

Abstract
Mechanical ventilation applies physical stresses to the tissues of the lung and thus may give rise to ventilator-induced lung injury (VILI), particular in patients with acute respiratory distress syndrome (ARDS). The most dire consequences of VILI result from injury to the blood-gas barrier. This allows plasma-derived fluid and proteins to leak into the airspaces where they flood some alveolar regions, while interfering with the functioning of pulmonary surfactant in those regions that remain open. These effects are reflected in commensurately increased values of dynamic lung elastance (EL), a quantity that in principle is readily measured at the bedside. Recent mathematical/computational modeling studies have shown that the way in which EL varies as a function of both time and positive end-expiratory pressure (PEEP) reflects the nature and degree of lung injury, and can even be used to infer the separate contributions of volutrauma and atelectrauma to VILI. Interrogating such models for minimally injurious regimens of mechanical ventilation that apply to a particular lung may thus lead to personalized approaches to the ventilatory management of ARDS.
Keywords: Over-distension, recruitment and derecruitment, computational model, surfactant function, acute respiratory distress syndrome (ARDS)
Introduction
Ventilator-induced lung injury (VILI) represents an ever-present danger for patients in respiratory failure, particular those with acute respiratory distress syndrome (ARDS) (1-3). This danger arises from the fact that mechanical ventilation is, first and foremost, a mechanical event; whereas the normal healthy lung is well able to stand the stresses and strains imposed by mechanical ventilation, it can mean irreparable and eventually fatal further damage for the already injured lung. Such an unfortunate outcome, however, follows not only from the reduced physical durability of the injured lung as a result of some infectious or chemical insult. Lung injury is invariably accompanied by alterations in the mechanical properties of the lung itself (4-6). Consequently, the microscale stresses applied to the parenchyma during ventilation of the injured lung are frequently greatly elevated compared to those experienced by the normal lung (7,8), with commensurate consequences for tissue integrity and the biologic processes that are invoked in the face of injury.
By the same token, altered lung mechanics can be highly indicative of, and indeed specific to, the nature and degree of lung injury (4). This means that ongoing monitoring of lung mechanical function during mechanical ventilation has the potential to warn caregivers of impending VILI, and even to serve as the feedback signal by which ventilation can be regulated so as to minimize VILI progression while continuing to provide life support (9). In order to serve such a function, however, it is necessary to have an understanding of the link between the stresses and strains of mechanical ventilation, reflected in measurable pressures and flows at the airway opening, and the underlying VILI mechanisms. Accordingly, the goal of this review is to bring these factors together in a way that provides a unifying framework for potentially optimizing the use of mechanical ventilation in managing the injured lung.
Alterations of lung mechanics in VILI
The lung is, by functional necessity, a readily deformable structure that expands to transport air along a branching airway system from the narrow tracheal opening to the enormous surface area of the blood-gas barrier. In so doing, the pressures generated to produce inspiration must overcome the flow resistance of the airway tree and the viscoelastic recoil of the parenchymal tissue and chest wall. A significant component of this recoil arises a consequence of surface tension forces at the alveolar air-liquid interface (10). The balance of forces involved is conventionally written in terms of a simple equation of motion based on the notion that, on the macroscale, the lung behaves like a single compartment:
| [1] |
where P(t) is driving pressure relative to that at functional residual capacity (FRC), is flow at the tracheal opening, V(t) is lung volume relative to FRC, RL is lung resistance (which includes both airway and tissue resistances), and EL is lung elastance (the inverse of lung compliance). The lung is, of course, vastly more complicated than this. Nevertheless, the simple model expressed by Eq. [1] captures much of its overall dynamic mechanical behavior during both normal spontaneous respiration and mechanical ventilation (11). From the perspective of lung mechanics, the most important consequences of VILI are reflected in its effects on EL, which is a measure of the elastic stiffness of the lung parenchyma. It must be remembered, however, that the tissues of the lung are highly viscoelastic, which means that a substantial (if not the major) component of RL arises from viscous dissipation of energy within the tissues of the respiratory system as opposed to streams of gas flowing along airway conduits (11).
There are a variety of microscale structures that contribute to the determination of organ-scale EL, and that can become altered in acute lung injury. One is the network of extracellular matrix proteins (mostly collagen and elastin) depicted in Figure 1, which provide the parenchyma with its structural integrity (12,13) and which can become stiffened as a result of the fibroproliferation that is a common sequellum of ARDS (14-16). This network occupies the interstitium of the blood-gas barrier, which is located between the endothelial and epithelial cell layers that provide most of the barrier function separating blood from the alveolar airspace. Also in this interstitial space are various cell types and fluid. Expansion of the fluid volume is a ubiquitous occurrence in the injured lung because blood plasma, often with associated plasma proteins, moves across the leaky endothelium to produce interstitial pulmonary edema (17). The resulting congestion of the interstitial space can lead to a stiffening of the parenchyma, and thus an elevation in EL, but studies of experimental hydrostatic edema have shown that this elevation, while certainly measureable, is relatively modest in clinical terms (18).
Figure 1.
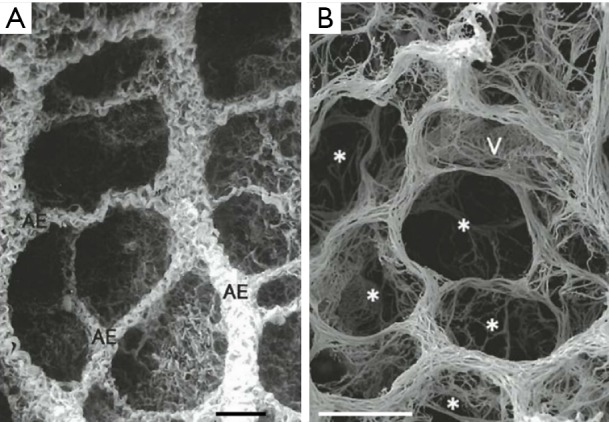
Scanning electron micrograph of the collagen fiber network in a rat lung (A) showing the alveolar entrances (AE) and helical collagen structure characteristic of low inflation levels. The elastic fibers network (B) crisscross at the point where three alveolar entrances (*) meet and are sparse in the alveolar septa. Reproduced with permission from Toshima et al. (12).
By far the most severe mechanical consequences of acute lung injury occur when edematous material arising from the vasculature breaches the epithelial barrier and accumulates in the airspaces. This potentially has two key consequences. One is simple displacement of air, which effectively reduces lung volume and so increases EL accordingly (19). The more important consequence, however, occurs when the edematous material interferes with the functioning of pulmonary surfactant in the aerated regions, which is readily accomplished by the plasma proteins via their competition at the air-liquid interface with surfactant (20-22). The resulting elevation in surface tension can dramatically increase EL and thus the pressure required to ventilate the lung. Elevated surface tension can also prevent inspiratory pressures from prying apart the walls of alveoli and airways that have come into apposition at the end of expiration (atelectasis) (23) or from eliminating plugs of fluid that have formed to occlude small airways (24-26). Lung regions that are isolated from ventilation in this way are said to be derecruited, and are thus unable to take part in gas exchange. Increased surface tension due to accumulation of plasma fluid and proteins thus increases EL both by causing derecruitment of lung units and by increasing the intrinsic stiffness of those units that remain open. The result is that the effective volume of the lung is reduced, so ventilation is delivered to what has been termed a ‘baby lung’ (19). The most important of these two effects, however, appears to be derecruitment, perhaps because lung injury is a heterogeneous process in which derecruitment occurs in those regions where surface tension is elevated while other regions are relatively spared.
The importance of derecruitment for ARDS and VILI is unequivocal, but what to do about it remains controversial. On the one hand, reversing derecruitment by applying larger inflation pressures to the lung would seem to make sense because this would increase the gas exchanging capacity of the lungs (4,27-29). On the other hand, increased inflation pressures have clear potential to cause physical damage to the lung tissues, particularly when the lung is already injured. Indeed, in some severely injured lungs, the regions that are derecruited, or flooded with edema, may be so recalcitrant to being reopened (30) that their recruitment would require pressures sufficient to cause immediate pneumothorax. Whether to try to recruit collapsed lung—the so-called “open lung approach”—or leave it derecruited is thus a key question that continues to vex the clinical community (31). Promising results with the open-lung approach in animal models have been obtained [e.g., (32)], but recent studies in patients have been inconclusive (33,34). It thus remains a puzzle as to why a strategy that appears to be based on such sound physiological principles should not be resoundingly successful in the clinic. Addressing this question begins with an understanding of the physical factors that determine recruitment and derecruitment.
The first-order view of recruitment and derecruitment is that they are functions of pressure (35-37). That is, a particular region of the lung, when closed, has a pressure, Po, above which it will open. Conversely, the same unit, when open, has a pressure, Pc, below which it will close. It is generally accepted that Pc < Po, implying there is a region of pressure, Pc < P < Po, within which closed lung will not open and open lung will not close. A distribution of values for PO and Pc throughout the lungs means that recruitment and derecruitment are continuous processes over some range of P (38,39). This gives rise to the notion that the pressures of mechanical ventilation should strive to exceed most of this range, or at least to exceed the range of values of Pc once the lung has been maximally recruited by a deep inflation (DI). This is not always possible since the ranges of Po and Pc may exceed pressures that can be safely applied, quite apart from the difficulties of reliably determining where these ranges lie for a given lung. Nevertheless, motivated by this notion, various strategies for identifying the “best positive end expiratory pressure (PEEP)” for a given injured lung have been devised based on analyses of measured pressure-volume relationships (40-43) or assessments of lung aeration from CT images (44,45). Such strategies have led to improvements in oxygenation, but the Holy Grail of reduced mortality has so far remained elusive (46).
One of the reasons why the clinical management of recruitment and derecruitment has been challenging may be that these phenomena are not, in fact, simply functions of pressure. They are also functions of time. In other words, once the pressure in a region of open lung descends to a level at which it may close, derecruitment does not happen instantaneously but rather manifests over some finite period of time. The reasons for this time dependence are complex in their intricate details but, broadly speaking, derecruitment involves fluid movement at the micro level. For example, the liquid layer coating the walls of a small airway is transported axially by a surface-tension induced Rayleigh-Plateau instability (47) into annular rings that eventually neck inward to occlude the airway lumen and draw the airway walls inward and expand the collapse region (48-52). Likewise, fluid displaced from a filled alveolus by inspired air takes time to flow back into and refill the alveolus once again. The processes involved in recruitment of closed lung units, which are roughly the reverse of those just described, also take time to manifest once pressures rise above the threshold at which recruitment can potentially occur. In vitro experiments and computational fluid dynamics have shown that the rates at which these processes take place are described by the dimensionless capillary number Ca = µU/γ that represents the balance between the viscosity of the lining fluid (µ), the surface tension at the air-liquid interface (γ), and the velocity (U) of a finger of air as it propagates along a collapsed tube (53-56). At the low Ca characteristic of the small airways and alveoli the surface tension forces dominate and viscous effects play a relatively minor role in the dynamics of recruitment and derecruitment.
The dynamic nature of recruitment and derecruitment means that the benefits of a recruitment maneuver are transient (27). The immediate benefits of recruiting the lung with a DI are evident in the reduction in EL that ensues. During a subsequent period of regular ventilation at a modest tidal volume, however, EL will gradually rise toward an elevated plateau. The level of this plateau and the rate at which it is approached are modest in a normal lung, but both can be dramatic in severe lung injury. In the latter case, studies using in vivo microscopy (57), lung stereology (58), and micro-CT in experimental animals (28) have shown that post-DI changes in EL correlate strongly and inversely with the fraction of open lung, while measurements of oxygenation support a similar conclusion in human subjects (59). It is not clear that derecruitment entirely explains the post-DI rise in EL in normal lungs, since this may also be due to transient changes in the distribution of surfactant and its surface tension lowering effects, or the gradual folding of alveolar septa (60), but the effect in this case is modest and thus unlikely to be of clinical importance. In any case, understanding the dynamics of recruitment and derecruitment is crucial for accurately predicting the long-term consequences of a given ventilation strategy, particularly if it involves recruitment maneuvers (61). The issue to be addressed regarding the use of recruitment maneuvers is thus not “if”, but “how often”.
Acute lung injury is invariably accompanied by increases in EL, which means that mechanical ventilation with a given minute ventilation results in commensurately increased stresses exerted on the parenchymal tissues. In other words, the same volume of inspired air is forced into an effectively smaller ‘baby lung’ (19), which leads to increased distension of the septal walls. This places the injured lung in danger of succumbing to a positive feedback cycle in which these elevated tissue stresses cause VILI, which worsens the stresses, which then accelerates the rate of injury development in a vicious cycle that is often ultimately fatal (62). There are currently no medical treatments for mitigating either the endothelial or epithelial leak that characterizes VILI, so the primary goal in managing the injured lung must be to ventilate in a way that minimizes VILI development in the hope that the patient’s own reparative machinery will prevail. Since EL is so dramatically affected by lung injury and its recruitment status, understanding what we can learn about VILI mechanisms from the way that EL changes over time in any given patient is crucial for designing optimal management strategies.
Making the most of EL as a marker of VILI requires some kind of mathematical model that allows it to be related in a quantitative fashion to relevant physiological mechanisms. At perhaps the simplest level, the amount of lung volume loss resulting from derecruitment can be equated inversely to the observed increase in EL (63). Exploiting this idea has led to a model of recruitment and derecruitment dynamics in which opening and closing of airspaces take place when pressure exceeds or falls below specified critical values, but with time delays that are accounted for using an empirical construct termed a virtual trajectory (37,38,61,64,65). Although not originally designed to represent any physical mechanism in particular, virtual trajectories are nevertheless reminiscent of the way that fluid plugs move along airways under the influence of an applied pressure until they have deposited enough of their material on the airway wall to finally disintegrate (25,66). In any case, models of distributions of airspaces built around this idea have been shown to describe experimental data rather accurately, and in particular to support the notion that increasing surface tension is the preeminent mechanical consequence of severe acute lung injury (38). Combined with representations of nonlinear tissue elasticity encompassing the idea of an injury threshold for overdistension, mathematical models appear to be usefully predictive of the relative contributions of volutrauma and atelectrauma in VILI (9,61).
Mechanisms of VILI
EL not only provides a means of monitoring the injury status of the lungs, it also mediates the generation of VILI by modulating the potentially injurious stresses and strains that are applied to the lung parenchymal tissues during mechanical ventilation. There are two apparently distinct biomechanical mechanisms by which these phenomena can cause VILI, known respectively as volutrauma and atelectrauma.
Volutrauma refers to the tissue damage caused by the excessive stretching of tissues that occurs when the lung is over-inflated, a concept that makes intuitive sense given that even an organ as expansile as the lung has a threshold volume above which structural failure ensues (67-71). Importantly, it is not high pressures per se that causes this type of injury, even though high pressures are required to produce large inflation volumes; experiments with chest strapping have demonstrated that VILI is reduced when high pressures are prevented from causing inflation, which is why the term barotrauma is no longer used in this context (69). The clinical success of protective ventilation strategies based on the use of low tidal volume (72) would seem to bear these ideas out.
Atelectrauma refers to the injury caused by the repetitive re-opening of closed lung units, something that can happen if regions of the lung become derecruited with each expiration and then recruited again during the next inspiration (5,65,73). Injury from atelectrauma is commonly ascribed to shear forces, which are those operating tangentially to the epithelium (i.e., in the axial direction relative to the airway) as apposed surfaces are peeled apart. However, in vitro studies (55,56,74) supported by detailed computational modeling (23,54,75-82) suggest that the culprit forces actually act normally to the surface (i.e., radial to the airway) and that cell injury is caused by the large axial gradient in these normal forces that occurs during the passage of the air-liquid interface (56) as depicted in Figure 2. Interestingly, injury and pressure gradients are more severe as the advancing air-liquid interface moves more slowly due to thinning of the residual fluid film left on the airway wall. In any case, it is clear that movement of high surface tension air-liquid interfaces over epithelial surfaces is highly damaging (77,83), and indeed can cause cellular necrosis after only a small number of passages.
Figure 2.
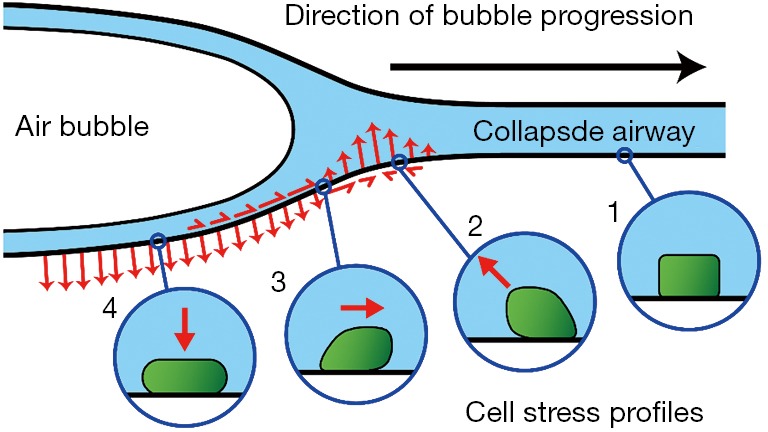
Hypothetical fluid-mechanical stress (red arrows) during the reopening of a collapsed, fluid-occluded (blue) airway as the air-liquid interface moves from left to right. The stress at the airway wall is applied to the epithelial lining (green insets) causing cell death. Adapted with permission from Bilek et al. (55).
A substantial amount of research in animal models of VILI during the 80s and 90s showed that VILI is accompanied by disruption of both the epithelial and endothelial barriers, and is correlated with the amount of pulmonary edema the accumulates as a result (69,84-86). Much of this earlier work focused on the role of over-distension, the importance of atelectrauma only becoming fully apparent somewhat later. In this regard, work from our laboratory in recent years has shown that initially normal mice do not develop VILI within 4 hours from over-ventilation alone; the animals either maintain normal lung function or, if tidal volume is excessive, succumb to a sudden pneumothorax (5,87). They also do not develop VILI when ventilated at zero PEEP provided tidal volume is also modest. Progressive VILI only develops when the mice are ventilated at zero PEEP simultaneously with a very large tidal volume (5). This combination of inspiratory and expiratory pressures leads to widespread disruption of the blood-gas barrier (Figure 3) and a progressive degeneration of lung mechanical function (65) due to accumulation of proteinaceous edema in the airspaces (87). In fact, under these conditions the rate of VILI development can be conveniently titrated by choice of tidal volume (65). Once VILI is underway, however, it continues to progress when ventilation is switched to either zero PEEP or high tidal volume alone (5), demonstrating that what is safe for the healthy lung may not be safe for a lung that is already injured.
Figure 3.

Scanning electron micrograph depicting an undamaged alveolar surface (right panel) and fragmented alveolar epithelium (left panel) caused by two hours of ventilation at high tidal volumes and zero end expiratory pressure. Reproduced with permission from Hamlington et al. (88).
There thus appears to be a complex relationship between over-distension, recruitment, and existing injury that determines whether a given mode of mechanical ventilation will further injure a given lung. Exactly what this relationship is remains a matter of ongoing research, but a specific question that arises in this context is why atelectrauma and volutrauma appear to be synergistic in the production of VILI in the normal lung. One possibility involves the role of so-called “stress concentrators”, which arise when an atelectatic region of the lung borders on a region of normal parenchyma (7,13,89,90). Because the atelectatic region is unable to receive any of the applied ventilation, so long as it remains derecruited, the adjacent normal region is at risk of being over-distended. In particular, at the border of these two regions the alveolar walls are stretched to an exaggerated degree by being constrained to adhere to the region of non-expansile atelectasis as illustrated in Figure 4. Strain and stress are thus concentrated in these alveolar walls, making them particularly susceptible to mechanical failure. Such failure, even at isolated locations throughout the lung, is sufficient to instigate the process of alveolar leak from the vasculature that starts the inexorable march toward full-blown VILI. Of course, this explanation requires the presence of some small degree of alveolar leak to begin the process, but this might be caused by levels of over-distension and repetitive recruitment that do not themselves lead to measurable changes in overall lung function (i.e., in EL). Stress concentrators, being inherently heterogeneous in nature, may also be more likely to exist in animals larger than mice and in humans where the effects of the gravitational gradient on regional differences in lung mechanics are more pronounced (91).
Figure 4.
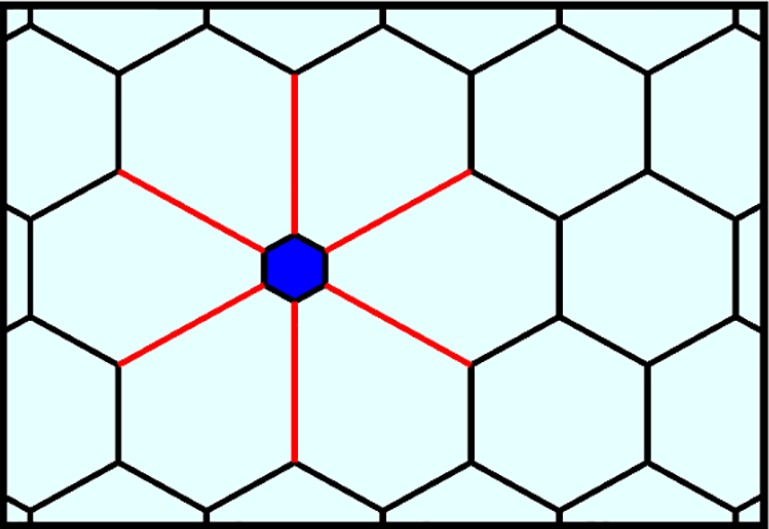
The collapse or flooding of an alveolus (blue) leads to increased distension of the adjacent septa (red).
Another possible explanation for volutrauma-atelectrauma synergy is suggested by our recent study of how VILI affects the size of the leak in the blood-gas barrier. By injecting three different sizes of fluorescently-labelled dextran molecules into mice having varying degrees of VILI and then observing the appearance of each molecule in bronchoalveolar lavage fluid (BALF) (Figure 5), we determined that the holes in the blood-gas barrier appear to follow a power-law distribution (92). That is, there are a large number of very small holes, a small number of large holes, and the intermediate sized holes number such that the histogram of hole sizes is approximately linear in a log-log plot. The significance of this finding lies in the fact that it can potentially be explained by a rich-get-richer mechanism. That is, whereby VILI begins with the generation of small holes, and this initial injury then progresses by the enlargement of these holes in a way that favors holes that are already large (e.g., large holes might correspond to weaker than average areas of the barrier that are particularly susceptible to becoming perforated). Taking this idea a step further leads to the notion that repetitive recruitment might be responsible for the initial creation of holes in the barrier because of its direct action on the epithelium, while subsequent hole enlargement is caused by over-distension. This theory (92) can only be considered speculative at present, but it offers a convenient explanation for why volutrauma and atelectrauma are required together to produce significant VILI, why volutrauma alone is able to exacerbate existing VILI, and why VILI is so difficult to deal with once it is underway. It also potentially explains recent exciting results using preemptive airway pressure release ventilation (APRV) in a large animal model of sepsis (57,93-96) showing that VILI is avoided provided that the duration of pressure release employed is extremely short (a small fraction of a second). The reason may be that this does not allow sufficient time during expiration for any airspaces to actually close (61), thereby eliminating the possibility for atelectrauma to occur. The result is maintenance of an intact alveolar epithelium and avoidance of airway edema despite severe septic shock in the rest of the animal (97). APRV applied in a similar manner in human patients is also showing promise as a means of avoiding the worst ravages of VILI in ARDS (98).
Figure 5.
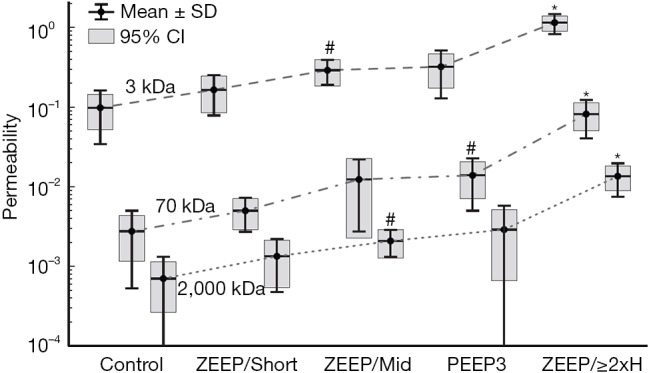
Permeability of the blood-gas barrier (ratio of fluorescent label in BALF versus serum) for three different sized dextran molecules following intravenous injection in mice. Control animals were not subjected to mechanical ventilation and ventilated animals received pressure-controlled ventilation with an end-inspiratory plateau pressure of 37.5 cmH2O. The ZEEP/Short group was ventilated with zero end-expiratory pressure for approximately 30 min. The ZEEP/Mid group was ventilated with zero PEEP for approximately 60 min. The PEEP3 group was ventilated with a positive end-expiratory pressure of 3 cmH2O for approximately 120 min. The ZEEP/2xH group was ventilated with zero PEEP until lung stiffness (H) had risen to twice its baseline value. Used with permission from Hamlington et al. (92).
Another important consideration is where the leak manifests in the blood-gas barrier. Computational modeling of a sheet of cells stitched together by tight junctions shows that the total area of leak that develops as the sheet is stretched is similar to the way that lung function worsens when failure occurs at the cell-cell junctions (99). On the other hand, extensive ultrastructural studies of the blood-gas barrier in animal models of VILI have revealed denudation and blebs in the endothelial and epithelial surfaces, indicating damage to the cells themselves (69,84,100-103). Such damage of the alveolar epithelial surface by high tidal volume ventilation at zero end-expiratory pressure is shown in Figure 3. Wherever these leaks occur, however, they are also presumably capable of being repaired to some extent; not all patients with ARDS and/or VILI die, so recovery from damage must be possible in some cases. It has been shown, for example, that tears in the cell membrane can spontaneously re-anneal in a matter of minutes (104). More profound injury involving cellular necrosis requires that Type II alveolar epithelial cells transdifferentiate to replace Type I epithelial cells and this process occurs over a much longer timescale (105) but is still well within the body’s reparative repertoire.
Finally, VILI has longer-term consequences than simply physical damage to and repair of the components of the blood-gas barrier. The upregulation of inflammatory mediators as a result of this damage can be extensive when injury is widespread and severe (106-111), and can itself have adverse downstream consequences for other parts of the body. Indeed, multi-organ failure resulting from an overwhelming inflammatory response of this nature, known as biotrauma (112), is frequently the ultimate cause of death in ARDS (16).
Mechanics-guided minimization of VILI
The current standard of care for mechanical ventilation in ARDS, derived from the landmark ARDSnet trial in 2000 (2), is essentially an exercise in open-loop control. That is, it employs the one-size-fits-all strategy of striving for a tidal volume of 6 mL/kg ideal body weight in all patients (72). Strong cases can be made for the consideration of other factors that have obvious and strong physiologic rationales, such as choice of PEEP and use of recruitment maneuvers. High-frequency oscillatory ventilation (113-116), variable tidal volume ventilation (117-121), or even liquid ventilation (122-124) have also been investigated extensively, all seemingly based on sound physiologic principles. Despite some positive effects on some physiologic outcomes such as oxygen, however, none has so far demonstrated a benefit in terms of the all-important outcome of reduced mortality. So-called protective (low-tidal volume) ventilation, along with the recent addition of prone positioning (125), thus continues to constitute the rather meagre underpinnings of evidence-based medicine in the management of ARDS. There nevertheless remains a general sentiment in the field that we can do better than this, particularly since the current ARDS mortality rate is still alarmingly high despite marked improvements over the past couple of decades.
One possible explanation for the failure of so many clinical trials of apparently well-founded approaches to ARDS management is the heterogeneity of the condition. ARDS patients vary greatly in the severity and regional distribution of their lung injury (126), the effectiveness with which recruitment maneuvers open collapsed regions of the lung (30), and the ease with which they can be oxygenated. It would seem somewhat obvious that this should be countered with management strategies that are tailored to the specific needs of the individual patient. These needs would have to be measured in each patient, of course, and then used to guide individualized treatment in a closed-loop paradigm. In fact, this is not a new idea, being best exemplified by attempts to determine optimal PEEP in a given patient from measurements of their lung pressure-volume relationships (127,128).
We recently generalized the concept of closed-loop control of mechanical ventilation of the injured lung by investigating some simple candidate injury cost functions with which to define the degree of VILI produced by a given regimen of mechanical ventilation (9). Figure 6 shows a predicted injury cost function based on the product of volutrauma and atelectrauma occurring during the respiratory cycle. Also shown are the positions of experimentally investigated combinations of tidal volume and PEEP that were shown to vary in their capacities to produce VILI in initially healthy mice. The injurious combinations are shown by black X’s while the safe combinations are indicated by white circles. These cost functions were defined in terms of a predictive mathematical model of the lung that estimates both tissue over-distension and repetitive recruitment based on applied pressures and flows. We have taken a similar approach in estimating intra-tidal recruitment and tissue distension from ongoing measurements of airway pressures and flow measured during specialized maneuvers designed to reveal these two phenomena at work (63). Specifically, observing how pressure increases as lung volume is raised to high levels reveals information about the volume-dependence of EL, which in turn can be related to putative thresholds of over-distension above which injury starts to occur. Conversely, observing how EL varies throughout the respiratory cycle reveals the extent to which recruitment of lung units may be occurring with each ventilated breath (129). A convenient way of gaining both types of information simultaneously is provided by the relationships between pressure, volume, and flow observed during variable tidal volume ventilation, since here both the time and amplitude dependence of the lung is being interrogated on an ongoing basis (130). Although this approach awaits clinical validation, in principle brief periods of variable tidal volume ventilation can be used to parameterize numerical models that are then used to predict the amount of volutrauma and atelectrauma generated during other forms of mechanical ventilation so as to determine which may be least injurious.
Figure 6.
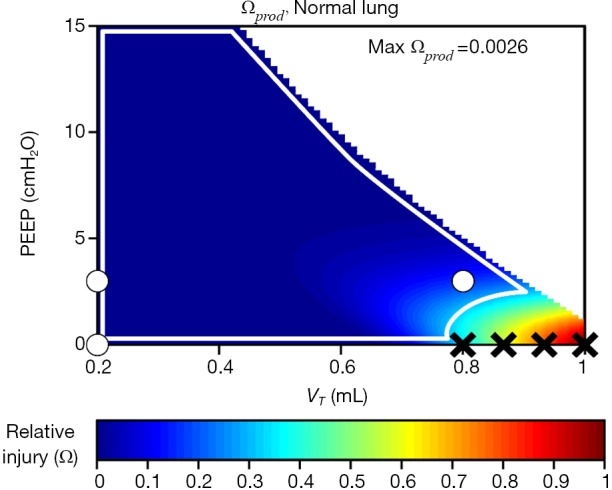
Predicted lung injury cost function for an initially healthy mouse consisting of the product of volutrauma and atelectrauma. Black ‘X’ indicates in vivo experiments that produced lung injury, white circles demarcate in vivo experiments that were non-injurious. The white line describes a ‘safe region’ of ventilation. Adapted from Hamlington et al. (92).
Conclusions
Lung mechanics reflected in EL and its changes over time contain much useful information about the injury status of the lung, particular when measurements are made in association with maneuvers that interrogate the behavior of the mechanical lung over sufficiently wide ranges of amplitude and time. These measurements afford the opportunity, in principle, to identify mechanical models of the lung that embody the essential mechanisms responsible for volutrauma and atelectrauma. Such models can then be used as virtual laboratories within which searches for optimal ventilation strategies can be made, opening the way to a rationalized approach to personalized management of ARDS.
Acknowledgements
Funding: This work was supported by the National Institutes of Health [grant numbers HL-124052 and R00 HL128944].
Footnotes
Conflicts of Interest: JH Bates is a member of the Advisory Board of and a minor shareholder in Oscillavent. BJ Smith has no conflicts of interest to declare.
References
- 1.Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 1998;157:294-323. 10.1164/ajrccm.157.1.9604014 [DOI] [PubMed] [Google Scholar]
- 2.International consensus conferences in intensive care medicine: Ventilator-associated Lung Injury in ARDS. This official conference report was cosponsored by the American Thoracic Society, The European Society of Intensive Care Medicine, and The Societe de Reanimation de Langue Francaise, and was approved by the ATS Board of Directors, July 1999. Am J Respir Crit Care Med 1999;160:2118-24. [DOI] [PubMed] [Google Scholar]
- 3.Amato MB, Meade MO, Slutsky AS, et al. Driving pressure and survival in the acute respiratory distress syndrome. N Engl J Med 2015;372:747-55. 10.1056/NEJMsa1410639 [DOI] [PubMed] [Google Scholar]
- 4.Allen G, Bates JH. Dynamic mechanical consequences of deep inflation in mice depend on type and degree of lung injury. J Appl Physiol (1985) 2004;96:293-300. [DOI] [PubMed] [Google Scholar]
- 5.Seah AS, Grant KA, Aliyeva M, et al. Quantifying the roles of tidal volume and PEEP in the pathogenesis of ventilator-induced lung injury. Ann Biomed Eng 2011;39:1505-16. 10.1007/s10439-010-0237-6 [DOI] [PubMed] [Google Scholar]
- 6.Smith BJ, Bates JHT. editors. The Relationship Between Nonlinear Lung Elastance And Ventilator-Induced Lung Injury. American Thoracic Society International Conference; 2013; Philadelphia, Pa. [Google Scholar]
- 7.Wu Y, Kharge AB, Perlman CE. Lung ventilation injures areas with discrete alveolar flooding, in a surface tension-dependent fashion. J Appl Physiol (1985) 2014;117:788-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pavone L, Albert S, DiRocco J, et al. Alveolar instability caused by mechanical ventilation initially damages the nondependent normal lung. Critical Care 2007;11:R104. 10.1186/cc6122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamlington KL, Smith BJ, Allen GB, et al. Predicting ventilator-induced lung injury using a lung injury cost function. J Appl Physiol (1985) 2016;121:106-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Radford EP. Recent studies of mechanical properties of mammalian lungs. In: Remington JW. editor. Tissue Elasticity. Washington DC: Waverly Press, 1957:177-90. [Google Scholar]
- 11.Bates J. Lung Mechanics. An Inverse Modeling Approach. Cambridge: Cambridge University Press, 2009. [Google Scholar]
- 12.Toshima M, Ohtani Y, Ohtani O. Three-dimensional architecture of elastin and collagen fiber networks in the human and rat lung. Arch Histol Cytol 2004;67:31-40. 10.1679/aohc.67.31 [DOI] [PubMed] [Google Scholar]
- 13.Wilson TA, Bachofen H. A model for mechanical structure of the alveolar duct. J Appl Physiol Respir Environ Exerc Physiol 1982;52:1064-70. [DOI] [PubMed] [Google Scholar]
- 14.Meduri GU, Belenchia JM, Estes RJ, et al. Fibroproliferative phase of ARDS. Clinical findings and effects of corticosteroids. Chest 1991;100:943-52. 10.1378/chest.100.4.943 [DOI] [PubMed] [Google Scholar]
- 15.Zapol WM, Trelstad RL, Coffey JW, et al. Pulmonary fibrosis in severe acute respiratory failure. Am Rev Respir Dis 1979;119:547-54. [DOI] [PubMed] [Google Scholar]
- 16.Montgomery AB, Stager MA, Carrico CJ, et al. Causes of mortality in patients with the adult respiratory distress syndrome. Am Rev Respir Dis 1985;132:485-9. [DOI] [PubMed] [Google Scholar]
- 17.Ware LB, Matthay MA. Clinical practice. Acute pulmonary edema. N Engl J Med 2005;353:2788-96. 10.1056/NEJMcp052699 [DOI] [PubMed] [Google Scholar]
- 18.Dellaca RL, Zannin E, Sancini G, et al. Changes in the mechanical properties of the respiratory system during the development of interstitial lung edema. Respir Res 2008;9:51. 10.1186/1465-9921-9-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gattinoni L, Pesenti A. The concept of "baby lung". Intensive care medicine 2005;31:776-84. 10.1007/s00134-005-2627-z [DOI] [PubMed] [Google Scholar]
- 20.Holm BA, Enhorning G, Notter RH. A Biophysical Mechanism by Which Plasma-Proteins Inhibit Lung Surfactant Activity. Chem Phys Lipids 1988;49:49-55. 10.1016/0009-3084(88)90063-1 [DOI] [PubMed] [Google Scholar]
- 21.Holm BA, Notter RH. Effects of hemoglobin and cell membrane lipids on pulmonary surfactant activity. J Appl Physiol (1985) 1987;63:1434-42. [DOI] [PubMed] [Google Scholar]
- 22.Günther A, Siebert C, Schmidt R, et al. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am J Respir Crit Care Med 1996;153:176-84. 10.1164/ajrccm.153.1.8542113 [DOI] [PubMed] [Google Scholar]
- 23.Halpern D, Gaver DP., III The influence of surfactant on the propagation of a semi-infinite bubble through a liquid filled compliant channel. J Fluid Mech 2012;698:125-59. 10.1017/jfm.2012.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Y, Anderson JC, Suresh V, et al. Effect of Gravity on Liquid Plug Transport Through an Airway Bifurcation Model. J Biomech Eng 2005;127:798-806. 10.1115/1.1992529 [DOI] [PubMed] [Google Scholar]
- 25.Fujioka H, Takayama S, Grotberg JB. Unsteady propagation of a liquid plug in a liquid-lined straight tube. Phys Fluids (1994) 2008;20:62104. [DOI] [PMC free article] [PubMed]
- 26.Halpern D, Gaver DP., 3rd The influence of surfactant on the propagation of a semi-infinite bubble through a liquid-filled compliant channel. J Fluid Mech 2012;698:125-59. 10.1017/jfm.2012.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen G, Lundblad LK, Parsons P, et al. Transient mechanical benefits of a deep inflation in the injured mouse lung. J Appl Physiol (1985) 2002;93:1709-15. [DOI] [PubMed] [Google Scholar]
- 28.Allen GB, Leclair T, Cloutier M, et al. The response to recruitment worsens with progression of lung injury and fibrin accumulation in a mouse model of acid aspiration. Am J Physiol Lung Cell Mol Physiol 2007;292:L1580-9. 10.1152/ajplung.00483.2006 [DOI] [PubMed] [Google Scholar]
- 29.Foti G, Cereda M, Sparacino ME, et al. Effects of periodic lung recruitment maneuvers on gas exchange and respiratory mechanics in mechanically ventilated acute respiratory distress syndrome (ARDS) patients. Intensive Care Med 2000;26:501-7. 10.1007/s001340051196 [DOI] [PubMed] [Google Scholar]
- 30.Ranieri VM, Eissa NT, Corbeil C, et al. Effects of positive end-expiratory pressure on alveolar recruitment and gas exchange in patients with the adult respiratory distress syndrome. Am Rev Respir Dis 1991;144:544-51. 10.1164/ajrccm/144.3_Pt_1.544 [DOI] [PubMed] [Google Scholar]
- 31.Marini JJ. Should We Embrace the "Open Lung" Approach? Crit Care Med 2016;44:237-8. 10.1097/CCM.0000000000001489 [DOI] [PubMed] [Google Scholar]
- 32.Santos A, Lucchetta L, Monge-Garcia MI, et al. The Open Lung Approach Improves Pulmonary Vascular Mechanics in an Experimental Model of Acute Respiratory Distress Syndrome. Crit Care Med 2017;45:e298-305. 10.1097/CCM.0000000000002082 [DOI] [PubMed] [Google Scholar]
- 33.Kacmarek RM, Villar J, Sulemanji D, et al. Open Lung Approach for the Acute Respiratory Distress Syndrome: A Pilot, Randomized Controlled Trial. Crit Care Med 2016;44:32-42. 10.1097/CCM.0000000000001383 [DOI] [PubMed] [Google Scholar]
- 34.Ferrando C, Soro M, Unzueta C, et al. Individualised perioperative open-lung approach versus standard protective ventilation in abdominal surgery (iPROVE): a randomised controlled trial. Lancet Respir Med 2018;6:193-203. 10.1016/S2213-2600(18)30024-9 [DOI] [PubMed] [Google Scholar]
- 35.Crotti S, Mascheroni D, Caironi P, et al. Recruitment and derecruitment during acute respiratory failure: a clinical study. Am J Respir Crit Care Med 2001;164:131-40. 10.1164/ajrccm.164.1.2007011 [DOI] [PubMed] [Google Scholar]
- 36.Broche L, Perchiazzi G, Porra L, et al. Dynamic Mechanical Interactions Between Neighboring Airspaces Determine Cyclic Opening and Closure in Injured Lung. Crit Care Med 2017;45:687-94. 10.1097/CCM.0000000000002234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bates JH, Irvin CG. Time dependence of recruitment and derecruitment in the lung: a theoretical model. J Appl Physiol 2002;93:705-13. 10.1152/japplphysiol.01274.2001 [DOI] [PubMed] [Google Scholar]
- 38.Massa CB, Allen GB, Bates JH. Modeling the dynamics of recruitment and derecruitment in mice with acute lung injury. J Appl Physiol 2008;105:1813-21. 10.1152/japplphysiol.90806.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hickling KG. The pressure-volume curve is greatly modified by recruitment. A mathematical model of ARDS lungs. Am J Respir Crit Care Med 1998;158:194-202. 10.1164/ajrccm.158.1.9708049 [DOI] [PubMed] [Google Scholar]
- 40.Talmor D, Sarge T, Malhotra A, et al. Mechanical ventilation guided by esophageal pressure in acute lung injury. N Engl J Med 2008;359:2095-104. 10.1056/NEJMoa0708638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pintado MC, de Pablo R, Trascasa M, et al. Individualized PEEP setting in subjects with ARDS: a randomized controlled pilot study. Respir Care 2013;58:1416-23. 10.4187/respcare.02068 [DOI] [PubMed] [Google Scholar]
- 42.Maggiore SM, Jonson B, Richard JC, et al. Alveolar derecruitment at decremental positive end-expiratory pressure levels in acute lung injury: comparison with the lower inflection point, oxygenation, and compliance. Am J Respir Crit Care Med 2001;164:795-801. 10.1164/ajrccm.164.5.2006071 [DOI] [PubMed] [Google Scholar]
- 43.Ward NS, Lin DY, Nelson DL, et al. Successful determination of lower inflection point and maximal compliance in a population of patients with acute respiratory distress syndrome. Crit Care Med 2002;30:963-8. 10.1097/00003246-200205000-00002 [DOI] [PubMed] [Google Scholar]
- 44.Malbouisson LM, Muller JC, Constantin JM, et al. Computed tomography assessment of positive end-expiratory pressure-induced alveolar recruitment in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;163:1444-50. 10.1164/ajrccm.163.6.2005001 [DOI] [PubMed] [Google Scholar]
- 45.Pelosi P, Rocco PR, de Abreu MG. Use of computed tomography scanning to guide lung recruitment and adjust positive-end expiratory pressure. Curr Opin Crit Care 2011;17:268-74. 10.1097/MCC.0b013e328344ddbc [DOI] [PubMed] [Google Scholar]
- 46.Brower RG, Lanken PN, MacIntyre N, et al. Higher versus Lower Positive End-Expiratory Pressures in Patients with the Acute Respiratory Distress Syndrome. N Engl J Med 2004;351:327-36. 10.1056/NEJMoa032193 [DOI] [PubMed] [Google Scholar]
- 47.Rayleigh L. On the capillary phenomena of jets. Proc R Soc Lond 1879;29:71-97. 10.1098/rspl.1879.0015 [DOI] [Google Scholar]
- 48.Heil M. Minimal Liquid Bridges in Non-Axisymmetrically Buckled Tubes. J Fluid Mech 1999;380:309-37. 10.1017/S0022112098003760 [DOI] [Google Scholar]
- 49.Heil M. Airway Closure: Occluding Liquid Bridges in Strongly Buckled Elastic Tubes. J Biomech Eng 1999;121:487-93. 10.1115/1.2835077 [DOI] [PubMed] [Google Scholar]
- 50.Heil M, White J. Airway closure: surface-tension-driven non-axisymmetric instabilities of liquid-lined elastic rings. J Fluid Mech 2002;462:79-109. 10.1017/S0022112002008613 [DOI] [Google Scholar]
- 51.Hazel AL, Heil M. Surface-tension-induced buckling of liquid-lined elastic tubes: a model for pulmonary airway closure. Proc Math Phys Eng Sci 2005;461:1847-68. 10.1098/rspa.2005.1453 [DOI] [Google Scholar]
- 52.White JP, Heil M. Three-dimensional instabilities of liquid-lined elastic tubes: A thin-film fluid-structure interaction model. Phys Fluids 2005;17:031506 10.1063/1.1862631 [DOI] [Google Scholar]
- 53.Gaver DP, 3rd, Samsel RW, Solway J. Effects of surface tension and viscosity on airway reopening. J Appl Physiol 1990;69:74-85. 10.1152/jappl.1990.69.1.74 [DOI] [PubMed] [Google Scholar]
- 54.Gaver DP, 3rd, Halpern D, Jensen OE, et al. The steady motion of a semi-infinite bubble through a flexible-walled channel. J Fluid Mech 1996;319:25-65. 10.1017/S0022112096007240 [DOI] [Google Scholar]
- 55.Bilek AM, Dee KC, Gaver DP., III Mechanisms of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. J Appl Physiol 2003;94:770-83. 10.1152/japplphysiol.00764.2002 [DOI] [PubMed] [Google Scholar]
- 56.Kay SS, Bilek AM, Dee KC, et al. Pressure gradient, not exposure duration, determines the extent of epithelial cell damage in a model of pulmonary airway reopening. J Appl Physiol 2004;97:269-76. 10.1152/japplphysiol.01288.2003 [DOI] [PubMed] [Google Scholar]
- 57.Kollisch-Singule M, Emr B, Smith B, et al. Mechanical Breath Profile of Airway Pressure Release Ventilation: The Effect on Alveolar Recruitment and Microstrain in Acute Lung Injury. JAMA Surg 2014;149:1138-45. 10.1001/jamasurg.2014.1829 [DOI] [PubMed] [Google Scholar]
- 58.Lutz D, Gazdhar A, Lopez-Rodriguez E, et al. Alveolar derecruitment and collapse induration as crucial mechanisms in lung injury and fibrosis. Am J Respir Cell Mol Biol 2015;52:232-43. 10.1165/rcmb.2014-0078OC [DOI] [PubMed] [Google Scholar]
- 59.Grasso S, Mascia L, Del Turco M, et al. Effects of recruiting maneuvers in patients with acute respiratory distress syndrome ventilated with protective ventilatory strategy. Anesthesiology 2002;96:795-802. 10.1097/00000542-200204000-00005 [DOI] [PubMed] [Google Scholar]
- 60.Gil J, Bachofen H, Gehr P, et al. Alveolar volume-surface area relation in air- and saline-filled lungs fixed by vascular perfusion. J Appl Physiol 1979;47:990-1001. 10.1152/jappl.1979.47.5.990 [DOI] [PubMed] [Google Scholar]
- 61.Smith BJ, Lundblad LK, Kollisch-Singule M, et al. Predicting the response of the injured lung to the mechanical breath profile. J Appl Physiol (1985) 2015;118:932-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Albert RK. The role of ventilation-induced surfactant dysfunction and atelectasis in causing acute respiratory distress syndrome. Am J Respir Crit Care Med 2012;185:702-8. 10.1164/rccm.201109-1667PP [DOI] [PubMed] [Google Scholar]
- 63.Smith BJ, Bates JH. Assessing the Progression of Ventilator-Induced Lung Injury in Mice. IEEE Trans Biomed Eng 2013;60:3449-57. 10.1109/TBME.2013.2267151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma B, Suki B, Bates JH. Effects of recruitment/derecruitment dynamics on the efficacy of variable ventilation. J Appl Physiol 2011;110:1319-26. 10.1152/japplphysiol.01364.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith BJ, Grant KA, Bates JH. Linking the Development of Ventilator-Induced Lung Injury to Mechanical Function in the Lung. Ann Biomed Eng 2013;41:527-36. 10.1007/s10439-012-0693-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fujioka H, Grotberg JB. The steady propagation of a surfactant-laden liquid plug in a two-dimensional channel. Phys Fluids 2005;17:082102 10.1063/1.1948907 [DOI] [Google Scholar]
- 67.Webb HH, Tierney DF. Experimental Pulmonary-Edema Due to Intermittent Positive Pressure Ventilation with High Inflation Pressures. Protection by Positive End-Expiratory Pressure. Am Rev Respir Dis 1974;110:556-65. [DOI] [PubMed] [Google Scholar]
- 68.Kolobow T, Moretti MP, Fumagalli R, et al. Severe Impairment in Lung-Function Induced by High Peak Airway Pressure during Mechanical Ventilation - an Experimental-Study. Am Rev Respir Dis 1987;135:312-5. [DOI] [PubMed] [Google Scholar]
- 69.Dreyfuss D, Soler P, Basset G, et al. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis 1988;137:1159-64. 10.1164/ajrccm/137.5.1159 [DOI] [PubMed] [Google Scholar]
- 70.Hernandez LA, Peevy KJ, Moise AA, et al. Chest Wall Restriction Limits High Airway Pressure-Induced Lung Injury in Young-Rabbits. J Appl Physiol (1985) 1989;66:2364-8. [DOI] [PubMed] [Google Scholar]
- 71.Carlton DP, Cummings JJ, Scheerer RG, et al. Lung Overexpansion Increases Pulmonary Microvascular Protein Permeability in Young Lambs. J Appl Physiol (1985) 1990;69:577-83. [DOI] [PubMed] [Google Scholar]
- 72.Brower RG, Matthay MA, Morris A, et al. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000;342:1301-8. 10.1056/NEJM200005043421801 [DOI] [PubMed] [Google Scholar]
- 73.Muscedere JG, Mullen JBM, Gan K, et al. Tidal Ventilation at Low Airway Pressures Can Augment Lung Injury. Am J Respir Crit Care Med 1994;149:1327-34. 10.1164/ajrccm.149.5.8173774 [DOI] [PubMed] [Google Scholar]
- 74.Yalcin HC, Perry SF, Ghadiali SN. Influence of airway diameter and cell confluence on epithelial cell injury in an in vitro model of airway reopening. J Appl Physiol 2007;103:1796-807. 10.1152/japplphysiol.00164.2007 [DOI] [PubMed] [Google Scholar]
- 75.Smith BJ, Gaver DP., 3rd The pulsatile propagation of a finger of air within a fluid-occluded cylindrical tube. J Fluid Mech 2008;601:1-23. 10.1017/S0022112008000360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Glindmeyer HW, 4th, Smith BJ, Gaver DP., 3rd In Situ Enhancement of Pulmonary Surfactant Function Using Temporary Flow Reversal. J Appl Physiol 2012;112:149-58. 10.1152/japplphysiol.00643.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dailey HL, Ricles LM, Yalcin HC, et al. Image-based finite element modeling of alveolar epithelial cell injury during airway reopening. J Appl Physiol 2009;106:221-32. 10.1152/japplphysiol.90688.2008 [DOI] [PubMed] [Google Scholar]
- 78.Jacob AM, Gaver DP., 3rd An investigation of the influence of cell topography on epithelial mechanical stresses during pulmonary airway reopening. Phys Fluids (1994) 2005;17:31502. 10.1063/1.1862642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heil M. Finite Reynolds number effects in the Bretherton problem. Phys Fluids 2001;13:2517-21. 10.1063/1.1389861 [DOI] [Google Scholar]
- 80.Hazel AL, Heil M. Three-dimensional airway reopening: The steady propagation of a semi-infinite bubble into a buckled elastic tube. J Fluid Mech 2003;478:47-70. 10.1017/S0022112002003452 [DOI] [Google Scholar]
- 81.Hazel AL, Heil M. Finite-Reynolds-Number Effects in Steady, Three-Dimensional Airway Reopening. J Biomech Eng 2006;128:573-8. 10.1115/1.2206203 [DOI] [PubMed] [Google Scholar]
- 82.Hazel AL, Heil M. The influence of gravity on the steady propagation of a semi-infinite bubble into a flexible channel. Phys Fluids 2008;20:092109 10.1063/1.2982520 [DOI] [Google Scholar]
- 83.Ghadiali SN, Gaver DP., 3rd Biomechanics of liquid-epithelium interactions in pulmonary airways. Respir Physiol Neurobiol 2008;163:232-43. 10.1016/j.resp.2008.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dreyfuss D, Basset G, Soler P, et al. Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am Rev Respir Dis 1985;132:880-4. [DOI] [PubMed] [Google Scholar]
- 85.John E, McDevitt M, Wilborn W, et al. Ultrastructure of the lung after ventilation. Br J Exp Pathol 1982;63:401-7. [PMC free article] [PubMed] [Google Scholar]
- 86.Vaneker M, Halbertsma FJ, van Egmond J, et al. Mechanical ventilation in healthy mice induces reversible pulmonary and systemic cytokine elevation with preserved alveolar integrity: an in vivo model using clinical relevant ventilation settings. Anesthesiology 2007;107:419-26. 10.1097/01.anes.0000278908.22686.01 [DOI] [PubMed] [Google Scholar]
- 87.Smith BJ, Bartolak-Suki E, Suki B, et al. Linking Ventilator Injury-Induced Leak across the Blood-Gas Barrier to Derangements in Murine Lung Function. Front Physiol 2017;8:466. 10.3389/fphys.2017.00466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hamlington KL, Dunn CM, Roy GS, et al. Linking Alveolar Epithelial Barrier Disruption To Function In Ventilator-Induced Lung Injury. Am J Respir Crit Care Med 2016;193:A4826. [Google Scholar]
- 89.Mead J, Takishima T, Leith D. Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol 1970;28:596-608. 10.1152/jappl.1970.28.5.596 [DOI] [PubMed] [Google Scholar]
- 90.Makiyama AM, Gibson LJ, Harris RS, et al. Stress concentration around an atelectatic region: A finite element model. Respir Physiol Neurobiol 2014;201:101-10. 10.1016/j.resp.2014.06.017 [DOI] [PubMed] [Google Scholar]
- 91.Wellman TJ, Winkler T, Costa EL, et al. Effect of local tidal lung strain on inflammation in normal and lipopolysaccharide-exposed sheep. Crit Care Med 2014;42:e491-500. 10.1097/CCM.0000000000000346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hamlington KL, Bates JH, Roy GS, et al. Alveolar Leak Develops by a Rich-Get-Richer Process 1 in Ventilator-Induced Lung Injury. PLoS One 2018;13:e0193934. 10.1371/journal.pone.0193934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Emr B, Gatto LA, Roy S, et al. Airway pressure release ventilation prevents ventilator-induced lung injury in normal lungs. JAMA Surg 2013;148:1005-12. 10.1001/jamasurg.2013.3746 [DOI] [PubMed] [Google Scholar]
- 94.Roy S, Habashi N, Sadowitz B, et al. Early airway pressure release ventilation prevents ARDS - a novel preventive approach to lung injury. Shock 2013;39:28-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roy SK, Emr B, Sadowitz B, et al. Preemptive application of airway pressure release ventilation prevents development of acute respiratory distress syndrome in a rat traumatic hemorrhagic shock model. Shock 2013;40:210-6. 10.1097/SHK.0b013e31829efb06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kollisch-Singule M, Jain S, Andrews P, et al. Effect of Airway Pressure Release Ventilation on Dynamic Alveolar Heterogeneity. JAMA Surg 2016;151:64-72. [DOI] [PubMed] [Google Scholar]
- 97.Kollisch-Singule M, Emr B, Smith BJ, et al. Airway Pressure Release Ventilation Reduces Conducting Airway Micro-Strain in Lung Injury. J Am Coll Surg 2014;219:968-76. 10.1016/j.jamcollsurg.2014.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Andrews PL, Shiber JR, Jaruga-Killeen E, et al. Early application of airway pressure release ventilation may reduce mortality in high-risk trauma patients: A systematic review of observational trauma ARDS literature. J Trauma Acute Care Surg 2013;75:635-41. 10.1097/TA.0b013e31829d3504 [DOI] [PubMed] [Google Scholar]
- 99.Hamlington KL, Ma B, Smith BJ, et al. Modeling the Progression of Epithelial Leak Caused by Overdistension. Cell Mol Bioeng 2016;9:151-61. 10.1007/s12195-015-0426-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Velazquez M, Weibel ER, Kuhn C, 3rd, et al. PET evaluation of pulmonary vascular permeability: a structure-function correlation. J Appl Physiol (1985) 1991;70:2206-16. [DOI] [PubMed] [Google Scholar]
- 101.Woods LW, Wilson DW, Schiedt MJ, et al. Structural and biochemical changes in lungs of 3-methylindole-treated rats. Am J Pathol 1993;142:129-38. [PMC free article] [PubMed] [Google Scholar]
- 102.Hirai KI, Witschi H, Cote MG. Electron microscopy of butylated hydroxytoluene-induced lung damage in mice. Exp Mol Pathol 1977;27:295-308. 10.1016/0014-4800(77)90002-8 [DOI] [PubMed] [Google Scholar]
- 103.Kapanci Y, Weibel ER, Kaplan HP, et al. Pathogenesis and reversibility of the pulmonary lesions of oxygen toxicity in monkeys. II. Ultrastructural and morphometric studies. Lab Invest 1969;20:101-18. [PubMed] [Google Scholar]
- 104.Gajic O, Lee J, Doerr CH, et al. Ventilator-induced cell wounding and repair in the intact lung. Am J Respir Crit Care Med 2003;167:1057-63. 10.1164/rccm.200208-889OC [DOI] [PubMed] [Google Scholar]
- 105.Jansing NL, McClendon J, Henson PM, et al. Unbiased Quantitation of Alveolar Type II to Alveolar Type I Cell Transdifferentiation during Repair after Lung Injury in Mice. Am J Respir Cell Mol Biol 2017;57:519-26. 10.1165/rcmb.2017-0037MA [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Park WY, Goodman RB, Steinberg KP, et al. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;164:1896-903. 10.1164/ajrccm.164.10.2104013 [DOI] [PubMed] [Google Scholar]
- 107.Wilson MR, Choudhury S, Goddard ME, et al. High tidal volume upregulates intrapulmonary cytokines in an in vivo mouse model of ventilator-induced lung injury. J Appl Physiol (1985) 2003;95:1385-93. [DOI] [PubMed] [Google Scholar]
- 108.Dolinay T, Kim YS, Howrylak J, et al. Inflammasome-regulated Cytokines Are Critical Mediators of Acute Lung Injury. Am J Respir Crit Care Med 2012;185:1225-34. 10.1164/rccm.201201-0003OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rich PB, Douillet CD, Hurd H, et al. Effect of ventilatory rate on airway cytokine levels and lung injury. J Surg Res 2003;113:139-45. 10.1016/S0022-4804(03)00195-1 [DOI] [PubMed] [Google Scholar]
- 110.Liu YY, Chiang CH, Chuang CH, et al. Spillover of Cytokines and Reactive Oxygen Species in Ventilator-Induced Lung Injury Associated With Inflammation and Apoptosis in Distal Organs. Respir Care 2014;59:1422-32. 10.4187/respcare.02992 [DOI] [PubMed] [Google Scholar]
- 111.Terragni PP, Rosboch G, Tealdi A, et al. Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med 2007;175:160-6. 10.1164/rccm.200607-915OC [DOI] [PubMed] [Google Scholar]
- 112.Tremblay LN, Slutsky AS. Ventilator-induced injury: From barotrauma to biotrauma. Proc Assoc Am Physicians 1998;110:482-8. [PubMed] [Google Scholar]
- 113.Derdak S, Mehta S, Stewart TE, et al. High-frequency oscillatory ventilation for acute respiratory distress syndrome in adults. Am J Respir Crit Care Med 2002;166:801-8. 10.1164/rccm.2108052 [DOI] [PubMed] [Google Scholar]
- 114.Krishnan JA, Brower RG. High-frequency ventilation for acute lung injury and ARDS. Chest 2000;118:795-807. 10.1378/chest.118.3.795 [DOI] [PubMed] [Google Scholar]
- 115.Chang HK. Mechanisms of gas transport during ventilation by high-frequency oscillation. J Appl Physiol Respir Environ Exerc Physiol 1984;56:553-63. [DOI] [PubMed] [Google Scholar]
- 116.Hamilton PP, Onayemi A, Smyth JA, et al. Comparison of conventional and high-frequency ventilation: oxygenation and lung pathology. J Appl Physiol Respir Environ Exerc Physiol 1983;55:131-8. [DOI] [PubMed] [Google Scholar]
- 117.Eschun GM, Lefevre GR, Graham MR, et al. Biologically variable ventilation prevents deterioration of gas exchange during prolonged anesthesia. Anesthesiology 1999;91:U516-U. [DOI] [PubMed]
- 118.Mutch WA, Harms S, Ruth Graham M, et al. Biologically variable or naturally noisy mechanical ventilation recruits atelectatic lung. Am J Respir Crit Care Med 2000;162:319-23. 10.1164/ajrccm.162.1.9903120 [DOI] [PubMed] [Google Scholar]
- 119.Arold SP, Mora R, Lutchen KR, et al. Variable tidal volume ventilation improves lung mechanics and gas exchange in a rodent model of acute lung injury. Am J Respir Crit Care Med 2002;165:366-71. 10.1164/ajrccm.165.3.2010155 [DOI] [PubMed] [Google Scholar]
- 120.Bellardine CL, Hoffman AM, Tsai L, et al. Comparison of variable and conventional ventilation in a sheep saline lavage lung injury model. Crit Care Med 2006;34:439-45. 10.1097/01.CCM.0000196208.01682.87 [DOI] [PubMed] [Google Scholar]
- 121.Thammanomai A, Hueser LE, Majumdar A, et al. Design of a new variable-ventilation method optimized for lung recruitment in mice. J Appl Physiol 2008;104:1329-40. 10.1152/japplphysiol.01002.2007 [DOI] [PubMed] [Google Scholar]
- 122.Clark LC, Gollan F. Survival of mammals breathing organic liquids equilibrated with oxygen at atmospheric pressure. Science 1966;152:1755-6. 10.1126/science.152.3730.1755 [DOI] [PubMed] [Google Scholar]
- 123.Hirschl RB, Pranikoff T, Wise C, et al. Initial experience with partial liquid ventilation in adult patients with the acute respiratory distress syndrome. JAMA 1996;275:383-9. 10.1001/jama.1996.03530290053037 [DOI] [PubMed] [Google Scholar]
- 124.Hirschl RB, Parent A, Tooley R, et al. Liquid ventilation improves pulmonary function, gas exchange, and lung injury in a model of respiratory failure. Ann Surg 1995;221:79-88. 10.1097/00000658-199501000-00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mancebo J, Fernandez R, Blanch L, et al. A multicenter trial of prolonged prone ventilation in severe acute respiratory distress syndrome. Am J Respir Crit Care Med 2006;173:1233-9. 10.1164/rccm.200503-353OC [DOI] [PubMed] [Google Scholar]
- 126.Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA 2012;307:2526-33. [DOI] [PubMed] [Google Scholar]
- 127.Ranieri VM, Giuliani R, Fiore T, et al. Volume-Pressure Curve of the Respiratory System Predicts Effects of Peep in Ards - Occlusion Versus Constant Flow Technique. Am J Respir Crit Care Med 1994;149:19-27. 10.1164/ajrccm.149.1.8111581 [DOI] [PubMed] [Google Scholar]
- 128.Terragni PP, Rosboch GL, Lisi A, et al. How respiratory system mechanics may help in minimising ventilator-induced lung injury in ARDS patients. Eur Respir J Suppl 2003;42:15s-21s. 10.1183/09031936.03.00420303 [DOI] [PubMed] [Google Scholar]
- 129.Mellenthin MM, Seong SA, Roy GS, et al. The Roles Of Volutrauma And Atelectrauma In Ventilator Induced Lung Injury Pathogenesis. Am J Respir Crit Care Med 2017;195:A7533. [Google Scholar]
- 130.Smith BJ, Bates JH. Variable Ventilation as a Diagnostic Tool for the Injured Lung. IEEE Trans Biomed Eng 2015;62:2106-13. 10.1109/TBME.2014.2315964 [DOI] [PMC free article] [PubMed] [Google Scholar]