

Abstract
Excessive fat deposition is a negative factor for poultry production because it reduces feed efficiency, increases the cost of meat production and is a health concern for consumers. We genotyped 497 birds from a Brazilian F2 Chicken Resource Population, using a high-density SNP array (600 K), to estimate the genomic heritability of fat deposition related traits and to identify genomic regions and positional candidate genes (PCGs) associated with these traits. Selection signature regions, haplotype blocks and SNP data from a previous whole genome sequencing study in the founders of this chicken F2 population were used to refine the list of PCGs and to identify potential causative SNPs. We obtained high genomic heritabilities (0.43–0.56) and identified 22 unique QTLs for abdominal fat and carcass fat content traits. These QTLs harbored 26 PCGs involved in biological processes such as fat cell differentiation, insulin and triglyceride levels, and lipid biosynthetic process. Three of these 26 PCGs were located within haplotype blocks there were associated with fat traits, five overlapped with selection signature regions, and 12 contained predicted deleterious variants. The identified QTLs, PCGs and potentially causative SNPs provide new insights into the genetic control of fat deposition and can lead to improved accuracy of selection to reduce excessive fat deposition in chickens.
Introduction
In the past, slow growth rates were a challenge in poultry production systems; consequently, intensive selection of this trait in elite great-grand parent lines has dramatically increased poultry productivity. Modern commercial broiler chickens are produced from crosses that have been simultaneously selected for rapid growth, increased meat-production, and improved carcass yield1,2. However, chickens selected for higher body weight might exhibit increased appetite and excessive energy consumption, which may lead to excessive fat accumulation3–5.
Excessive fat deposition in chickens is a negative factor for meat production because it reduces feed efficiency and the value of the carcass6. Therefore, understanding the genetic architecture, uncovering genomic regions, and finding positional candidate genes (PCGs) associated with fat deposition related traits could be helpful in breeding programs.
A total of 200 quantitative trait loci (QTL) have been reported for abdominal fat weight, 139 for abdominal fat percentage and 11 for carcass fat content7. Previous QTL studies, performed in the same population evaluated in this study (Embrapa F2 Chicken Resource Population) using 128 microsatellite markers for 22 autosomal chromosomes, mapped QTLs for abdominal fat traits8–10 and carcass fat traits9, but the genomic locations of these QTL had large confidence intervals. Genotyping of animals using high-density marker arrays can help the identification of genomic regions with smaller intervals11, which, in turn, facilitates the identification of candidate genes.
Many studies have been performed to identify candidate genes for fat deposition traits in chicken12–15, but the discovery of a causal mutation is still a challenge. In a study with chicken, candidate genes for egg production and feed efficiency were identified by genome-wide association study (GWAS), and this information was integrated with selection signature results highlighting important biological processes and causal variants16. However, to the best of our knowledge, no integrative studies were performed to identify potential causal genes and mutations for fat deposition regulation.
Thus, the aims of this study were to estimate genomic heritability for different fat deposition related traits and identify genomic regions and PCGs in an F2 population from a cross between a broiler line and an egg-layer line. We also integrated these results to refine our list of candidate genes using selection signature regions and SNP data information previously obtained from whole genome sequence of the parental generation evaluated in this study.
Results
Genotyping and quality control
From the 529 genotyped chickens, 40 were removed before GWAS analysis. Of these, 12 were removed because their genotypes did not pass quality control (DishQC ≥0.82 and call rate ≥90% filter), and 28 did not have complete phenotypic data. As a result, 489 F2 chickens were used for the association analysis.
From the 580,961 SNPs originally available on the chicken SNP array, 399,693 SNPs segregating in the F2 population were kept for further analysis. A total of 4,304 were removed due to low minor allele frequency (MAF ≤0.02), and 23,603 SNPs located in sex chromosomes and unmapped linkage groups were also removed. After these filtering criteria, 371,786 SNPs from the autosomal chromosomes (GGA1-28) remained for the GWAS analysis. The average genotype density per chromosome was 541 SNPs/Mbp, with the lowest density being observed on GGA2 (297 SNPs/Mbp), and the highest density on GGA21 (816 SNPs/Mbp).
Descriptive Statistics
The number of animals, mean and standard deviations, variance components and estimated genomic heritabilities are presented in Table 1. Genomic heritability values ranged from 0.43 for carcass fat content (CFC) to 0.56 for abdominal fat percentage (ABFP).
Table 1.
Descriptive statistics, variance components and genomic heritability.
| Trait | N | Average ± SDa | Genetic variance | Residual variance | Total variance | Genomic heritabilityb |
|---|---|---|---|---|---|---|
| ABF | 476 | 15.60 ± 7.26 | 11.481 | 10.301 | 21.782 | 0.47 |
| ABFP | 476 | 1.56 ± 0.60 | 0.133 | 0.169 | 0.302 | 0.56 |
| CFC | 451 | 145.35 ± 40.52 | 212.237 | 159.809 | 372.047 | 0.43 |
| CFCDM | 451 | 39.75 ± 4.63 | 8.404 | 9.859 | 18.263 | 0.54 |
ABF: abdominal fat weight in grams; ABFP: abdominal fat percentage; CFC: carcass fat content in grams; CFCDM: carcass fat content on dry matter basis.
aMeans and standard errors.
bGenomic heritability estimated with a Bayes B model.
Genome-wide association analysis (GWAS)
The genomic windows associated with fat traits are detailed in Table 2. Twenty-two significant unique 1 Mb windows (based on genome position) present on GGA1, 2, 7, 15, 20, 27 and 28 were identified. The posterior probability of association (PPA), as described by Onteru et al.17, ranged from 0.41 to 0.85 for these regions, and the percentage of genetic variance explained by the windows ranged from 0.53 to 1.71.
Table 2.
Characterization of 1 Mb genomic windows associated with abdominal fat and carcass fat content traits in the Embrapa F2 Chicken Resource Population.
| Trait | GGA_Mba | Genome interval (start – end position)a | N° of SNP/window | % genetic variance explained | PPAb | SNP IDc | Model frequency |
|---|---|---|---|---|---|---|---|
| ABF | 1_52 | 52,000,127–52,998,004 | 387 | 1.23 | 0.56 | rs313050579 | 0.0108 |
| 1_53 | 53,002,697–53,997,943 | 282 | 1.32 | 0.56 | rs312317108 | 0.0158 | |
| 1_54 | 54,001,671–54,998,619 | 257 | 0.75 | 0.42 | rs15271198 | 0.0173 | |
| 1_179 | 179,001,074–179,999,169 | 411 | 0.67 | 0.54 | rs13557213 | 0.0111 | |
| 2_30 | 30,004,050–30,999,519 | 315 | 0.63 | 0.47 | rs317553502 | 0.0138 | |
| 2_61 | 61,003,805–61,992,322 | 290 | 0.94 | 0.52 | rs13619262 | 0.0527 | |
| 2_62 | 62,001,908–62,998,786 | 307 | 1.23 | 0.56 | rs314667858 | 0.0253 | |
| 27_3 | 3,000,222–3,996,811 | 820 | 0.84 | 0.77 | rs315719114 | 0.0097 | |
| 28_4 | 4,004,758–4,964,406 | 629 | 0.53 | 0.68 | rs314073448 | 0.0038 | |
| ABFP | 2_61 | 61,003,805–61,992,322 | 290 | 0.71 | 0.57 | rs13619262 | 0.0272 |
| 2_62 | 62,001,908–62,998,786 | 307 | 0.66 | 0.47 | rs14193698 | 0.0193 | |
| 7_35 | 35,001,761–35,996,723 | 386 | 0.58 | 0.58 | rs312894632 | 0.0106 | |
| 28_0 | 23,942–999,295 | 829 | 1.09 | 0.80 | rs316394502 | 0.0512 | |
| 28_3 | 3,000,142–3,988,940 | 621 | 0.82 | 0.73 | rs15251024 | 0.0146 | |
| CFC | 1_53 | 53,002,697–53,997,943 | 282 | 0.91 | 0.51 | rs314857319 | 0.0083 |
| 1_168 | 168,005,668–168,997,872 | 318 | 0.56 | 0.41 | rs312378109 | 0.0093 | |
| 1_169 | 169,001,420–169,999,438 | 346 | 0.82 | 0.44 | rs315077363 | 0.0109 | |
| 1_170 | 170,002,808–170,999,129 | 446 | 0.98 | 0.48 | rs13973557 | 0.0223 | |
| 1_171 | 171,000,120–171,999,874 | 407 | 0.91 | 0.52 | rs315852521 | 0.0142 | |
| 1_175 | 175,003,078–175,996,880 | 405 | 1.28 | 0.52 | rs313574684 | 0.0246 | |
| 7_35 | 35,001,761–35,996,723 | 386 | 0.73 | 0.52 | rs314947533 | 0.0125 | |
| 7_36 | 36,000,235–36,898,384 | 257 | 0.58 | 0.44 | rs312848275 | 0.0158 | |
| 15_9 | 9,002,743–9,999,015 | 639 | 0.82 | 0.64 | rs316091564 | 0.0637 | |
| 15_10 | 10,001,717–10,999,147 | 577 | 0.71 | 0.63 | rs13528818 | 0.0156 | |
| 28_4 | 4,004,758–4,964,406 | 629 | 0.82 | 0.75 | rs314212680 | 0.0069 | |
| CFCDM | 1_105 | 105,000,541–105,997,476 | 383 | 1.16 | 0.60 | rs13916775 | 0.0154 |
| 1_175 | 175,003,078–175,996,880 | 405 | 1.49 | 0.60 | rs313574684 | 0.0206 | |
| 1_179 | 179,001,074–179,999,169 | 411 | 0.89 | 0.53 | rs317863254 | 0.0121 | |
| 7_35 | 35,001,761–35,996,723 | 386 | 0.95 | 0.58 | rs16614131 | 0.0170 | |
| 7_36 | 36,000,235–36,898,384 | 257 | 1.23 | 0.57 | rs312848275 | 0.0257 | |
| 20_12 | 12,000,087–12,998,691 | 562 | 0.54 | 0.67 | rs739732531 | 0.0204 | |
| 28_3 | 3,000,142–3,988,940 | 621 | 0.76 | 0.62 | rs315921612 | 0.0176 | |
| 28_4 | 4,004,758–4,964,406 | 629 | 1.71 | 0.85 | rs313086976 | 0.0181 |
ABF: abdominal fat weight in grams; ABFP: abdominal fat percentage; CFC: carcass fat content in grams; CFCDM: carcass fat content on dry matter basis.
aMap position based on Gallus_gallus-5.0, NCBI assembly.
bPosterior probability of association (PPA) as described by Onteru et al.17.
cSNP within the window with the highest model frequency.
Many SNPs were fitted simultaneously in the model (see Methods), and due to high linkage disequilibrium, the QTL effect will be distributed across these markers, shrinking the SNPs effects at individual loci18. In order to identify the SNPs within the windows most probably linked to the QTL, we selected those with highest model frequency (Table 2), as adopted by Van Goor et al.19,20. The characterization of those SNPs including their effect, frequency in the population and frequency in the founder lines (TT and CC) is available in Supplementary Spreadsheet S1.
Carcass fat content exhibited the highest number of significant QTLs (Fig. 1), followed by ABF, CFCDM and ABFP (Supplementary Figs S1, S2 and S3). For CFC, the region that explained the largest amount of genetic variation was on GGA1 (175 Mb), with 1.28% of the genetic variance. For ABF, the region was on GGA1 (53 Mb), with 1.32% of the genetic variance. For CFCDM, it was on GGA28 (4 Mb), with 1.71% of the genetic variance, and for ABFP, it was on GGA28 (0 Mb), with 1.09% of the genetic variance.
Figure 1.
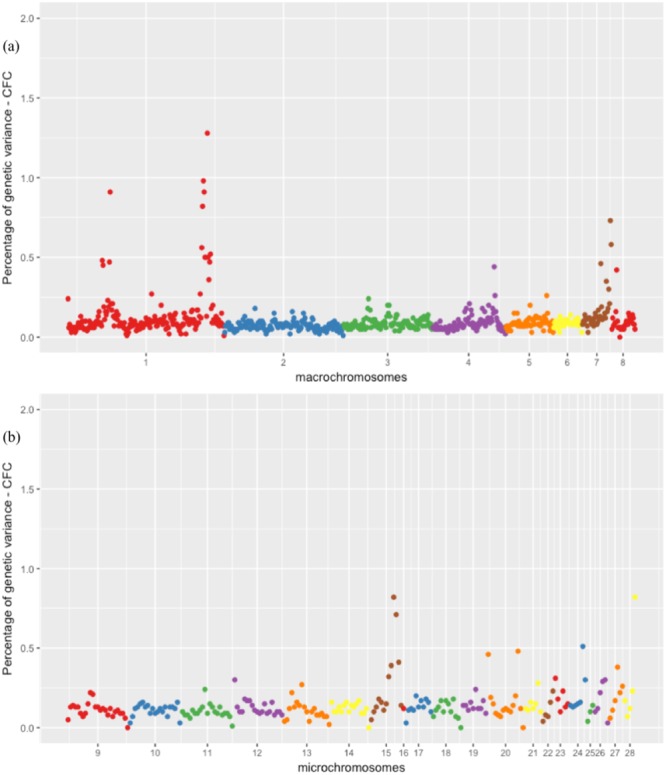
Manhattan plot of the posterior means of the percentage of genetic variance explained by each 1 Mb SNP window across the 28 autosomal chromosomes for CFC (carcass fat content in grams): (A) genomic windows located on macrochromosomes, and (B) genomic windows located on microchromosomes.
Adjacent significant windows were considered as likely representing the same QTL, and their respective percentages of genetic variance explained were summed. For abdominal fat traits, three adjacent windows on GGA1 (52–54 Mb) were associated and cumulatively accounted for 3.3% of the genetic variance for ABF, and two adjacent windows on GGA2 (61–62 Mb) cumulatively accounted for 2.17% and 1.37% of the genetic variance for ABF and ABFP, respectively (Table 2).
For carcass fat content traits, four adjacent windows on GGA1 (168–171 Mb) were associated and cumulatively accounted for 3.27% of the genetic variance for CFC; two adjacent windows on GGA7 (35–36 Mb) cumulatively accounted for 2.18% and 1.31% of the genetic variance for CFC and CFCDM, respectively; two adjacent windows on GGA15 (9-10 Mb) cumulatively accounted for 1.53% of the genetic variance for CFC; and two adjacent windows on GGA28 (3-4 Mb) cumulatively accounted for 2.47% of the genetic variance for CFCDM (Table 2).
Co-located QTLs (the same QTL associated with abdominal fat and carcass fat content traits) were identified on GGA1 at 53 Mb, GGA1 at 179 Mb, GGA7 at 35 Mb and on GGA28 (3-4 Mb).
Overlap with previously reported QTLs
The 22 QTL regions identified in our study overlapped with 56 previously published QTLs for fatness traits mapped in different populations. The QTL on GGA28 at 0 Mb is novel (Table 3).
Table 3.
Overlap of QTLs identified in our study with previously fatness QTLs reported from the Chicken QTLdb7.
| GGA (Mb) | Known QTLs for fatness traitsa |
|---|---|
| 1 (52) | ABF (#6806, #12478); ABFP (#3350); SCFT (#14359); CFWID (#14360); VISAT (#17319); SCNF (#17325); TWF (#17332) |
| 1 (53) | ABF (#6806, #6869, #9665, #12478); ABFP (#3350, #12590); SCFT (#14359); CFWID (#14360); VISAT (#17319); SCNF (#17325); TWF (#17332); CFCDM (#17117) |
| 1 (54) | SCFT (#14359); CFWID (#14360); VISAT (#17319); SCNF (#17325); TWF (#17332); CFCDM (#17117) |
| 1 (105) | ABF (#6858, #12466, #14361); ABFP (#14362); SCFT (#14359); VISAT (#17319); SCNF (#17325); TWF (#17332); CFC (#17119) |
| 1 (168-172)b | ABF (#7010, #66054); ABFP (#7011); TWF (#17332); CFC (#17129); CFCDM (#17120, #17130); SFWT (#1856) |
| 1 (175)b | ABF (#7010); CFC (#17129); CFCDM (#17120, #17130); SFWT (#1856) |
| 1 (179)b | ABF (#7010); CFC (#17129); CFCDM (#17120, #17130); SFWT (#1856) |
| 2 (30) | VISAT (#17320); SCNF (#17326, #17327); TWF (#17333) |
| 2 (61-63) | VISAT (#17320); SCNF (#17326, #17327); TWF (#17333) |
| 7 (35) | ABF (#2167) |
| 7 (36) | ABF (#2167) |
| 15 (9-11)b | ABF (#2347, #9451, #12631); ABFP (#9450); CFC (#17122); CFCDM (#17121); FATDIS (#12645) |
| 20 (12) | ABF (#19476, #30881); ABFP (#19477, #30882) |
| 27 (3)b | ABF (#11809, #11817, #66072); ABFP (#3354, #11820, #11934); CFC (#17126, #17135); CFCDM (#17125); IF (#3360) |
| 28 (0) | — |
| 28 (3) | ABF (#2417, #12632); SFWT (#2418, #12641) |
| 28 (4) | ABF (#2417, #12632); SFWT (#2418, #12641) |
ABF: abdominal fat weight; ABFP: abdominal fat percentage; CFC: carcass fat content; CFCDM: carcass fat content on dry matter basis; CFWID: cingular fat width; FATDIS: fat distribution; SCFT: subcutaneous fat thickness (fat thickness under skin); SCNF: subcutaneous neck fat weight (subcutaneous neck adipose tissue); SFWT: skin fat weight; TWF: total white fat weight (total white adipose tissue); VISAT: visceral fat weight (visceral adipose tissue weight).
aPreviously known QTLs were reported by QTL ID numbers available at Chicken QTLdb7 – release 33.
bIndicates that this genomic window overlaps with known QTLs mapped in the same population9,10.
Five of the detected QTLs overlapped with known QTLs for ABF, CFC and CFCDM mapped in the same F2 population9,10 used herein (Table 3). These known QTLs covered 1.9 Mb (#11817 and #11809), 23.1 Mb (#17129), 2.7 Mb (#17122), and 23.1 Mb (#17130 and #17120).
Positional candidate genes, overlap with selection signature regions, haplotype blocks and screening of sequencing SNPs
From the 22 QTLs identified, 14 contained PCGs selected based on Gene Ontology terms related to fat deposition from literature records. Among these, four PCGs overlapped with selection signature regions (Table 4; Supplementary Fig. S4.) identified in a previous study from our group21. In that previous study, our group reported a genome-wide characterization of regions under selection based on the Fst method applied to the sequencing variants of 14 broilers and 14 layers (founders of Embrapa F2 Chicken Resource Population)21. One PCG (CRY1) was located 1.5 kb from a selection signature region (Table 4, Supplementary Fig. S4).
Table 4.
Genomic windows associated with fat deposition traits that harbor positional candidate genes.
| GGA (Mb) | PCGa | Ensembl gene IDb | Number of SNPsc | SNP density (SNPs/kb)d |
|---|---|---|---|---|
| 1 (52) | MB | ENSGALG00000012541 | 221 | 60 |
| 1 (53)e | CRY1 g | ENSGALG00000012638 | 675 | 20 |
| 1 (54)e | CHST11 f | ENSGALG00000030607 | 2593 | 17 |
| 1 (168) | HTR2A | ENSGALG00000016992 | 535 | 21 |
| RB1 | ENSGALG00000016997 | 1673 | 20 | |
| 1 (170) | FOXO1 | ENSGALG00000017034 | 835 | 13 |
| 1 (175) | SLC7A1 | ENSGALG00000017085 | 559 | 30 |
| 2 (30) | IL6 | ENSGALG00000010915 | 161 | 60 |
| 7 (36)e | NR4A2 f | ENSGALG00000012538 | 1961 | 27 |
| GPD2 f | ENSGALG00000012543 | 628 | 15 | |
| 15 (9) | PLA2G1B | ENSGALG00000020989 | 145 | 88 |
| SIRT4 | ENSGALG00000007244 | 314 | 71 | |
| 15 (10) | SELM | ENSGALG00000025972 | 166 | 82 |
| 20 (12) | DOK5 | ENSGALG00000007786 | 663 | 19 |
| 28 (0) | SLC1A6 | ENSGALG00000000558 | 554 | 25 |
| ANGPTL4 | ENSGALG00000000619 | 305 | 43 | |
| RAB11B | ENSGALG00000000613 | 384 | 30 | |
| 28 (3) | STK11 | ENSGALG00000040008 | 738 | 22 |
| GDF3 | ENSGALG00000003161 | 103 | 49 | |
| TM6SF2 | ENSGALG00000029015 | 214 | 65 | |
| SLC25A42 | ENSGALG00000002621 | 239 | 35 | |
| SLC5A5 | ENSGALG00000041932 | 244 | 54 | |
| SLC39A3 | ENSGALG00000020582 | 170 | 116 | |
| 28 (4)e | PIK3R2 | ENSGALG00000003428 | 275 | 15 |
| INSR f | ENSGALG00000040758 | 578 | 17 | |
| SLC35E1 | ENSGALG00000003794 | 103 | 22 |
aPositional candidate genes.
bEnsembl gene ID based on Galgal5 (Ensembl Genes 90 Database).
cNumber of SNPs annotated on the PCG.
dSNP density in the respective PCG.
eIndicates that this genomic window overlaps with a selection signature region21.
fIndicates that the positional candidate gene was annotated within a selection signature region.
gIndicates that this positional candidate gene was located 1.5 kb from a selection signature region.
In order to demonstrate further support for our findings, we checked the distribution of SNPs model frequencies within the genomic windows and their overlap with selection signature regions. Manhattan plots for ABF, CFC and CFCDM are presented in Fig. 2.
Figure 2.

Manhattan plot of the SNP model frequencies distribution within each significant genomic window showing overlap with selection signature regions for: (a) abdominal fat weight (ABF); (b) carcass fat content (CFC); (c) carcass fat content on dry matter basis (CFCDM). The X-axis denotes the significant SNP window represented by the number of the respective chromosome and the Y-axis shows the model frequencies from Bayes B analysis.
Those SNPs with the highest model frequency within the large effect genomic windows are likely to be in linkage disequilibrium with the candidate genes and causative mutation. Thus, the detection of haplotype blocks was performed and three PCGs were located within haplotypes blocks that harbored the SNPs with the highest model frequency within the associated genomic window. The characterization of the genomic windows, haplotype blocks and the overlapped PCGs are shown in Table 5. All of the haplotype blocks detected and the harboring SNPs with the highest model frequency in each associated genomic window can be found in Supplementary Spreadsheet S2.
Table 5.
Characterization of the genomic windows and their respective haplotype blocks that encompass PCGs for fat deposition.
| GGA_Mba | Traits associated | SNP IDb | Haplotype blocksc | ||
|---|---|---|---|---|---|
| Start-End positiona | Size (kb) | PCGsd | |||
| 7_36 | CFC, CFCDM | rs312848275 | 36,163,395–36,333,047 | 169.653 | NR4A2, GPD2 |
| 28_4 | CFCDM | rs313086976 | 4,111,155–4,174,053 | 62.899 | INSR |
CFC: carcass fat content in grams; CFCDM: carcass fat content on dry matter basis.
aMap position based on Gallus_gallus-5.0, NCBI assembly.
bSNP within the window with the highest model frequency.
cHaplotype block that harbor the SNP with the highest model frequency within the genomic window.
dPositional candidate genes located within the haplotype blocks.
Using the SNP data generated from the whole genome sequence of the founders of this population, we observed that 15,036 SNPs were located in the 26 PCGs. The annotation of these SNPs are in Fig. 3.
Figure 3.
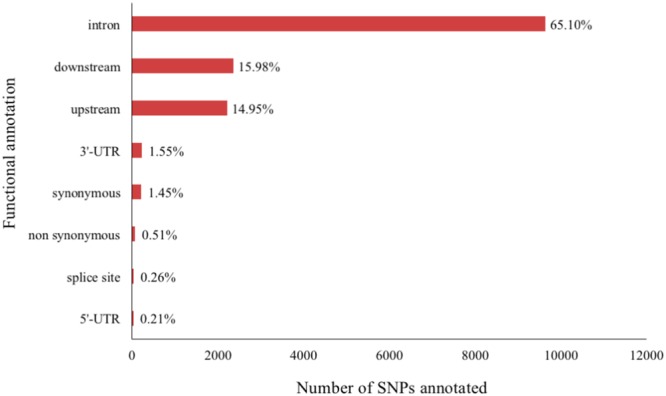
Functional annotation of unique SNPs identified in 26 PCGs. The percentage was estimated based on the total number of SNPs annotated in 26 PCGs.
We also looked for potentially deleterious and high impact sequence SNPs, which may affect protein function or result in truncated proteins, based on SIFT score estimates and Ensembl prediction. Twelve potentially deleterious SNPs were identified in eight PCGs (Table 6), but none of the mutations in our PCGs were annotated as having high impact.
Table 6.
Characterization of deleterious SNPs identified in eight PCGs.
| Associated Gene Name | SNP ID | GGA | Genome positiona | SIFT scoreb | Amino acid changed |
|---|---|---|---|---|---|
| FOXO1 | g.170581941 > C/T | 1 | 170,581,941 | deleterious low confidencec (0.01) | Pro/Leu |
| NR4A2 | g.36224286 > C/T | 7 | 36,224,286 | deleterious (0) | Val/Met |
| g.36225242 > G/T | 36,225,242 | deleterious (0) | Arg/Ser | ||
| g.36225278 > C/T | 36,225,278 | deleterious (0.01) | Val/Met | ||
| SIRT4 | rs316192467 | 15 | 9,435,175 | deleterious (0.02) | Ala/Thr |
| DOK5 | g.12473540 > C/A | 20 | 12,473,540 | deleterious (0.02) | Lys/Asn |
| ANGPTL4 | g.846035 > G/A | 28 | 846,035 | deleterious (0.03) | Ser/Phe |
| TM6SF2 | g.3553753 > T/C | 28 | 3,553,753 | deleterious low confidencec (0.01) | Leu/Pro |
| rs315426765 | 3,554,427 | deleterious (0.05) | Leu/Phe | ||
| rs741325985 | 3,554,836 | deleterious (0.04) | Val/Met | ||
| SLC39A3 | rs316529053 | 28 | 3,285,533 | deleterious (0) | Arg/His |
| SLC35E1 | g.4327417 > G/A | 28 | 4,327,417 | deleterious (0.05) | Gly/Arg |
aPosition based on Gallus_gallus 5.0 assembly.
bSIFT (Sorting Intolerant From Tolerant) score.
cDeleterious low confidence: little sequence diversity in this position affecting the substitution model and consequently, means of conservation value and the confidence of the prediction74.
Discussion
Genomic heritability
Genomic heritability was estimated using relationships inferred from high-density SNP panel genotypes instead of pedigree-based relationships. The use of close relatives and a higher density of SNPs may lead to a better genomic prediction with less bias22,23 than can be achieved using pedigree-based relationships.
For the evaluated traits, pedigree-based heritability estimates were found in the literature. In a study with 1,069 purebred full-sib male chickens, Chen et al.24 reported heritability estimates for ABF and ABFP of 0.62 and 0.24, respectively. Zerehdaran et al.25 reported heritability estimates for ABF and ABFP of 0.62 ± 0.09 and 0.71 ± 0.09, respectively, using 3,278 chickens from a meat-type population after nine generations of intercrossing. A higher value was reported for ABF (0.82) using the records of 300 chickens from a commercial female grandparent stock26, and could be explained by the fact that female broiler chickens generally deposit more abdominal fat than male broiler chickens4,27.
In the same population (Embrapa F2 Chicken Resource Population), pedigree-based heritability for ABF was estimated at 0.33 ± 0.19 in the F2-CTCT generation (layer males crossed with broiler females) and 0.82 ± 0.3 in the F2-TCTC generation (broiler males crossed with layer females)28. Considering the carcass fat content traits, heritability estimates for CFC expressed in percentage of wet carcass was 0.53 ± 0.10, and the heritability for fat percentage in dry-matter basis (CFCDM) was 0.55 ± 0.10, using records of 3,422 chickens3.
Genomic heritability estimates for ABF (0.33) and ABFP (0.31) were reported in one recent study performed with 1,408 chickens from a Brazilian broiler population under multiple trait selection29. Our genomic heritability estimates (Table 1) for fat deposition related traits were high (greater than 0.43), similar to the pedigree-based heritability reported for the same traits in the same F2 population. Therefore, a high proportion of the total variance for fat deposition traits is explained by genetic variance22, and selection against fat deposition can achieve good results in chicken.
Genome-wide association (GWAS)
We observed that genetic variation explained by each 1 Mb genomic window ranged from 0.53 to 3.3% depending on the trait analyzed. Cumulatively, the significant windows associated with ABF, ABFP, CFC and CFCDM explained 8.14%, 3.28%, 9.12% and 8.73% of the genetic variance, respectively. Previous QTL mapping studies, using microsatellites markers in the same experimental population evaluated here, detected QTLs explaining 6.65%, 12.18%, 9.9% and 11% of the phenotypic variance for ABF, ABFP, CFC and CFCDM, respectively9,10, corroborating our findings. It is important to highlight that most of the genomic windows explained <0.53% of the genetic variance for the traits analyzed in our study (Supplementary Spreadsheet S3) and these percentages were not considered.
We performed the GWAS studies in a F2 experimental population generated from a cross between a white-egg type (layer, CC) line and a meat-type (broiler, TT) line30. These lines have a different background and previously underwent multi-trait selection for different traits for several generations, exhibiting considerable differences in growth and carcass traits8,31. According to Campos et al.10, chickens from this broiler line exhibit 15 fold more fat deposition than hens from the layer line reared as broilers.
Almost all the QTLs detected overlapped with previously reported QTLs associated with fat deposition traits in this or in other populations, confirming and refining previous results. The QTL located in the first bases of GGA28 was novel and might be a population specific QTL. Additionally, the QTL located on GGA27 at 3 Mb associated with ABF overlapped with a QTL previously mapped in the TT Reference population for chicken skin weight and percentage, used as indicators of subcutaneous fat29. The TT Reference population originated after 17 generations of multiple trait selection on birds from the broiler line (TT) used in the cross to obtain the Embrapa F2 Chicken Resource Population32. Thus, our findings indicate that even after many generations of multiple trait selection, the QTL located on GGA27 still has an effect on fat deposition whereas the other QTLs detected herein were fixed in the TT Reference population.
We compared the size of the QTL regions obtained in this study with a previous one that used 128 microsatellite markers in the same population9,10. For example, the QTL for ABF on GGA27 at 3 Mb overlapped with two known QTLs mapped to a region of 1.9 Mb (#11817 and #11809). The high-density SNP panel narrowed this region down to a 1 Mb interval, thus allowing a more focused search for candidate genes.
The QTLs mapped on GGA1 (at 53 and 179 Mb), GGA7 (35 Mb), and GGA28 (3-4 Mb) were associated with both abdominal fat and carcass fat content traits, indicating that they might be pleiotropic or reflect multiple causal mutations in the same QTLs.
We checked the overlap of our QTLs associated with fat deposition related traits and previously mapped QTLs for body weight at 35 (BW35) and also 41 (BW41) days-of-age mapped in the same F2 population8,33–35. From the 22 QTLs detected, 17 did not overlap with QTLs for body weight traits as expected since we included BW42 as a covariate in our analysis. The inclusion of BW42 as a covariate allowed the detection of 17 QTLs associated with fat deposition traits but with no phenotypic association with body weight traits. The five QTLs that overlapped with BW traits may have pleiotropic effects.
Positional candidate genes for fat deposition
Positional candidate genes were selected based on their associated Gene Ontology (GO) terms and literature information. From the 26 PCGs selected, 17 showed GO terms for fat cell differentiation, insulin and triglycerides levels among other processes involved in fat deposition (Table 7).
Table 7.
List of PCGs that exhibited GO terms related to lipid metabolic processes.
| Gene | Gene Ontology terms |
|---|---|
| CRY1 | response to insulin, lipid storage, glucose homeostasis |
| HTR2A | positive regulation of fat cell differentiation |
| RB1 | regulation of lipid kinase activity |
| FOXO1 | cellular response to insulin stimulus, insulin receptor signaling pathway, negative regulation of fat cell differentiation, glucose homeostasis |
| IL6 | positive regulation of B cell activation |
| NR4A2 | fat cell differentiation |
| GPD2 | oxidation-reduction process, gluconeogenesis, glycerol-3-phosphate dehydrogenase activity |
| PLA2G1B | phospholipid metabolic process, lipid catabolic process, lipid metabolic process |
| SIRT4 | negative regulation of insulin secretion, positive regulation of lipid biosynthetic process |
| SELM | adipose tissue development |
| DOK5 | insulin receptor binding |
| ANGPTL4 | triglyceride homeostasis, negative regulation of lipoprotein lipase activity |
| RAB11B | insulin secretion involved in cellular response to glucose stimulus |
| STK11 | negative regulation of lipid biosynthetic process, glucose homeostasis |
| TM6SF2 | regulation of lipid metabolic process |
| PIK3R2 | cellular response to insulin stimulus, insulin receptor signaling pathway, cellular glucose homeostasis |
| INSR | cellular response to insulin stimulus, insulin binding, insulin-activated receptor activity, insulin-like growth factor receptor binding, insulin-like growth factor I binding, insulin-like growth factor II binding, insulin receptor substrate binding, insulin receptor signaling pathway, insulin receptor complex, glucose homeostasis, positive regulation of glucose import |
From the genes related to lipid metabolic processes described in Table 7, eight were annotated with GO terms for insulin synthesis, secretion and regulation, including the IL6 gene that exhibited a GO term for positive regulation of pancreatic β cell activation (responsible for synthesizing and secreting insulin36). Increases in insulin levels in chicken may affect expression of genes related to glucose and lipid metabolism37, consequently affecting fat accumulation. Based on these facts, we considered the genes annotated with GO terms related to insulin as PCGs for lipid metabolism and fat deposition regulation in chicken.
Several genes, namely PLA2G1B, SELM, DOK5, HTR2A, and GDF3 have been previously associated with obesity38–42. The PLA2G1B gene harbored a SNP associated with fat accumulation and distribution in humans38. In mice, the knock-out of SELM gene resulted in elevated white adipose tissue deposition39. In the DOK5 gene, genetic variants were associated with obesity in North Indian patients40. In the HTR2A gene, polymorphisms were associated with central adiposity in a study with humans41. Mice that were GDF3 deficient exhibited a modest reduction in adiposity42. These studies corroborate the selection of these genes as PCGs for fat deposition.
Four genes were located within selection signature regions previously identified21 in founders of the F2 population used in this study: CHST11, NR4A2, GPD2 and INSR (Supplementary Fig. S4), indicating that SNPs in these genes exhibit frequency differences between the parental lines, and may be associated with fat deposition. Thus, these genes exhibits additional evidences to support their selection as candidates, even considering that the selection signature regions did not overlap with the SNPs with the highest model frequency in each window (Fig. 2); in most cases, are located nearby. Moreover, NR4A2, GPD2 and INSR were located within haplotype blocks that harbored the SNP with the highest model frequency within the genomic window associated, providing additional information in support of the PCGs.
The CHST11 gene is associated with lipid metabolism, and its expression affects lipid accumulation in adipocytes43. The NR4A2 gene encodes a member of the steroid-thyroid hormone-retinoid receptor superfamily. NR4A receptors regulate hepatic glucose44, and consequently, lipid metabolism. Additionally, this gene influences retinoid signaling44–46, and although the mechanisms have still not been clearly elucidated, it is known that retinoid plays an important role in lipid metabolism47.
The protein encoded by GPD2 gene acts on the mitochondrial membrane, and its expression may affect gluconeogenesis and glucose homeostasis48. In a study with mice, Brown et al.49 reported a reduction of 40% in the weight of white adipose tissue in the individuals with knocked-out GPD2. NR4A2 and GPD2 genes overlapped with one selection signature region (Supplementary Fig. S4), indicating that selection possibly affected the frequency of SNPs in both genes. These SNPs may be associated with glucose homeostasis and lipid metabolism.
The INSR gene plays an important role in insulin signaling50, and as mentioned before, insulin levels affect lipogenesis, and consequently, lipid accumulation. Additionally, the INSR gene overlapped with a selection signature region previously reported (Supplementary Fig. S4) with genetic variants mainly fixed in the broiler line21. These findings indicate that selection could affected the frequency of SNPs in this PCG leading to changes in lipogenesis and lipid accumulation in chickens.
In humans, CRY1 is expressed in subcutaneous abdominal and visceral fat depots51. In chicken, the CRY1 gene is located 1.5 kb from a selection signature region previously identified in chicken by our group13 (Supplementary Fig. S4). Genetic variants located in this gene may have their frequencies affected by selection52.
Seven other genes (MB, SLC7A1, SLC1A6, SLC25A42, SLC5A5, SLC39A3 and SLC35E1) were not annotated with GO terms related to fat deposition but were considered PCGs for fat deposition for the reasons explained below. Myoglobin (MB) expression was associated with fatty acid metabolism in a study with mice53. Members of the solute carrier (SLC) family encodes membrane-bound transporters54, and one of these has been associated with obesity in humans55,56. Additionally, the SLC1A6 gene belongs to the SLC1 family that regulates glutamate transport, and in liver cells, this amino acid is a precursor of fatty acid biosynthesis57. In a study with rats, mice and rabbits, Collin et al.58 demonstrated that glutamate transporter activity might regulate energy balance. Energy balance is directly related with fatty acid biosynthesis and consequently, fat storage.
Some positional candidate genes were considered based on their function on lipid metabolism in mammals, because no information was available in chickens. Although many studies reported that the function of some genes can be different in chickens compared to mammals59–65, Li et al.59 reported differentially expressed genes consistent with conservation of lipid metabolism and adipogenesis processes in chicken and mammal. We performed an investigation for potential candidate genes based on GO terms and literature information, and we considered those genes as candidates for fat deposition regulation in chickens. We suggest further functional studies to validate our findings.
SNPs annotated in PCGs genes
The density of sequence SNPs in our PCGs ranged from 13 to 116 SNPs/kb, with an average of 40 SNPs/kb. Previous studies in chicken reported average density of SNPs across the entire genome ranging from 5 to 78 SNPs/kb50,66, corroborating our findings. The top three genes with the highest SNP density were SELM, PLA2G1B and SLC39A3 (Table 4). These genes should be thoroughly investigated, since this variability may be affecting fat deposition in the F2 Chicken Resource Population.
Genetic variants in PCGs that overlapped with selection signature regions may exhibit polymorphisms responsible for phenotypic variation67–69. Moreover, haplotype blocks that harbor the SNP with the highest model frequency can carry the causative mutation70,71. Thus, the genetic variants annotated in potentially functional gene regions from CRY1, CHST11, NR4A2, GPD2 and INSR are important candidates for further association and functional studies.
Approximately 65% of the sequence SNPs detected in our PCGs were located in intronic regions (Fig. 3). Intronic SNPs can modulate gene expression, and consequently, affect the phenotype72,73. However, they are commonly deemed as potentially neutral. The other 35% of the SNPs detected were annotated in functional gene regions: up/downstream from the gene, 3’and 5’-UTRs, exons (synonymous and non-synonymous), and splice sites (Fig. 3).
Variants in coding regions can be related to phenotypic variation, and, more specifically, non-synonymous variants imply amino acid changes74. Changes in amino acids can potentially affect protein function. To predict whether SNPs in coding regions are deleterious or not (may affect the protein function), we predicted the SIFT score as described in the Methods section.
Twelve potentially deleterious SNPs were identified in eight PCGs (Table 6), and most of them are located in PCGs involved in lipogenesis, levels of triglycerides and obesity. Moreover, two of these genes exhibited more than one deleterious mutation: NR4A2, involved in the regulation of hepatic glucose affecting lipid metabolism, and TM6SF2, involved in the regulation of triglyceride levels in the liver. Changes in the function of these genes may affect fat deposition in chicken. All deleterious mutations are important candidates for further association and functional studies.
From the 12 potentially deleterious SNPs, only rs315426765 SNP on the TM6SF2 gene was present in the Affymetrix genotyping array. However, that SNP was removed after genotyping quality control due to low MAF. The search for potential candidate SNPs identified previously from whole genome sequence data in similar chicken populations allowed us to identify a great number of SNPs, which are not present in the Affymetrix genotyping array, improving our chances to identify potentially causative mutations.
In summary, our study identified 22 unique QTLs for abdominal fat and carcass fat content, and approximately 40% of the QTLs detected were considered novel QTLs for the traits analyzed. The 22 QTLs detected harbored 26 PCGs that were involved in biological processes of fat deposition. Three of these 26 PCGs were located within haplotype blocks that were associated with fat traits and five of these 26 genes overlapped with selection signature regions. From the total number of SNPs annotated in PCGs, approximately 35% were in functional regions, and from those 12 were predicted to be deleterious variants. The NR4A2 gene is a strong candidate for fat deposition regulation in chicken, since it is within a QTL for carcass fat content traits, is in linkage disequilibrium with the SNP with the highest model frequency, is under selection in the founder lines, and contains three potential causal SNPs.
GWAS using a high density of SNPs allowed us to map QTLs with better resolution than previously done with microsatellite markers, thus facilitating the search for PCGs. The integration of haplotype blocks detection, selection signature regions and potentially deleterious SNPs allowed us to refine the list of PCGs for fat deposition traits. The PCGs identified, especially those within the haplotype blocks, overlapped with selection signature regions and harboring genetic variants located on potentially functional gene regions, are strong candidates for selection in poultry breeding programs aiming to improve the accuracy of selection and reduce excessive fat deposition. Further functional validation studies could be helpful to understand the role of the candidate genes and genetic variants associated with fat deposition regulation.
Methods
All experimental protocols related to animal experimentation in this study were performed in agreement with resolution number 010/2012 approved by Embrapa Swine and Poultry Ethics Committee on Animal Utilization to ensure compliance with international guidelines for animal welfare.
Chicken Population
A total of 529 chickens from the Embrapa F2 Chicken Resource Population were genotyped (28 parental chickens from layer and broiler lines; 5 chickens from F1 and 496 chickens from the F2-TCTC generations). This F2 population is the result of crosses between two closed parental lines: a broiler line (called TT) and a layer line (called CC). The TT line had been selected for higher body weight, feed conversion, carcass and breast yield, viability, fertility, hatchability, reduction of abdominal fat weight and metabolic syndromes. The CC line had been selected for egg production, egg weight, feed conversion, viability, sexual maturity, fertility, hatchability, egg quality and low body weight. More details about the Embrapa F2 Chicken Resource Population were described by Rosário et al.30. All the birds from the Embrapa F2 Chicken Resource Population were reared as broilers with free access to water and a corn and soybean meal-based diet33. More details of the diet content can be found on Nones et al.9.
This F2-TCTC generation was previously used to map numerous QTLs for performance, carcass and chemical component traits8–10,31,33,75 using microsatellite markers. For the study reported in this paper, selection of families for genotyping and GWAS was based on F1 males that appeared to be heterozygous for QTL effects reported in previous studies.
Phenotype measurement
A total of 502 chickens from the F2 population were slaughtered and eviscerated at 42 days of age, after 6 h of fasting to avoid contamination of the carcass during slaughter and post-slaughter processing. The body weight at 42 days of age (BW42) was measured and carcasses were stored at 4 °C for 6 h. Then, the carcass and its cuts were weighed. Abdominal fat was removed from chilled carcass for weighing (abdominal fat weight, ABF). Abdominal fat percentage (ABFP) was calculated dividing ABF by BW42 and multiplying by 10010.
Fat (ether extract) was measured by near-infrared reflectance spectroscopy (NIRS) and estimated as percentage according to the weight of each sample (250 g of ground and homogenized carcass)9. Carcass fat content weight (CFC) was estimated by multiplying the percentage in the sample by BW42. Carcass fat content on dry matter basis (CFCDM) was estimated dividing sample fat by carcass dry matter content and multiplying by 100. More details about trait measurement can be found in Campos et al.10, Nunes et al.3 and Nones et al.9.
DNA extraction, genotyping and quality control
Genomic DNA was extracted from blood samples with DNAzol® protocol. After extraction, DNA integrity was evaluated on agarose gel (1%), quantified in spectrophotometer NanoDrop® (Thermo Fisher Scientific), and diluted to the final concentration of 20 ng.µL−1. Diluted genomic DNA was prepared for genotyping following Affymetrix protocols and genotyped with the 600 K Axiom Chicken Genotyping Array. That array comprises SNPs chosen to be segregating for different chicken lines66.
Quality control analysis and genotype calling were performed with Affymetrix Power Tools v1.17.0 (APT). Samples that exhibited DishQC ≥0.82 and call rate ≥90% were kept for further analysis. Among those high-quality samples, the most accurate and polymorphic marker SNPs, with call rate ≥98% and minor allele frequency (MAF) ≥2%, were kept for further analysis. In this step, R scripts from the package SNPolisher were used. SNPs located in the sex chromosomes, unmapped linkage groups, without genomic annotation and those that were monomorphic were removed from the analysis. SNP annotation was based on the Gallus_gallus-5.0 chicken assembly (NCBI). Missing genotypes were replaced by their average covariate value at that locus76.
Descriptive statistics and heritability
The mean and standard deviation of each phenotype were calculated using in-house scripts in R software (http://www.r-project.org/). The estimation of variance components was performed using a Bayes C model in GenSel software18. The resultant posterior means of the variance components were used as priors in a Bayes B model to estimate genomic heritability for each trait using GenSel. Sex and hatch were included as fixed effects in the model, and BW42 was used as a fixed covariate for ABF and CFC.
Genome-wide association analysis
The SNPs retained after quality control were used in the GWAS analysis with a Bayesian approach, performed with the GenSel software18. In the first step, a Bayes C model was used to estimate the genetic and residual variances, and these values were used to run a Bayes B model, as performed by Cesar et al.76. The Bayes B model samples the effects of SNPs assuming that some fraction of their effects are zero and with unequal variance of each effect77. The mathematical model presented below was used in the association analysis:
In this model, y represents the vector of phenotypic values; X is the incidence matrix for fixed effects; b is the vector of fixed effects; k is the number of SNPs; aj is the column vector representing the SNP as a covariate in locus j coded with the number of B alleles; βj is the random substitution effect for locus j assumed to be normally distributed N (0, σ2βi) when δj = 1 but βj = 0 when δj = 0, with δj being a random variable 0/1 indicating the absence (with probability π) or presence (with probability 1-π) of the locus j in the model, and e is the vector of residual effects assumed to be normally distributed N (0, σ2e). Sex and hatch were included as fixed effects in the model, and BW42 was used as a fixed covariate for ABF and CFC.
We assumed π = 0.9988 in the Bayes B models and obtained 41,000 Markov Chain Monte Carlo (MCMC) samples with the first 1,000 samples being discarded. A map file was used to position the SNPs into 943 non-overlapping windows of 1 Mb. We adopted 1 Mb non-overlapping windows in our study in agreement with other recent GWAS studies in chickens reported in the literature using genomic prediction methodology19,20,78,79. Many SNPs were fitted simultaneously in the model and due to high linkage disequilibrium, the QTL effect can be distributed across these markers18, and these previous studies showed that the 1 Mb windows can capture the effects.
Each window is expected to explain 0.106% of the genetic variance (100%/943) based on an infinitesimal model20, and windows that explained five times more than the expected value (0.53%) were considered to be biologically significant. Thus, we selected only biologically significant windows to characterize and identify PCGs. Additionally, within each significant window (QTLs), we selected the SNP most frequently included in the model.
Overlap with previously mapped QTLs
We checked the overlaps of all genomic windows detected with QTLs previously mapped in chicken7, using Chicken QTLdb - release 33, accessed in September, 2017. We used the search tool in Chicken QTLdb website, using our QTL coordinates based on Gallus_gallus-5.0 chicken genome assembly. Previously mapped QTLs overlapped were reported by their respective QTL ID numbers. The genomic windows that did not overlap with previously annotated QTL regions were considered novel discoveries.
Identification of candidate genes, overlap with selection signature regions, haplotype blocks and SNP screening
The goal of this study was to investigate PCGs based on literature information to provide new insights for further studies, and also to better understand the genetic architecture of fat deposition traits. In this context, the list of annotated genes within each associated genomic window were searched using NCBI and OMIM databases, the BioMart tool and literature to find GO terms and biological processes related to abdominal fat, lipid metabolism, fat content, and fat deposition. For all the analyses, we considered the gene annotation from Ensembl Genes 90 Database and the Gallus_gallus-5.0 (NCBI) chicken genome assembly.
To refine the list of candidate genes, we compared our list of PCGs against selection signature regions identified in a previous study with 28 parental chickens from the two lines that generated the F2 population analyzed in our study21. That study used whole genome sequence to identify genetic variants and applied the Fst method21,80 to estimate the divergence between populations and identify regions under selection (TT vs. CC lines). We used the CrossMap tool (http://crossmap.sourceforge.net/) to convert selection signature coordinates (from Gallus_gallus-4.0) to the Gallus_gallus-5.0 chicken genome assembly (NCBI).
We also compared our list of PCGs against the haplotype blocks that harbored the SNPs with the highest model frequency within each associated genomic window. PLINK v.1.981 software was used to detect the haplotype blocks, with default parameters.
Additionally, to identify potential candidate genetic variants for fat deposition in chicken, we performed a screening of SNPs located in PCGs, using the same dataset of sequencing SNPs used to detect the selection signature regions (all the 13 million SNPs from NGS data identified were submitted to dbSNP-NCBI with the handle “LBA_ESALQ”)21.
In order to refine our list of PCG variants, we searched for genetic variants predicted as deleterious and high impact. To predict whether SNPs in coding regions are deleterious or not (may affect the protein function), we obtained the SIFT (sorting intolerant from tolerant) scores. That score is an assessment of the level of conservation in homologous protein sequences82 implemented by the VEP tool83. SIFT scores were predicted for all non-synonymous and stop codon (gained/lost) variants located in the PCGs.
The prediction of high impact SNPs was also performed using the VEP tool83 that provides an estimation of the putative impact of the variant classified as high impact, i.e. annotating all the mutations annotated as transcript ablation, splice acceptor, splice donor, stop gained, frameshift, stop loss, start lost and transcript amplification, mutations that may cause protein truncation, loss of function or trigger nonsense mediated decay (http://www.ensembl.org/info/genome/variation/predicted_data.html).
Electronic supplementary material
Acknowledgements
This study was supported by thematic project 2014/08704-0 from São Paulo Research Foundation (FAPESP). The Embrapa F2 Chicken Resource Population was funded by the Embrapa/PRODETAB project no. 038-01/01 (Brazilian Government). The authors are grateful to Ricardo Brassaloti (University of Sao Paulo) for help in genotyping the chickens. G.C.M. Moreira received fellowships from FAPESP, grants 14/21380-9 and 16/00569-1. C. Boschiero received a fellowship from the program Science Without Borders - CNPq, grant 370620/2013-5. T.F. Godoy received fellowship from FAPESP, grant 15/00616-7. F. Pértille received fellowship from FAPESP, grant 16/20440-3. L.L. Coutinho is recipient of productivity fellowship from CNPq. The authors would like to acknowledge the collaborative efforts among EMBRAPA, University of São Paulo, and Iowa State University.
Author Contributions
G.C.M.M., C.B., M.C.L., J.M.R. and L.L.C. conceived the idea of this research and participated in the experimental design. G.C.M.M., C.B., T.F.G., D.J.G. performed data analysis. G.C.M.M. drafted the manuscript. G.C.M.M., C.B., A.S.M.C., J.M.R., T.F.G., F.P., M.C.L., A.S.A.M.T.M., D.J.G. and L.L.C. collaborated with interpretation, discussion and writing of the manuscript. M.C.L., J.M.R. and L.L.C. provided the experimental environment, phenotype and data analysis support. All authors have read and approved the final manuscript.
Data Availability
All SNPs reported (identified by sequencing) were submitted to dbSNP-NCBI with the handle “LBA_ESALQ”. The datasets used and/or analysed during the current study (genotypes and phenotypes) are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-34364-0.
References
- 1.Wang SZ, et al. Quantitative trait loci associated with body weight and abdominal fat traits on chicken chromosomes 3, 5 and 7. Genet. Mol. Res. 2012;11:956–965. doi: 10.4238/2012.April.19.1. [DOI] [PubMed] [Google Scholar]
- 2.Fouad AM, El-Senousey HK. Nutritional factors affecting abdominal fat deposition in poultry: a review. Asian-Australasian J. Anim. Sci. 2014;27:1057–68. doi: 10.5713/ajas.2013.13702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nunes BDN, et al. Genetic parameters for body weight, carcass chemical composition and yield in a broiler-layer cross developed for QTL mapping. Genet. Mol. Biol. 2011;34:429–34. doi: 10.1590/S1415-47572011005000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leenstra FR. Effect of age, sex, genotype and environment on fat deposition in broiler chickens—A review. Worlds. Poult. Sci. J. 1986;42:12–25. doi: 10.1079/WPS19860002. [DOI] [Google Scholar]
- 5.Havenstein GB, Toelle VD, Nestor KE, Bacon WL. Estimates of Genetic Parameters in Turkeys.: 2. Body Weight and Carcass Characteristics. Poult. Sci. 1988;67:1388–1399. doi: 10.3382/ps.0671388. [DOI] [PubMed] [Google Scholar]
- 6.Jennen DGJ, et al. Detection and localization of quantitative trait loci affecting fatness in broilers. Poult. Sci. 2004;83:295–301. doi: 10.1093/ps/83.3.295. [DOI] [PubMed] [Google Scholar]
- 7.Hu Z-L, Park CA, Wu X-L, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013;41:D871–D879. doi: 10.1093/nar/gks1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nones K, et al. Mapping QTLs on chicken chromosome 1 for performance and carcass traits in a broiler x layer cross. Anim. Genet. 2006;37:95–100. doi: 10.1111/j.1365-2052.2005.01387.x. [DOI] [PubMed] [Google Scholar]
- 9.Nones K, et al. Quantitative trait loci associated with chemical composition of the chicken carcass. Anim. Genet. 2012;43:570–576. doi: 10.1111/j.1365-2052.2012.02321.x. [DOI] [PubMed] [Google Scholar]
- 10.Campos RLR, et al. Quantitative trait loci associated with fatness in a broiler-layer cross. Anim. Genet. 2009;40:729–36. doi: 10.1111/j.1365-2052.2009.01910.x. [DOI] [PubMed] [Google Scholar]
- 11.Georges M. Mapping, fine mapping, and molecular dissection of quantitative trait Loci in domestic animals. Annu. Rev. Genomics Hum. Genet. 2007;8:131–62. doi: 10.1146/annurev.genom.8.080706.092408. [DOI] [PubMed] [Google Scholar]
- 12.Larkina TA, et al. [Expression profiling of candidate genes for abdominal fat mass in domestic chicken Gallus gallus] Genetika. 2011;47:1140–4. [PubMed] [Google Scholar]
- 13.Resnyk CW, et al. Transcriptional analysis of abdominal fat in genetically fat and lean chickens reveals adipokines, lipogenic genes and a link between hemostasis and leanness. BMC Genomics. 2013;14:557. doi: 10.1186/1471-2164-14-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Resnyk CW, et al. RNA-Seq Analysis of Abdominal Fat in Genetically Fat and Lean Chickens Highlights a Divergence in Expression of Genes Controlling Adiposity, Hemostasis, and Lipid Metabolism. PLoS One. 2015;10:e0139549. doi: 10.1371/journal.pone.0139549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moreira GCM, et al. Variant discovery in a QTL region on chromosome 3 associated with fatness in chickens. Anim. Genet. 2015;46:141–147. doi: 10.1111/age.12263. [DOI] [PubMed] [Google Scholar]
- 16.Liu Z, Sun C, Qu L, Wang K, Yang N. Genome-Wide Detection of Selective Signatures in Chicken through High Density SNPs. PLoS One. 2016;11:e0166146. doi: 10.1371/journal.pone.0166146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onteru SK, et al. Whole Genome Association Studies of Residual Feed Intake and Related Traits in the Pig. PLoS One. 2013;8:e61756. doi: 10.1371/journal.pone.0061756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garrick DJ, Fernando RL. Implementing a QTL Detection Study (GWAS) Using Genomic Prediction Methodology. in. Methods in molecular biology (Clifton, N.J.) 2013;1019:275–298. doi: 10.1007/978-1-62703-447-0_11. [DOI] [PubMed] [Google Scholar]
- 19.Van Goor A, et al. Identification of quantitative trait loci for body temperature, body weight, breast yield, and digestibility in an advanced intercross line of chickens under heat stress. Genet. Sel. Evol. 2015;47:96. doi: 10.1186/s12711-015-0176-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Goor A, et al. Quantitative trait loci identified for blood chemistry components of an advanced intercross line of chickens under heat stress. BMC Genomics. 2016;17:287. doi: 10.1186/s12864-016-2601-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boschiero C, et al. Genome-wide characterization of genetic variants and putative regions under selection in meat and egg-type chicken lines. BMC Genomics. 2018;19:83. doi: 10.1186/s12864-018-4444-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de los Campos G, Sorensen D, Gianola D. Genomic Heritability: What Is It? PLOS Genet. 2015;11:e1005048. doi: 10.1371/journal.pgen.1005048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai H-Y, et al. Genomic prediction of host resistance to sea lice in farmed Atlantic salmon populations. Genet. Sel. Evol. 2016;48:47. doi: 10.1186/s12711-016-0226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen JL, Zhao GP, Zheng MQ, Wen J, Yang N. Estimation of Genetic Parameters for Contents of Intramuscular Fat and Inosine-5′-Monophosphate and Carcass Traits in Chinese Beijing-You Chickens<xref ref-type = "fn" rid = "FN1"><sup>1</sup></xref>. Poult. Sci. 2008;87:1098–104. doi: 10.3382/ps.2008-00147. [DOI] [PubMed] [Google Scholar]
- 25.Zerehdaran S, Vereijken ALJ, van Arendonk JAM, van der Waaijt EH. Estimation of genetic parameters for fat deposition and carcass traits in broilers. Poult. Sci. 2004;83:521–5. doi: 10.1093/ps/83.4.521. [DOI] [PubMed] [Google Scholar]
- 26.Cahaner A, Nitsan Z. Evaluation of simultaneous selection for live body weight and against abdominal fat in broilers. Poult. Sci. 1985;64:1257–63. doi: 10.3382/ps.0641257. [DOI] [PubMed] [Google Scholar]
- 27.Tůmová, E. & Teimouri, A. Review fat deposition in the broiler chicken: a review. 41, 0–0 (2010).
- 28.Faveri JC, et al. Parâmetros genéticos e efeitos de sexo e cruzamento recíproco sobre características de interesse econômico em aves F2. Arq. Bras. Med. Veterinária e Zootec. 2016;68:716–724. doi: 10.1590/1678-4162-8446. [DOI] [Google Scholar]
- 29.Moreira GCM, et al. A genome-wide association study reveals novel genomic regions and positional candidate genes for fat deposition in broiler chickens. BMC Genomics. 2018;19:374. doi: 10.1186/s12864-018-4779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosário MFdo, Ledur MC, Moura ASAMT, Coutinho LL, Garcia AAF. Genotypic characterization of microsatellite markers in broiler and layer selected chicken lines and their reciprocal F1s. Sci. Agric. 2009;66:150–158. doi: 10.1590/S0103-90162009000200002. [DOI] [Google Scholar]
- 31.Moura ASAMT, et al. Quantitative trait loci with sex-specific effects for internal organs weights and hematocrit value in a broiler-layer cross. J. Appl. Genet. 2016;57:215–224. doi: 10.1007/s13353-015-0325-2. [DOI] [PubMed] [Google Scholar]
- 32.Marchesi J. A. P., Buzanskas M. E., Cantão M. E., Ibelli A. M. G., Peixoto J. O., Joaquim L. B., Moreira G. C. M., Godoy T. F., Sbardella A. P., Figueiredo E. A. P., Coutinho L. L, Munari D. P., Ledur M. C. Relationship of runs of homozygosity with adaptive and production traits in a paternal broiler line. animal. 2017;12(06):1126–1134. doi: 10.1017/S1751731117002671. [DOI] [PubMed] [Google Scholar]
- 33.Ambo M, et al. Quantitative trait loci for performance traits in a broiler x layer cross. Anim. Genet. 2009;40:200–8. doi: 10.1111/j.1365-2052.2008.01824.x. [DOI] [PubMed] [Google Scholar]
- 34.Felício AM, et al. Identification and association of polymorphisms in CAPN1 and CAPN3 candidate genes related to performance and meat quality traits in chickens. Genet. Mol. Res. 2013;12:472–482. doi: 10.4238/2013.February.8.12. [DOI] [PubMed] [Google Scholar]
- 35.Boschiero C, et al. Association of IGF1 and KDM5A polymorphisms with performance, fatness and carcass traits in chickens. J. Appl. Genet. 2013;54:103–112. doi: 10.1007/s13353-012-0129-6. [DOI] [PubMed] [Google Scholar]
- 36.Rachdi L, et al. Dyrk1A induces pancreatic β cell mass expansion and improves glucose tolerance. Cell Cycle. 2014;13:2221–2229. doi: 10.4161/cc.29250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji B, et al. Transcriptomic and metabolomic profiling of chicken adipose tissue in response to insulin neutralization and fasting. BMC Genomics. 2012;13:441. doi: 10.1186/1471-2164-13-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson SG, et al. Linkage and potential association of obesity-related phenotypes with two genes on chromosome 12q24 in a female dizygous twin cohort. Eur. J. Hum. Genet. 2006;14:340–348. doi: 10.1038/sj.ejhg.5201551. [DOI] [PubMed] [Google Scholar]
- 39.Pitts MW, et al. Deletion of Selenoprotein M Leads to Obesity without Cognitive Deficits. J. Biol. Chem. 2013;288:26121–26134. doi: 10.1074/jbc.M113.471235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabassum R, et al. Evaluation of DOK5 as a susceptibility gene for type 2 diabetes and obesity in North Indian population. BMC Med. Genet. 2010;11:35. doi: 10.1186/1471-2350-11-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halder I, Muldoon MF, Ferrell RE, Manuck SB. Serotonin Receptor 2A (HTR2A) Gene Polymorphisms Are Associated with Blood Pressure, Central Adiposity, and the Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2007;5:323–30. doi: 10.1089/met.2007.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen JJ, et al. Deficiency of growth differentiation factor 3 protects against diet-induced obesity by selectively acting on white adipose. Mol. Endocrinol. 2009;23:113–23. doi: 10.1210/me.2007-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tasdelen I, Berger R, Kalkhoven E. PPARγ Regulates Expression of Carbohydrate Sulfotransferase 11 (CHST11/C4ST1), a Regulator of LPL Cell Surface Binding. PLoS One. 2013;8:e64284. doi: 10.1371/journal.pone.0064284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han Y-F, Cao G-W. Role of nuclear receptor NR4A2 in gastrointestinal inflammation and cancers. World J. Gastroenterol. 2012;18:6865–73. doi: 10.3748/wjg.v18.i47.6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Volakakis N, Joodmardi E, Perlmann T. NR4A orphan nuclear receptors influence retinoic acid and docosahexaenoic acid signaling via up-regulation of fatty acid binding protein 5. Biochem. Biophys. Res. Commun. 2009;390:1186–1191. doi: 10.1016/j.bbrc.2009.10.116. [DOI] [PubMed] [Google Scholar]
- 46.Mucida D, et al. Reciprocal TH17 and Regulatory T Cell Differentiation Mediated by Retinoic Acid. Science (80-.). 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 47.Bonet ML, Ribot J, Palou A. Lipid metabolism in mammalian tissues and its control by retinoic acid. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. 2012;1821:177–189. doi: 10.1016/j.bbalip.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 48.Madiraju AK, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown LJ, et al. Normal thyroid thermogenesis but reduced viability and adiposity in mice lacking the mitochondrial glycerol phosphate dehydrogenase. J. Biol. Chem. 2002;277:32892–32898. doi: 10.1074/jbc.M202408200. [DOI] [PubMed] [Google Scholar]
- 50.Rubin C-J, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–91. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- 51.Gómez-Abellán P, et al. Sexual Dimorphism in Clock Genes Expression in Human Adipose Tissue. Obes. Surg. 2012;22:105–112. doi: 10.1007/s11695-011-0539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hancock AM, Rienzo A. Di. Detecting the Genetic Signature of Natural Selection in Human Populations: Models, Methods, and Data. Annu. Rev. Anthropol. 2008;37:197–217. doi: 10.1146/annurev.anthro.37.081407.085141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hendgen-Cotta UB, et al. A novel physiological role for cardiac myoglobin in lipid metabolism. Sci. Rep. 2017;7:43219. doi: 10.1038/srep43219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He L, Vasiliou K, Nebert DW. Analysis and update of the human solute carrier (SLC) gene superfamily. Hum. Genomics. 2009;3:195–206. doi: 10.1186/1479-7364-3-2-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Durand E, et al. Polymorphisms in the amino acid transporter solute carrier family 6 (neurotransmitter transporter) member 14 gene contribute to polygenic obesity in French Caucasians. Diabetes. 2004;53:2483–6. doi: 10.2337/diabetes.53.9.2483. [DOI] [PubMed] [Google Scholar]
- 56.Suviolahti E, et al. The SLC6A14 gene shows evidence of association with obesity. J. Clin. Invest. 2003;112:1762–72. doi: 10.1172/JCI200317491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Newsholme P, et al. Glutamine and glutamate as vital metabolites. Brazilian J. Med. Biol. Res. = Rev. Bras. Pesqui. medicas e Biol. 2003;36:153–63. doi: 10.1590/S0100-879X2003000200002. [DOI] [PubMed] [Google Scholar]
- 58.Collin M, et al. Plasma membrane and vesicular glutamate transporter mRNAs/proteins in hypothalamic neurons that regulate body weight. Eur. J. Neurosci. 2003;18:1265–78. doi: 10.1046/j.1460-9568.2003.02840.x. [DOI] [PubMed] [Google Scholar]
- 59.Li H, et al. Transcriptome profile of liver at different physiological stages reveals potential mode for lipid metabolism in laying hens. BMC Genomics. 2015;16:763. doi: 10.1186/s12864-015-1943-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wiskocil R, et al. Coordinate regulation of two estrogen-dependent genes in avian liver. Proc. Natl. Acad. Sci. USA. 1980;77:4474–8. doi: 10.1073/pnas.77.8.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirchgessner TG, et al. Regulation of chicken apolipoprotein B: cloning, tissue distribution, and estrogen induction of mRNA. Gene. 1987;59:241–51. doi: 10.1016/0378-1119(87)90332-5. [DOI] [PubMed] [Google Scholar]
- 62.Hermier D, Catheline D, Legrand P. Relationship between hepatic fatty acid desaturation and lipid secretion in the estrogenized chicken. Comp. Biochem. Physiol. A. Physiol. 1996;115:259–64. doi: 10.1016/0300-9629(96)00057-6. [DOI] [PubMed] [Google Scholar]
- 63.Mason TM. The Role of Factors that Regulate the Synthesis and Secretion of Very-Low-Density Lipoprotein by Hepatocytes. Crit. Rev. Clin. Lab. Sci. 1998;35:461–487. doi: 10.1080/10408369891234246. [DOI] [PubMed] [Google Scholar]
- 64.Claire D’Andre H, et al. Identification and characterization of genes that control fat deposition in chickens. J. Anim. Sci. Biotechnol. 2013;4:43. doi: 10.1186/2049-1891-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bergen WG, Mersmann HJ. Comparative Aspects of Lipid Metabolism: Impact on Contemporary Research and Use of Animal Models. J. Nutr. 2005;135:2499–2502. doi: 10.1093/jn/135.11.2499. [DOI] [PubMed] [Google Scholar]
- 66.Kranis A, et al. Development of a high density 600 K SNP genotyping array for chicken. BMC Genomics. 2013;14:59. doi: 10.1186/1471-2164-14-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Simoni Gouveia JJ, da Silva MVGB, Paiva SR, de Oliveira SMP. Identification of selection signatures in livestock species. Genet. Mol. Biol. 2014;37:330–42. doi: 10.1590/S1415-47572014000300004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bamshad M, Wooding SP. Signatures of natural selection in the human genome. Nat. Rev. Genet. 2003;4:99–111. doi: 10.1038/nrg999. [DOI] [PubMed] [Google Scholar]
- 69.Nielsen R, Hellmann I, Hubisz M, Bustamante C, Clark AG. Recent and ongoing selection in the human genome. Nat. Rev. Genet. 2007;8:857–868. doi: 10.1038/nrg2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wong GK-S, et al. A genetic variation map for chicken with 2.8 million single-nucleotide polymorphisms. Nature. 2004;432:717–22. doi: 10.1038/nature03156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng C, et al. A cis-regulatory mutation of PDSS2 causes silky-feather in chickens. PLoS Genet. 2014;10:e1004576. doi: 10.1371/journal.pgen.1004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shastry BS. SNPs: Impact on Gene Function and Phenotype. Methods in molecular biology (Clifton, N.J.) 2009;578:3–22. doi: 10.1007/978-1-60327-411-1_1. [DOI] [PubMed] [Google Scholar]
- 73.Pinsonneault JK, et al. Intronic SNP in ESR1 encoding human estrogen receptor alpha is associated with brain ESR1 mRNA isoform expression and behavioral traits. PLoS One. 2017;12:e0179020. doi: 10.1371/journal.pone.0179020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 75.Baron EE, et al. QTL for percentage of carcass and carcass parts in a broiler x layer cross. Anim. Genet. 2011;42:117–124. doi: 10.1111/j.1365-2052.2010.02105.x. [DOI] [PubMed] [Google Scholar]
- 76.Cesar AS, et al. Genome-wide association study for intramuscular fat deposition and composition in Nellore cattle. BMC Genet. 2014;15:39. doi: 10.1186/1471-2156-15-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rolf MM, et al. Comparison of Bayesian models to estimate direct genomic values in multi-breed commercial beef cattle. Genet. Sel. Evol. 2015;47:23. doi: 10.1186/s12711-015-0106-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolc A, et al. Genome-wide association analysis and genetic architecture of egg weight and egg uniformity in layer chickens. Anim. Genet. 2012;43:87–96. doi: 10.1111/j.1365-2052.2012.02381.x. [DOI] [PubMed] [Google Scholar]
- 79.Wolc A, et al. Genome-wide association study for egg production and quality in layer chickens. J. Anim. Breed. Genet. 2014;131:173–182. doi: 10.1111/jbg.12086. [DOI] [PubMed] [Google Scholar]
- 80.Weir BS, Cockerham CC. Estimating F-Statistics for the Analysis of Population Structure. Evolution (N. Y). 1984;38:1358. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 81.Purcell S, et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McLaren W, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–70. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All SNPs reported (identified by sequencing) were submitted to dbSNP-NCBI with the handle “LBA_ESALQ”. The datasets used and/or analysed during the current study (genotypes and phenotypes) are available from the corresponding author on reasonable request.