

Abstract
We report a mild and efficient electrochemical protocol to access a variety of vicinally C–O and C–N difunctionalized compounds from simple alkenes. Detailed mechanistic studies revealed a distinct reaction pathway from those previously reported for TEMPO-mediated reactions. In this mechanism, electrochemically generated oxoammonium ion facilitates the formation of azidyl radical via a charge-transfer complex with azide, TEMPO–N3. DFT calculations together with spectroscopic characterization provided a tentative structural assignment of this charge-transfer complex. Kinetic and kinetic isotopic effect studies revealed that reversible dissociation of TEMPO–N3 into TEMPO• and azidyl precedes the addition of these radicals across the alkene in the rate-determining step. The resulting azidooxygenated product could then be easily manipulated for further synthetic elaborations. The discovery of this new reaction pathway mediated by the TEMPO+/TEMPO• redox couple may expand the scope of aminoxyl radical chemistry in synthetic contexts.

INTRODUCTION
Since its discovery in 1959,1 tetramethylpiperidine-1-oxyl (TEMPO•), a stable aminoxyl radical, has shown broad utility in the fields of chemistry and biochemistry research.2 TEMPO• can undergo one-electron redox processes, granting access to three discrete oxidation states (Scheme 1). This behavior makes TEMPO• and related aminoxyls highly versatile agents in the organic synthesis of small molecules and polymers.3
Scheme 1. Accessible Oxidation States of TEMPO via Single-Electron Oxidation and Reduction Processes.

aThis value was reported in ref 4 and measured in an aqueous buffer.
The mechanism of action of TEMPO• and related aminoxyl radicals in organic transformations primarily relies on four key attributes. In one scenario, the electrophilic nature of TEMPO+, the oxidized form of TEMPO•, is used in the activation of nucleophilies such as alcohols5,6 and amines7 toward oxidation (Scheme 2A). Alternatively, the free radical character of TEMPO• is exploited in the abstraction of H atoms in weak C–H bonds8 (Scheme 2B) and the reversible trapping and liberation of transient carbon-centered radicals9 (Scheme 2C). In particular, the latter mode of action serves as the basis of nitroxide-mediated radical polymerizations (NMP).10 Finally, TEMPO• can serve as a mild single-electron oxidant, while TEMPO− reacts as a strong single-electron reductant; these features have been exploited in the conversion of organometallic compounds11 (Scheme 2D) and hypervalent iodine species,12 respectively, to the corresponding radicals.
Scheme 2.

Existing and New Reaction Modes of TEMPO
Given our research interests in synthetic electrochemistry and the attractive electrochemical features of aminoxyls, we aim to develop new synthetically useful transformations mediated by these persistent radicals. In particular, we are interested in exploring the chemical space of the TEMPO+/TEMPO• redox couple because its application in synthetic contexts remains rare13 when compared with TEMPO+/TEMPO− and TEMPO•/TEMPO− redox couples. Based on its reduction potential, TEMPO+ is a stronger oxidant than TEMPO• itself (see Scheme 1) and can in principle engage nucleophiles that are less potent than main group organometallic compounds. This hypothesis is evident from the alcohol oxidation activity (see Scheme 2A) but is currently limited to the two-electron oxidation regime (i.e., use of the TEMPO+/TEMPO− couple).6 In principle, by exploring the function of TEMPO+ as a single-electron oxidant, we could expand the scope of this aminoxyl radical’s redox chemistry and advance its limits beyond C–C and C–O bond-forming processes (Scheme 2E).
Furthermore, the mechanistic insights into TEMPO-mediated reactions have lagged behind their development. Elegant and thorough mechanistic investigations exist but are largely limited to systems such as catalytic alcohol oxidation5 and NMP.14 Detailed studies on TEMPO-mediated radical transformations will aid in the development of new and better reagents, enrich the reaction chemistry of aminoxyl radicals, and advance fundamental knowledge in radical chemistry.
While studying electrochemical diazidation reactions,15,16 we uncovered a new electrochemical transformation in which the electrolysis of a mixture of tert-butylstyrene (1a), TEMPO•, and NaN3 led to the regioselective formation of a vicinally azidooxygenated product (2a) (Scheme 3). Herein, we describe our effort to elucidate the mechanism responsible for the observed reactivity using electrochemical, experimental physical organic, and computational methods. The aggregate data obtained accord with a new reaction pathway for the formation of C–N bonds mediated by the TEMPO+/TEMPO• redox couple. Analysis of the computational and spectroscopic data supports the formation of an unprecedented charge-transfer complex from TEMPO+ and N3− that serves as a key reaction intermediate. The mechanistic insights may guide the design of new radical reactions promoted by TEMPO• and related aminoxyl radicals.
Scheme 3.
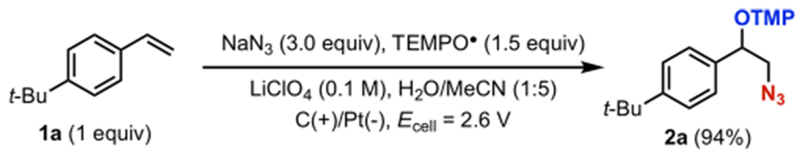
Electrochemical Azidooxygenation of Alkenes
RESULTS AND DISCUSSION
A. Reaction Development.
From a synthetic point of view, vicinal aminoalcohol-type structures frequently exist in bioactive natural products and synthetic pharmaceuticals.17 As such, we investigated the substrate scope and found that our electrochemical azidooxygenation protocol is applicable to a wide variety of alkenes beyond styrenes (Scheme 4A). Aliphatic alkenes with different substitution patterns are all compatible with the described method. Common functional groups including ketones, aldehydes, carboxylic acids, amides, and alkyl bromides remain intact under the electrochemical conditions. Cyclic alkenes were converted to the corresponding difunctionalized products in moderate to high stereoselectivity favoring the trans configuration. Stilbene was transformed to a mixture of diastereomeric compounds in ca. 1:1 ratio; the two products were readily separated via column chromatography, and the structure of the syn isomer was confirmed via X-ray crystallography.
Scheme 4. Substrate Scope of the Azidooxygenation Reaction.

aReaction conditions: alkene (0.2 mmol, 1 equiv), TEMPO• (0.3 mmol, 1.5 equiv), NaN3 (0.6 mmol, 3 equiv), MeCN (3.5 mL), H2O (0.3 mL) at 22 °C, cell voltage (Ecell) = 2.6 V, under N2. bUnder air. cReaction was conducted at 40 °C. dEcell = 2.2 V with 2 equiv of NaN3. eWith 1 equiv of TEMPO• and 1 equiv of NaN3. fCuSO4, phenylacetylene, sodium ascorbate, tBuOH, and water. gmCPBA and DCM. h(1) Pd/C and H2; (2) Boc2O; (3) Zn and HOAc.
The sole existing method available for obtaining the same types of products emerged from an elegant seminal work by Studer and co-worker.18 However, this protocol requires the use of a hypervalent iodine-derived azide source and a highly reducing NaTEMPO complex, which limits the functional group compatibility and the broad use of this method in organic synthesis. In comparison, our reaction employs readily available reagents, proceeds under milder conditions, and offers a substantially broader substrate scope. For example, our protocol features the use of functional groups including ketone (2e), carboxylic acid (2g), aldehyde (2i), alkyl bromide (2j), alcohol (2x; see Scheme 9), and sulfide (2y) groups, which represents an important synthetic advancement. In addition, the previous method requires 3 equiv each of NaTEMPO and the azido-hypervalent iodine. In contrast, while our substrate scope studies were conducted using 1.5 equiv of TEMPO• and 3 equiv of NaN3, we demonstrated that the amount of the reagents can be reduced to 1 equiv each without significantly affecting the efficiency of the reaction (76% yield with 2m).
Scheme 9.

Expanded Substrate Scope to Include Oxidatively Labile Functional Groups
We further expanded the scope of the reaction to feature a completely aqueous buffer medium (Scheme 4c). By replacing LiClO4 and MeCN with a pH 7 phosphate buffer solution, we were able to observe the same reactivity with a select number of substrates, albeit with diminished yields. This feature is likely difficult with the chemical conditions reported previously, as NaTEMPO is reportedly sensitive to moisture. These promising results, taken together with the broad use of organic azides and TEMPO in biological labeling studies, indicate that our reaction may provide a new chemical tool for studying biological processes.
When compared to other existing catalytic reactions that convert alkenes to vicinal amino alcohols,19 our new method displays complementary scope, regioselectivity, and stereoselectivity. For instance, the classical Os-catalyzed aminohydroxylation encounters problematic regioselectivity with certain types of alkenes, while sterically hindered substrates frequently undergo undesirable dihydroxylation.20 Another elegant example is the Fe-catalyzed aminooxygenation using oxaziridines.19c However, this protocol has primarily been applied to activated alkenes (e.g., styrenes and conjugate dienes) and shows decreased efficiency with unactivated alkenes. It is also noteworthy that both aforementioned methods are stereospecific, with both O and N groups added to the same side of the C═C bond. In contrast, our reaction is applicable to a wide range of activated and unactivated alkenes and provides exclusive regioselectivity with all substrates studied; the latter of which can be explained by the intrinsic relative stability of carbon-centered radicals. Our electrochemical method is also nonstereospecific and gives moderate to high dr with cyclic alkenes favoring the anti-addition products.
An attractive feature of the products from this transformation lies in the versatile and often orthogonal reactivity of the azido and aminooxy groups, which leads to a diverse array of vicinally difunctionalized organic structures upon additional synthetic elaborations (for examples, see Scheme 4B and ref 18).
B. Radical Clock Experiments.
The mild reaction conditions and potential synthetic utility of this methodology prompted us to undertake a detailed mechanistic investigation. Radical clock experiments using cyclopropyl-substituted alkenes 1u and 1v confirmed our hypothesis that radical intermediates are involved in the observed reactivity (Scheme 5). Upon formation of the C–N3 bond, the vicinal carbon-centered radical induces the rupture of the three-membered ring prior to capture by the aminoxyl radical. However, when diallyl ether was subjected to the standard reaction conditions, no cyclization product was observed, whereas the monoazidooxygenated product was isolated in 42% yield. These findings suggest that the radical cross coupling between the carbon-centered radical intermediate and TEMPO• happens at a rate between 106 and 1011 s–1, consistent with previous reports (109 M–1 s–1).21 Further supporting the stepwise radical mechanism, cis-stilbene underwent nonstereospecific addition to produce a pair of diastereomeric products with the same dr.
Scheme 5.

Radical Clock Experimental Results
We considered two plausible mechanistic scenarios for the electrochemical generation of N3• or its equivalent before alkene addition (Scheme 6). Pathway 1a involves the direct anodic oxidation of N3− [Ep/2 = 0.45 V vs ferrocenium/ferrocene (Fc/Fc+);22 Figure 1] to N3·. Alternatively, in pathway 1b, TEMPO•, with its significantly lower reduction potential (E1/2 = 0.25 V), will first lose an electron on the anode to form the corresponding oxoammonium ion (TEMPO+).23 TEMPO+ will then oxidize NaN3 either through the formation of a TEMPO–azide adduct (I; innersphere mechanism) or through direct electron transfer from N3− to form free N3• (outersphere mechanism).24
Scheme 6.

Plausible Mechanistic Pathways for the Formation of the Azidyl Radical or Its Equivalent
Figure 1.

Cyclic voltammograms of TEMPO•, N3−, and their mixture.
Data from chemical and electrochemical experiments exclude pathway 1a and support pathway 1b as a possible reaction mechanism. First, reaction of TEMPO+ClO4− (independently synthesized), NaN3, and indene (1m) in the absence of applied potential afforded 66% of the vicinally difunctionalized product 2m. In addition, controlled potential electrolysis revealed that the reaction occurs smoothly at 0.35 V (vs Fc/Fc+), which is sufficient to oxidize TEMPO• to TEMPO+, but insufficient to drive direct oxidation of N3− (see Figure 1). Both results indicate that direct oxidation of N3− to N3” is unnecessary and unlikely to account for the observed reactivity.
In pathway 1b, the outer-sphere oxidation of N3− by TEMPO+ is unlikely due to the highly unfavorable energetics of this electron-transfer process, indicated by the 200 mV potential difference between the donor (N3−) and acceptor (TEMPO+). Experimental evidence detailed in the following section strongly support an inner-sphere mechanism involving the formation of a TEMPO–N3 adduct (I).
C. Identification of a Charge-Transfer Complex.
Voltammetric and spectroscopic data revealed the formation of a new TEMPO–N3 adduct upon oxidation of TEMPO•.25,26 The TEMPO+/TEMPO• redox couple appeared on the cyclic voltammogram as a highly reversible wave at 0.25 V in MeCN, but the addition of TBAN3 resulted in a significant cathodic shift of the redox wave (Figure 1). This shift likely arose from the reversible formation of a discrete and stable complex between TEMPO+ and N3−. The magnitude of the potential shift follows a Nernstian dependence on [N3−] with a slope of 62 mV/dec (Figure 2), indicating a 1:1 stoichiometry of the complex.
Figure 2.

Cyclic voltammograms of TEMPO• in the presence ofN3− (top), showing a Nernstian dependence of the reduction potential on [N3−] (bottom). The curve depicts a least-squares fit to the function f(x) = ax + b: a = −62 ± 2; b = 210 ± 2; R2 = 0.990.
UV–vis absorption experiments provide further evidence for the formation of the TEMPO–N3 adduct (I). TEMPO• and TEMPO+ display UV–vis features with λmax at 446 and 465 nm, respectively (Figure 3).27 The addition of NaN3 to the TEMPO+ solution led to the appearance of a new, intense absorption peak at 380 nm.28 The resulting solution exhibited a dark brown color at high concentrations and orange at low concentrations (Figure 4), the latter of which was also observed under electrochemical reaction conditions immediately upon application of an anodic potential.
Figure 3.

UV–vis absorption spectra of TEMPO• (8.6 mM), TEMPO+ClO4− (8.6 mM), and the TEMPO–N3 complex (0.2 mM) in MeCN at room temperature (ca. 22 °C).
Figure 4.

Color of the MeCN solution of TEMPO–N3 adduct (right) in comparison to TEMPO• (left) and TEMPO+ (center).
The TEMPO–N3 adduct proved sufficiently stable for various voltammetric and spectroscopic analyses. However, it slowly decomposes at room temperature to form TEMPO•, as detected by electron spin resonance spectroscopy (ESR; see Figure S30) and presumably N3”. This nitrogen-centered radical is known to undergo rapid dimerization followed by fragmentation to form N2 (vide infra).29 The half-life of I is ca. 7 min as measured by the disappearance of the UV absorbance at 380 nm.30
The identity of this new TEMPO–N3 adduct was further analyzed using a variety of experimental and computational techniques. A UV–vis absorption titration of a solution of TBAN3 with TEMPO+ClO4− resulted in a linear correlation between absorbance and molar ratio (Figure 5). Upon reaching 1:1 stoichiometry, the addition of more TEMPO+ salt to the mixture resulted in a rapid decrease of the absorbance at 380 nm because TEMPO+ catalytically decomposes the TEMPO–N3 adduct through a currently unknown mechanism. To complete the titration, we added additional TBAN3 to the 1:1 mixture of TBAN3 and TEMPO+ClO4− and observed no further change in the absorption intensity at 380 nm. These data confirmed the 1:1 stoichiometry of the complexation event and further indicated that the formation of I, although reversible, is effectively quantitative.
Figure 5.

Titration experiments showing the 1:1 stoichiometry of the TEMPO–N3 adduct.
DFT calculations were then carried out to predict the structure of adduct I. Previous studies showed that TEMPO+ can form complexes by reacting at either O or N of the oxoammonium group31 depending on the identity of the reacting partner and the reaction medium. We computed the structure and energy of the corresponding O- and N-adducts of N3− using B3LYP/def2-TZVP32 with the polarizable continuum solvation model in MeCN (I1 and I2, respectively; Figure 6). Intriguingly, we also found two additional energetically viable structures (I3 and I4), both displaying a five-membered cyclic scaffold resulting from a complexation event akin to a [3+2] cycloaddition between N═O and N3−. The terminal N–N and N–O distances are both about 2.5 Å, substantially longer than that of a typical chemical bond, indicating that this adduct is more of a charge-transfer complex than a covalent adduct. I4 appeared most energetically favorable among all plausible structures examined. Structural and energetic agreement across a series of DFT methods (see table in Figure 6), including methods consistent with previous studies of metal-free charge-transfer complexes,33 offers strong evidence for the validity of these models.
Figure 6.
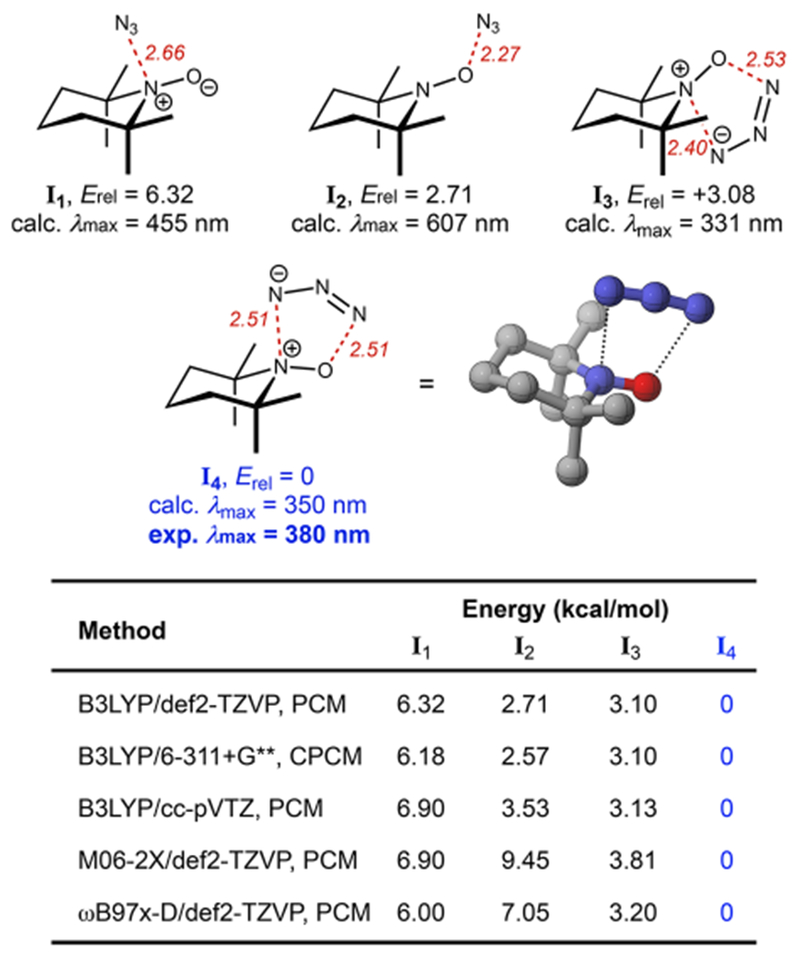
Energetically viable structures of the TEMPO–N3 complex predicted by DFT calculation. All energies are reported in terms of the ground state energy corrected for zero-point energy (E + ZPE).
To further confirm our structural assignment, we predicted the UV–vis spectrum for each structure (I1–I4) using time-dependent DFT (TDDFT). To assess the accuracy of our computational method in predicting the spectroscopic data of structurally related compounds, we used TEMPO• and TEMPO+ as references. The maximum absorbance predicted for TEMPO• and TEMPO+ was at 439 and 466 nm, respectively. These data are consistent with experimental measurements (446 and 465 nm, respectively; see Figure 3). When comparing the experimentally measured absorbance maximum of 380 nm to the predicted value for each viable structure, we found that I4 is again the most likely structure. DFT also estimated a high molar extinction coefficient of I4 of 13 100 M−1 cm−1, which is on par with the experimental value (5174 M−1 cm−1)34 and substantially more intense than that of either free TEMPO (~2 M−1 cm−1) or the oxoammonium (~1 M−1 cm−1).35 We also computed structures with H+ or Na+ bound to various forms of the adduct, but none appear energetically viable or predict spectroscopic features consistent with the experimental data.
The structure of adduct I was also supported by NMR data. When formed with azide labeled at a terminal position [(15N═N═N)−], adduct I showed a 15N resonance of 244.66 ppm in MeCN-d3, a dramatic chemical shift change from 99.34 ppm for the free azide in D2O (Figure 7). Computationally, the complex in MeCN has a chemical shift of 255.71 ppm (when 15N is bound to the N terminus of the oxoammonium) and 233.85 ppm (when 15N is bound to the O terminus of the oxoammonium), while the free sodium azide has a chemical shift of 88.15 ppm.36 The observed chemical shift of the complex is in between the two predicted values, suggesting that there is rapid scrambling of the two terminal azides. This finding is consistent with the reversible formation of the adduct from TEMPO+ and N3−.
Figure 7.

NMR spectra of the TEMPO–N3 complex in MeCN (top) and NaN3 in D2O (bottom).
D. Mechanism of Reaction between TEMPO–N3 and Alkenes.
We then investigated the reaction between this TEMPO–N3 adduct and the alkene substrate. Three plausible mechanisms are proposed for this reaction (Scheme 7). In pathway 2a, TEMPO–N3 undergoes polar addition across the C═C bond in either a concerted or a stepwise fashion to yield the difunctionalized product. Alternatively, in pathway 2b, TEMPO–N3 directly delivers the N3 group onto the alkene in a manner akin to metal-mediated radical group transfer.37 This process results in the formation of TEMPO• and carbon-centered radical II, which subsequently cross couple to furnish the desired product. Finally, in pathway 2c, TEMPO–N3 first decomposes into TEMPO• and azidyl radical (N3•), upon which the sequential addition of these two open-shell species across the alkene yields the desired azidooxygenated product. Based on the information from the radical clock experiments (see Scheme 5), pathway 2a can be conclusively ruled out.
Scheme 7.

Plausible Mechanistic Pathways for the Reaction between TEMPO–N3 and Alkenes
Pathway 2b requires transition of adduct I from a closed-shell ground state to an open-shell excited state (singlet or triplet) prior to transferring N3• to the alkene. However, the first singlet excited state of complex I is predicted by DFT calculation to be 3.56 eV more expensive than the ground state. Therefore, this spin-unpaired singlet state is thermally inaccessible. The triplet state, which requires intersystem crossing from the singlet state, is thus also inaccessible. Analysis of other plausible isomers of I in Figure 6 revealed that none of these structures possess a thermally available excited state at room temperature. Further, we conducted an experiment under standard electrochemical conditions with the exclusion of light and observed minimal changes in efficiency in the azidooxygenation reaction. This finding indicates that photoexcitation is not necessary for the observed reactivity. Taken together, pathway 2b is an unlikely mechanism for the radical addition of complex I to the alkene.
Kinetics experiments were carried out to further distinguish between pathways 2b and 2c. The TEMPO–N3 adduct alone is metastable under ambient conditions with a decomposition half-life of ca. 7 min. This decomposition results in the formation of TEMPO•, as detected by ESR (see SI) and presumably N2 from the dimerization of N3•.25 Notably, upon the addition of an excess of indene (1m) at room temperature, the UV absorbance peak at 380 nm disappears instantaneously (<5 s).
Using a less reactive alkene, 2-methyl-4-phenylbutene (1l), we studied the kinetics of the radical addition of TEMPO–N3 (I) to 1l using UV–vis spectroscopy. The experiment was carried out by mixing TEMPO+ClO4− and TBAN3 (used in lieu of NaN3 to ensure full solubility in the reaction medium) in MeCN followed by immediate addition of 1l prior to spectrum collection at 380 nm. This reaction shows a first-order dependence on [1l] (Figure 8) and zero-order on [N3−] (Figure 9). The rate vs [1l] plot shows a nonzero intercept due to the competing, alkene-independent decomposition of I (vide infra). When the amount of 1l is greater than 6 equiv with respect to TEMPO+, saturation kinetics behavior was observed (see SI). Exogenous TEMPO• inhibits this reaction and an inverse first-order rate dependence was observed on [TEMPO•] (Figure 10). The rate vs 1/[TEMPO•] plot exhibits a small, nonzero intercept, which likely also arises from the competing decomposition pathway that unproductively consumes the TEMPO–N3 adduct.
Figure 8.

Plot of initial rates versus concentration of alkene 1l for the reaction between adduct I (TEMPO+ClO4− + TBAN3) and alkene 1l, showing first-order rate dependence on [1l]. The data point corresponding to [1l] = 0 represents the rate of decomposition of I in the absence of 1l. Error bars represent the standard deviation of three independent measurements. The curve depicts an unweighted least-squares fit to the function f(x) = ax + b: a = 2.09 ± 0.08; b = 0.73 ± 0.04; R2 = 0.993.
Figure 9.

Plot of initial rates versus concentration of N3− for the reaction between adduct I (TEMPO+ClO4− + TBAN3) and alkene 1l, showing zero-order rate dependence on [N3−]. Error bars represent the standard deviation of three independent measurements. The curve depicts an unweighted least-squares fit to the function f(x) = ax + b: a = −0.04 ± 0.01; b = 0.78 ± 0.02; R2 = 0.63.
Figure 10.

Plot of initial rates versus concentration of TEMPO• for the reaction between adduct I (TEMPO+ClO4− + TBAN3) and alkene 1l, showing inverse first-order rate dependence on [TEMPO•]. Error bars represent the standard deviation of three independent measurements. The curve depicts an unweighted least-squares fit to the function f(x) = ax−1 + b: a = 0.0345 ± 0.0008; b = 0.057 ± 0.002; R2 = 0.998.
The inhibition effect of TEMPO• on the reaction rate was also studied under conditions that resemble the electrochemical reaction (Figure 11). At high [alkene] ([1l] = 50 mM), two kinetic regimes were observed when altering the concentration of exogenous [TEMPO•]. At very low [TEMPO•] ([TEMPO•] < 0.25 mM, or 1/[TEMPO•] > 4), the lack of rate dependence on [TEMPO•] was evident. At higher [TEMPO•] ([TEMPO•] > 0.25 mM, or 1/[TEMPO•] < 4), which is the case under synthetic electrochemical conditions,38 inverse first-order rate dependence was again observed.
Figure 11.

Plot of initial rates versus concentration of TEMPO• for the reaction between adduct I (TEMPO+ClO4− + TBAN3) and alkene 1l (50 mM). Saturation kinetics were observed when TEMPO• concentration is less than 0.25 mM.
These kinetic data are summarized in the following rate equation:
| (1) |
The rate equation is fully consistent with the proposed pathway 2c (Scheme 7). Using steady-state approximation, the following rate equation can be constructed for this mechanistic scenario.
| (2) |
In eq 1, the zero-order rate dependence on [N3−] indicates that the formation of TEMPO–N3 is quantitative, in agreement with our previous titration experiment (see Figure 5). The inverse first-order rate dependence on [TEMPO•] at synthetically relevant [TEMPO•] suggests that the homolysis of TEMPO–N3 is reversible and that the subsequent addition of N3· to alkene 1l is rate-determining (k–1[TEMPO•] ≫ k2[alkene]). The saturation kinetics at very low [TEMPO•] and high [alkene] (k−1[TEMPO•] ≪ k2[alkene]) is also consistent with the proposed mechanism.
E. Mechanism of the Decomposition of TEMPO–N3 in the Absence of an Alkene.
As previously discussed, adduct I undergoes slow decomposition in the absence of an alkene. We hypothesize that this process also occurs through the reversible homolytic fragmentation of I into TEMPO• and N3•. Based on previous reports, the resultant N3• will undergo facile dimerization followed by irreversible decomposition to form N2.27 Kinetic data reveal that this process is inhibited by exogenous TEMPO• (see SI), which is consistent with our hypothesis. However, the reaction rate does not follow a simple inverse first-order dependence on [TEMPO•], likely as a result of the complex mechanism underlying the decomposition of N3•.26,27 This dimerization–decomposition side reaction is significantly slower than the desired alkene addition presumably as a result of the low concentration of N3• in the reaction medium.
F. Full Mechanism of the Electrochemical Azidooxygenation.
The voltammetric, kinetic, spectroscopic, and computational data presented above lead to a full understanding of the mechanism for the electrochemical azidooxygenation reaction (Scheme 8). Upon oxidation of TEMPO• to TEMPO+, the complexation of N3− with TEMPO+ takes place to furnish the charge-transfer adduct I. This complexation event is reversible but quantitative (association constant Ka ≫ 1). This metastable intermediate will then undergo uphill and reversible fragmentation, resulting in the formation of TEMPO• and azidyl radical. The transient N-centered radical can dimerize to form N2 as an undesired side reaction. In the presence of an alkene, however, the addition of N3• to the C═C π-bond outcompetes the decomposition pathway to yield carbon-centered radical II.39 Finally, the capture of II by TEMPO generates the difunctionalized product to complete the reaction.
Scheme 8.
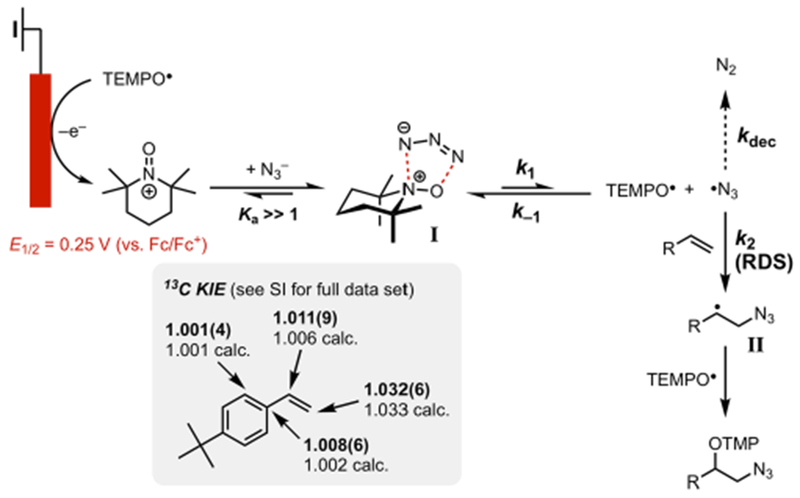
Mechanism of the Electrochemical Azidooxygenation Reaction
The addition of N3• to the alkene is the slowest chemical step post-electron transfer. Natural abundance 13C kinetic isotope effects (KIEs) further support the assignment of the rate-determining step (RDS) (see Scheme 8). A significant primary KIE (3%) was observed at the terminal carbon connected to the azide moiety. The computationally predicted value (1.033) of an azidyl addition to the terminal position of tert-butylstyrene is consistent with the experimentally measured value of 1.032(6). In addition, on using cis-stilbene as a substrate, upon 50% conversion, the alkene was recovered in complete cis conformation, thereby showing that the N3• addition to the alkene is irreversible and thus the RDS.
An alternative mechanistic scenario includes the formation of N3• and TEMPO• through an endergonic outersphere electron transfer (+4.6 kcal/mol) from N3− to TEMPO+. In this scenario, the TEMPO–N3 complex serves as an off-cycle reservoir of azidyl radical. This mechanism is in principle plausible and cannot be kinetically differentiated from the proposed scenario in Scheme 8. However, this outer-sphere electron transfer is unlikely the predominant pathway. As is evident from the UV–vis titration (see Figure 5) and cyclic voltammetry data (see Figure 1), the formation of TEMPO–N3 from TEMPO+ and N3− is instantaneous and quantitative; this process is essentially barrierless and outcompetes the outer-sphere electron transfer from N3− to TEMPO+. From the TEMPO–N3 resting state, it is highly unlikely that TEMPO–N3 completely dissociates into discrete ions before electron transfer. In fact, outer-sphere electron transfer between two molecules frequently occurs when the redox pair is within a few angstroms.40 Taken together, we propose a concerted inner-sphere mechanism for the generation of TEMPO• and N3• that comprises simultaneous dissociation of I and electron transfer from N3− to TEMPO+.
G. Mechanism-Guided Expansion of Substrate Scope.
The quantitative formation of the TEMPO–N3 adduct as a result of the strong association between N3− and the oxoammonium suggests that functional groups that are typically incompatible with TEMPO-mediated oxidation conditions may be tolerated in our electrochemical azidooxygenation reaction. For instance, alcohols and sulfides have been shown to undergo oxidation to carbonyl compounds and sulfoxides, respectively, in the presence of TEMPO+.5,41 However, owing to the lower nucleophilicity of alcohols and sulfides compared with N3−, the formation of adduct I will result in a very low concentration of TEMPO+ when N3− is in excess.
Indeed, alkenes with pendant hydroxyl or sulfide groups proved suitable substrates and were transformed to the azidooxygenated products in high yield (Scheme 9). In the case of alcohol 1×, monitoring the reaction progress and controlling the reaction time are crucial to preventing the side reaction that oxidizes the hydroxyl group. Currently, amines and thiols are not compatible with the azidooxygenation conditions.
CONCLUSIONS
We described the development of an electrochemical protocol for the regioselective synthesis of vicinal azidooxygenated products from alkenes. Through detailed mechanistic studies, we uncovered a new mode of reactivity of TEMPO in which the TEMPO+/TEMPO• redox couple promotes the single-electron oxidation of N3− to N3• toward C–N bond formation with an alkene. This oxidation event is mediated by TEMPO–N3, a newly observed charge-transfer complex formed from TEMPO+ and N3−. Although the direct structural elucidation of this complex proved challenging, DFT calculations combined with NMR and UV–vis spectroscopic data provided a tentative structural assignment. The most viable structure displays an intriguing molecular scaffold resulting from the cycloaddition between N3− and the N═O motif of TEMPO+. Kinetic and computational studies depict a detailed mechanistic scheme of the electrochemical reaction. The mechanism involving the formation and subsequent reaction of the TEMPO–N3 adduct guided the further expansion of the reaction scope to include alkenes with oxidatively sensitive functional groups. This charge-transfer complex exhibits a new class of reactivity not reported for TEMPO+. Current work is focused on applying this new mechanistic pathway enabled by the TEMPO+/TEMPO• redox cycle for the development of new transformations mediated by aminoxyl radicals.
Supplementary Material
ACKNOWLEDGMENTS
Financial support to S.L. was provided by Cornell University, the Atkinson Center for a Sustainable Future, and NSF (CHE-1751839). K.M.L. gratefully acknowledges the Alfred P. Sloan Foundation and NIGMS (R35GM124908) for support. This study made use of the Cornell Center for Materials Research Shared Facilities supported by NSF MRSEC (DMR-1719875), the NMR facility supported by the NSF (CHE-1531632), and National Biomedical Research Center for AdvanCed ESR Technology (ACERT) laboratory supported by the NIGMS (P41GM103521). G.S.S. is grateful for an NSF Graduate Fellowship (DGE-1650441); A.S. is grateful for an ACS SURF Award. We thank Dr. Ivan Keresztes for help with NMR spectral analysis, Dr. Samantha MacMillan for help with X-ray crystallography analysis, Dr. Bo Chen for discussions on the DFT calculation, and Dr. Ke-Yin Ye for experimental assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b06744.
Experimental procedures and characterization data (PDF)
Crystallographic data for compound 2o (also deposited in the Cambridge Structural Database: CCDC1843443) (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).Lebedev OL; Kazarnovsky SN Tr. Khim. Khim. Tekhnol 1959, 3, 649. [Google Scholar]
- (2).(a) For representative recent reviews, see: Tebben L; Studer A Angew. Chem., Int. Ed 2011, 50, 5034. [DOI] [PubMed] [Google Scholar]; (b) Wertz S; Studer A Green Chem 2013, 15, 3116. [Google Scholar]; (c) Nutting JE; Rafiee MR; Stahl SS Chem. Rev 2018, 118, 4834. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Leadbeater NE; Bobbitt J Aldrichim. Acta 2014, 47, 65. [Google Scholar]
- (3).Greszta D; Matyjaszewski K Macromolecules 1996, 29, 7661. [Google Scholar]
- (4).Kato Y; Shimizu Y; Yijing L; Unoura K; Utsumi H; Ogata T Electrochim. Acta 1995, 40, 2799. [Google Scholar]
- (5).(a) For some examples, see: Anelli PL; Biffi C; Montanari F; Quici S J. Org. Chem 1987, 52, 2559. [Google Scholar]; (b) Hoover J; Stahl SS J. Am. Chem. Soc 2011, 133, 16901. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hoover JM; Ryland BL; Stahl SS J. Am. Chem. Soc 2013, 135, 2357. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bailey WF; Bobbitt JM J. Org. Chem 2007, 72, 4504. [DOI] [PubMed] [Google Scholar]; (e) Ganem BJ Org. Chem 1975, 40, 1998. [Google Scholar]; (f) Hickey D; Milton R; Chen D; Sigman M; Minteer S ACS Catal 2015, 5, 5519. [Google Scholar]; (g) Semmelhack MF; Chou CS; Cortes DA J. Am. Chem. Soc 1983, 105, 4492. [Google Scholar]; (h) de Nooy AEJ; Besemer AC Tetrahedron 1995, 51, 8023. [Google Scholar]; (i) Bobbitt JM; Brückner C; Merbouch N Org. React 2009, 74, 103. [Google Scholar]
- (6).The oxidation of alcohol can also go through a single-electron pathway relying on the TEMPO/TEMPO− redox cycle in the presence of a copper catalyst. See: Badalyan A; Shannon SS Nature 2016, 535, 406 A mechanistic study shows that this reaction is mediated by a [Cu]–TEMPO complex in which the bound TEMPO behaves like TEMPO+. [DOI] [PubMed] [Google Scholar]; See: Walroth RC; Miles KC; Lukens JT; MacMillan SN; Stahl SS; Lancaster KM J. Am. Chem. Soc 2017, 139, 13507. [DOI] [PubMed] [Google Scholar]
- (7).(a) For examples of enamine addition to a TEMPO+ equivalent (Lewis acid-activated TEMPO), see: Van Humbeck J; Simonovich S; Knowles R; MacMillan DWC J. Am. Chem. Soc 2010, 132, 10012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Koike T; Akita M Chem. Lett 2009, 38, 166. [Google Scholar]; (c) Sibi MP; Hasegawa MJ Am. Chem. Soc 2007, 129, 4124. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) For examples of amine oxidation, see: Semmelhack MF; Schmid CR J. Am. Chem. Soc 1983, 105, 6732. [Google Scholar]; (e) Sonobe T; Oisaki K; Kanai M Chem. Sci 2012, 3, 3249. [Google Scholar]; (f) Wu Y; Yi H; Lei A ACS Catal 2018, 8, 1192. [Google Scholar]
- (8).(a) Babiarz JE; Cunkle GT; DeBellis AD; Eveland D; Pastor SD; Shum SP J. Org. Chem 2002, 67, 6831. [DOI] [PubMed] [Google Scholar]; (b) Coseri S; Ingold KU Org. Lett 2004, 6, 1641. [DOI] [PubMed] [Google Scholar]
- (9).(a) Beckwith ALJ; Bowry VW; Ingold KU J. Am. Chem. Soc 1992, 114, 4983. [Google Scholar]; (b) Sobek J; Martschke R; Fischer H J. Am. Chem. Soc 2001, 123, 2849. [DOI] [PubMed] [Google Scholar]; (c) Gentry EC; Rono LJ; Hale ME; Matsuura R; Knowles RR J. Am. Chem. Soc 2018, 140, 3394. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xu F; Zhu L; Zhu S; Yan X; Xu H-C Chem. - Eur. J 2014, 20, 12740. [DOI] [PubMed] [Google Scholar]; (e) Fuller PH; Kim J-W; Chemler SR J. Am. Chem. Soc 2008, 130, 17638. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) For a related work on reversible Ti radical capture by TEMPO, see: Huang K-W; Han JH; Musgrave CB; Waymouth RM Organometallics 2006, 25, 3317. [Google Scholar]
- (10).Nicolas J; Guillaneuf Y; Lefay C; Bertin D; Gigmes D; Charleux B Prog. Polym. Sci 2013, 38, 63. [Google Scholar]
- (11).(a) Maji MS; Pfeifer T; Studer A Angew. Chem., Int. Ed 2008, 47, 9547. [DOI] [PubMed] [Google Scholar]; (b) Vogler T; Studer A Org. Lett 2008, 10, 129. [DOI] [PubMed] [Google Scholar]; (c) Lam PYS; Vincent G; Clark CG; Deudon S; Jadhav PK Tetrahedron Lett 2001, 42, 3415. [Google Scholar]; (d) Liwosz T; Chemler SR Synlett 2015, 26, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Li Y; Studer A Angew. Chem., Int. Ed 2012, 51, 8221. [DOI] [PubMed] [Google Scholar]
- (13).(a) Krishna M; Grahame D; Samuni A; Mitchell J; Russo A Proc. Natl. Acad. Sci. U. S. A 1992, 89, 5537. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) TEMPO+/TEMPO has been used for the single-electron oxidation of superoxide and reduction of peroxyl. See:Griesser M; Shah R; Van Kessel AT; Zilka O; Haidasz EA; Pratt DA J. Am. Chem. Soc 2018, 140, 3798 TEMPO+ has also been proposed to mediate a radical cyclization reaction (ref 9d) via a single-electron pathway, although little evidence was provided to support such a mechanism. [DOI] [PubMed] [Google Scholar]
- (14).(a) For examples, see: Knoop CA; Studer A J. Am. Chem. Soc 2003, 125, 16327. [DOI] [PubMed] [Google Scholar]; (b) Hawker CJ; Bosman AW; Harth E Chem. Rev 2001, 101, 3661. [DOI] [PubMed] [Google Scholar]; (c) Puts RD; Sogah DY Macromolecules 1996, 29, 3323. [Google Scholar]; (d) Moad G; Rizzardo E Macromolecules 1995, 28, 8722. [Google Scholar]
- (15).Fu N; Sauer G; Saha A; Loo A; Lin S Science 2017, 357, 575. [DOI] [PubMed] [Google Scholar]
- (16).(a) For representative reviews on synthetic electrochemistry, see: Moeller KD Tetrahedron 2000, 56, 9527. [Google Scholar]; (b) Yoshida J; Kataoka K; Horcajada R; Nagaki A Chem. Rev 2008, 108, 2265. [DOI] [PubMed] [Google Scholar]; (c) Francke R; Little RD Chem. Soc. Rev 2014, 43, 2492. [DOI] [PubMed] [Google Scholar]; (d) Yan M; Kawamata Y; Baran PS Chem. Rev 2017, 117, 13230. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Möhle S; Zirbes M; Rodrigo E; Gieshoff T; Wiebe A; Waldvogel SR Angew. Chem., Int. Ed 2018, 57, 6018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Mitsudo K; Kurimoto Y; Yoshioka K; Suga S Chem. Rev 2018, 118, 5985–5999. [DOI] [PubMed] [Google Scholar]; (g) Tang S; Liu Y; Lei A Chem 2018, 4, 27. [Google Scholar]
- (17).(a) For a review, see: Bergmeier SC Tetrahedron 2000, 56, 2561. [Google Scholar]; (b) Liu G; Stahl SS J. Am. Chem. Soc 2006, 128, 7179. [DOI] [PubMed] [Google Scholar]; (c) Lu D-F; Zhu C-L; Jia Z-X; Xu H J. Am. Chem. Soc 2014, 136, 13186. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hemric BN; Shen K; Wang Q J. Am. Chem. Soc 2016, 138, 5813. [DOI] [PubMed] [Google Scholar]; (e) Sun X; Li X; Song S; Zhu Y; Liang Y-F; Jiao N J. Am. Chem. Soc 2015, 137, 6059. [DOI] [PubMed] [Google Scholar]; Also see ref 9e.
- (18).Zhang B; Studer A Org. Lett 2013, 15, 4548. [DOI] [PubMed] [Google Scholar]
- (19).(a) For examples, see: Bruncko M; Schlingloff G; Sharpless KB Angew. Chem., Int. Ed. Engl 1997, 36, 1483. [Google Scholar]; (b) Michaelis DJ; Shaffer CJ; Yoon TP J. Am. Chem. Soc 2007, 129, 1866. [DOI] [PubMed] [Google Scholar]; (c) Williamson KS; Yoon TP J. Am. Chem. Soc 2010, 132, 4570. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Intramolecular examples: Hemric BN; Shen K; Wang QJ Am. Chem. Soc 2016, 138, 5813. [DOI] [PubMed] [Google Scholar]; (e) Fuller PH; Kim JW; Chemler SR J. Am. Chem. Soc 2008. 130, 17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Muñiz K Chem. Soc. Rev 2004, 33, 166–174. [DOI] [PubMed] [Google Scholar]
- (21).Newcomb M Tetrahedron 1993, 49, 1151 Comparison of these first-order and second-order rate constants can be made at a given reaction concentration (ca. 0.06 M). [Google Scholar]
- (22).(a) The potential of N3−/N3• has been measured in aqueous media. See: Ram S; Stanbury DM J. Phys. Chem 1986, 90, 3691. [Google Scholar]; (b) Alfassi ZB; Harriman A; Huie RE; Mosseri S; Neta P J. Phys. Chem 1987, 91, 2120. [Google Scholar]
- (23).(a) For examples, see: Hickey DP; McCammant MS; Giroud F; Sigman MS; Minteer SD J. Am. Chem. Soc 2014, 136, 15917. [DOI] [PubMed] [Google Scholar]; (b) Rafiee M; Miles KC; Stahl SS J. Am. Chem. Soc 2015, 137, 14751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) TEMPO has been used for the generation of azidyl radical via different mechanisms. See: Wang T; Jiao N J. Am. Chem. Soc 2013, 135, 11692. [DOI] [PubMed] [Google Scholar]; (b) Magnus P; Lacour J; Evans PA; Roe MB; Hulme C J. Am. Chem. Soc 1996, 118, 3406. [Google Scholar]; Also see ref 18.
- (25).The formation of an adduct between TEMPO and azidyl was postulated as a side reaction of the alkene functionalization in ref 18.
- (26).(a) A charge-transfer complex between TEMPO+ and ClO2− was reported in the context of alcohol oxidation: Furukawa K; Shibuya M; Yamamoto Y Org. Lett 2015, 17, 2282. [DOI] [PubMed] [Google Scholar]; (b) Charge-transfer complexes between TEMPO and quinone derivatives have also been described: Nakatsuji S; Takai A; Nishikawa K; Morimoto Y; Yasuoka N; Suzuki K; Enokid T; Anzai H Chem. Commun 1997, 275. [Google Scholar]
- (27).These experimentally measured values are consistent with literature report. See: Hase Y; Ito E; Shiga T; Mizuno F; Nishikoori H; Iba H; Takechi K Chem. Commun 2013, 49, 8389. [DOI] [PubMed] [Google Scholar]
- (28).This spectroscopic feature at 380 nm was observed in a previous study when azidyl generated from pulse radiolysis was mixed with TEMPO, although the speciation was not discussed in this previous study. See: Samuni A; Goldstein S; Russo A; Mitchell J; Krishna M; Neta P J. Am. Chem. Soc 2002, 124, 8719. [DOI] [PubMed] [Google Scholar]
- (29).(a) Hayon E; Simic M J. Am. Chem. Soc 1970, 92, 7486. [DOI] [PubMed] [Google Scholar]; (b) Workentin M; Wagner B; Negri F; Zgierski M; Lusztyk J; Siebrand W; Wayner D J. Phys. Chem 1995, 99, 94. [Google Scholar]
- (30).Although the decay of adduct I should in theory follow the first-order behavior, the generation of TEMPO, which inhibits the reaction (see section below), results in deviation from first-order reaction kinetics. The half-life reported here gives a rough estimate of the stability of the complex.
- (31).(a) For examples, see: Wang X; Ye Y; Zhang S; Feng J; Xu Y; Zhang Y; Wang J J. Am. Chem. Soc 2011, 133, 16410. [DOI] [PubMed] [Google Scholar]; (b) Türkyilmaz F; Kehr G; Li J; Daniliuc C; Tesch M; Studer A; Erker G Angew. Chem., Int. Ed 2016, 55, 1470. [DOI] [PubMed] [Google Scholar]; Also see refs 5 and 12.
- (32).(a) For examples of B3LYP/def2-TZVP being used in the study of charge-transfer complexes, see: Marsh BM; Zhao J; Garand E Phys. Chem. Chem. Phys 2015, 17, 25786. [DOI] [PubMed] [Google Scholar]; (b) Baumeier B; Andrienko D; Rohlfing MJ Chem. Theory Comput 2012, 8, 2790. [DOI] [PubMed] [Google Scholar]
- (33).(a) Steinmann SN; Piemontesi C; Delachat A; Corminboeuf C J. Chem. Theory Comput 2012, 8, 1629. [DOI] [PubMed] [Google Scholar]; (b) Bauzá A; Ramis R; Frontera A J. Phys. Chem. A 2014, 118, 2827. [DOI] [PubMed] [Google Scholar]
- (34).The extinction coefficient is highly dependent on the medium. As such, the computational data with the PCM solvation model can only provide an approximation for this value. It is further known that extinction coefficients are poorly predicted by default TDDFT methods. However, the ratio between experimental:experimental and calculated:calculated molar extinction coefficients for different UV peaks exhibiting a difference of less than a factor of 2 has been used as evidence supporting the accuracy of TDDFT UV–vis prediction. This is true of the results presented herein. For a relevant reference, see: Sundholm D Chem. Phys. Lett 1999, 302, 480. [Google Scholar]
- (35).DFT-predicted values are 8 M−1 cm−1 for TEMPO and 4 M−1 cm−1 for TEMPO+. See ref 25 for experimental data on the extinction coefficients of TEMPO and TEMPO+.
- (36).The NMR prediction was made using the gauge-independent atomic orbital (GIAO) method with the Tao–Perdew–Staroverov–Scuseria hybrid functional and a polarization continuum model. See SI for details.
- (37).(a) For some recent examples of metal-mediated azidyl transfer, see: Huang X; Bergsten T; Groves J J. Am. Chem. Soc 2015, 137, 5300. [DOI] [PubMed] [Google Scholar]; (b) Sharma A; Hartwig J Nature 2015, 517, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yuan Y-A; Lu D-F; Chen Y-R; Xu H Angew. Chem., Int. Ed 2016, 2, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]; Also see ref 15.
- (38).Under synthetic conditions, TEMPO–N3 is generated electrochemically in small doses, whereas TEMPO is in large access in comparison.
- (39).The kinetics of azidyl addition to alkenes has been studied using time-resolved UV–vis and laser flash photolysis. See: Workentin M; Wagner B; Lusztyk J; Wayner D J. Am. Chem. Soc 1995, 117, 119. [Google Scholar]
- (40).Hupp JT; Weaver MJ J. Phys. Chem 1984, 88, 1463. [Google Scholar]
- (41).(a) Siedlecka R; Skarzewski J Synlett 1996, 8, 757. [Google Scholar]; (b) Velusamy S; Kumar A; Saini R; Punniyamurthy T Tetrahedron Lett 2005, 46, 3819. [Google Scholar]; (c) Huang J; Li S; Wang Y Tetrahedron Lett 2006, 47, 5637. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.