

Epithelial cells work together to maintain a tight barrier, yet can turn over rapidly by cell division and death. To help accomplish this feat, epithelia eject cells fated to die by a process called cell extrusion. To extrude, a cell produces and emits the lipid sphingosine-1-phosphate (S1P), which binds to its cognate receptor S1P2 in cells neighboring it to form a intercellular, basolateral, contractile actomyosin ring that squeezes the cell out of the epithelium [1,2]. In this manner, cells triggered to undergo apoptosis [2,3] or, more commonly, supernumerary live cells that later die by anoikis [4], are eliminated without disrupting the barrier [3–7]. Here, we review current cell extrusion literature focusing on its mechanism and signaling, and also highlight emerging new roles for extrusion in driving cell competition and tumor suppression.
One of the most important roles for extrusion is to maintain constant epithelial cell densities by mechanically matching the number of cells that die to those that divide. In vertebrates, mechanical force links cell division with cell death, as crowding triggers apical extrusion of live cells through the stretch-activated channel Piezo1 [4]. Previous work suggested that crowding also drives live cells to basally extrude or delaminate in Drosophila notum [8]. However, recent work from Levayer et al. shows that crowding causes cells to first undergo apoptosis which, in turn, drives delamination [9]. Here, inhibition of the apoptotic pathway by diap1 or p35 overexpression or homozygous loss of hid, grim, and reaper blocks delamination in crowded regions. Additionally, rasV12-overexpressing cells crowd wildtype cells up to three cell diameters away and force their delamination, suggesting that overall tissue crowding causes cell delamination (Fig. 1c) [9]. Differential results in these two systems may be due to differences in the strength of promoters used to over-express DIAP [8,9]. In other epithelia at different times during Drosophila development, cells activate apoptosis before delaminating, suggesting that flies regulate extrusion of supernumary cells using different signaling pathways to vertebrates [10]. An important reason for this may be due to the fact that extrusion of live cells basally is risky if cells do not later die; for instance, cancer cells or stem cells may use this mechanism to escape their primary epithelial sites and invade or differentiate, respectively. In fact, live neuroblast cells delaminate from the neuroepithelium before becoming neurons [11]. Thus, activating cell death simultaneously with basal extrusion could ensure that supernumery cells are eliminated.
Figure 1. The driving forces on and fates of apoptotic and live extruding cells.
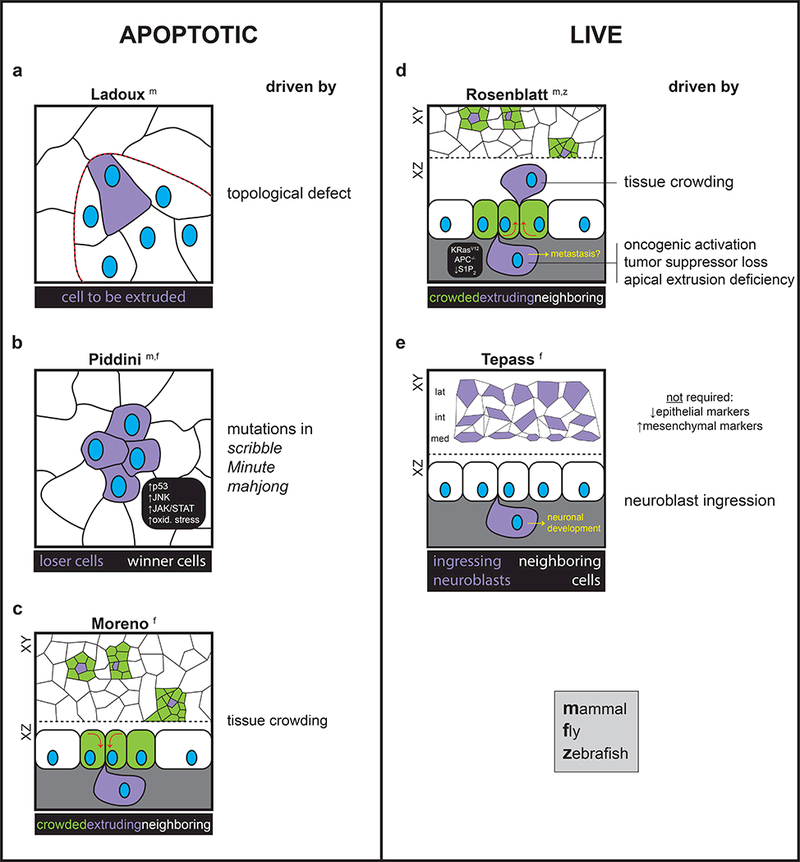
Depending on species, cell context, and neighboring cell status, cells bound to extrude can have several different fates. (a,b) Mammalian cells at the leading edge of a comet-shaped topological defect (a) or harboring “loser” mutations (b) are destined to die and extrude apically. (c) Conversely, cells in the fly notum that are to be eliminated experience crowding forces and delaminate (i.e. extrude basally). In this context, cells necessarily commit to apoptosis, in contrast to a previous report (ref. 4). (d) In mammalian cell cultures, mouse gut epithelia, and the developing zebrafish epidermis, cells that experience crowding forces extrude apically and later die by anoikis. Conversely, cells that harbor oncogenic mutations, lose tumor suppressors, or have deficiencies in apical extrusion machinery extrude basally and may live, revealing a novel path to metastasis. (e) Fly neuroblasts ingress (i.e. extrude basally; lateral, intermediate, and medial neuroblasts are shown) in an EMT-independent fashion, and later develop into mature neurons
While crowding within epithelia drives some cells to extrude, what causes a specific cell within a crowded region to extrude is not well understood. Several recent papers shed light on important signaling and mechanical forces that contribute to one cell extruding. Using Madin-Darby canine kidney (MDCK) epithelial cells grown to confluence on micropatterned, functionalized substrates, Saw et al. found that epithelial cells can behave like nematic liquid crystals (liquids comprised of molecules oriented in a crystal like pattern) that align along their long axes, with cell extrusions occur at sites of patterning defects to relieve cell strain (Fig. 1a) [12]. At these defect sites, extruding cells were not locally crowded but instead the result of perpendicular single-cell collisions. Using elegant experiments where they confined cell growth and movement by plating on matrices of different shapes, they could increase or decrease these defects and collisions, and the extrusion rates, accordingly. Sites of single-cell defects had increased cytoplasmic yes-associated protein (YAP) and caspase-3 activation [12], in contrast with live cell extrusions that result from whole epithelial sheet crowding [4,8]. Future work will need to examine if single-cell perpendicular collisions are at the heart of live cell extrusions in crowded regions of tissues, or if this represents another pathway to eliminate cells causing patterning defects in the otherwise regular epithelial fabric that coats organs.
Cell packing density can also result in different modes of extrusion. In a loosely packed environment, dynamic, large-scale cell movements lead to transient increases in local cell density where extrusions occur [13]. At lower densities, some cells can extrude by their neighboring cells’ crawling into replace where a cell exits. Although epithelia in vivo exist at high density, in cases where they experience wounds or excessive cell death, this different mechanism would ensure that no gaps form in the epithelial barrier. As cells grow and density increases in mature epithelia, cell movement decreases and the numbers and rates of cell extrusions increase, this time occurring via formation and contraction of the actomyosin cable in surrounding cells, as described previously [2,13]. Here, recent work has found a role for coronin1B in reorganizing actomyosin in adherens junctions at steady-state and during apoptotic cell extrusion. Depletion of E-cadherin or coronin1B compromises the integrity of adherens junctions and consequently hampers cell extrusion [14].
Just as cell crowding due to excess cells can cause cell extrusion and death, cell stretch due to sparse cell populations can drive rapid cell division through a mechanism that also depends on Piezo1 [15]. Stretch, induced mechanically or by wounding a MDCK monolayer, triggers rapid cell division by Piezo1-dependent activation of a single spike of calcium, which in turn, activates ERK1/2 phosphorylation and cyclin B transcription in cells poised in G2. Loss of Piezo1 in MDCK cells or zebrafish markedly decreased cell divisions. While Piezo1 activates calcium to control the two opposing processes of cell division and extrusion, the type of force is critical for the outcome: stretch triggers cell division, not extrusion; and crowding triggers extrusion, not division (Fig. 1d). Interestingly, the threshold of mechanical strain is ~1.6-fold in both cases [4,15]. How Piezo1 activates calcium to control two opposing processes is not entirely clear; however, calcium activation alone is sufficient to drive cells poised in G2 to enter mitosis, whereas extrusion requires both calcium and crowding [15].
Neural development
While earlier work demonstrated that basal extrusion of cells within the Drosophila neuroectoderm gives rise to neuroblasts [16], little was known about the mechanism driving neuroblast delamination (or ingression). Recent work by Simões et al. shows that during this ingression, fly neuroblasts lose their apical surfaces anisotropically, with anterior-posterior junctions shrinking before dorsal-ventral junctions, due to polarized myosin distribution, causing a ratchet-like apical constriction of ingressing neuroblasts (Fig. 1e) [11]. Downregulation of epithelial markers such as DEcad and upregulation of mesenchymal markers such as Snail family members are not required for ingression, suggesting that classical mechanisms proposed for epithelial-to-mesenchymal transition (EMT) are dispensable for ingression. Lastly, they show that neuroblasts ingress non-autonomously: experimentally reducing tension in neighboring non-ingressing cells (NICs) by laser ablation of apical junctions hastens ingression, while increasing tension by converting NICs to neuroblasts through Delta or Notch depletion delays it [11].
Cell competition
Cell competition is a process whereby fit cells eliminate less fit cells to improve tissue health. While the genetic drivers of this process have been known for over 40 years, the mechanism for this process has eluded researchers. Now, recent studies suggest that cell extrusion could be a conserved mechanism for removing loser cells during cell competition. Chiba et al. showed that constitutively active YAP mutant cells act as losers, which get apically extruded by surrounding wildtype cells [17]. The TEAD and PDZ domains of YAP, necessary for nuclear localization and gene activation, are required for this phenotype [18–20]. Interestingly, genetic or chemical inhibition of vimentin abrogates the ability of the neighboring cells to extrude YAP-expressing cells, suggesting a role for intermediate filaments in measuring cell fitness. Finally, using co-cultures of different oncogene-expressing or wildtype cells, the authors determined an increasing order of competitiveness, thus: v-Src < KRasV12 < YAP (5SA) < wildtype [17].
Additionally, MDCK cells depleted of the cell polarity gene scribble (scribKD) lose to wildtype cells through extrusion in response to mechanical forces, rather than soluble factors. Wagstaff et al. showed that scribKD cells grow to a lower density threshold than wild type cells: on their own they reach a sparser, less compacted steady state density than wild type cells; however, when grown mosaically with wild type cells, they become compacted and extrude (Fig. 1b) [21]. In these cases, wildtype cells corral and then compact scribKD cells in an E-cadherin-dependent manner. Compaction activates p53 transcription in scribKD cells, which is necessary and sufficient for their apoptotic extrusion. p53 induction is ROCK- and p38 kinase-dependent, demonstrating a new mechanical mechanism for activating p53 [21].
Other work from the same group revealed a new role for oxidative stress pathways in loser cells in fly wing imaginal discs. In two different classes of loser cells, either ribosomal mutant alleles of Minute or the cell polarity-associated complex component mahjong, cells experience oxidative stress and an increase of JNK and JAK/STAT activation (Fig. 1b). During competition, the JNK pathway restricts the growth of prospective loser cells whereas JAK/STAT activation drives rapid growth in winner cells. Oxidative stress confers loser status via transcription mediated through Nrf2 [22]. Interestingly, a fly suppressor screen for scrib deficiency-induced loser status identified the Slit-Robo pathway, known for its role in angiogenesis and axon guidance. Vaughen and Igaki showed that indeed deficiencies in slit, robo or ena rescued elimination of scrib−/− eye disc cells, and that the Slit-Robo pathway acts downstream of JNK pathway activation, adding further resolution of the signaling pathway controlling cell loss [23].
Dysregulation of extrusion in disease
Brain defects from excess extrusion.
Germline knockout of Alix leads to mice with hydrocephaly, smaller cerebrum and hippocampus, and poor epithelial barrier function within the choroid plexus, causing loss of cerebral spinal fluid, compared to control. Further inspection of the choroid plexus epithelia suggests that the poor barrier results from high levels of apical extrusion accompanied by extensive blebbing of the microvilli and abnormal cilia. The extrusion defects and poor barrier of Alix−/− mice appear to result from aberrant actomyosin assembly and mislocalization of tight junction proteins at the basolateral membrane, which disrupts proper reformation of cell-cell junctions post-extrusion [24].
Aberrant extrusion and cancer.
In cells where certain oncogenes are induced, a program called epithelial defense against cancer (EDAC) causes their expulsion through a process that resembles extrusion but is driven by a separate signaling and mechanism than that occurring in wild-type cells [25–27]. EDAC is an important tumor suppressor mechanism similar to cell competition that can help preserve tissue fitness by eliminating cells with aberrant growth signaling. However, other types of oncogenic mutations can persist in cells and instead drive tumor formation and progression, mainly by switching their direction of extrusion.
As described above, most epithelial cells in vertebrates extrude apically while still alive, whereas in Drosophila, cells trigger extrusion basally as they undergo apoptosis, except when delaminating stem cells (Fig. 1). Importantly, oncogenic or tumor suppressor mutations can hijack the direction that cells extrude in both vertebrates and in flies. Tamori et al. showed that fly wing disc cells without tumor suppressor genes like lgl or scrib form tumors—but only in “hotspots,” where JAK/STAT levels are high and structures within the basal membrane and underlying ECM prevent basal extrusion and apoptosis. Instead, cells extrude apically and accumulate into tumor-like masses [28]. Strikingly, RhoGEF2, the fly ortholog of p115 RhoGEF, necessary for apical extrusion in mammals [25], is critical for basal extrusion in flies: when it and the tumor suppressors are absent, ectopic JAK/STAT activation drives apical extrusion and tumorigenesis [28].
Similarly, basal extrusion can be exploited by oncogenic mechanisms to promote cancer in vertebrates, enabling transformed cells to invade and metastasize throughout the body [29]. Pancreatic cancers unilaterally downregulate S1P2, the receptor required for apical extrusion (Fig. 1d) [30]. In pancreatic cancer cells, or in cell lines or zebrafish lacking S1P2, cells that cannot extrude apically instead form cell masses that are chemo-resistant, cause poor epithelial barriers, and instead basally extrude. Moreover, labeled pancreatic cancer cells grown orthotopically in nude mice form large metastatic tumors. Rescue of S1P2 in these cells is sufficient to abrogate both tumor growth and metastases [30].
Two other mutations that drive aggressive tumor types, adenomatous polyposis coli (APC) truncation and KRasV12, also hijack apical extrusion (Fig. 1d). During apical extrusion, APC and microtubules reorient basally, targeting p115 RhoGEF-mediated Rho activation to contract actomyosin basally and extrude cells apically, disrupts this microtubule targeting, causing actin already at apical junctions to contract and extrude cells basally [31,32]. KrasV12, an oncogenic KRas mutation that drives a class of aggressive tumors (pancreatic, lung, and colon cancers [33]) shifts extrusion from apical to basal by degrading the S1P ligand of the S1P2 receptor via increased autophagy. Simply blocking autophagy either genetically or with chloroquine rescues S1P accumulation and apical cell extrusion, suggesting a potential therapeutic approach for shifting cells from a potentially invasive fate to an apoptotic one [34]. Moreover, p120 catenin may also contribute to this aberrant extrusion signaling [35]. Hendley et al. showed that human pancreatic tumors express low levels of p120 catenin, compared to PanIN lesions. Deletion of p120 catenin in a KRasD12- pancreatic cancer mouse model reduces S1P signaling via upregulation of the NF-κΒ pathway, resulting in poorer survival, more basally extruded cells and increased barrier function defects and inflammation [35]. These studies suggest that invasion of tumor cells may depend on mechanisms independent of classically inferred EMT signaling. Additionally, EMT is also dispensable for lung and pancreatic cancer metastasis in mice [36,37]. Future work will need to determine if basally extruded cells drive the distant metastases linked to the increased mortality (Fig. 1d).
In summary, recent studies have highlighted new ways that mechanical forces can drive signaling important for both live and apoptotic cell extrusion. It is unclear why vertebrates tend to extrude cells apically while those in developing Drosophila do so basally. However, Drosophila genetics could offer further insight to our understanding of the signaling that drives basal extrusion, a point on which little is known. It is particularly noteworthy that shifting the direction that cells normally extrude can impact malignancy in both Drosophila and vertebrates. Interestingly, the finding that basal extrusion can cause invasion of both tumor and stem cells in ways that do not implicate normal EMT mechanisms suggest new horizons for our understanding of both metastasis and development. Further work will need to define if the two processes are mutually exclusive and their exact contributions to cancer progression.
Acknowledgements:
An National Institute of Health R01GM102169 and Howard Hughes Faculty Scholar Award 55108560 to J. R. supported this work.
Footnotes
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gu Y, Forostyan T, Sabbadini R, Rosenblatt J: Epithelial cell extrusion requires the sphingosine- 1-phosphate receptor 2 pathway. J Cell Biol 2011, 193:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenblatt J, Raff MC, Cramer LP: An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol 2001, 11:1847–1857. [DOI] [PubMed] [Google Scholar]
- 3.Andrade D, Rosenblatt J: Apoptotic regulation of epithelial cellular extrusion. Apoptosis 2011, 16:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, Rosenblatt J: Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 2012, 484:546–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frisch SM, Francis H: Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 1994, 124:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilmore AP, Owens TW, Foster FM, Lindsay J: How adhesion signals reach a mitochondrial conclusion--ECM regulation of apoptosis. Curr Opin Cell Biol 2009, 21:654–661. [DOI] [PubMed] [Google Scholar]
- 7.Reddig PJ, Juliano RL: Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev 2005, 24:425–439. [DOI] [PubMed] [Google Scholar]
- 8.Marinari E, Mehonic A, Curran S, Gale J, Duke T, Baum B: Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding. Nature 2012, 484:542–545. [DOI] [PubMed] [Google Scholar]
- 9.**.Levayer R, Hauert B, Moreno E: Cell mixing induced by myc is required for competitive tissue invasion and destruction. Nature 2015. In agreement with a previous report (ref. 4), this study shows that tissue crowding in the fly notum is sufficient to induce delamination. However, here the authors show conclusively that caspase activation precedes and is required for delamination. [DOI] [PubMed] [Google Scholar]
- 10.Muliyil S, Krishnakumar P, Narasimha M: Spatial, temporal and molecular hierarchies in the link between death, delamination and dorsal closure. Development 2011, 138:3043–3054. [DOI] [PubMed] [Google Scholar]
- 11.**.Simoes S, Oh Y, Wang MFZ, Fernandez-Gonzalez R, Tepass U: Myosin II promotes the anisotropic loss of the apical domain during Drosophila neuroblast ingression. J Cell Biol 2017, 216:1387–1404. The authors show that ingressing fly neuroblasts lose apical surface anisotropically, with myosin driving ratchet-like contractions. Additionally, ingression does not require classic EMT signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.**.Saw TB, Doostmohammadi A, Nier V, Kocgozlu L, Thampi S, Toyama Y, Marcq P, Lim CT, Yeomans JM, Ladoux B: Topological defects in epithelia govern cell death and extrusion. Nature 2017, 544:212–216. This report demonstrates that epithelia can act as nematic liquid crystals, where stochastic patterning defects cause apical extrusions to correct the regular patterning. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kocgozlu L, Saw TB, Le AP, Yow I, Shagirov M, Wong E, Mege RM, Lim CT, Toyama Y, Ladoux B: Epithelial Cell Packing Induces Distinct Modes of Cell Extrusions. Curr Biol 2016, 26:2942–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michael M, Meiring JCM, Acharya BR, Matthews DR, Verma S, Han SP, Hill MM, Parton RG, Gomez GA, Yap AS: Coronin 1B Reorganizes the Architecture of F-Actin Networks for Contractility at Steady-State and Apoptotic Adherens Junctions. Dev Cell 2016, 37:58–71. [DOI] [PubMed] [Google Scholar]
- 15.**.Gudipaty SA, Lindblom J, Loftus PD, Redd MJ, Edes K, Davey CF, Krishnegowda V, Rosenblatt J: Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature 2017, 543:118–121. Mechanical stretch drives mitosis through the stretch-activated ion channel Piezo1, which is also responsible for driving crowding-induced apical extrusion (ref. 3). Piezo1 is, thus, a master regulator of homeostatic cell numbers in epithelia, depending on the type of force it measures. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartenstein V, Younossi-Hartenstein A, Lekven A: Delamination and division in the Drosophila neurectoderm: spatiotemporal pattern, cytoskeletal dynamics, and common control by neurogenic and segment polarity genes. Dev Biol 1994, 165:480–499. [DOI] [PubMed] [Google Scholar]
- 17.*.Chiba T, Ishihara E, Miyamura N, Narumi R, Kajita M, Fujita Y, Suzuki A, Ogawa Y, Nishina H: MDCK cells expressing constitutively active Yes-associated protein (YAP) undergo apical extrusion depending on neighboring cell status. Sci Rep 2016, 6:28383 This report demonstrates that cells expressing constitutively active YAP act as losers and are apically extruded by their wildtype neighbors, and that cells follow a hierarchy of loser/winner phenotype depending on the oncogene expressed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oka T, Remue E, Meerschaert K, Vanloo B, Boucherie C, Gfeller D, Bader GD, Sidhu SS, Vandekerckhove J, Gettemans J, et al. : Functional complexes between YAP2 and ZO-2 are PDZ domain-dependent, and regulate YAP2 nuclear localization and signalling. Biochem J 2010, 432:461–472. [DOI] [PubMed] [Google Scholar]
- 19.Oka T, Sudol M: Nuclear localization and pro-apoptotic signaling of YAP2 require intact PDZ- binding motif. Genes Cells 2009, 14:607–615. [DOI] [PubMed] [Google Scholar]
- 20.Shimomura T, Miyamura N, Hata S, Miura R, Hirayama J, Nishina H: The PDZ-binding motif of Yes-associated protein is required for its co-activation of TEAD-mediated CTGF transcription and oncogenic cell transforming activity. Biochem Biophys Res Commun 2014, 443:917–923. [DOI] [PubMed] [Google Scholar]
- 21.**.Wagstaff L, Goschorska M, Kozyrska K, Duclos G, Kucinski I, Chessel A, Hampton-O’Neil L, Bradshaw CR, Allen GE, Rawlins EL, et al. : Mechanical cell competition kills cells via induction of lethal p53 levels. Nat Commun 2016, 7:11373 The authors report that scrib-depleted cells are corralled and compressed by their wildtype neighbors, leading to their apical extrusion and death. Further, mechanical activation of p53 in loser cells is both required and sufficient for elimination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.*.Kucinski I, Dinan M, Kolahgar G, Piddini E: Chronic activation of JNK JAK/STAT and oxidative stress signalling causes the loser cell status. Nat Commun 2017, 8:136. The authors report that Minute and mahjong mutations cause upregulation in the JNK, JAK/STAT, and oxidative stress pathways and that these pathways potentiate loser status. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaughen J, Igaki T: Slit-Robo Repulsive Signaling Extrudes Tumorigenic Cells from Epithelia. Dev Cell 2016, 39:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campos Y, Qiu X, Gomero E, Wakefield R, Horner L, Brutkowski W, Han YG, Solecki D, Frase S, Bongiovanni A, et al. : Alix-mediated assembly of the actomyosin-tight junction polarity complex preserves epithelial polarity and epithelial barrier. Nat Commun 2016, 7:11876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogan C, Dupre-Crochet S, Norman M, Kajita M, Zimmermann C, Pelling AE, Piddini E, Baena- Lopez LA, Vincent JP, Itoh Y, et al. : Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol 2009, 11:460–467. [DOI] [PubMed] [Google Scholar]
- 26.Kajita M, Fujita Y: EDAC: Epithelial defence against cancer-cell competition between normal and transformed epithelial cells in mammals. J Biochem 2015, 158:15–23. [DOI] [PubMed] [Google Scholar]
- 27.Kajita M, Sugimura K, Ohoka A, Burden J, Suganuma H, Ikegawa M, Shimada T, Kitamura T, Shindoh M, Ishikawa S, et al. : Filamin acts as a key regulator in epithelial defence against transformed cells. Nat Commun 2014, 5:4428. [DOI] [PubMed] [Google Scholar]
- 28.*.Tamori Y, Suzuki E, Deng WM: Epithelial Tumors Originate in Tumor Hotspots, a Tissue- Intrinsic Microenvironment. PLoS Biol 2016, 14:e1002537 The authors reveal that fly epithelia contain tumor “hotspots,” where rigid basal membrane and ECM and active JAK/STAT signaling promote apical extrusion, accumulation, and tumor formation in cells that have lost tumor suppressors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slattum GM, Rosenblatt J: Tumour cell invasion: an emerging role for basal epithelial cell extrusion. Nat Rev Cancer 2014, 14:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.**.Gu Y, Shea J, Slattum G, Firpo MA, Alexander M, Mulvihill SJ, Golubovskaya VM, Rosenblatt J: Defective apical extrusion signaling contributes to aggressive tumor hallmarks. Elife 2015, 4:e04069 Disruption of apical extrusion by loss of S1P2 receptor, as occurs in all pancreatic cancers, causes cell masses, poor barrier function and an increase in basal extrusion, which may drive the invasiveness of these tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slattum G, McGee KM, Rosenblatt J: P115 RhoGEF and microtubules decide the direction apoptotic cells extrude from an epithelium. J Cell Biol 2009, 186:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall TW, Lloyd IE, Delalande JM, Nathke I, Rosenblatt J: The tumor suppressor adenomatous polyposis coli controls the direction in which a cell extrudes from an epithelium. Mol Biol Cell 2011, 22:3962–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernandez-Medarde A, Santos E: Ras in cancer and developmental diseases. Genes Cancer 2011, 2:344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slattum G, Gu Y, Sabbadini R, Rosenblatt J: Autophagy in oncogenic K-Ras promotes basal extrusion of epithelial cells by degrading S1P. Curr Biol 2014, 24:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.**.Hendley AM, Wang YJ, Polireddy K, Alsina J, Ahmed I, Lafaro KJ, Zhang H, Roy N, Savidge SG, Cao Y, et al. : p120 Catenin Suppresses Basal Epithelial Cell Extrusion in Invasive Pancreatic Neoplasia. Cancer Res 2016, 76:3351–3363. The authors report that p120 catenin is downregulated in human pancreatic cancers. p120 catenin deletion in a KRasD12 pancreatic cancer mouse model leads to decreased S1P signaling and increased basal extrusion, both dependent on NF-kB signaling. p120−/− mice in this model also have increased formation of premalignant PanIN lesions, cachexia, and a dramatic decrease in survival. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al. : Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527:472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R: Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527:525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]