

A chromosome 6p24 locus harboring the phosphatase and actin regulator 1 (PHACTR1) gene has one of the strongest genome-wide association study signals for coronary artery disease (CAD)1 and other vascular phenotypes. The variant in this locus with the strongest CAD association is rs9349379, located in an intron of PHACTR1.2 A previous report established an rs9349379-PHACTR1 expression quantitative trait locus (eQTL) in human coronary artery samples, with the European major allele (A) associated with higher PHACTR1 expression than the minor allele (G).2 Querying of the Genotype-Tissue Expression Project portal reveals strong rs9349379-PHACTR1 eQTLs with the same directionality in three human vascular tissues—tibial artery, coronary artery, and aorta (Figure [A]). The rs9349379 A allele conferred binding of myocyte enhancer factor-2 transcription factors in human umbilical vein endothelial cell nuclear extracts, binding that was disrupted by the G allele.2 Deletion of a 34-bp sequence around one of the rs9349379 A alleles in the HUES 9 human embryonic stem cell line (homozygous major) with CRISPR-Cas9, followed by differentiation into endothelial cells, resulted in lower PHACTR1 expression, consistent with but not proving that the A allele confers vascular-specific enhancer activity for PHACTR1.2
Figure. PHACTR1 and EDN1 gene expression in vascular tissues and cells.
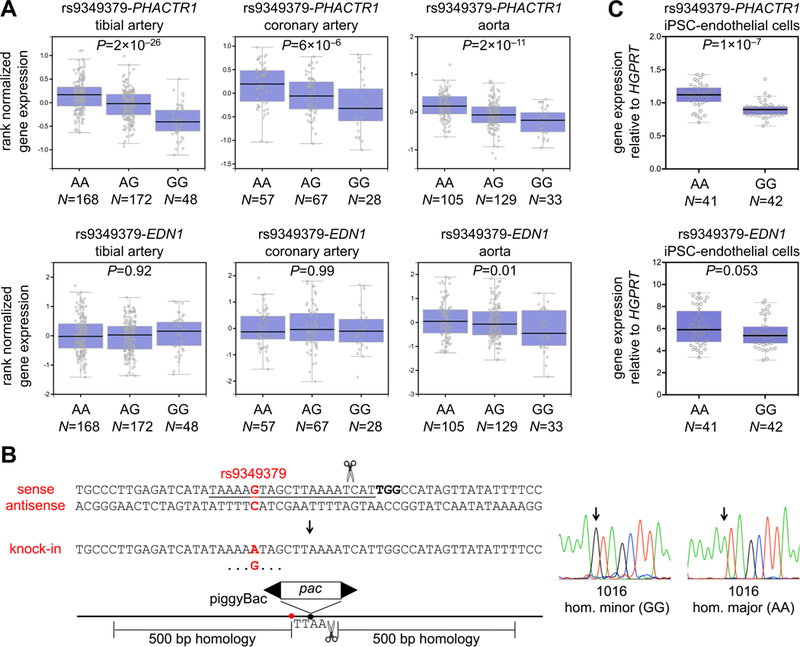
(A) Gene expression in three human vascular tissue types stratified by rs9349379 genotype. The Tukey box plots were generated by the Genotype-Tissue Expression Project portal (https://www.gtexportal.org/). (B) Homozygous knock-in of the rs9349379 major allele (A) or minor allele (G) using CRISPR-Cas9 and a targeting vector with puromycin resistance encoded within a scarless-excision piggyBac transposon. The protospacer is underlined, the protospacer-adjacent motif is bolded, and the position of rs9349379 is indicated in red. Sanger sequencing electropherograms of successfully targeted clones are shown. (C) Gene expression in isogenic rs9349379 homozygous major (AA) and homozygous minor (GG) human induced pluripotent stem cell (iPSC)-derived endothelial cells. Expression levels relative to HGPRT (reference gene) were quantified by the 2−ΔΔCt method. Data are displayed as Tukey box plots with all individual data points shown. P-values were calculated with Mann-Whitney U tests.
A subsequent report contradicted these findings.3 CRISPR-Cas9 deletion of an 88-bp sequence around both rs9349379 A alleles in HUES 9 cells (homozygous major) or both rs9349379 G alleles in DiPS 1016SevA (“1016”) induced pluripotent stem cells (homozygous minor), followed by differentiation into endothelial cells, resulted in unchanged PHACTR1 expression.3 Rather, there was higher expression of the endothelin-1 (EDN1) gene, 600 kb away from rs9349379, in the deleted cells of either background (AA or GG). Of note, these experiments do not permit attribution of the EDN1 expression change to rs9349379 allelic variation, since the variant itself was not specifically altered. In a more directed experiment with CRISPR-Cas9 editing of the HUES 66 embryonic stem cell line (heterozygous) to generate isogenic rs9349379 homozygous major (AA) and homozygous minor (GG) lines, followed by differentiation into endothelial cells, there was lower EDN1 expression and unchanged PHACTR1 expression in AA cells.3 Notwithstanding that these newer data were inconsistent with the prior data, a model wherein altered EDN1 expression is the mechanism by which rs9349379 allelic variation modulates CAD risk and other vascular phenotypes was proposed.3
In light of these contradictory studies, and mindful of past reports of the failure of human pluripotent stem cell-based models to replicate tissue eQTLs,4 we sought to replicate the experiment performed with isogenic rs9349379 homozygous major (AA) and homozygous minor (GG) lines, using a larger sample size (6 clones of each genotype instead of 3 clones each3). Starting with 1016 cells (homozygous minor, GG), we used CRISPR-Cas9 to introduce a piggyBac transposon harboring a puromycin selection cassette into a TTAA site near rs9349379 (Figure [B]) via homology-directed repair, using previously described techniques.5 In one targeting, the repair template had G at the site of rs9349379 (preserving the original genotype), whereas in a parallel targeting, the repair template had A (changing the genotype). In either targeting, several clones were identified with homozygous knock-in of the transposon and the rs9349379 allele. After each targeting, each set of clones with the same genotype was pooled and treated with piggyBac transposase as previously described,5 resulting in scarless removal of the puromycin selection cassettes. We expanded 6 clones of each genotype (GG or AA) and differentiated them in parallel into endothelial cells as previously described,2 with 7 biological replicates per clone.
We used the same quantitative reverse transcriptase-polymerase chain reaction reagents as the previous experiment3 to measure PHACTR1, EDN1, and HGPRT (reference gene) expression. Homozygous major (AA) endothelial cells had significantly higher PHACTR1 expression (22%, P=1×10−7) compared to homozygous minor (GG) endothelial cells, consistent with the A allele conferring enhancer activity for PHACTR1 and with previous rs9349379-PHACTR1 eQTL findings in vascular tissues (Figure [C]). In contrast, we observed no significant difference in EDN1 expression between alternative genotypes, with a trend towards higher expression in AA endothelial cells (rather than significantly lower expression, as observed in the previous experiment3).
Thus, while our experiment did not replicate an endothelial rs9349379-EDN1 eQTL, it did confirm previously observed vascular rs9349379-PHACTR1 eQTLs and support rs9349379 as the causal variant. Reasons for discrepancies between our experiment and the experiment we attempted to replicate could include: the use of human pluripotent stem cell lines of different genetic backgrounds; differences in endothelial cell differentiation; differences in the genome-editing approach (the previous experiment’s clones were each generated with two successive rounds of CRISPR-Cas9-mediated homology-directed repair); or simply the play of chance. We note that the Genotype-Tissue Expression Project portal shows no significant rs9349379-EDN1 eQTLs in three vascular tissues (Figure [A]). In light of our current results, we feel that it is premature to conclude that EDN1 is a causal gene responsible for the chromosome 6p24 CAD association, in preference to PHACTR1 being a causal gene.
Acknowledgments
Sources of Funding: This work was supported by an American Heart Association Postdoctoral Fellowship (X.W.) and NIH grants R01-HL118744 and R01-GM104464 (K.M.).
Footnotes
Data Sharing: The data and cell lines that support the findings of this study are available from the corresponding authors upon reasonable request.
Disclosures: None.
References:
- 1.Myocardial Infarction Genetics Consortium. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet 2009;41:334–341. 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beaudoin M, et al. Myocardial infarction-associated SNP at 6p24 interferes with MEF2 binding and associates with PHACTR1 expression levels in human coronary arteries. Arterioscler Thromb Vasc Biol 2015;35:1472–1479. 10.1161/ATVBAHA.115.305534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta RM, et al. A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell 2017;170:522–533. 10.1016/j.cell.2017.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, et al. Interrogation of the atherosclerosis-associated SORT1 (sortilin 1) locus with primary human hepatocytes, induced pluripotent stem cell-hepatocytes, and locus-humanized mice. Arterioscler Thromb Vasc Biol 2018;38:76–82. 10.1161/ATVBAHA.117.310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pashos EE, et al. Large, diverse population cohorts of hiPSCs and derived hepatocyte-like cells reveal functional genetic variation at blood lipid-associated loci. Cell Stem Cell 2017;20:558–570. 10.1016/j.stem.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]