Gut microbiota, metabolites and host immunity. Nat Rev Immunol 16:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serino M, Blasco-Baque V, Nicolas S, Burcelin R. 2014. Far from the eyes, close to the heart: Dysbiosis of gut microbiota and cardiovascular consequences. Curr Cardiol Rep 16:540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Wang J, Huang F, Lv X, Ma M, Du Y. 2014. Augmented atherogenesis in apoe-null mice co-exposed to polychlorinated biphenyls and 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol 276:136–146. [DOI] [PubMed] [Google Scholar]
- Siegmund B 2010. Interleukin-18 in intestinal inflammation: Friend and foe? Immunity 32:300–302. [DOI] [PubMed] [Google Scholar]
- Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa-Boyle B, et al. 2015. Gut microbiota-dependent trimethylamine n-oxide (tmao) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res 116:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier M, Shih DM, Burrows AC, Ferguson D, Gromovsky AD, Brown AL, et al. 2015. The tmao-generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Nishimura N, Kuo V, Fiehn O, Shahbaz S, Van Winkle L, et al. 2011. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein e−/− mice. Arterioscler Thromb Vasc Biol 31:1260–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Nichols RG, Correll J, Murray IA, Tanaka N, Smith PB, et al. 2015. Persistent organic pollutants modify gut microbiota-host metabolic homeostasis in mice through aryl hydrocarbon receptor activation. Environ Health Perspect 123:679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shen D, Fang Z, Jie Z, Qiu X, Zhang C, et al. 2013. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS One 8:e71108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. 2016. Gut microbial metabolite tmao enhances platelet hyperreactivity and thrombosis risk. Cell 165:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.