

Abstract
Blood platelets can interact with bacteria, possibly leading to platelet activation, cytokine and microparticle release and immune signalling. Besides, bacteria can also affect the platelet RNA content. We investigated the impact of non-pathogenic K12 and pathogenic O18:K1 Escherichia (E.) coli strains on platelet activation, RNA expression patterns, and selected proteins. Depending on bacteria concentration, contact of platelets with E. coli K12 lead to an increase of P-selectin (24–51.3%), CD63 (15.9–24.3%), PAC-1 (3.8–14.9%) and bound fibrinogen (22.4–39%) on the surface. E. coli O18:K1 did not affect these markers. Sequencing analysis of total RNA showed that E. coli K12 caused a significant concentration change of 103 spliced mRNAs, of which 74 decreased. For the RNAs of HMBS (logFC = +5.73), ATP2C1 (logFC = −3.13) and LRCH4 (logFC = −4.07) changes were detectable by thromboSeq and Tuxedo pipelines. By Western blot we observed the conversion of HMBS protein from a 47 kDA to 40 kDa product by E. coli K12, O18:K1 and by purified lipopolysaccharide. While ATP2C1 protein was released from platelets, E. coli either reduced the secretion or broke down the released protein making it undetectable by antibodies. Our results demonstrate that different E. coli strains influence activation, RNA and protein levels differently which may affect platelet-bacteria crosstalk.
Introduction
Blood platelets are anucleated cells, derived from megakaryocytes in the bone marrow. They are continuously shed into and cleared from the blood stream maintaining a high abundance with 150–400 × 109 cells per litre of whole blood.
Platelets have an important role in haemostasis and thrombosis. They are packed with organelles, granules, RNA and proteins, which they primarily receive from their precursor cells. They have the capability to sequester proteins and RNA while in circulation1,2. It has been shown that platelets have a rich repertoire of RNAs, including ribosomal, circular and micro RNAs3–5, and ~9500 messenger RNAs6. Platelets contain a functional cellular machinery and have the capability to splice pre-mRNA into its mature form7. It has been shown that activation of splicing can be induced by lipopolysaccharide (LPS)8, thrombin9 or a septic environment10. Furthermore, cancer11,12 and cardiovascular diseases13 can influence the platelet RNA profile. Upon thrombin activation, translation of certain spliced RNAs to proteins has been reported14,15, which proved the presence of a translational machinery in platelets.
Besides haemostasis, platelets are important for humoral as well as cellular immune responses. They are able to interact with bacteria, which may result in their activation, aggregation, release of granules and platelet-leukocyte complex formation16–19. Recently it has been shown that platelets can act as cellular scavengers; they collect deposited bacteria and recruit phagocytes to boost the inflammatory reaction20. The interaction of platelets with Gram-positive bacteria, such as Staphylococcus aureus and Streptococcus sanguinis, has been extensively studied. It has been shown that both strains are able to trigger platelet aggregation21,22, S. sanguinis can induce cytokine release from platelets23, whereas S. aureus enhances thrombocytopenia24. The crosstalk between platelets and Gram-negative bacteria is less well characterized, although it has been shown that Gram-negative Escherichia coli25,26, Helicobacter pylori27–30 and Klebsiella pneumoniae31–33 have an activating or - in some cases - aggregating effect on platelets. E. coli are commensal bacteria of the gastrointestinal tract in humans and rarely cause disease. However, clones with specific virulence attributes exist which are able to induce clinical syndromes such as enteric disease, urinary tract infections and sepsis34. Some E. coli strains interact with platelets via the LPS ligand TLR435, FcγRIIA or integrin complex αIIbβ336,37. Little is known about the molecular consequences of the interactions between platelets and E. coli, and how they influence RNA or protein expression patterns or release of platelet contents.
We investigated the effects of a non-pathogenic (K12) and a pathogenic (O18:K1) E. coli strain on human platelets. We found that contact with E. coli K12 increases the activation markers P-selectin and CD63 on the platelet surface as well as PAC-1 and fibrinogen binding, while the pathogenic E. coli O18:K1 did not affect these markers. By next generation RNA sequencing, we found that the two E. coli strains affected different spliced platelet RNAs (mRNAs). Using two bioinformatics pipelines for analysis of RNA fingerprints we identified significant effects of E. coli on the mRNAs HMBS, ATP2C1 and LRCH4. To see whether these three proteins were present in platelets and were influenced by the bacteria, we analysed HMBS, ATP2C1 and LRCH4 in platelet lysates and releasates by Western blot and ELISA.
Results
E. coli K12 affects platelet activation
The effect of non-pathogenic (K12) and pathogenic (O18:K1) E. coli strains on platelet activation was measured by flow cytometry analyses of P-selectin and CD63 expression (Fig. 1), as well as the fibrinogen binding capacity of platelets (Fig. 2).
Figure 1.

P-selectin and CD63 expression on platelets after co-incubation with bacteria or platelet activators. P-selectin (a,b) or CD63 (c,d) were measured on the platelet surface by flow cytometry after gating for the presence of CD41. Platelets incubated without bacteria (PLT) and E. coli K12 or O18:K1 co-incubated platelets (platelet-bacteria ratios 1:1, 1:5, 1:10) were analysed at zero hours (a,c) and after three hours (b,d) incubation. The activating effect of bacteria was compared to platelet activation by TRAP, ADP or LPS after 15 minutes or three hours incubation time. The data represents percentages (mean ± standard error of the mean) from 3–6 independent experiments. Activation was compared to PLT controls, significance levels are: *p < 0.05, **p < 0.01, and ***p < 0.001. TRAP, thrombin receptor activating peptide 6; ADP, adenosine diphosphate; LPS, lipopolysaccharide.
Figure 2.

PAC-1 antibody and fibrinogen binding on platelet surface after co-incubation with E. coli K12 or O18:K1. PAC-1 antibody (a,b) and fibrinogen (c,d) binding were measured on the platelet surface by flow cytometry after gating for the presence of CD61. Platelets incubated without bacteria (PLT) and E. coli K12 or O18:K1 co-incubated platelets (platelet-bacteria ratios 1:1, 1:5, 1:10) were analysed at zero hours (a,c) and after three hours (b,d). TRAP treated platelets served as a positive control after 15 minutes or three hours incubation time. The data represents percentages (mean ± standard error of the mean) from three independent experiments. Activation was compared to PLT controls, significance levels are: *p < 0.05, **p < 0.01, and ***p < 0.001. TRAP, thrombin receptor activating peptide 6.
We observed an increase of P-selectin (24–51.3%, n = 3, p < 0.05) on the platelet’s surface exposed to E. coli K12 for either zero or three hours incubation compared to platelets not incubated with bacteria (Fig. 1a,b). P-selectin expression correlated with the number of bacteria added. The effect by E. coli K12 was rapid and reached the levels achieved by platelet activators TRAP and ADP already few minutes after addition of the bacteria (zero hours, 1:5 platelet to bacteria ratio).
Similarly, CD63 also increased on the surface of platelets incubated with E. coli K12 (15.9–24.3%, n = 3, p < 0.05) compared to platelets not exposed to bacteria. Apparently, this effect was independent of the bacteria concentration (Fig. 1c,d).
Furthermore, we observed that E. coli K12 rapidly induced fibrinogen binding to the platelet surface (22.4–39%, n = 3, p < 0.05) compared to control platelets (Fig. 2c,d).
Exposure of platelets to E. coli O18:K1 for zero or three hours did not induce a measurable increase of either P-selectin or CD63 on the platelet surface (n = 3) (Fig. 1a–d), and only insignificantly affected fibrinogen (n = 3) (Fig. 2c,d).
The responses of platelets to LPS are still controversial in spite of many attempts for clarification38,39. We observed that LPS was only able to induce a minor increase of surface P-selectin after three hours incubation (non-significant).
Also low number of E. coli K12 and O18:K1 per platelet (i.e. 10:1 platelet to bacteria ratio) did not increase platelet surface P-selectin or CD63 on co-incubated platelets versus control conditions (data not shown).
E. coli K12 induces activation of the integrin αIIbβ3 receptor
It is essential for platelet aggregation that integrin αIIb and integrin β3 form an active complex (integrin αIIbβIII) to which fibrinogen can bind. The complex formation is detected via binding a PAC-1 antibody, what we evaluated on platelets incubated without or with E. coli bacteria.
We observed that platelets exposed to E. coli K12 showed PAC-1 positivity already after few minutes (zero hours) of contact between platelets and E. coli K12 what corresponded to the number of bacteria added (3.8–8.6%, n = 3, non-significant) (Fig. 2a). The antibody binding was increased after three hours incubation in samples containing a 5-fold or 10-fold bacteria excess over platelets (8.4–14.9%, n = 3, p < 0.05) (Fig. 2b).
E. coli O18:K1 did not trigger the binding of PAC-1 antibody (n = 3) (Fig. 2a,b).
Importantly, both bacteria strains were alive, but did not show measurable growth after three hours incubation in SSP + buffer at room temperature (sub-optimal conditions for bacteria growth) compared to zero hours. The presence of washed platelets did not significantly affect the number of E. coli K12 or O18:K1 colonies (data not shown).
E. coli K12 differentially alters platelet mRNAs
It has been shown that LPS and a septic environment may affect platelet RNA splicing8,10. We measured the influence of external platelet activation by E. coli strains on platelet mRNA repertoire by high-throughput mRNA-sequencing.
We incubated platelets isolated from healthy individuals (n = 4) with both E. coli strains in 1:1 platelet to bacteria ratio. Platelets of the same healthy individuals incubated without bacteria served as controls. Total RNA was isolated and subjected to RNA-sequencing. We included a poly-A-tailed oligo-dT amplification protocol to minimize the contribution of bacterial RNA to the platelet RNA profiles. We used thromboSeq pipeline11,12 to analyse our data. First, an average 9.1 × 106 reads in total per sample (n = 12, (SD) = 1.8 × 106) was observed, from which approximately 6.5 × 106 (n = 12, (SD) = 1.6 × 106) mapped to the human reference genome. We allowed for maximum ten mismatches per 100 base pairs RNA-sequencing reads between our reads and the human reference genome. Of these, 1.2 × 106 corresponded to intron-spanning domains (n = 12, (SD) = 2.6 × 105) (Supplementary Table S1). We specifically investigated intron-spanning RNA reads to uncover the potentially spliced RNA repertoire. We filtered for low abundant RNAs (logCPM >3) and removed those from the dataset. The RNA-sequencing profiles of platelets correlated to previously published platelet RNA-sequencing datasets (Supplementary Fig. S2)3,6,40.
After E. coli K12 exposure 3072 platelet mRNAs were identified with high abundance, whereas 3095 mRNAs were detected after E. coli O18:K1 exposure of platelets. Thus, incubation of platelets from four healthy donors with E. coli K12 (Fig. 3) or E. coli O18:K1 (Supplementary Fig. S3) (1:1 platelet to bacteria ratio, three hours incubation) influenced platelet RNAs.
Figure 3.

Changes of mRNAs in platelets after exposure to E. coli K12. Analysis with thromboSeq pipeline. (a) Heatmap of platelet mRNA profiles of control (grey) and E. coli K12 co-incubated platelets (green) after three hours incubation in 1:1 platelet-bacteria ratio. (b) Volcano plot of mRNAs of control versus E. coli K12 (1:1) exposed platelets. RNAs of which the concentrations changed significantly after E. coli K12 co-incubation are labelled in red (p < 0.05). (c) Gene ontology (GO) analysis of mRNAs with significantly changed concentrations after E. coli K12 exposure (p < 0.05) using DAVID functional annotation analysis (cluster enrichment score >1). (d) Logarithm 2 fold change (logFC) and normalized counts (norm. counts) of HMBS, ATP2C1 and LRCH4 RNAs in platelets exposed to E. coli K12 bacteria compared to control platelets. Cl., Cluster.
By ANOVA likelihood-ratio test for detection of differentially expressed RNAs, we could show that exposure of platelets to E. coli K12 affected the concentration of 103 spliced RNAs statistically significantly (n = 4 individuals’ platelets incubated without E. coli, n = 4 individuals’ platelets incubated with E. coli K12, p < 0.05). The level of 29 RNAs was increased (+3.77 average logarithm 2 fold change (logFC)), and of 74 RNAs decreased (−4.98 average logFC) (Fig. 3b). The changes allowed a clear differentiation between pre- and post- E. coli K12 contact (p < 0.001 by Fischer’s exact test) (Fig. 3a).
Interestingly, only 21 RNAs which were affected overlapped between E. coli K12 and O18:K1 profiles (Supplementary Fig. S3d).
Gene ontology (GO) functional annotation analysis using Database for Annotation, Visualization and Integrated Discovery (DAVID) was performed for all RNAs with significantly changed concentration (p < 0.05) (Fig. 3c, Supplementary Tables S3, S4). We found that incubation of platelets with E. coli K12 caused an enrichment of RNAs with the following functional characteristics: involved in splicing (Cluster 4, 15 RNAs), cell-cell adhesion (Cluster 5 with 7 RNAs), related to Golgi apparatus (Cluster 2, 11 members) and ubiquitin related processes (Cluster 1 with 13 and Cluster 3 with 5 RNAs) (cluster enrichment score >1).
When we tested the effects of E. coli K12 on platelet RNA concentrations with the Tuxedo RNA-sequencing processing41–43 pipeline, we observed significant concentration changes of RNAs, particularly hydroxymethylbilane synthase (HMBS), encoding for the protein porphobilinogen deaminase, an enzyme contributing to heme biosynthesis; ATPase secretory pathway Ca2+ transporting 1 (ATP2C1), a protein involved in calcium ion transport in a magnesium-dependent manner; and leucine rich repeats and calponin homology domain containing 4 (LRCH4), translated to a protein with leucine-rich repeats (LRR) involved in ligand binding in both pipelines (Fig. 3d).
We could validate the presence of HMBS, ATP2C1 and LRCH4 in platelets by RT- PCR analysis. Even though RT-PCR is a semi-quantitative method, we could confirm the same trend of changes of the tested platelet RNAs induced by E. coli K12 that we observed by RNA-seqencing. Using human-sequence specific RT-PCR primers, we made sure that the changes are independent of potential HMBS, ATP2C1, and LRCH4 production by bacteria (data not shown).
HMBS RNA44 as well as ATP2C1 RNA and protein45,46 have been reported to be present in platelets, but their functions remain largely unclear. The presence of LRCH4 had not been described in platelets yet. Until now, the effect of E. coli on these RNAs in platelets has not been investigated. Our data indicates that exposure of platelets to E. coli K12 results in an increase of HMBS (logFC = +5.73), and a decrease of ATP2C1 (logFC = −3.13) and LRCH4 (logFC = −4.07) RNA.
Effect of E. coli on HMBS, ATP2C1 and LRCH4 proteins in platelets
Alterations of platelet mRNA may influence the platelet protein content as well.
By Western blot analysis we could demonstrate the presence of HMBS, ATP2C1 and LRCH4 proteins in platelets before and after E. coli K12 exposure (in 1:1, 1:5 and 1:10 platelet to bacteria ratios) (Fig. 4a, Supplementary Fig. S4).
Figure 4.

Western blot and ELISA analysis of HMBS and ATP2C1. (a) Cell lysates were applied to SDS-PAGE gels under reducing conditions. HMBS and ATP2C1 protein were detected in platelets using a polyclonal antibody. Incubation of platelets with E. coli K12 and E. coli O18:K1 in 1:5 or 1:10 platelet-bacteria ratios converted HMBS 47 kDa form to a 40 kDa protein. ATP2C1 was not affected. GAPDH was used as a loading control. (b) The releasates of the platelet-bacteria mix were collected after centrifugation (500 g, 10 minutes without break). HMBS, ATP2C1 and LRCH4 levels were measured by ELISA. Data represents the mean of three independent experiments (n = 3). ATP2C1 was detectable in platelet supernatants, while HMBS and LRCH4 proteins were either not released from platelets or in concentrations below the detection level of the ELISA (data not shown). BDL, below detection limit. Western blot results are representative image of three replications. The same exposure was applied equally across the entire image. The original pictures of the full-length western blots can be found in Supplementary Fig. S7a, b, c, d, g, h.
We detected no visible concentration difference or molecular weight change of ATP2C1 and LRCH4 in platelets after E. coli K12 exposure. In contrast, when present in 5-fold or 10-fold excess over platelets, E. coli K12 converted HMBS from a 47 kDa form to a 40 kDa molecular weight protein.
We also evaluated the effect of E. coli O18:K1 (Fig. 4a, Supplementary Fig. S4) as well as of the platelet agonists TRAP, ADP and LPS on the HMBS, LRCH4 and ATP2C1 proteins (Fig. 5, Supplementary Fig. S5). We saw that E. coli O18:K1 and LPS converted HMBS to the 40 kDa form. Interestingly, LPS had little influence on platelet activation of washed platelets (Fig. 1). Neither E. coli O18:K1, nor the three platelet activators had any effect on the protein levels or molecular weight of ATP2C1 or LRCH4.
Figure 5.
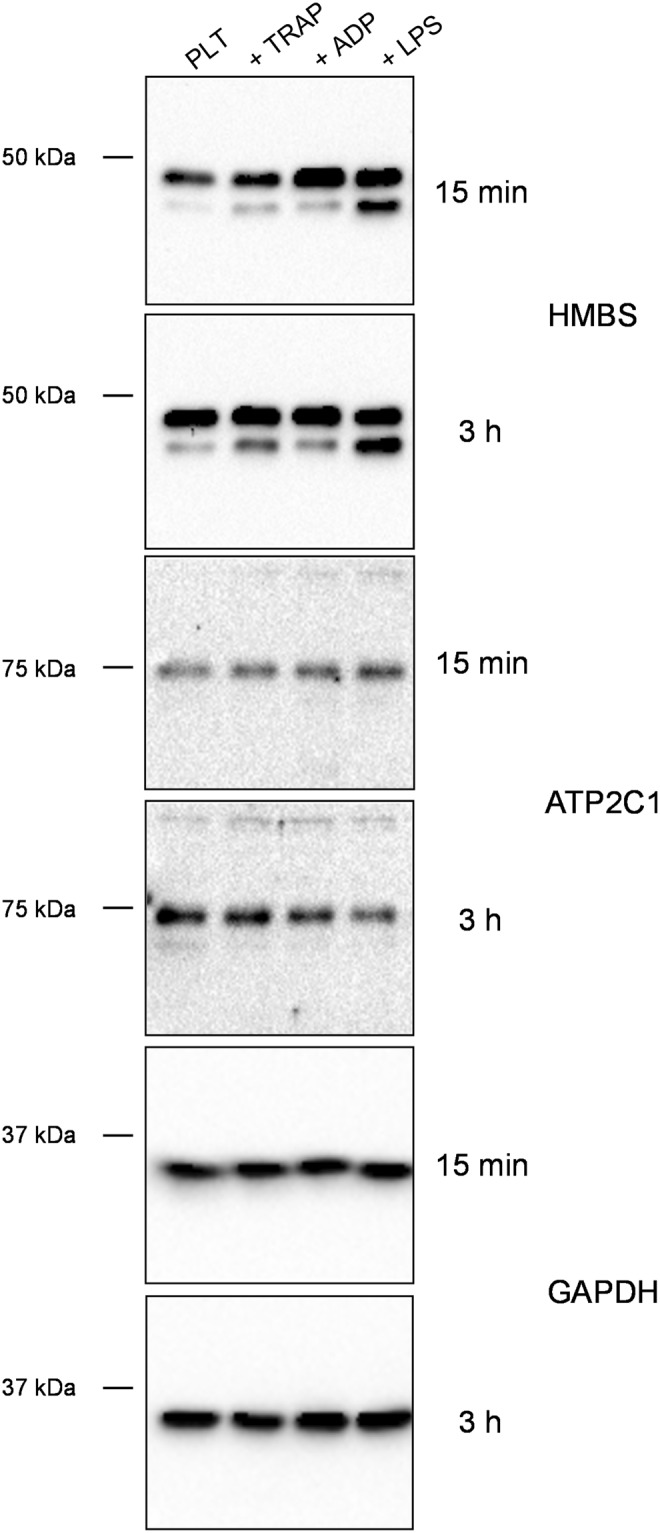
Effect of platelet activators on HMBS and ATP2C1 proteins. Electrophoretic migration behaviour of HMBS and ATP2C1 was analysed in platelets treated with different activators. The molecular weight of HMBS in platelets was changed by LPS similar to the changes caused by E. coli K12 and O18:K1. TRAP and ADP had no effect on the molecular weights of HMBS and ATP2C1. TRAP, thrombin receptor activating peptide 6; ADP, adenosine diphosphate; LPS, lipopolysaccharide. Western blots show representative images of three replicative experiments. The same exposure was applied equally across the entire image. The original pictures of the full-length western blots can be found in Supplementary Fig. S7i, j, k, l, m, n, o, p.
ATP2C1 seems to be released from platelets; E. coli exposure seems to reduce the release
It is known, that platelets can sequester1,2 and release47,48 RNA and proteins. By ELISA analysis of the supernatant obtained after incubation of platelets in buffer for zero and three hours, we observed ~0.5 nanogram/milliliter of ATP2C1 protein in the supernatant (Fig. 4b). This potential release of ATP2C1 seems to be reduced by the interaction of platelets with E. coli K12 and O18:K1, especially when measured at three hours post-incubation. HMBS or LRCH4 could not be detected in the supernatant of platelets or platelets exposed to bacteria with the applied ELISA assays (data not shown).
Discussion
Platelets are able to interact with different bacteria directly via platelet surface receptors such as P-selectin, glycoproteins49 and TLR450, indirectly through plasma proteins51, or via bacteria released exotoxins52. The interaction with E. coli can be mediated through FcγRIIa, integrin αIIbβ336 and shiga toxin53 or LPS via TLR454, however the latter one seems to be dependent on the surface structure of the bacteria.
We observed that platelet exposure to E. coli K12 led to an increased surface expression of the activation markers P-selectin and CD63. Presumably, platelets release contents of α- granules and dense bodies upon interaction with this bacteria strain. Furthermore, E. coli K12 also triggers PAC-1 antibody and fibrinogen binding to integrin αIIbβ3 on platelets in a bacteria concentration dependent manner. The presence of active integrin αIIbβ3 on the platelet surface is a prerequisite for platelet aggregation. Interestingly, the pathogenic E. coli O18:K1 was not able to induce an increase of the platelet surface activation markers and it also did not significantly affect integrin αIIbβ3 activity.
A possible explanation for the difference between the pathogenic and non-pathogenic E. coli may be found in the surface of the two strains. E. coli strains carrying an O side-chain on the surface besides the lipid A and core oligosaccharides55 are denoted as “smooth”, like the pathogenic E. coli O18:K1. In contrast, “rough” strains lack the O-antigen, such as the non-pathogenic E. coli K12. In vitro studies show differences in the activity of the two types of LPS, with “rough” LPS activating a broader range of cells with higher efficiency compared to the “smooth” form. It has been proven, that macrophages are able to respond to “rough” LPS and lipid A, but not to “smooth” LPS56. The “smooth” form requires CD14 to activate immune cells57. Possibly, the same holds true for platelets in vitro. Studying the contribution of plasma proteins to platelet - E. coli interaction would be an interesting future aspect to consider.
In in vivo comparisons, several studies showed an involvement of the complement system in LPS - induced platelet granular release and accumulation. It was found that O-antigen is important, but is not the only virulence factor influencing platelet activation57–59. The fact that in spite of its ability to activate platelets E. coli K12 is non-pathogenic while O18:K1 cannot activate washed platelets but is pathogenic may be associated with the sensitivity to the complement system. While E. coli K12 is a complement sensitive strain, E. coli O18:K1 is complement resistant and uropathogenic. The O antigen on the surface reduces activation of the alternative complement pathway, and the K1 antigen of this strain inhibits the classical complement pathway60,61. The complement resistance may contribute to the survival of the bacteria strain in the blood stream.
We showed that both E. coli strains were able to induce alterations in the platelet RNA repertoire. The E. coli K12 exposure lead to a decreased expression of 74 out of 103 RNAs in comparison to platelets not co-incubated with E. coli K12.
DAVID GO functional annotation analyses revealed an enrichment of groups of RNAs with common functional characteristics in platelets after incubation with E. coli K12 (cluster enrichment score >1) (Fig. 3c). In Cluster 2, Golgi apparatus and membrane related RNAs were enriched. It is known, that in platelets Golgi elements support selective modification and secretion of molecules, like enzymes or sugar nucleotides62. It has been shown that blocking of the Golgi apparatus may result in decreased CD40L expression63. ATP2C1, of which the RNA concentration is decreased by the presence of E. coli, is found in this cluster. ATP2C1 has a role in Ca2+/Mn2+ transport and membrane trafficking, localized on the Golgi apparatus. It has been reported that a deficiency of the protein induces defects in secretory pathways64. Interestingly, RNAs annotated to ubiquitination processes, GPVI activation and platelet survival were also affected65–67. Platelets with an impaired secretion system cannot be activated and cannot release microbicidal components or activate other immune cells. This may result in improved survival of bacteria in the blood stream. Cluster 4 comprises 15 RNAs involved in mRNA splicing. Perhaps, platelet - E. coli K12 interaction influences splicing and translation of proteins affecting RNA splicing, however that needs further investigations. Interestingly, RNAs annotated to cell-cell adhesion (Cluster 5) were also enriched. This suggests that cell adhesion mechanisms occurring prior to platelet activation19 may be induced by E. coli. A number of other possibly relevant RNAs were also affected.
The analysis of platelet RNA after exposing platelets to bacteria for three hours provides an interesting insight into RNA concentration changes. However, including further time points (e.g. 15 minutes, one hour) will reveal more information about the expression kinetics of different RNA subsets. Besides, inter-individual variation might result in differential platelet reactivity. Kinetic analysis might also add knowledge on the differences experienced between individuals (Fig. 3d).
It is known that changes in RNA concentrations are not always paralleled by a change of expression of the corresponding proteins68. Possibly, a reduced level of a spliced RNA indicates either translation to protein, or release of this RNA into the supernatant, or degradation of the RNA. Therefore, we evaluated whether HMBS, ATP2C1 and LRCH4 proteins were present and/or affected in platelet lysates and supernatants after exposure to E. coli strains.
In humans, two isoforms of HMBS enzyme have been described: a ubiquitously expressed “housekeeping” form69 and a shorter erythroid form which is apparently only present in erythropoetic cells70. The role of HMBS in platelets remains so far unclear. The two isoforms are transcribed from a single gene, but encoded by two different, alternatively spliced mRNAs. Surprisingly, we did not observe the two alternatively spliced mRNAs, but identified two protein forms in platelets. Incubation of platelets with E. coli or with LPS very quickly converts the larger into the shorter form. We can only speculate that HMBS may be relevant for platelet-bacteria interaction via the LPS pathway.
Interestingly, we found that ATP2C1 was released from platelets incubated without bacteria (~0.5 ng/ml). This release happened almost immediately after the start of the incubation, but decreased in platelets co-incubated with E. coli K12 and O18:K1. The decrease was more pronounced in the presence of a higher amount of both bacteria strains (1:5 and 1:10 platelet to bacteria ratio). The effect of E. coli on the release of ATP2C1 could represent a defence mechanism, and it cannot be excluded that similar effects may exist for other platelet-derived proteins. The reduced levels of ATP2C1 RNA in the platelets (Fig. 3d) together with the reduced protein levels in the supernatant could also indicate an active translation of ATP2C1 protein and retention in the platelets in the presence of bacteria, even though the qualitative Western blot analysis did not show major differences of this protein in platelets incubated without or with E. coli. To clarify what happens to ATP2C1 RNA, protein and its secretion in the presence of bacteria will require further studies.
Our results show that the changes of platelet RNA levels induced by bacteria cannot be directly translated to the pathogenicity status of bacteria. Non-pathogenic strains which do not trigger sepsis or bacteraemia, can still cause changes in platelet RNA and protein profiles, as we could show for E. coli K12; and even though the pathogenic E. coli O18:K1 was not able to induce an increase of platelet surface activation markers, it significantly affected platelet RNAs and certain proteins. We suspect that the altered RNA and protein expressions could affect platelets or other immune cells. Further investigations regarding platelet RNA and protein analysis will be necessary for a more detailed understanding of platelet function in bacterial infections, for instance urinary tract infection or sepsis.
Methods
The study was approved by the Ethics Committee of the Medical University of Vienna and all research was performed according to the ethical guidelines.
Isolation of platelets
Citrated blood of healthy donors was used to isolate platelets. The blood was collected at the Department of Transfusion Medicine (Medical University of Vienna), and all donors gave written informed consent. Platelet rich plasma (PRP) was prepared using centrifugation at 150 g for 15 minutes at room temperature. Since immune-depletion of platelet suspension is not suitable for RNA profiling, PRP was applied to an Optiprep (Axis-Shield, Oslo, Norway) density gradient and spun at 350 g for 15 minutes to decrease the leukocyte number of the suspension. After harvesting the platelet layer, cells were washed with HEPES-Tyrode buffer (10 mM HEPES, 137 mM NaCl, 2.8 mM KCl, 1 mM MgCl2,12 mM NaHCO3, 0.4 mM Na2HPO4, 5.5 mM glucose, and 0.35% bovine serum albumin [BSA]). Centrifugation was performed without break, and before every centrifugation step, 400 nM prostaglandin I2 (PGI2) (Sigma-Aldrich, Munich, Germany) was added to prevent platelet activation. Washed platelets were resuspended in SSP + buffer (Macopharma SA, Turcoing, France) and kept on a see-saw shaker (10 rpm) for one hour before starting the experiments. Platelet and leukocyte counts were determined on a Sysmex XE-2100 (Sysmex, Kobe, Japan) instrument.
After optimizing the isolation protocol, the leukocyte contamination (CD45 positivity) of platelet suspensions was tested with qPCR. Leukocyte contamination was determined using a standard curve prepared with platelets spiked with a different number of leukocytes. Occasional tests of the platelet suspensions showed less than 1 leukocyte/105 platelets, which was considered adequate for RNA sequencing. All samples contained less than 15% P-selectin positive platelets after isolation.
Incubation of washed platelets with bacteria
Two different strains of E. coli bacteria were used: the non-pathogenic E. coli K12 C600 (purchased from the Coli Genetic Stock Centre; Yale, CT, USA) and the uropathogenic E. coli O18:K1 (patient isolate, gift from S. Knapp; Medical University of Vienna). The bacteria were grown in lysogeny broth (LB) (Sigma-Aldrich) at 37 °C and 160 rpm until they reached an OD600 nm of 0.5–0.9. To determine the number of bacteria in the suspensions, OD measurements were performed in suspensions in which the number of colony forming units (CFU)/ml was known. With these data, standard curves were prepared. The bacteria cells were pelleted by centrifugation at 18 000 × g for 10 minutes and washed with SSP + buffer. After washing, the platelet pellet was resuspended in SSP + and used for in vitro exposure to the bacteria in 1:1, 1:5 or 1:10 platelet-bacteria ratio. Incubation was done on a see-saw shaker (10 rpm) at room temperature.
In order to monitor the bacteria numbers and physiological status during incubation, platelets, platelets co-incubated with E. coli K12 or O18:K1 (in 1:1, 1:5 and 1:10 platelet to bacteria ratio) and bacteria alone as controls (same number of bacteria as mixed with platelets) were plated on LB agar plates after appropriate dilution at zero hours and three hours. The agar plates were incubated overnight on 37 °C and the number of colonies was counted. The CFU/ml were calculated and the different time points and conditions were compared to each other.
Platelet analysis by flow cytometry
Platelets were identified by expression of CD41 (APC, Clone: HIP8) or CD61 (FITC, Clone VI-PL2). To determine possible platelet activation following incubation with bacteria, P-selectin (from alpha granules) and CD63 (from dense bodies) expression on the platelet surface was measured by flow cytometry. To test integrin αIIbβ3 activation on the platelet surface PAC-1 antibody and antibody-labelled fibrinogen were used. At baseline (zero hours) and after three hours incubation the platelet-bacteria mixtures (5 × 106 platelets in 1:1, 1:5 or 1:10 platelet to bacteria ratios) were fixed with 0.5% formalin (Roth, Karlsruhe, Germany), mixed with antibodies against CD41 (APC, Clone: HIP8), P-selectin (PE, Clone: AK4) and CD63 (FITC, Clone: H5C6) (BioLegend, San Diego, CA, USA) for 30 minutes and again fixed with 1% formalin. For the PAC-1 (FITC, Clone: PAC-1) (BioLegend, San Diego, CA, USA) and fibrinogen (from human plasma, Alexa Fluor 647 Conjugate) (Thermo Fisher Scientific, Waltham, MA, USA) assays, the staining procedure was performed without the first fixation step gating for CD61 (FITC, Clone VI-PL2) (BioLegend, San Diego, CA, USA) positive platelets during the measurement. All samples were measured after staining using a BD FACSCalibur flow cytometer. Analysis was performed with the FlowJo (TreeStar, Ashland, OR, USA) software, and visualized by Graph Pad Prism version 5.01. for Windows (GraphPad Software, La Jolla, CA, USA. www.graphpad.com).
The following platelet activators were used as positive controls: thrombin receptor-activating peptide 6 (TRAP, 14.5 µM; Roche, Penzberg, Germany), adenosine diphosphate (ADP, 5 µM; Roche) and lipopolysaccharide O111:B4 (LPS, 3 µg/ml; Sigma-Aldrich).
RNA isolation
Incubation of washed platelets with bacteria (5 × 108–109 platelets in 1:1 platelet to bacteria ratio, for three hours) was followed by centrifugation of the samples at 500 g for 10 minutes. The pellets were used for RNA isolation with a standard phenol-chloroform procedure. Briefly, centrifuged cells were resuspended in 500 µl Trizol reagent (Invitrogen, Life Technologies, Carlsbad, CA, USA), and added to Phase lock tubes (QuantaBio, Beverly, MA, USA) together with chloroform. All centrifugation steps were performed as previously described26. The RNA pellet was air-dried and resuspended in 40 µl RNAse free water.
RNA-sequencing library preparation and sequencing
Preparation of RNA samples for sequencing was performed as described by Best at al.12. RNA quality and quantity was tested using the RNA 6000 Picochip (Bioanalyzer 2100, Agilent, Santa Clara, CA, USA) (Supplementary Fig. S1). For cDNA synthesis and amplification ~600 pg total platelet RNA was added to the SMARTer Ultra Low RNA Kit for Illumina Sequencing v3 (Clontech, Mountain View, CA, USA). The bacteria co-incubated platelet samples contained higher RNA concentrations, which reflected bacterial RNA contribution to the total RNA. To make sure that we applied the same amount of platelet RNA from samples incubated without or with bacteria, we corrected for the bacteria RNA content in our co-incubated samples and added ~1200 pg total RNA to the SMARTer preparation from the bacteria exposed samples. The chosen amount of total RNA was based on exemplary sequencing results. Following a cDNA quality test on a DNA High Sensitivity chip (Agilent), cDNA samples were sonicated (Covaris Inc., Woburn, MA, USA) for nucleic acid shearing. DNA was labelled with the Truseq Nano DNA Sample Prep Kit (Illumina, San Diego, CA, USA). The quality check was performed on DNA 7500 chips (Agilent). Then, a 100 bp Single-Read sequencing on the Illumina Hiseq. 2500 platform was carried out.
Quantification and statistical analysis of RNA-sequencing data with thromboSeq pipeline
The FASTQ-files of the raw RNA-sequencing data were processed using a standardized, platelet optimized RNA-sequencing alignment pipeline as previously described11,12. Trimmomatic (version 0.22) was used to clip the sequencing adapters and perform 5′-end quality trimming71. Mapping to the human reference genome (hg19) was performed with STAR (version 2.3.0, number of mismatches allowed per read: 10)72, and HTseq (version 0.6.1) was applied to summarize the intron-spanning reads. R (version 3.3.0) and R-studio (version 0.99.902) were used to perform normalization, statistical and analytical analyses, particularly using the edgeR package73. Gene ontology (GO) analysis was done with Database for Annotation, Visualization and Integrated Discovery (DAVID) 6.8 functional cluster annotation74. Platelet specific functional analyses of RNA-sequencing data was performed with PlateletWeb database75.
RNA-sequencing analysis with Tuxedo pipeline
RNA-sequencing analysis was also performed with the Tuxedo pipeline. For each sample, RNA-sequencing reads passing vendor quality filtering was aligned to the hg38 reference genome assembly with the TopHat2 (version 2.1.1)41. Cufflinks (version 2.1.1)43 was used for transcriptome assembly, customary including novel transcript structures, on the basis of the reference transcriptome and spliced read alignments, as well as raw transcript quantification. Differential expression was performed with Cuffdiff (included in Cufflinks version 2.1.1)42 and R scripts were used to perform quality assessment and further refine analysis results. The results were compared to the data produced by thromboSeq pipeline.
Western blot protein analysis
Following incubation (without or with bacteria in 1:1, 1:5 or 1:10 platelet to bacteria ratios or activators) platelets were pelleted via centrifugation (at 500 g for 10 minutes, without break) and lysed with RIPA buffer containing 2% SDS (Sigma-Aldrich) and 100 µl HaltTM Protease Inhibitor Single-Use Cocktail (Thermo Scientific, Boston, MA, USA). The total protein concentration was determined by bicinchonic acid protein assay (Thermo Scientific) and we controlled how significantly bacteria cells would influence it. We did not observe significant contributions of the bacteria to the measured total protein concentration in samples incubated without or with bacteria. Therefore we were confident that we could use an aliquot of 18 µg protein for electrophoresis on 8% polyacrylamide gel SDS-PAGE for all platelet samples. Gels were blotted as described before26. The blots were incubated overnight with primary anti-human antibodies (rabbit polyclonal ATP2C1 (1:750), LRCH4 (1:500), HMBS (1:1000) or GAPDH (1:600) from Atlas Antibodies, Bromma, Sweden). Goat anti-rabbit (H + L) horseradish peroxidase (HRP) conjugate (1:50000; BioRad, Hercules, CA, USA) served as secondary antibody. Signal development was performed by ECL West Pico and West Femto detection system (Thermo Scientific); imaging was done with a Bio-Rad Imaging System. The molecular weights of HMBS, ATP2C1 and LRCH4 proteins in platelets were compared to the molecular weights of the proteins in HeLa cells (Supplementary Fig. S6).
Enzyme-linked immunosorbent assay (ELISA)
Platelet releasates were collected from platelets incubated with buffer or bacteria (108 – 5 × 108 platelets in 1:1, 1:5 or 1:10 platelet to bacteria ratios) and used for ELISA analyses. Commercially available kits detecting HMBS, ATP2C1 and LRCH4 (DlDevelop, Wuxi, China) proteins were applied. Aliquots of 100 µl undiluted supernatants were analysed. The protocols provided by the manufacturers were followed.
The datasets generated during the current study are available in the Gene Expression Omibus (GEO) database repository (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114710).
Electronic supplementary material
Acknowledgements
We want to thank M. Schuster, F. Schischlik (CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna), A. Assinger and her team, particularly J. B. Kral, W. C. Schrottmaier and M. Salzmann (Medical University of Vienna) and G. Leitner (University Clinic for Blood Group Serology and Transfusion Medicine, Vienna) for their scientific and technical support and helpful discussions. The project was funded by the Austrian Science Fund (FWF) PhD program ‘Cell Communication in Health and Disease’, Project APW01205FW_07.
Author Contributions
A.V.F. designed and performed experiments, analysed the data and wrote the manuscript. M.G.B. provided help in data analysis and contributed to writing of the manuscript. W.A. van der H. performed some platelet-bacteria co-incubation experiments and flow cytometry analysis together with A.V.F. A.V. helped with in silico data analysis. H.V. performed sample processing for RNA-sequencing. Q. de M. provided access to bacteria workspace and material. T.W. supervised the RNA work and analysed data. C.M. designed and supervised the study, analysed data and contributed to writing of the manuscript. All authors were involved in experiment design, data interpretation and provided critical comments on the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-34473-w.
References
- 1.Nilsson RJA, et al. Blood platelets contain tumor-derived RNA biomarkers. Blood. 2011;118:3680–3683. doi: 10.1182/blood-2011-03-344408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klement GL, et al. Platelets actively sequester angiogenesis regulators. Blood. 2009;113:2835–2842. doi: 10.1182/blood-2008-06-159541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowley JW, et al. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118:101–112. doi: 10.1182/blood-2011-03-339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landry P, et al. Existence of a microRNA pathway in anucleate platelets. Nat. Struct. Mol. Biol. 2009;16:961–966. doi: 10.1038/nsmb.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alhasan Abd A., Izuogu Osagie G., Al-Balool Haya H., Steyn Jannetta S., Evans Amanda, Colzani Maria, Ghevaert Cedric, Mountford Joanne C., Marenah Lamin, Elliott David J., Santibanez-Koref Mauro, Jackson Michael S. Circular RNA enrichment in platelets is a signature of transcriptome degradation. Blood. 2015;127(9):e1–e11. doi: 10.1182/blood-2015-06-649434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bray PF, et al. The complex transcriptional landscape of the anucleate human platelet. BMC Genomics. 2013;14:1. doi: 10.1186/1471-2164-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denis MM, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122:379–91. doi: 10.1016/j.cell.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shashkin PN, Brown GT, Ghosh A, Marathe GK, McIntyre TM. Lipopolysaccharide is a direct agonist for platelet RNA splicing. J. Immunol. 2008;181:3495–3502. doi: 10.4049/jimmunol.181.5.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwertz H, et al. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J. Exp. Med. 2006;203:2433–2440. doi: 10.1084/jem.20061302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rondina MT, et al. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J. Thromb. Haemost. 2011;9:748–58. doi: 10.1111/j.1538-7836.2011.04208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Best MG, et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell. 2015;28:666–676. doi: 10.1016/j.ccell.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Best MG, et al. Swarm Intelligence-Enhanced Detection of Non-Small-Cell Lung Cancer Using Tumor-Educated Platelets. Cancer Cell. 2017;32:238–252.e9. doi: 10.1016/j.ccell.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eicher JD, et al. Characterization of the platelet transcriptome by RNA sequencing in patients with acute myocardial infarction. Platelets. 2015;7104:1–10. doi: 10.3109/09537104.2015.1083543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weyrich AS, et al. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc. Natl. Acad. Sci. 1998;95:5556–5561. doi: 10.1073/pnas.95.10.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mills EW, Green R, Ingolia NT. Slowed decay of mRNAs enhances platelet specific translation. Blood. 2017;129:e38–e48. doi: 10.1182/blood-2016-08-736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deppermann C, Kubes P. Platelets and infection. Semin. Immunol. 2016;28:536–545. doi: 10.1016/j.smim.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Shannon O. Platelet interaction with bacterial toxins and secreted products. Platelets. 2015;7104:1–7. doi: 10.3109/09537104.2015.1014471. [DOI] [PubMed] [Google Scholar]
- 18.Kerrigan SW, Cox D. Platelet-bacterial interactions. Cell. Mol. Life Sci. 2010;67:513–523. doi: 10.1007/s00018-009-0207-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arman M, et al. Amplification of bacteria-induced platelet activation is triggered by FcγRIIA, integrin αiIbβ3, and platelet factor 4. Blood. 2014;123:3166–3174. doi: 10.1182/blood-2013-11-540526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaertner F, et al. Migrating Platelets Are Mechano-scavengers that Collect and Bundle Bacteria. Cell. 2017;171:1368–1382.e23. doi: 10.1016/j.cell.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Bayer AS, et al. Staphylococcus aureus induces platelet aggregation via a fibrinogen-dependent mechanism which is independent of principal platelet glycoprotein IIb/IIIa fibrinogen-binding domains. Infect. Immun. 1995;63:3634–41. doi: 10.1128/iai.63.9.3634-3641.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herzberg MC, Brintzenhofe KL, Clawson CC. Aggregation of human platelets and adhesion of Streptococcus sanguis. Infect. Immun. 1983;39:1457–1469. doi: 10.1128/iai.39.3.1457-1469.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cognasse F, et al. Streptococcus sanguinis - induced cytokine and matrix metalloproteinase-1 release from platelets. BMC Immunol. 2014;15:1–5. doi: 10.1186/1471-2172-15-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gafter-Gvili A, et al. Thrombocytopenia in Staphylococcus aureus bacteremia: Risk factors and prognostic importance. Mayo Clin. Proc. 2011;86:389–396. doi: 10.4065/mcp.2010.0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watson CN, et al. Human platelet activation by Escherichia coli: roles for FcγRIIA and integrin αIIbβ3. Platelets. 2016;27:535–540. doi: 10.3109/09537104.2016.1148129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arbesu I, Bucsaiova M, Fischer MB, Mannhalter C. Platelet-borne complement proteins and their role in platelet–bacteria interactions. J. Thromb. Haemost. 2016;14:2241–2252. doi: 10.1111/jth.13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeh JJ, et al. P-selectin-dependent platelet aggregation and apoptosis may explain the decrease in platelet count during Helicobacter pylori infection. Blood. 2010;115:4247–4253. doi: 10.1182/blood-2009-09-241166. [DOI] [PubMed] [Google Scholar]
- 28.Satoh K, et al. VacA, the vacuolating cytotoxin of Helicobacter pylori, binds to multimerin 1 on human platelets. Thromb. J. 2013;11:23. doi: 10.1186/1477-9560-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuwana M. Helicobacter pylori -associated immune thrombocytopenia: Clinical features and pathogenic mechanisms. World J. Gastroenterol. 2014;20:714–723. doi: 10.3748/wjg.v20.i3.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frydman GH, Davis N, Beck PL, Fox JG. Helicobacter pylori Eradication in Patients with Immune Thrombocytopenic Purpura: A Review and the Role of Biogeography. Helicobacter. 2015;20:239–251. doi: 10.1111/hel.12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shibazaki M, et al. Complement-dependent accumulation and degradation of platelets in the lung and liver induced by injection of lipopolysaccharides. Infect. Immun. 1999;67:5186–5191. doi: 10.1128/iai.67.10.5186-5191.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Stoppelaar SF, et al. Thrombocytopenia impairs host defense in gram-negative pneumonia-derived sepsis in mice. Blood. 2014;124:3781–3790. doi: 10.1182/blood-2014-05-573915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Claushuis TAM, et al. Nbeal2 Deficiency Increases Organ Damage but Does Not Affect Host Defense During Gram-Negative Pneumonia-Derived Sepsis. Arterioscler. Thromb. Vasc. Biol. ATVBAHA. 2018;118:311332. doi: 10.1161/ATVBAHA.118.311332. [DOI] [PubMed] [Google Scholar]
- 34.Kaper JB, Nataro JP, Mobley HLT. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 35.Ståhl AL, et al. Lipopolysaccharide from enterohemorrhagic Escherichia coli. binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. 2006;108:167–176. doi: 10.1182/blood-2005-08-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watson CN, et al. Human platelet activation by Escherichia coli: roles for FcγRIIA and integrin αIIbβ3. Platelets. 2016;7104:1–6. doi: 10.3109/09537104.2016.1148129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moriarty RD, Cox A, McCall M, Smith SGJ, Cox D. Escherichia coli induces platelet aggregation in an FcγRIIa‐dependent manner. J. Thromb. Haemost. 2016;14:797–806. doi: 10.1111/jth.13226. [DOI] [PubMed] [Google Scholar]
- 38.Berthet J, et al. Human platelets can discriminate between various bacterial LPS isoforms via TLR4 signaling and differential cytokine secretion. Clin. Immunol. 2012;145:189–200. doi: 10.1016/j.clim.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 39.Ward JR, et al. Agonists of toll-like receptor (TLR) 2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb. Haemost. 2005;94:831–838. [PubMed] [Google Scholar]
- 40.Hrdlickova, B. et al. Expression profiles of long non-coding RNAs located in autoimmune disease-associated regions reveal immune cell-type specificity. Genome Med. 88 (2014). [DOI] [PMC free article] [PubMed]
- 41.Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions 0–9, 10.1101/000851 (2013). [DOI] [PMC free article] [PubMed]
- 42.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun L, Mao G, Rao AK. Association of CBFA2 mutation with decreased platelet PKC-theta and impaired receptor-mediated activation of GPIIb-IIIa and pleckstrin phosphorylation: Proteins regulated by CBFA2 play a role in GPIIb-IIIa activation. Blood. 2004;103:948–954. doi: 10.1182/blood-2003-07-2299. [DOI] [PubMed] [Google Scholar]
- 45.Lewandrowski U, et al. Platelet membrane proteomics: A novel repository for functional research. Blood. 2009;114:10–20. doi: 10.1182/blood-2009-02-203828. [DOI] [PubMed] [Google Scholar]
- 46.Nassa G, et al. Splicing of platelet resident pre-mRNAs upon activation by physiological stimuli results in functionally relevant proteome modifications. Sci. Rep. 2018;8:498. doi: 10.1038/s41598-017-18985-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kirschbaum M, et al. Horizontal RNA transfer mediates platelet-induced hepatocyte proliferation. Blood. 2015;126:798–806. doi: 10.1182/blood-2014-09-600312. [DOI] [PubMed] [Google Scholar]
- 48.Coppinger, J. A. et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions Characterization of the proteins released from activated platelets leads to localization of novel platelet 103, 2096–2104 (2012). [DOI] [PubMed]
- 49.Plummer C, et al. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br. J. Haematol. 2005;129:101–109. doi: 10.1111/j.1365-2141.2005.05421.x. [DOI] [PubMed] [Google Scholar]
- 50.Clark SR, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 51.Kerrigan SW, et al. A role for glycoprotein Ib in Streptococcus sanguis -induced platelet aggregation. Blood. 2002;100:509–516. doi: 10.1182/blood.V100.2.509. [DOI] [PubMed] [Google Scholar]
- 52.Ahn ER, et al. Platelet activation in Helicobacter pylori-associated idiopathic thrombocytopenic purpura: Eradication reduces platelet activation but seldom improves platelet counts. Acta Haematol. 2006;116:19–24. doi: 10.1159/000092343. [DOI] [PubMed] [Google Scholar]
- 53.Ståhl Anne-lie, Sartz Lisa, Nelsson Anders, Békássy Zivile D., Karpman Diana. Shiga Toxin and Lipopolysaccharide Induce Platelet-Leukocyte Aggregates and Tissue Factor Release, a Thrombotic Mechanism in Hemolytic Uremic Syndrome. PLoS ONE. 2009;4(9):e6990. doi: 10.1371/journal.pone.0006990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matus V, et al. Human platelet interaction with E. coli O111 promotes tissue-factor-dependent procoagulant activity, involving Toll like receptor 4. PLoS One. 2017;12:1–12. doi: 10.1371/journal.pone.0185431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pupo E, Lindner B, Brade H, Schromm AB. Intact rough- and smooth-form lipopolysaccharides from Escherichia coli separated by preparative gel electrophoresis exhibit differential biologic activity in human macrophages. FEBS J. 2013;280:1095–1111. doi: 10.1111/febs.12104. [DOI] [PubMed] [Google Scholar]
- 56.Jiang Z, et al. CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 57.Huber M, et al. R-form LPS, the master key to the activation of TLR4/MD-2-positive cells. Eur. J. Immunol. 2006;36:701–711. doi: 10.1002/eji.200535593. [DOI] [PubMed] [Google Scholar]
- 58.Zanoni I, et al. Similarities and differences of innate immune responses elicited by smooth and rough LPS. Immunol. Lett. 2012;142:41–47. doi: 10.1016/j.imlet.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 59.Zhao L, et al. LPS-induced platelet response and rapid shock in mice: Contribution of O-antigen region of LPS and involvement of the lectin pathway of the complement system. Blood. 2002;100:3233–3239. doi: 10.1182/blood-2002-01-0252. [DOI] [PubMed] [Google Scholar]
- 60.Devine, D. A. & Roberts, A. P. K1, K5 and 0 antigens of Escherichia coli in relation to serum killing via the classical and alternative complement pathways. 41, 139–144 (1994). [DOI] [PubMed]
- 61.Pluschke, G., Mayden, I. J. a N., Achtman, M. & Levine, R. P. Role of the capsule and the O antigen in resistance of O18:K1 Escherichia coli to complement-mediated killing. 42, 907–913 (1983). [DOI] [PMC free article] [PubMed]
- 62.Yadav S, et al. Golgi proteins in circulating human platelets are distributed across non-stacked, scattered structures. Platelets. 2017;28:400–408. doi: 10.1080/09537104.2016.1235685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu W., Xu D., Tu R. & Zhiping, H. Morphology of platelet Golgi apparatus and their significance after acute cerebral infarction 2134–43, 10.3969/j.issn.1673-5374.2013.23.003 (2013). [DOI] [PMC free article] [PubMed]
- 64.Ramos-Castañeda J, et al. Deficiency of ATP2C1, a Golgi Ion Pump, Induces Secretory Pathway Defects in Endoplasmic Reticulum (ER)-associated Degradation and Sensitivity to ER Stress. J. Magn. Reson. 2013;236:47–56. doi: 10.1016/j.jmr.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shao M, et al. E3 ubiquitin ligase CHIP interacts with C-type lectin-like receptor CLEC-2 and promotes its ubiquitin-proteasome degradation. Cell. Signal. 2016;28:1530–1536. doi: 10.1016/j.cellsig.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 66.Dangelmaier CA, et al. Rapid ubiquitination of Syk following GPVI activation in platelets. Blood. 2005;105:3918–3924. doi: 10.1182/blood-2004-09-3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chatterjee M, et al. SDF-1o: Induces differential trafficking of CXCR4-CXCR7 involving cyclophilin A, CXCR7 ubiquitination and promotes platelet survival. FASEB J. 2014;28:2864–2878. doi: 10.1096/fj.14-249730. [DOI] [PubMed] [Google Scholar]
- 68.McRedmond JP, et al. Integration of Proteomics and Genomics in Platelets. Mol. Cell. Proteomics. 2004;3:133–144. doi: 10.1074/mcp.M300063-MCP200. [DOI] [PubMed] [Google Scholar]
- 69.Raich N, et al. Molecular cloning and complete primary sequence of human erythrocyte porphobilinogen deaminase. Nucleic Acids Res. 1986;14:5955–5968. doi: 10.1093/nar/14.15.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grandchamp B, et al. Tissue-specific expression of porphobilinogen deaminase: Two isoenzymes from a single gene. Eur. J. Biochem. 1987;162:105–110. doi: 10.1111/j.1432-1033.1987.tb10548.x. [DOI] [PubMed] [Google Scholar]
- 71.Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dobin A, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dennis G, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:R60. doi: 10.1186/gb-2003-4-9-r60. [DOI] [PubMed] [Google Scholar]
- 75.Boyanova D., Nilla S., Birschmann I., Dandekar T., Dittrich M. PlateletWeb: a systems biologic analysis of signaling networks in human platelets. Blood. 2011;119(3):e22–e34. doi: 10.1182/blood-2011-10-387308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.