

Abstract
It has been well recognized that exposure to stress can lead to the onset of psychosocial disorders such as depression. While there are a number of antidepressant therapies currently available and despite producing immediate neurochemical alterations, they require weeks of continuous use in order to exhibit antidepressant efficacy. Moreover, up to 30% of patients do not respond to typical antidepressants, suggesting that our understanding of the pathophysiology underlying stress-induced depression is still limited. In recent years inflammation has become a major focus in the study of depression as several clinical and preclinical studies have demonstrated that peripheral and central inflammatory mediators, including interleukin (IL)-1β, are elevated in depressed patients. Moreover, it has been suggested that inflammation and particularly neuroinflammation may be a direct and immediate link in the emergence of stress-induced depression due to the broad neural and glial effects that are elicited by proinflammatory cytokines. Importantly, individual differences in inflammatory reactivity may further explain why certain individuals exhibit differing susceptibility to the consequences of stress. In this review article, we discuss sources of individual differences such as age, sex and coping mechanisms that are likely sources of distinct changes in stress-induced neuroimmune factors and highlight putative sources of exaggerated neuroinflammation in susceptible individuals. Furthermore, we review the current literature of specific neural and glial mechanisms that are regulated by stress and inflammation including mitochondrial function, oxidative stress and mechanisms of glutamate excitotoxicity. Taken together, the impetus for this review is to move towards a better understanding of mechanisms regulated by inflammatory cytokines and chemokines that are capable of contributing to the emergence of depressive-like behaviors in susceptible individuals.
Keywords: stress susceptibility, neuroinflammation, depression, microglia, glutamate
Introduction
Depression is considered to be one of the most debilitating diseases in the United States (Almeida, 2005) and has been globally recognized as a significant source of disability (Reddy, 2010). The prevalence of depression has been steadily increasing over the last 10 years from 6.6% to 7.3% in adults and 8.7%–12.7% in adolescents (Weinberger et al., 2017). While there are a number of available antidepressant therapies, many like the selective serotonin re-uptake inhibitor citalopram, are only 33% effective in producing full remission of depressive symptoms (Trivedi et al., 2006). Moreover, up to 30% of depressed patients are resistant to traditional antidepressant therapies (Joffe et al., 1996; Al-Harbi, 2012). These data strongly suggest that the pathophysiology underlying the emergence of depression is variable between individuals and is still largely unclear. It was first noted that activation of the immune system impacted psychiatric functioning back in 1927 when Julius Wagner-Jauregg won the Nobel Prize for this seminal observation. Since this initial discovery, there has been a striking increase in the number of publications on the topic of inflammation related depression (Loftis et al., 2010). These studies have demonstrated that certain subpopulations of depressed patients exhibit greater levels of interleukin (IL)-6 and C reactive protein (CRP) in the plasma (Irwin and Miller, 2007) and cerebrospinal fluid (Sasayama et al., 2013; Devorak et al., 2015). Importantly, this vast body of literature has also established a causal link between inflammation and depression. Several clinical studies demonstrated that chronic administration of the cytokines interferon (INF)-α and IL-2 as chemotherapeutics were capable of inducing depression in a large number of patients (Denicoff et al., 1987; Renault et al., 1987). Moreover, it should be noted that individuals with inflammatory diseases such as irritable bowel disease, allergic rhinitis and rheumatoid arthritis (Cuffel et al., 1999; Stauder and Kovács, 2003; Katon et al., 2004; Marrie et al., 2017) as well as cardiovascular disease (Anda et al., 1993; Riba et al., 2011; Huffman et al., 2013) are at increased risk of developing psychiatric comorbidities.
Beyond immune diseases as a risk factor for psychiatric disorders, it has been well established that exposure to stress can also serve as an independent risk factor for the emergence of psychosocial disorders. While there are many different types of stress, social stressors such as bullying, abuse, isolation, witnessing traumatic events, or taking care of a terminally ill loved one are the most common types of stress encountered by people (Almeida, 2005). Importantly, it has been shown that exposure to social stress can not only produce increases in markers of inflammatory activity (Slavich et al., 2010; Allen et al., 2017) but can also augment underlying inflammatory disorders including allergic responses (Sandberg et al., 2000; Liu et al., 2002; Kiecolt-Glaser et al., 2008). However, preclinical and clinical studies have shown that there is considerable individual variability in the behavioral and inflammatory consequences induced by stress exposure resulting in the emergence of resilient and susceptible subpopulations. Specifically, it has been shown that greater inflammatory responses to stress are associated with greater negative affect in humans (Dickerson et al., 2009) and promote the development of depressive-like behaviors in rodents (Wohleb et al., 2013, 2014a,b; Hodes et al., 2014; Wood et al., 2015; Finnell and Wood, 2016; Finnell et al., 2017a,b, 2018). These stress-induced inflammatory effects are known to extend well beyond the immediate response to stress such that late phase inflammatory responses are also enhanced following social stress exposure (Kiecolt-Glaser et al., 2008; Deak et al., 2017). These late phase inflammatory effects have been tied to the emergence of chronic elevations of inflammatory factors through the recruitment and sensitization of inflammatory competent cell types including peripherally derived T cells (Janeway et al., 2001; Hansen et al., 2004) and microglia (Badoer, 2010).
Activation or sensitization of microglia, the resident immune cells of the brain, is of particular relevance to depression as a recent clinical study showed for the first time that depressed patients exhibit significant increases in translocator protein density, a marker of activated microglia (Setiawan et al., 2015). Under normal resting conditions, microglia exhibit a highly ramified morphology that is associated with monitoring and maintenance of the neural cell microenvironment (Nimmerjahn et al., 2005; Kettenmann et al., 2011). In response to a stress or immune challenge, these cell types take on an ameboid morphology that is associated with a reactive inflammatory state (Gemma and Bachstetter, 2013; Brites and Fernandes, 2015) resulting in the release of a number of different effectors including cytokines and chemokines (Brites and Fernandes, 2015). In this way reactive microglia are known to propagate inflammatory signals throughout the brain (Fruhbeis et al., 2013). However, the discrete neural mechanisms that may be impacted by the release of cytokines and chemokines in susceptible individuals remains unclear. Therefore, the focus of this review is to first provide an overview of the sources of individual differences in stress and inflammatory responses and second, to highlight discrete neural and glial mechanisms that are regulated by inflammatory effectors that may contribute to the emergence of behavioral dysfunction associated with a depressive-like state. Great focus has been placed on clinical and preclinical studies documenting the effects of social stress. However, other modalities of stress are discussed in instances where literature using social stress models is lacking.
Sources of Individual Differences in Inflammatory Stress Responses
Prior to beginning a discussion on the discrete neural mechanisms that may underlie the emergence of inflammatory related depressive-like behavior, it is critical to understand how individual factors such as age, sex and inherent differences in personality or coping may differentially impact the inflammatory system thereby contributing to stress susceptibility or resiliency.
Age
Stress susceptibility is well known to change across the lifespan. Importantly, life stages in which the brain is undergoing significant alterations, such as neural development and maturation in the young and senescence in the elderly (Graham et al., 2006), are associated with heightened susceptibility to the consequences of stress exposure. Much like stress susceptibility, immune function is also known to change across the lifespan. In general, innate and adaptive immune function decreases as individuals age (Lord et al., 2001; Gomez et al., 2005), resulting in dysregulated inflammatory responses to stress or immune challenges (Lord et al., 2001). For example, studies in rodents have indicated that aged rats do not develop inflammatory tolerance to repeated lipopolysaccharide (LPS) injections as is observed in younger rats (Li et al., 2009). Moreover, LPS inflammatory reactivity has also been shown to be greater in middle-aged mice compared with young mice (Kohman et al., 2010). This increase in inflammatory reactivity in aged animals has also been demonstrated in the brain as a result of natural microglial shifts towards a “primed” phenotype (Barrientos et al., 2015). Heightened inflammatory sensitivity in aging populations, termed inflammatory senescence, also extends to the inflammatory response to stress. Specifically, it has been shown that transient stressors more commonly produce maladaptive inflammatory responses in the elderly compared to younger individuals (Segerstrom and Miller, 2004). Moreover, exposure to stress can also accelerate the process of inflammatory senescence. This assumption is supported by a prospective clinical study which determined that older adults serving as care givers exhibited a four-fold faster elevation in resting plasma IL-6 over a 6-year period compared to age-matched non-caregivers (Kiecolt-Glaser et al., 2003). While clinical studies assessing stress responsivity in aging populations are relatively limited, it is well recognized that social stress and particularly social isolation is extremely common especially for those living in retirement communities. This is of particular importance as approximately 15% of elderly individuals living in retirement communities exhibit significant depressive symptomatology and are more likely to exhibit suicidal tendencies (Fiske et al., 2009). Based on the strong role that stress-induced inflammation is suggested to play in the emergence of depressive-like behavioral states, it is possible that inflammatory senescence may represent a putative mechanism underlying the emergence of depression in aged populations.
Younger populations on the other hand generally exhibit greater resilience to immune challenges while simultaneously exhibiting enhanced behavioral susceptibility to stress. At a cursory glance these effects seem to be opposing. However, these data do not consider the detrimental effects that inflammation produces in the developing organism. Specifically, it has been shown that stress (Bath et al., 2016) and inflammation (Johnson and Kaffman, 2018) at early developmental stages can significantly alter the function, maturation and proliferation of neurons and glia. Moreover, exposure to early life stress is known to promote shifts in the function of immune cells that are resistant to alterations later in life (Lubach et al., 1995), suggesting that early life stress results in long-term reprogramming of the immune system. This assumption has been verified by several studies demonstrating that early life stress not only increases the susceptibility to developing autoimmune deficiencies (Capitanio and Lerche, 1991) but also produces sensitization to subsequent immune challenges (Graham et al., 2006; Roque et al., 2014). Importantly, these shifts in immune function are known to persist for several years (Graham et al., 2006) and has the potential to persist into adulthood (Harry and Kraft, 2012; Delpech et al., 2016). This long-term reprogramming of the immune system has been suggested to underlie the emergence of depressive episodes in younger populations as subsequent stress exposures can produce augmented and poorly regulated physiological responses (Brown et al., 1977).
Sex
Over the last two decades special attention has been paid to understanding the putative contribution of sex, and more specifically gonadal hormones, to the consequences of stress exposure. This research interest was facilitated by several clinical reports that documented that women are more likely to be diagnosed with depression compared with men (Weissman and Klerman, 1992; Gallo et al., 1993; Kessler et al., 1993; Hankin et al., 1998). This two-fold increased risk is known to emerge at the onset of puberty, persists into adulthood (Kessler et al., 1993; Hankin et al., 1998; Nolen-Hoeksema, 2001), and ends following menopause (Kessler et al., 1993; Hankin et al., 1998) strongly suggesting that ovarian hormones may mediate this enhanced stress susceptibility in females. It is important to note that under non-stress conditions, ovarian hormones have consistently been suggested to confer protection because ovariectomy increases depressive-like behaviors (Li et al., 2014). However, when gonadally-intact and ovariectomized female mice are exposed to repeated stress, ovariectomy confers protection against stress-induced depressive-like behavior (LaPlant et al., 2009). Ovarian hormones, like androgens in males, exert control over a number of physiological systems including inflammation (Villa et al., 2016). This is of particular importance as women exhibit greater inflammatory-induced depressive behaviors following an acute endotoxin challenge compared to men (Moieni et al., 2015). Importantly, this ovarian hormone mediated control over inflammatory systems has also been reported in preclinical models demonstrating that female mice exhibit a greater number of microglia that also exhibit more reactive morphology in brain areas associated with emotional regulation (Schwarz et al., 2012). Moreover, when estrogen is administered in vivo and microglia are subsequently cultured, microglia with prior estrogen treatment are sensitized to LPS stimulation (Loram et al., 2012). However, it should be noted that the effect of estrogen on microglia have also been demonstrated to suppress cytokine release, but only when estrogen is applied ex vivo to microglial cells in culture (Dimayuga et al., 2005; Loram et al., 2012).
One of the most common forms of social stress conducted in the laboratory setting is the resident intruder paradigm of social defeat originally developed by Miczek (1979). Social defeat capitalizes on the protection and defense of territory. This model of social stress has proven to be very effective in males and readily produces anxiety- and depressive-like behaviors in the intruder rats (Wood et al., 2010, 2013, 2015; Chaijale et al., 2013; Patki et al., 2013; Finnell et al., 2017a). However, running social defeat in female rats can be difficult and requires either a lactating female resident (Jacobson-Pick et al., 2013) or modification of the male resident with DREADDs to induce heightened aggression via activation of the ventromedial hypothalamus (Takahashi et al., 2017). Recently a new modification to the resident intruder paradigm has also been conducted in which aggression by the male resident was induced following the application of male odorants to the female intruders (Harris et al., 2018). Exposure to this particular modality of social stress (i.e., defeat by a male resident) in female rats has produced incongruent results (Haller et al., 1999; Huhman et al., 2003; Shimamoto et al., 2011; Trainor et al., 2011; Holly et al., 2012; Greenberg et al., 2013, 2015; Jacobson-Pick et al., 2013; Ver Hoeve et al., 2013; Takahashi et al., 2017; Harris et al., 2018). In contrast, findings from the Trainor lab have consistently demonstrated that female California mice display greater sensitivity to the behavioral and molecular consequences to social defeat stress compared with males (Trainor et al., 2011; Greenberg et al., 2013, 2015; Duque-Wilckens et al., 2018). These species dependent effects of social defeat stress in females may underscore the ethological relevance of this stress modality. Unlike female rats which demonstrate territorial aggression only during the lactation period, female California mice inherently demonstrate territorial aggression. These data suggest that the physical interaction of social defeat may be more ethologically relevant in female/male California mice and male rats compared with female rats. This assumption is further validated by studies demonstrating that female rats exhibit greater sensitivity to social isolation/instability compared with social defeat (Haller et al., 1999).
With this in mind, a new model of social stress has recently emerged that combines the olfactory, auditory and visual exposure of social defeat without requiring the physical interaction of defeat. Using this vicarious witness stress model originally developed for use in male mice by Warren et al. (2013), we have shown that intact female rats demonstrate greater sensitivity to the inflammatory, cardiovascular and behavioral consequences of witness stress exposure compared to ovariectomized female rats (Finnell et al., 2018). We have further demonstrated that this enhanced and prolonged behavioral and physiological sensitivity to the consequences of witness stress is not exhibited to the same extent in male rats (Finnell et al., 2017b). While this is still a relatively new model of stress, others have also been able to demonstrate similar behavioral sensitivity of intact female mice to this vicarious witness stress exposure (Iniguez et al., 2018), suggesting that female susceptibility to witness stress may be conserved across species. In humans, bearing witness to a major stressor is one type of event that can elicit post traumatic stress disorder (PTSD). Therefore, it should be noted that similar to findings in depressed patients, PTSD in the clinical setting is also associated with a significant shift in immune reactivity (reviewed in Segerstrom and Miller, 2004). Interestingly, this immune reactivity differs between men and women with men exhibiting a general under-expression of inflammatory related genes of collected CD14+ monocytes while women exhibit an upregulation of pathways associated with inflammatory activation (Neylan et al., 2011). Several comprehensive reviews have recently been published regarding enhanced stress sensitivity and increased risk of mood disorders in females (Goel and Bale, 2009; Bangasser and Wicks, 2017; Bangasser and Wiersielis, 2018; Wickens et al., 2018). Moving forward it will be critical to further validate whether stress sensitive mechanisms in females are mediated in part by inflammatory processes.
Personality and Coping
It has long been recognized that there is wide variability in the way people process and assess stressful situations (Lupien et al., 2007). This may be driven by the individual’s cognitive interpretation (Lupien et al., 2007; Nicolai et al., 2013) as well as the behavioral coping mechanism that is adopted during the stress exposure. In general, coping strategies are broadly classified into two categories termed passive and active. Passive coping strategies include avoidance, seeking excessive reassurance, withdrawal and substance abuse (Cambron et al., 2009; Cairns et al., 2014). In contrast, active coping strategies include problem solving, seeking support, exercising and engaging in adaptive processes (Cairns et al., 2014). It is understood that the coping response adopted by an individual will vary depending on the type and severity of the stress exposure. However, it has been suggested that individuals who more readily adopt active coping strategies are more likely to be resilient to the behavioral and physiological consequences of stress compared to those who more readily adopt passive coping strategies (Kendler et al., 1991). Importantly, coping responses have also been shown to play a large role in the inflammatory outcomes of stress. For example, individuals who more readily adopt passive coping strategies exhibit greater plasma concentrations of IL-6 following a 3 min simulated public speaking challenge compared with individuals that adopt active coping strategies (Carroll et al., 2011). Additionally, feelings of helplessness during stress exposure are associated with sensitized immune responses to a common allergen and promote greater release of IL-6 from stimulated primary blood leucocytes (Kiecolt-Glaser et al., 2009). These data suggest that feelings of helplessness or uncontrollability could promote sensitization of inflammatory pathways that can be amplified by stress exposure (Chen et al., 2009). Although it is impossible to truly assess the emotional state of an animal, a number of preclinical studies demonstrated that both coping (Koolhaas et al., 1999, 2007; Sih et al., 2004; Bell, 2007; Wood et al., 2015; Finnell and Wood, 2016) and stressor controllability (Gray and Cooney, 1982; Frank et al., 2007; Christianson et al., 2009; Arakawa et al., 2014) are large factors in the susceptibility for developing stress-induced behavioral and inflammatory dysfunction. Several recent reviews have also been published on the topic of stress coping and inflammatory outcomes (Maier and Watkins, 2005; Koolhaas et al., 2007; Wood, 2014; Finnell and Wood, 2016; Wood et al., 2017).
Brain Areas Associated With Stress Susceptibility and Resiliency
There are a number of brain regions that have been implicated in the emergence of stress-induced behavioral dysfunction that are discussed throughout this review. Several extensive reviews have been published on this topic, for example (McEwen and Gianaros, 2010). However, to highlight the importance of the brain regions described herein, we have included a brief overview of the brain areas that are associated with social processing and stress responses.
Prefrontal Cortex
The prefrontal cortex works to integrate the social, emotional and cognitive aspects of behavior (Satpute and Lieberman, 2006). Dysfunction within the prefrontal cortex in humans has been associated with the emergence of socially inappropriate behaviors, apathy, inflexibility and isolation (Barrash et al., 2000). In addition to producing overall shifts in social behavior, the prefrontal cortex has also been implicated in stress-induced coping strategies (Robinson et al., 2015). Similar associations between prefrontal cortex activation and susceptibility to the consequences of stress have also been demonstrated in rodents. Utilizing chronic social defeat stress in mice, it was shown that individual susceptibility to the behavioral effects of chronic social defeat (i.e., social avoidance) was directly associated with the activity of the prefrontal cortex (Kumar et al., 2014). Moreover, Kumar et al. (2014) went on to demonstrate that prefrontal cortex reactivity during a pre-stress forced interaction test was predictive of individual stress susceptibility following chronic social defeat. In the context of emotional regulation and threat assessment, the prefrontal cortex serves as a top down inhibitory regulator of the amygdala and hypothalamus (Mujica-Parodi et al., 2017). Several clinical and preclinical studies have consistently reported that stress-induced behavioral deficits are often associated with dendritic atrophy, loss of synapses, and altered prefrontal cortex connectivity (Radley and Morrison, 2005; Banasr et al., 2007; Drevets et al., 2008; Radley et al., 2008; Ota et al., 2014). These morphological and physiological alterations of prefrontal cortex neurons may therefore result in disinhibition of downstream signaling targets including the amygdala.
Hippocampus
Although largely known for its role in declarative memory, the hippocampus has also been implicated in social and emotional episodic memories (Dolcos et al., 2017). Through its connectivity and bottom-up signaling with the amygdala, the hippocampus is responsible for the encoding and retrieval of emotionally laden memories (Dolcos et al., 2017). In addition, the hippocampus is also critical for the re-encoding and extinction of these memories. Exposure to chronic unpredictable restraint stress was shown to produce reductions in several hippocampal sub regions including CA1, CA3 and the dentage gyrus (Schoenfeld et al., 2017). Reductions of hippocampal volume in response to stress have been associated with both dendritic atrophy (Watanabe et al., 1992; Wood et al., 2004; Eiland and McEwen, 2012) and reduced neurogenesis (Simon et al., 2005; Jayatissa et al., 2006; Mitra et al., 2006; Schoenfeld et al., 2017). Interestingly, preclinical studies using social defeat in mice have indicated that defeat-induced reductions of neurogenesis within the hippocampus is associated with stress susceptibility (Tse et al., 2014) and mice demonstrating stress resiliency exhibited an increase of hippocampal neurogenesis by approximately 4% (Tse et al., 2014).
Amygdala
The amygdala is best known for its role in fear responses. For example animals with lesions of the amygdala exhibit a disinhibition of fear responses and a significant increase in prosocial behavior (Kluver and Bucy, 1997). Chronic stress is also known to produce significant structural and functional effects within the amygdala that are highly dependent upon the type and duration of the stressor (Wilson et al., 2015). Moreover, it is now well recognized that depression and anxiety are both associated with amygdala hyperactivity (Drevets, 2000; Sheline et al., 2001). While it is currently unclear how active and passive stress coping strategies are associated with amygdala activity, it has been shown using rodent models that resilient individuals exhibit a number of stress-induced adaptations that may inhibit over activation of the amygdala (Silveira Villarroel et al., 2018).
Bed Nucleus of the Stria Terminalis
Considered to be a part of the extended amygdala, the bed nucleus of the stria terminalis (BNST) is best known for its involvement in behaviors associated with social bonding (Coria-Avila et al., 2014), aggression (Nelson and Trainor, 2007), mating (Coria-Avila et al., 2014) and stress-induced cardiovascular function (Crestani et al., 2013; Oliveira et al., 2015). Interestingly, fMRI studies in humans have determined that the BNST is also involved in the generation of anticipatory anxiety to unpredictable noxious stimuli (Straube et al., 2007; Alvarez et al., 2011; Yassa et al., 2012). Although it is currently unclear how stress affects BNST function in humans, studies using rodent models have determined that exposure to stress results in enhanced BNST activation (Kollack-Walker et al., 1997; Martinez et al., 1998). It is also important to note that the BNST is a sexually dimorphic brain region that has been shown to play a critical role in the consequences of social defeat exposure in male and female monogamous California mice. Following social defeat exposure, Greenberg et al. (2013) showed that female California mice not only demonstrated greater social avoidance but also exhibited greater brain derived neurotrophic factor in the BNST compared to their defeated male counterparts.
Nucleus Accumbens
The Nucleus Accumbens (NAc) is largely studied in the field of addiction due to its role in motivation and reward. However, the NAc is quickly gaining attention in the field of stress and depression due to its potential involvement in the development of anhedonia (Di Chiara et al., 1999; Yadid et al., 2001). The NAc is predominantly inhibitory, releasing γ-aminobutylic acid (GABA) in the ventral tegmental area, thereby exerting control over cortical dopamine (Shirayama and Chaki, 2006). fMRI studies conducted in humans have shown that patients suffering from major depressive disorder exhibit altered activation of the NAc during reward anticipation and outcome compared to healthy controls (Misaki et al., 2016). Importantly, the NAc has also been implicated in coping behaviors. Use of active coping behaviors was associated with increases in NAc activity while passive coping was association with reductions in NAc activity (Levita et al., 2012).
Sources of Stress-Induced Neuroinflammation
While it is clear that neuroinflammatory processes may be a critical link in the pathogenesis of stress-related psychiatric disorders in certain subpopulations of patients, it is important to understand which stress sensitive processes are capable of promoting neuroinflammation. The two most likely mechanisms of increased neuroinflammation include stress-induced activation and sensitization (i.e., priming) of microglia and stress-induced disruption of the blood brain barrier (BBB; see Figure 1).
Figure 1.

Schematic highlighting key sources of stress-induced neuroinflammation. Stress exposure is known to promote shifts in microglial morphology from a highly ramified “resting” state to an ameboid M1 proinflammatory state. In addition to directly stimulating the release of cytokines, activation of microglial glucocorticoid receptors (GCRs) also results in priming of inflammatory responses. This process can occur directly through activation of the NLRP3 inflammasome or indirectly by promoting the release of reactive oxygen species (ROS) from mitochondria which results in the oxidation of high mobility group box -1 (HMGB-l). Once released, HMGB-1 and proinflammatory cytokines such as interleukin (IL)-1β can act on toll like receptor 4 (TLR 4) on the surface of microglia to further stimulate the NLRP3 signaling cascade. Another significant source of stress-induced neuroinflammation is the breakdown of the blood brain barrier (BBB). In pre-stress conditions endothelial cells tightly adhere to one another, blocking the flow of circulating cytokines to the brain. However, in response to stress exposure, tight junctions between these endothelial cells break down allowing for peripheral cytokines and inflammatory cells to penetrate into the brain. This process is known to be facilitated by plasma vascular endothelial growth factor (VEFG)-164, endothelial claudin-5 (CLDN-5) and microglia released matrix metalloproteinase-9 (MMP-9). *Designate non-neuronal and non-glial origins.
Microglial Activation and Priming
Microglia are considered to be highly adaptive cell types as they are capable of transitioning between pro-inflammatory (M1) and anti-inflammatory (M2) states. However, in response to stress a greater number of microglia exhibit the proinflammatory M1 phenotype (Tang et al., 2018). This change in morphology can be stimulated by activation of glucocorticoid receptors (GCRs; Ros-Bernal et al., 2011; Liu et al., 2016) found on the cell surface of microglia, suggesting that stress-induced release of cortisol (in humans) and corticosterone (in rodents) could promote this shift to an M1 microglial state. The involvement of M1 type microglia in stress-induced neuroinflammation has further been supported by studies utilizing the tetracycline analog minocycline. Minocycline, traditionally used as an antibiotic, is well documented to inhibit the polarization of microglia to an M1 proinflammatory phenotype (Kobayashi et al., 2013). Moreover, use of minocycline in conjunction with inescapable foot shock (Blandino et al., 2006) and chronic mild stress (Wang et al., 2018) have shown that inhibition of the M1 microglial phenotype, and subsequent suppression of proinflammatory cytokine release, protects against the development of stress-induced depressive- and anxiety-like responses in rodents. Notably, minocycline is now being evaluated as a putative treatment for bipolar depressive disorder in humans. A very recent clinical trial demonstrated that daily doses of minocycline was capable of producing anti-depressant effects in 90% of study participants (Murrough et al., 2018). While more information is required about the putative anti-inflammatory effects minocycline may have in these patients, these studies in combination provide clear evidence for the involvement of M1 microglia in the emergence of depressive symptomatology.
In addition to promoting the release of cytokines from microglia (Nair and Bonneau, 2006; Kreisel et al., 2014), stress is also capable of sensitizing microglia such that a subsequent stress or immune challenge produces a faster and more robust neuroinflammatory response (Frank et al., 2012, 2018; Fonken et al., 2016). Importantly, glucocorticoid signaling is one factor that has been shown to initiate this phase of stress-induced neuroinflammatory sensitization termed microglial priming. One potential mechanism by which this priming may occur is though the dysregulation of the danger, damage and disease signal high mobility group box-1 (HMGB-1). In response to stress, the membrane glycoprotein CD200 and its receptor (CD200R) exhibit significant down regulation at both the genomic and protein levels (Frank et al., 2018). Notably CD200R is almost exclusively expressed on microglia (Koning et al., 2009) and is known to regulate proinflammatory signaling by constitutively inhibiting myloid cells (Gorczynski, 2005). Loss of CD200 and CD200R in rats exposed to inescapable foot shock was further associated with enhancement of HMGB-1 and increased gene expression of IL-1β, tumor necrosis factor (TNF)-α and nuclear factor kappa (NFκ)B (Frank et al., 2018). This increased expression of proinflammatory genes by HMGB-1 has been directly linked to the activation of the nucleotide-binding oligomerization domain-like receptor (NLRP3) inflammasome (Weber et al., 2015). In addition to the noted effects on gene expression, HMGB-1 activation of the NLRP3 inflammasome can further potentiate proinflammatory signaling by enhancing the cleavage of proIL-1β to IL-1β via activation of caspase-1 (Yan et al., 2012). However, it is important to note that this proinflammatory capacity of HMGB-1 is strongly tied to the redox state of the protein. In its fully reduced state, HMGB-1 promotes chemotaxis but lacks the ability to promote proinflammatory signaling. Alternatively, the oxidized state of HMGB-1, designated by the formation of disulfide linkages, is capable of potentiating proinflammatory signaling as discussed above but lacks chemotactic abilities (Yang et al., 2012). Although the majority of studies assessing the involvement of HMGB-1 in microglial priming have come from studies using inescapable foot shock (Yang et al., 2012; Weber et al., 2015), chronic unpredictable stress (Franklin et al., 2018), and single prolonged stress (Lai et al., 2018), exposure to social stressors such as social defeat is known to enhance the intracellular concentration of reactive oxygen species (ROS; see section Oxidative Stress/Reactive Oxygen Species). Therefore, it is highly plausible that HMGB-1 may also contribute to the emergence of social stress-induced behavioral deficits.
These stress-induced alterations in the morphology and reactivity of microglia requires several hours to manifest and are evident for up to 72 h following the termination of stress, a time point at which peripheral cytokine responses are no longer detected (Tynan et al., 2010; Kopp et al., 2013; Deak et al., 2017). These data nicely parallel findings indicating that the development of depressive-like behaviors following a robust inflammatory challenge occurs over a period of several hours and persist well beyond 24 h (Capuron et al., 2002; Dantzer et al., 2008). Moreover, preclinical studies using social defeat and vicarious witness stress have demonstrated that repeated stress exposure is capable of enhancing resting neuroinflammation that persists for at least 5 days following the final stress exposure, a time at which depressive-like behavior is evident (Finnell et al., 2017a,b). Importantly these studies determined that despite elevations in neuroinflammation and depressive-like behavior 5 days following the final stress exposure, resting peripheral inflammation had returned to baseline comparable to non-stressed controls (Finnell et al., 2017a). The importance of central inflammation in the emergence of stress-induced depressive-like behavior has been further substantiated by studies outlining the effectiveness of centrally administered IL-1 receptor antagonist in inhibiting social defeat-induced depressive-like behavior (Wood et al., 2015). Similar antidepressant-like effects were demonstrated with the use of resveratrol, a natural anti-inflammatory. Importantly, these effects were only achieved by the highest dose, which was the only dose to effectively prevent the neuroinflammatory response to social defeat (Finnell et al., 2017a). These data strongly suggest that stress likely promotes the emergence of an M1 microglial phenotype which may directly underlie stress and inflammatory-induced behavioral dysfunction.
Blood Brain Barrier Disruption
While cells within the brain are robustly capable of producing a major source of neuroinflammation, cytokines circulating in the blood can also serve as a source to increase neuroinflammation. The BBB, in part a meshwork of specialized endothelial cells along blood vessels surrounding the brain, serves the purpose of regulating entry and export of cytokines (and other substances) between the peripheral circulation and the brain. In a healthy brain, cytokines are considered to be too large and hydrophilic to passively diffuse across the BBB (Banks, 2005). However, the IL-1 family, TNF and IL-6 exhibit distinct and saturable transport mechanisms to effectively pass from the blood to the brain (Banks et al., 1989, 1991). Moreover, pro-inflammatory cytokines can disrupt the integrity of the BBB (Muramatsu et al., 2012). As such, circulating inflammation may initiate a cascade that enhances the flow of inflammatory factors from the circulation into the brain, further exacerbating neuroinflammation. This concept was demonstrated in an elegant study that showed that microglia initiated the recruitment of IL-1β producing monocytes to the brain and stimulated brain endothelial IL-1R1 (McKim et al., 2018). This study went on to further demonstrate that microglial depletion prevented monocyte recruitment and inhibited the development of anxiety in socially defeated mice.
It is tempting to suggest that the link between diseases characterized by peripheral inflammation including cardiovascular disease, rheumatoid arthritis, etc., and the striking increased risk of major depression in these patients (Anda et al., 1993; Huffman et al., 2013; Marrie et al., 2017) may be driven by an impaired BBB and exaggerated neuroinflammation. In addition, social stress exposure, another risk factor for psychiatric disorders is recognized to increase the release of circulating proinflammatory cytokines in animals and humans (Pace et al., 2006; Hodes et al., 2014; Wood et al., 2015; Quinn et al., 2018). While circulating cytokine levels typically return to baseline following cessation of a single acute social stressor (Cheng et al., 2015), preclinical models generating a stress-induced depressive-like phenotype achieved by repeated exposure to social defeat stress demonstrate persistent enhancement in peripheral inflammatory sensitivity (Hodes et al., 2014; Finnell et al., 2017a). In line with the deleterious role of pro-inflammatory cytokines on the integrity of the BBB, recent reports have identified the role of social stress on various factors known to disrupt the BBB. For example, male rats that demonstrate susceptibility to social defeat stress as evidenced by passive coping responses during social defeat and development of depressive-like behaviors, selectively demonstrated enhanced BBB permeability in the ventral hippocampus (Pearson-Leary et al., 2017) while the active coping resilient subset of rats did not. Moreover, administration of the proinflammatory cytokine vascular endothelial growth factor-164 increased permeability of the BBB and was shown to induce vulnerability in socially defeat rats (Pearson-Leary et al., 2017). Stress-induced BBB disruption has also been documented in a mouse model of social defeat, whereby the susceptible subset of male mice also demonstrated stress-induced suppression of claudin-5, an endothelial cell-specific tight junction protein, in the NAc and the hippocampus as compared with controls or the resilient subset of mice. Moreover, BBB permeability was also confirmed in the susceptible subset of mice (Menard et al., 2017). Importantly, these studies further established suppressed claudin-5 expression in post mortem tissue from the NAc of depressed patients. Taken together, disruption of the BBB is a likely susceptibility mechanism driving increased neuroinflammation and social stress-induced behavioral dysfunction in animals, and may contribute to psychopathology in humans.
Other proteins are likely targets for stress-induced increases in BBB permeability and include HMGB-1 and matrix metalloproteinase-9 (MMP-9). For example, HMGB-1 is upregulated by social defeat stress (Finnell et al., 2017b) and beyond its role in neuroinflammatory priming, is also involved in BBB dysfunction. This role for HMGB-1 is supported by studies demonstrating that administration of monoclonal antibody to HMGB-1 protects against ischemia-induced BBB disruption in rats (Zhang et al., 2011), and in humans anti-HMGB1 monoclonal antibody improves the BBB integrity of patients with Alzheimers disease (Festoff et al., 2016). Together these findings clearly define a role for HMGB-1 in BBB dysfunction that could precipitate stress-related psychiatric dysfunction. Furthermore, inflammatory factors including HMGB-1 (Qiu et al., 2010) also stimulate the release of MMP-9, a zymogen that breaks down the BBB, from infiltrating leukocytes and microglia to contribute to endothelial damage (Crocker et al., 2006) and BBB leakage (Seo et al., 2013). MMP-9 protein expression is elevated in peripheral tissues and serum by social defeat stress (Stelzhammer et al., 2015; Wu et al., 2015). While this has yet to be documented in the brain following social defeat, central MMP-9 has been shown to be regulated by fear learning (Ganguly et al., 2013) and lends support to the possibility that MMP-9 may be a putative target by which social stress could lead to BBB disruption.
Central Mechanisms Conferring Risk or Resilience to Stress That Are Regulated by Neuroinflammation
Acute stress is well recognized to stimulate the release of proinflammatory cytokines from microglia (Blandino et al., 2006, 2013) and repeated stress exposure is capable of producing enduring increases in neuroinflammation in stress sensitive brain regions (Voorhees et al., 2013; Wohleb et al., 2013; Wood et al., 2015; Finnell et al., 2017a,b). While evidence links an inflammatory state with a depressive phenotype, our understanding of exactly which neuromodulatory systems are acted upon by inflammatory cytokines and chemokines that serve to increase stress susceptibility is in its infancy. Several reviews have been published on the impact that neuroinflammation has on the metabolism of the neurotransmitters serotonin, dopamine and glutamate and therefore, while clearly relevant to the pathophysiology of depression, this topic will not be covered here (see Miller et al., 2013). Herein, we will focus on the potential role of neuroinflammation on mitochondrial dysfunction and oxidative stress as well as glutamate neurotransmission or excitotoxicity (see Figure 2).
Figure 2.
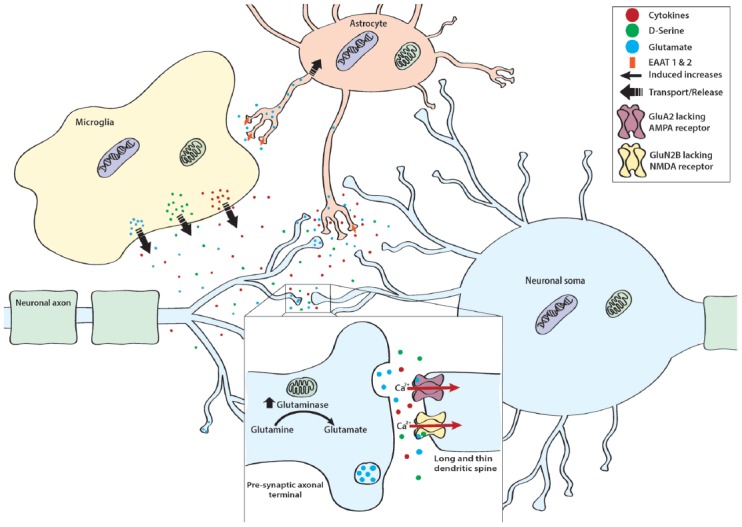
Mechanisms of stress-induced cytotoxicity. In addition to releasing cytokines, Ml type proinflammatory microglia release a variety of neurotransmitters, co-agonists, and neuromodulators such as glutamate and its co-agonist D-serine. Normally, excess glutamate is taken up by excitatory amino acid transporter (EAAT) 1 and 2 found on astrocytic processes. However, in proinflammatory conditions and in the presence of excess glutamate, EAAT 1 and 2 are down regulated, thereby resulting in excess glutamate within the synaptic cleft. Importantly, neurons also contribute to stress-induced enhancements of glutamatergic tone. This is thought to occur as stress exposure enhances mitochondrial glutaminase, the enzyme responsible for converting glutamine to glutamate. In addition to enhancing excitatory tone, stress also sensitizes neurons to the excitatory effect of glutamate. Specifically, stress promotes the expression of GluA2 lacking α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and GLUN2B lacking NMDA receptors. These receptor subtypes allow for calcium (Ca2+) to freely pass into the cell thereby enhancing the depolarizing effect of glutamate. This cumulative increase in excitatory tone is particularly detrimental for dendritic spines that exhibit a long and thin morphology, as these spines are more sensitive to the degenerative effects induced by glutamatergic excitotoxicity.
Mitochondrial Dysfunction
Mitochondria play a critical role in cellular energy metabolism and supply the large energy demand required by the brain, especially under stressful conditions. The inner membrane of mitochondria houses the electron transport chain, which is made up of five protein complexes. Three of these respiratory chain complexes (I, III and IV) pump protons throughout the inner membrane generating the proton gradient that is ultimately responsible for synthesizing adenosine triphosphate (ATP) at complex V. Mitochondria are responsible for producing the vast majority of the ATP in neurons and in particular within presynaptic terminals mitochondrial ATP is required for synaptic ion homeostasis and phosphorylation (Mattson et al., 2008). There is mounting evidence that patients with psychiatric disorders demonstrate mitochondrial abnormalities at the functional level. For example, positron emission tomography studies of brain glucose metabolism have identified reduced glucose utilization in the brains of depressed patients (Videbech, 2000). Moreover, mitochondrial ATP production was also reduced in depressed patients (Gardner et al., 2003). While the cause of this mitochondrial dysfunction is not understood, it is noteworthy to consider the findings that proinflammatory cytokines can impair mitochondrial function. For example, physiologically relevant levels of TNF-α can induce mitochondrial dysfunction; low (post-stroke) levels of TNF-α rapidly reduce mitochondrial function as indicated by increased caspase 8 activity and a decrease in mitochondrial membrane potential (Doll et al., 2015). This effect was shown to signal through TNF-R1 selectively and highlights the role that proinflammatory cytokines may play in mitochondrial dysfunction. Beyond the capability of neuroinflammation to induce mitochondrial dysfunction, it is interesting to note that reducing activity of mitochondria within microglia amplifies the NLRP3 inflammasome and IL-1β release (Sarkar et al., 2017). Taken together these studies demonstrate a striking relationship between neuroinflammation and mitochondrial dysfunction.
While no studies to date have directly evaluated the role that stress-induced release of proinflammatory cytokines has on mitochondrial function, it has been demonstrated in various stress paradigms that repeated stress exposure has dramatic effects on mitochondria. For example, chronic immobilization stress and chronic mild stress inhibit the activity of the respiratory chain complexes within the rat brain cortex (Madrigal et al., 2001; Rezin et al., 2008) and was shown to reduce hippocampal Na+, K+-ATPase activity (Gamaro et al., 2003). Moreover, chronic mild stress has been shown to reduce respiration rates of mitochondria located in the mouse hippocampus, cortex and hypothalamus (Gong et al., 2011). This study also confirmed that stress significantly impacted the ultrastructure of mitochondria (Gong et al., 2011), which are features of mitochondria in presynaptic neurons that have been coupled to changes in synaptic strength (Cserép et al., 2018). Moreover, there is evidence to suggest that distinct differences in mitochondrial function regulate an anxiety-like phenotype. For example, rats that exhibited high anxiety-like behavioral tendencies also demonstrated reduced expression of mitochondrial complex I and II proteins and decreased respiratory capacity and ATP (Hollis et al., 2015). Surprisingly, however, there is a paucity of studies evaluating the impact of social defeat stress on brain mitochondria and even further lack of studies determining whether the vast stress-induced changes in mitochondrial function are driven by stress-induced proinflammatory cytokines.
Oxidative Stress/Reactive Oxygen Species (ROS)
Active neurons exhibit high rates of oxygen consumption and as a result, produce large amounts of ROS (Halliwell, 1992). Mitochondria are the energy powerhouse of the cell and represent the largest source of ROS production in addition to monoamine oxidase and nitric oxide synthase. While ROS play a role in several critical neuronal functions such as neuronal plasticity and learning and memory (reviewed in: Massaad and Klann, 2011), the large amounts of ROS are tightly regulated by an antioxidant system. Under conditions where this system becomes unbalanced, a deleterious buildup of ROS is linked to stress-related psychiatric pathology in clinical studies and is demonstrated to occur in stress-related preclinical studies (de Oliveira et al., 2007; Salim et al., 2010, 2011; Lindqvist et al., 2017). Because mitochondria play a critical role in the production and metabolism of ROS, mitochondrial dysfunction is directly related to increased oxidative stress (Mattson et al., 2008). In line with evidence discussed above that TNF-α reduces mitochondrial function, ROS are also dose dependently increased by treatment with either TNF-α or IL-6 (Rochfort et al., 2014). Social defeat stress has also been shown to induce ROS in stress-related brain regions, and moreover ROS have been shown to play a permissive role in the anxiety-like behavior following social defeat in rats (Solanki et al., 2017). Interestingly, rats demonstrating a high anxiety-like phenotype also exhibit increased ROS production within the NAc (Hollis et al., 2015). Finally, lending evidence to the role for ROS in the pathogenesis of psychiatric disorders in humans, depressed patients not only exhibited elevated markers of inflammation and the oxidative stress marker F2-isoprostanes, but compared to individuals who readily respond to antidepressants, non-responders had higher levels of both oxidative stress markers and inflammation (Strawbridge et al., 2015; Vaváková et al., 2015; Lindqvist et al., 2017). Taken together, it is clear that proinflammatory cytokines are capable of shifting the balance of ROS production/elimination from a healthy balance towards maladaptive. However, it is yet to be determined whether stress-induced ROS and subsequent anxiety- and depressive-like behavior is initiated by proinflammatory cytokines and chemokines.
Glutamate Neurotransmission and Excitotoxicity
The involvement of glutamate has also become an area of interest in the etiology of depression. For example, heightened excitability of hippocampal neurons may underlie the loss of glutamatergic pyramidal neurons in depressed patients (Rajkowska et al., 2005) and evidence from human postmortem tissue has identified alterations in excitatory amino acid transporters (EAATs) 1 and 2 and glutamine synthetase (Rajkowska and Stockmeier, 2013). Moreover, it has been shown that ketamine, a noncompetitive NMDA antagonist (Anis et al., 1983), is capable of producing long lasting antidepressant effects (Berman et al., 2000) even in patients that demonstrate resistance to traditional antidepressant therapies (Zarate et al., 2006). Importantly, the inhibitory action of ketamine requires the presence of open NMDA channels (MacDonald et al., 1987) and can remain bound to NMDA receptors even after the channels have closed (Huettner and Bean, 1988), providing pharmacological validity to these prolonged treatment effects. Several preclinical models have demonstrated that exposure to stress can result in abnormalities in glutamate signaling. For example, 8 weeks of social isolation has been shown to enhance the expression of both NR2A and NR2B subunits within the hippocampus (Chang et al., 2015). Stress-induced increases in these NMDA receptor subunits within the hippocampus are known to not only enhance the intensity of excitatory postsynaptic potentials (Chang et al., 2015) but are also associated with the emergence of aggression, anxiety- and depressive-like behaviors in rodents (Costa-Nunes et al., 2014; Chang et al., 2015). However, it is important to note that these alterations in NMDA receptor subunits following stress exposure are brain region specific. Within the NAc, mice exposed to chronic social defeat that also demonstrate behavioral susceptibility, exhibit long term reductions of NR2B subunit (Jiang et al., 2013). The loss of NR2B subunits significantly impacted the synaptic function of NAc neurons by promoting an increase in long-term depression (Jiang et al., 2013). Interestingly, this study went on to determine that treatment with Fluoxetine, a selective serotonin re-uptake inhibitor, was capable of reversing the effects of defeat stress in susceptible mice such that the molecular profiles within the NAc were nearly identical to mice demonstrating resilience to the effects of social defeat (Jiang et al., 2013).
Stress-induced alterations of NMDA receptors are not the only putative source of glutamatergic excitotoxicity in the brain. For example, unpredictable stress exposure has been documented to produce similar alterations in the subunit composition of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors such that stress exposed rodents demonstrated greater expression of GluR1 subunits (Hubert et al., 2014). Moreover, exposure to the unpredictable stress paradigm resulted in a shift in AMPA receptor distribution such that a greater number of AMPA receptors were found on dendritic spines (Hubert et al., 2014).This seemingly minor shift is well known to produce functionally relevant alterations in neuronal signaling. AMPA receptors which express the GluR2 subunit are impermeable to extracellular Ca2+ due to an arginine block. Therefore, loss of GluR2 subunits enhances the signaling strength of AMPA receptors by enhancing the magnitude of the elicited depolarization following AMPA receptor stimulation (Isaac et al., 2007). Taken together, these findings suggest that exposure to unpredictable stress may result in significant remodeling of dendritic spines to vastly increase their sensitivity to excitatory stimuli. These effects, similar to those demonstrated in microglia, require at least 24 h following the cessation of stress to become evident suggesting that these alterations are largely driven by alterations in gene expression (Nasca et al., 2015) and are not associated with the immediate stress response. Interestingly, GluR2 subunits are also known to shift across the lifespan. In rodents it has been shown that GluR2 steadily increases from birth until adulthood. However, this composition of AMPA receptors does not remain stable and does decrease such that 70-week-old rodents exhibit significant decline in both protein and mRNA for the GluR2 subunits within the hippocampus (Pandey et al., 2015). While it is unclear if inflammatory senescence and enhanced inflammatory reactivity is associated with this shift in GluR2 subunits, these data suggest that a natural loss of GluR2 may contribute to the enhanced risk of mood disorders in aging populations.
A number of studies have directly implicated neuroinflammation and microglial processes in the emergence of glutamatergic excitotoxicity (Faust et al., 2010; Diamond and Volpe, 2012). Most directly, glutamate can be released from activated microglia (Barger et al., 2007) or neurons following stimulation with cytokines such as IL-1β in a dose dependent manner (Zhu et al., 2006). In addition, cytokines released by neighboring microglia are capable of acting upon neurons to increase neuronal glutaminase (Ye et al., 2013), a mitochondrial enzyme responsible for the conversion of glutamine to glutamate (Zhao et al., 2012). Importantly, TNF-α-induced increases in glutaminase have been tied to the induction of ROS (Wang K. et al., 2017), demonstrating the functional overlap that exists in these stress and inflammatory sensitive systems. In addition to stimulating the release of glutamate, microglia can actively synthesize and release D-serine (Wu et al., 2004). D-serine is a co-agonist for NMDA receptors and strikingly exhibits a three-fold greater affinity for the receptor compared with glycine (Matsui et al., 1995). Several studies have demonstrated that exposure to social defeat stress in mice is capable of enhancing D-serine that is associated with anxiety- and depressive-like behaviors (Wang J. et al., 2017; Dong et al., 2018). Moreover, genetic deletion of D-serine was capable of conferring resilience to mice exposed to chronic social defeat (Dong et al., 2018). While it is currently unclear if these defeat-induced increases in D-serine are driven by defeat-induced proinflammatory cytokines or activation of microglia, it has been shown that administration of nonsteroidal anti-inflammatories such as mefenamic acid (Armagan et al., 2012a,b), acetaminophen, and naproxen (Armagan et al., 2012a) are capable of inhibiting D-serine.
Importantly, the role of stress and inflammation in glutamatergic excitotoxicity extends beyond glutamate receptors and their ligands. A number of studies have further demonstrated microglial involvement in glutamate accumulation in the extracellular space. Specifically, microglial stimulation with IL-1β (Ye et al., 2013) or TNF-α (Takeuchi et al., 2006; Ye et al., 2013) promotes the release of microglial glutamate. Under normal resting non-stress conditions, the brain has a number of mechanisms in place to manage excess synaptic glutamate. One such method is astrocyte mediated uptake via EAAT1 and EAAT2 in humans and glutamate–aspartate transporter (GLAST) and glutamate transporter 1 (GLT1) in rodents (Bezzi et al., 2004; Furuta et al., 2005). However, this protective mechanism has been shown to fail in instances where glutamate accumulation resulted from stimulation of microglia. Specifically, accumulation of glutamate in astrocytes results in a compensatory downregulation of EAAT1 (Takaki et al., 2012). Although preclinical studies assessing the role of GLAST and GLT1 in social stress-induced behavioral dysfunction has not been assessed, clinical studies have demonstrated altered expression of EAAT1 and 2 within brains of depressed patients (Miguel-Hidalgo et al., 2010; Rajkowska and Stockmeier, 2013). Together these data indicate that cytokine activation of microglia may result in a complex dysregulation of glutamate neuronal transmission by both enhancing local glutamate synthesis, stimulating glutamate release, and indirectly resulting in a downregulation of receptors involved in the maintenance of extra synaptic glutamate.
Remodeling of Excitatory Synaptic Terminals
In addition to modifying the release, synthesis and uptake of glutamate, stress and inflammation are known to alter the structure of excitatory synaptic terminals. Specifically, it has been shown that chronic stress results in the loss of dendritic spines in areas such as the prefrontal cortex (Goldwater et al., 2009). This loss of spines is directly associated with the emergence of anxiety- and depressive-like behaviors (Qiao et al., 2016). Stress has further been postulated to contribute to these effects by modulating a number of factors including the synthesis and release of MMP-9. In addition to promoting disruptions in the BBB (see “Blood Brain Barrier Disruption” section), MMP-9 is also involved in synaptic plasticity and remodeling of dendritic spines (Wang et al., 2008). In the presence of MMP-9, dendritic spines reshape from a short and round to a long and thin morphology (Michaluk et al., 2011). These long and thin spines are suggested to be less effective in conducting excitatory signals as they restrict Ca2+ flow (Ebrahimi and Okabe, 2014). Moreover, the thin and elongated spines also demonstrate greater vulnerability to the damaging cellular consequences of stress exposure (Radley et al., 2008; Bloss et al., 2011). In this manner, MMP-9-induced remodeling of dendritic spines may reduce neuronal excitability and promote the loss of dendritic spines. While clinical studies documenting the role of MMP-9 in the emergence of stress-induced depression are lacking, preclinical studies have demonstrated that exposure to social defeat results in elevations of the cytokine IL-1α and MMP-9. Importantly, these findings were most pronounced in susceptible rodents (Stelzhammer et al., 2015). These effects of social defeat on dendritic spines is not limited to MMP-9. Within the NAc inhibition of κB kinase (IκK) has also been shown to promote the formation of long and thin spines in animals exposed to social defeat (Christoffel et al., 2012). Moreover, this study found a trend to suggest that a greater number of long and thin spines was negatively associated with social interaction which could be reversed by inhibiting IκK (Christoffel et al., 2012). This same group later showed that chronic exposure to social defeat was also associated with an increase in the number of immature stubby spines in the NAc (Christoffel et al., 2015). In accordance with their previous findings, a larger number of stubby spines was associated with the emergence of social avoidance (Christoffel et al., 2015).
In the developing brain, microglia are well known to contribute to the remodeling of neuronal synapses through a process termed synaptic pruning (reviewed in Lenz and Nelson, 2018). Synaptic pruning has been described as a phagocytic event where immature and highly active synapses are permanently removed. It was originally suggested that this occurred via microglial engulfment of dendritic spines. However, a study by Weinhard et al. (2018) demonstrated that although microglia did contact dendritic spines, they did not completely engulf the dendritic spines for elimination. Instead it was found that microglia participate in a process termed trogocytosis in which only a small portion of the dendritic spine is phagocytosed (Weinhard et al., 2018). This process of trogocytosis also stimulates the formation of new long and thin filopodia shaped spines (Weinhard et al., 2018). While studies determining the involvement of microglial pruning in the consequences of stress exposure is unknown, it is probable that similar phagocytic processes could occur as a consequence of stress exposure.
Conclusion
Prospective studies have clearly linked inflammatory related disorders with increased risk of depression. Moreover, several clinical studies support the notion that neuroinflammation is associated with depressive symptomatology. However, our understanding of the mechanisms that are impacted by neuroinflammation, especially in the context of social stressors is at its infancy. Gaining a better understanding of neuroinflammatory-mediated adaptations that occur during stress and are capable of producing psychopathology will be a great advance in understanding the role of neuroinflammation in the etiology of depressive and anxiety disorders. Beyond the recognized effects of inflammatory cytokines on neurotransmitter and neuropeptide systems, inflammation may likely regulate susceptibility to social stress by altering the BBB, sensitizing microglia, producing mitochondrial dysfunction and oxidative stress as well as contributing to glutamate toxicity. This review highlights these cytokine-sensitive mechanisms that are favorably positioned to contribute to pathology, yet in many cases their direct regulation by inflammatory cytokines in the context of social stress has not been determined and will represent a great advance to the etiology of stress-induced psychiatric disorders.
Author Contributions
SW researched and wrote a considerable amount of the review article (2.5–3 of the 5 sections) and edited the rest of the review written by JF. JF researched and wrote a large amount of this review article (2.5 of the 5 sections), and revised the document as suggested.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. The authors were supported by research funding from the National Institute of Mental Health (1R01MH113892-01A1 to SW), the American Heart Association (15SDG22430017 to SW, 17PRE33670106 to JF), the Veterans Administration (I21 BX002085 to SW), and the Brain Behavior Research Foundation NARSAD Young Investigator Grant (26809 to SW).
References
- Al-Harbi K. S. (2012). Treatment-resistant depression: therapeutic trends, challenges and future directions. Patient Prefer. Adherence 6, 369–388. 10.2147/PPA.s29716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen A. P., Kennedy P. J., Dockray S., Cryan J. F., Dinan T. G., Clarke G. (2017). The trier social stress test: principles and practice. Neurobiol. Stress 6, 113–126. 10.1016/j.ynstr.2016.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida D. M. (2005). Resilience and vulnerability to daily stressors assessed via diary methods. Curr. Dir. Psychol. Sci. 14, 64–68. 10.1111/j.0963-7214.2005.00336.x [DOI] [Google Scholar]
- Alvarez R. P., Chen G., Bodurka J., Kaplan R., Grillon C. (2011). Phasic and sustained fear in humans elicits distinct patterns of brain activity. Neuroimage 55, 389–400. 10.1016/j.neuroimage.2010.11.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anda R., Williamson D., Jones D., Macera C., Eaker E., Glassman A., et al. (1993). Depressed affect, hopelessness and the risk of ischemic heart disease in a cohort of U.S. adults. Epidemiology 4, 285–294. 10.1097/00001648-199307000-00003 [DOI] [PubMed] [Google Scholar]
- Anis N. A., Berry S. C., Burton N. R., Lodge D. (1983). The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br. J. Pharmacol. 79, 565–575. 10.1111/j.1476-5381.1983.tb11031.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa K., Arakawa H., Hueston C. M., Deak T. (2014). Effects of the estrous cycle and ovarian hormones on central expression of interleukin-1 evoked by stress in female rats. Neuroendocrinology 100, 162–177. 10.1159/000368606 [DOI] [PubMed] [Google Scholar]
- Armagan G., Kanit L., Yalcin A. (2012a). Effects of non-steroidal antiinflammatory drugs on D-serine-induced oxidative stress in vitro. Drug Chem. Toxicol. 35, 393–398. 10.3109/01480545.2011.633086 [DOI] [PubMed] [Google Scholar]
- Armagan G., Turunc E., Kanit L., Yalcin A. (2012b). Neuroprotection by mefenamic acid against D-serine: involvement of oxidative stress, inflammation and apoptosis. Free Radic. Res. 46, 726–739. 10.3109/10715762.2012.669836 [DOI] [PubMed] [Google Scholar]
- Badoer E. (2010). Microglia: activation in acute and chronic inflammatory states and in response to cardiovascular dysfunction. Int. J. Biochem. Cell Biol. 42, 1580–1585. 10.1016/j.biocel.2010.07.005 [DOI] [PubMed] [Google Scholar]
- Banasr M., Valentine G. W., Li X.-Y., Gourley S. L., Taylor J. R., Duman R. S. (2007). Chronic unpredictable stress decreases cell proliferation in the cerebral cortex of the adult rat. Biol. Psychiatry 62, 496–504. 10.1016/j.biopsych.2007.02.006 [DOI] [PubMed] [Google Scholar]
- Bangasser D. A., Wicks B. (2017). Sex-specific mechanisms for responding to stress. J. Neurosci. Res. 95, 75–82. 10.1002/jnr.23812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangasser D. A., Wiersielis K. R. (2018). Sex differences in stress responses: a critical role for corticotropin-releasing factor. Hormones 17, 5–13. 10.1007/s42000-018-0002-z [DOI] [PubMed] [Google Scholar]
- Banks W. A. (2005). Blood-brain barrier transport of cytokines: a mechanism for neuropathology. Curr. Pharm. Des. 11, 973–984. 10.2174/1381612053381684 [DOI] [PubMed] [Google Scholar]
- Banks W. A., Kastin A. J., Durham D. A. (1989). Bidirectional transport of interleukin-1 alpha across the blood-brain barrier. Brain Res. Bull. 23, 433–437. 10.1016/0361-9230(89)90185-8 [DOI] [PubMed] [Google Scholar]
- Banks W. A., Ortiz L., Plotkin S. R., Kastin A. J. (1991). Human interleukin (IL) 1 alpha, murine IL-1 alpha and murine IL-1 beta are transported from blood to brain in the mouse by a shared saturable mechanism. J. Pharmacol. Exp. Ther. 259, 988–996. [PubMed] [Google Scholar]
- Barger S. W., Goodwin M. E., Porter M. M., Beggs M. L. (2007). Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J. Neurochem. 101, 1205–1213. 10.1111/j.1471-4159.2007.04487.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrash J., Tranel D., Anderson S. W. (2000). Acquired personality disturbances associated with bilateral damage to the ventromedial prefrontal region. Dev. Neuropsychol. 18, 355–381. 10.1207/s1532694205Barrash [DOI] [PubMed] [Google Scholar]
- Barrientos R. M., Kitt M. M., Watkins L. R., Maier S. F. (2015). Neuroinflammation in the normal aging hippocampus. Neuroscience 309, 84–99. 10.1016/j.neuroscience.2015.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bath K. G., Manzano-Nieves G., Goodwill H. (2016). Early life stress accelerates behavioral and neural maturation of the hippocampus in male mice. Horm. Behav. 82, 64–71. 10.1016/j.yhbeh.2016.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A. M. (2007). Evolutionary biology: animal personalities. Nature 447, 539–540. 10.1038/447539a [DOI] [PubMed] [Google Scholar]
- Berman R. M., Cappiello A., Anand A., Oren D. A., Heninger G. R., Charney D. S., et al. (2000). Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354. 10.1016/s0006-3223(99)00230-9 [DOI] [PubMed] [Google Scholar]
- Bezzi P., Gundersen V., Galbete J. L., Seifert G., Steinhäuser C., Pilati E., et al. (2004). Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat. Neurosci. 7, 613–620. 10.1038/nn1246 [DOI] [PubMed] [Google Scholar]
- Blandino P., Jr., Barnum C. J., Deak T. (2006). The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1β responses to stress. J. Neuroimmunol. 173, 87–95. 10.1016/j.jneuroim.2005.11.021 [DOI] [PubMed] [Google Scholar]
- Blandino P., Jr., Hueston C. M., Barnum C. J., Bishop C., Deak T. (2013). The impact of ventral noradrenergic bundle lesions on increased IL-1 in the PVN and hormonal responses to stress in male sprague dawley rats. Endocrinology 154, 2489–2500. 10.1210/en.2013-1075 [DOI] [PubMed] [Google Scholar]
- Bloss E. B., Janssen W. G., Ohm D. T., Yuk F. J., Wadsworth S., Saardi K. M., et al. (2011). Evidence for reduced experience-dependent dendritic spine plasticity in the aging prefrontal cortex. J. Neurosci. 31, 7831–7839. 10.1523/JNEUROSCI.0839-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brites D., Fernandes A. (2015). Neuroinflammation and Depression: microglia activation, extracellular microvesicles and microRNA dysregulation. Front. Cell. Neurosci. 9:476. 10.3389/fncel.2015.00476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G. W., Harris T., Copeland J. R. (1977). Depression and loss. Br. J. Psychiatry 130, 1–18. 10.1192/bjp.130.1.1 [DOI] [PubMed] [Google Scholar]
- Cairns K. E., Yap M. B., Pilkington P. D., Jorm A. F. (2014). Risk and protective factors for depression that adolescents can modify: a systematic review and meta-analysis of longitudinal studies. J. Affect. Disord. 169, 61–75. 10.1016/j.jad.2014.08.006 [DOI] [PubMed] [Google Scholar]
- Cambron J. M., Acitelli L. K., Pettit J. W. (2009). Explaining gender differences in depression: an interpersonal contingent self-esteem perspective. Sex Roles 61, 751–761. 10.1007/s11199-009-9616-6 [DOI] [Google Scholar]
- Capitanio J. P., Lerche N. W. (1991). Psychosocial factors and disease progression in simian AIDS: a preliminary report. AIDS 5, 1103–1106. 10.1097/00002030-199109000-00007 [DOI] [PubMed] [Google Scholar]
- Capuron L., Ravaud A., Neveu P. J., Miller A. H., Maes M., Dantzer R. (2002). Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol. Psychiatry 7, 468–473. 10.1038/sj.mp.4000995 [DOI] [PubMed] [Google Scholar]
- Carroll J. E., Low C. A., Prather A. A., Cohen S., Fury J. M., Ross D. C., et al. (2011). Negative affective responses to a speech task predict changes in interleukin (IL)-6. Brain Behav. Immun. 25, 232–238. 10.1016/j.bbi.2010.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaijale N. N., Curtis A. L., Wood S. K., Zhang X.-Y., Bhatnagar S., Reyes B. A. S., et al. (2013). Social Stress engages opioid regulation of locus coeruleus norepinephrine neurons and induces a state of cellular and physical opiate dependence. Neuropsychopharmacology 38, 1833–1843. 10.1038/npp.2013.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-H., Hsiao Y.-H., Chen Y.-W., Yu Y.-J., Gean P.-W. (2015). Social isolation-induced increase in NMDA receptors in the hippocampus exacerbates emotional dysregulation in mice. Hippocampus 25, 474–485. 10.1002/hipo.22384 [DOI] [PubMed] [Google Scholar]
- Chen E., Miller G. E., Walker H. A., Arevalo J. M., Sung C. Y., Cole S. W. (2009). Genome-wide transcriptional profiling linked to social class in asthma. Thorax 64, 38–43. 10.1136/thx.2007.095091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Jope R. S., Beurel E. (2015). A pre-conditioning stress accelerates increases in mouse plasma inflammatory cytokines induced by stress. BMC Neurosci. 16:31. 10.1186/s12868-015-0169-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson J. P., Thompson B. M., Watkins L. R., Maier S. F. (2009). Medial prefrontal cortical activation modulates the impact of controllable and uncontrollable stressor exposure on a social exploration test of anxiety in the rat. Stress 12, 445–450. 10.1080/10253890802510302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffel D. J., Golden S. A., Heshmati M., Graham A., Birnbaum S., Neve R. L., et al. (2012). Effects of inhibitor of κB kinase activity in the nucleus accumbens on emotional behavior. Neuropsychopharmacology 37, 2615–2623. 10.1038/npp.2012.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffel D. J., Golden S. A., Walsh J. J., Guise K. G., Heshmati M., Friedman A. K., et al. (2015). Excitatory transmission at thalamo-striatal synapses mediates susceptibility to social stress. Nat. Neurosci. 18, 962–964. 10.1038/nn.4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coria-Avila G. A., Manzo J., Garcia L. I., Carrillo P., Miquel M., Pfaus J. G. (2014). Neurobiology of social attachments. Neurosci. Biobehav. Rev. 43, 173–182. 10.1016/j.neubiorev.2014.04.004 [DOI] [PubMed] [Google Scholar]
- Costa-Nunes J., Zubareva O., Araújo-Correia M., Valença A., Schroeter C. A., Pawluski J. L., et al. (2014). Altered emotionality, hippocampus-dependent performance and expression of NMDA receptor subunit mRNAs in chronically stressed mice. Stress 17, 108–116. 10.3109/10253890.2013.872619 [DOI] [PubMed] [Google Scholar]
- Crestani C. C., Alves F. H., Gomes F. V., Resstel L. B., Correa F. M., Herman J. P. (2013). Mechanisms in the bed nucleus of the stria terminalis involved in control of autonomic and neuroendocrine functions: a review. Curr. Neuropharmacol. 11, 141–159. 10.2174/1570159X11311020002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker S. J., Milner R., Pham-Mitchell N., Campbell I. L. (2006). Cell and agonist-specific regulation of genes for matrix metalloproteinases and their tissue inhibitors by primary glial cells. J. Neurochem. 98, 812–823. 10.1111/j.1471-4159.2006.03927.x [DOI] [PubMed] [Google Scholar]
- Cserép C., Pósfai B., Schwarcz A. D., Dénes A. (2018). Mitochondrial ultrastructure is coupled to synaptic performance at axonal release sites. eNeuro 5:ENEURO.0390-17.2018. 10.1523/ENEURO.0390-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuffel B., Wamboldt M., Borish L., Kennedy S., Crystal-Peters J. (1999). Economic consequences of comorbid depression, anxiety and allergic rhinitis. Psychosomatics 40, 491–496. 10.1016/s0033-3182(99)71187-4 [DOI] [PubMed] [Google Scholar]
- Dantzer R., O’Connor J. C., Freund G. G., Johnson R. W., Kelley K. W. (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. 10.1038/nrn2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak T., Kudinova A., Lovelock D. F., Gibb B. E., Hennessy M. B. (2017). A multispecies approach for understanding neuroimmune mechanisms of stress. Dialogues Clin. Neurosci. 19, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpech J.-C., Wei L., Hao J., Yu X., Madore C., Butovsky O., et al. (2016). Early life stress perturbs the maturation of microglia in the developing hippocampus. Brain Behav. Immun. 57, 79–93. 10.1016/j.bbi.2016.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira M. R., Silvestrin R. B., Mello E. S. T., Moreira J. C. (2007). Oxidative stress in the hippocampus, anxiety-like behavior and decreased locomotory and exploratory activity of adult rats: effects of sub acute vitamin A supplementation at therapeutic doses. Neurotoxicology 28, 1191–1199. 10.1016/j.neuro.2007.07.008 [DOI] [PubMed] [Google Scholar]
- Denicoff K. D., Rubinow D. R., Papa M. Z., Simpson C., Seipp C. A., Lotze M. T., et al. (1987). The neuropsychiatric effects of treatment with interleukin-2 and lymphokine-activated killer cells. Ann. Intern. Med. 107, 293–300. 10.7326/0003-4819-107-2-293 [DOI] [PubMed] [Google Scholar]
- Devorak J., Torres-Platas S. G., Davoli M. A., Prud’homme J., Turecki G., Mechawar N. (2015). Cellular and molecular inflammatory profile of the choroid plexus in depression and suicide. Front. Psychiatry 6:138. 10.3389/fpsyt.2015.00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G., Loddo P., Tanda G. (1999). Reciprocal changes in prefrontal and limbic dopamine responsiveness to aversive and rewarding stimuli after chronic mild stress: implications for the psychobiology of depression. Biol. Psychiatry 46, 1624–1633. 10.1016/s0006-3223(99)00236-x [DOI] [PubMed] [Google Scholar]
- Diamond B., Volpe B. T. (2012). A model for lupus brain disease. Immunol. Rev. 248, 56–67. 10.1111/j.1600-065X.2012.01137.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson S. S., Gable S. L., Irwin M. R., Aziz N., Kemeny M. E. (2009). Social-evaluative threat and proinflammatory cytokine regulation: an experimental laboratory investigation. Psychol. Sci. 20, 1237–1244. 10.1111/j.1467-9280.2009.02437.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimayuga F. O., Reed J. L., Carnero G. A., Wang C., Dimayuga E. R., Dimayuga V. M., et al. (2005). Estrogen and brain inflammation: effects on microglial expression of MHC, costimulatory molecules and cytokines. J. Neuroimmunol. 161, 123–136. 10.1016/j.jneuroim.2004.12.016 [DOI] [PubMed] [Google Scholar]
- Dolcos F., Katsumi Y., Weymar M., Moore M., Tsukiura T., Dolcos S. (2017). Emerging directions in emotional episodic memory. Front. Psychol. 8:1867. 10.3389/fpsyg.2017.01867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll D. N., Rellick S. L., Barr T. L., Ren X., Simpkins J. W. (2015). Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J. Neurochem. 132, 443–451. 10.1111/jnc.13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C., Zhang J. C., Ren Q., Ma M., Qu Y., Zhang K., et al. (2018). Deletion of serine racemase confers D-serine -dependent resilience to chronic social defeat stress. Neurochem. Int. 116, 43–51. 10.1016/j.neuint.2018.03.008 [DOI] [PubMed] [Google Scholar]
- Drevets W. C. (2000). Neuroimaging studies of mood disorders. Biol. Psychiatry 48, 813–829. 10.1016/S0006-3223(00)01020-9 [DOI] [PubMed] [Google Scholar]
- Drevets W. C., Price J. L., Furey M. L. (2008). Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct. Funct. 213, 93–118. 10.1007/s00429-008-0189-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque-Wilckens N., Steinman M. Q., Busnelli M., Chini B., Yokoyama S., Pham M., et al. (2018). Oxytocin receptors in the anteromedial bed nucleus of the stria terminalis promote stress-induced social avoidance in female california mice. Biol. Psychiatry 83, 203–213. 10.1016/j.biopsych.2017.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi S., Okabe S. (2014). Structural dynamics of dendritic spines: molecular composition, geometry and functional regulation. Biochim. Biophys. Acta 1838, 2391–2398. 10.1016/j.bbamem.2014.06.002 [DOI] [PubMed] [Google Scholar]
- Eiland L., McEwen B. S. (2012). Early life stress followed by subsequent adult chronic stress potentiates anxiety and blunts hippocampal structural remodeling. Hippocampus 22, 82–91. 10.1002/hipo.20862 [DOI] [PubMed] [Google Scholar]
- Faust T. W., Chang E. H., Kowal C., Berlin R., Gazaryan I. G., Bertini E., et al. (2010). Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc. Natl. Acad. Sci. U S A 107, 18569–18574. 10.1073/pnas.1006980107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festoff B. W., Sajja R. K., van Dreden P., Cucullo L. (2016). HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J. Neuroinflammation 13:194. 10.1186/s12974-016-0670-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnell J. E., Lombard C. M., Melson M. N., Singh N. P., Nagarkatti M., Nagarkatti P., et al. (2017a). The protective effects of resveratrol on social stress-induced cytokine release and depressive-like behavior. Brain Behav. Immun. 59, 147–157. 10.1016/j.bbi.2016.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnell J. E., Lombard C. M., Padi A. R., Moffitt C. M., Wilson L. B., Wood C. S., et al. (2017b). Physical versus psychological social stress in male rats reveals distinct cardiovascular, inflammatory and behavioral consequences. PLoS One 12:e0172868. 10.1371/journal.pone.0172868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnell J. E., Muniz B. L., Padi A. R., Lombard C. M., Moffitt C. M., Wood C. S., et al. (2018). Essential role of ovarian hormones in susceptibility to the consequences of witnessing social defeat in female rats. Biol. Psychiatry 84, 372–382. 10.1016/j.biopsych.2018.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnell J. E., Wood S. K. (2016). Neuroinflammation at the interface of depression and cardiovascular disease: evidence from rodent models of social stress. Neurobiol. Stress 4, 1–14. 10.1016/j.ynstr.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiske A., Wetherell J. L., Gatz M. (2009). Depression in older adults. Annu. Rev. Clin. Psychol. 5, 363–389. 10.1146/annurev.clinpsy.032408.153621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken L. K., Weber M. D., Daut R. A., Kitt M. M., Frank M. G., Watkins L. R., et al. (2016). Stress-induced neuroinflammatory priming is time of day dependent. Psychoneuroendocrinology 66, 82–90. 10.1016/j.psyneuen.2016.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M. G., Baratta M. V., Sprunger D. B., Watkins L. R., Maier S. F. (2007). Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav. Immun. 21, 47–59. 10.1016/j.bbi.2006.03.005 [DOI] [PubMed] [Google Scholar]
- Frank M. G., Fonken L. K., Annis J. L., Watkins L. R., Maier S. F. (2018). Stress disinhibits microglia via down-regulation of CD200R: a mechanism of neuroinflammatory priming. Brain Behav. Immun. 69, 62–73. 10.1016/j.bbi.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M. G., Thompson B. M., Watkins L. R., Maier S. F. (2012). Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav. Immun. 26, 337–345. 10.1016/j.bbi.2011.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin T. C., Wohleb E. S., Zhang Y., Fogaca M., Hare B., Duman R. S. (2018). Persistent increase in microglial RAGE contributes to chronic stress-induced priming of depressive-like behavior. Biol. Psychiatry 83, 50–60. 10.1016/j.biopsych.2017.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruhbeis C., Frohlich D., Kuo W. P., Kramer-Albers E. M. (2013). Extracellular vesicles as mediators of neuron-glia communication. Front. Cell. Neurosci. 7:182. 10.3389/fncel.2013.00182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta A., Takashima S., Yokoo H., Rothstein J. D., Wada K., Iwaki T. (2005). Expression of glutamate transporter subtypes during normal human corticogenesis and type II lissencephaly. Dev. Brain Res. 155, 155–164. 10.1016/j.devbrainres.2005.01.005 [DOI] [PubMed] [Google Scholar]
- Gallo J. J., Royall D. R., Anthony J. C. (1993). Risk factors for the onset of depression in middle age and later life. Soc. Psychiatry Psychiatr. Epidemiol. 28, 101–108. 10.1007/BF00801739 [DOI] [PubMed] [Google Scholar]
- Gamaro G. D., Streck E. L., Matte C., Prediger M. E., Wyse A. T., Dalmaz C. (2003). Reduction of hippocampal Na+, K+-ATPase activity in rats subjected to an experimental model of depression. Neurochem. Res. 28, 1339–1344. 10.1023/A:1024988113978 [DOI] [PubMed] [Google Scholar]
- Ganguly K., Rejmak E., Mikosz M., Nikolaev E., Knapska E., Kaczmarek L. (2013). Matrix metalloproteinase (MMP) 9 transcription in mouse brain induced by fear learning. J. Biol. Chem. 288, 20978–20991. 10.1074/jbc.m113.457903 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gardner A., Johansson A., Wibom R., Nennesmo I., Von Dobeln U., Hagenfeldt L., et al. (2003). Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 76, 55–68. 10.1016/s0165-0327(02)00067-8 [DOI] [PubMed] [Google Scholar]
- Gemma C., Bachstetter A. D. (2013). The role of microglia in adult hippocampal neurogenesis. Front. Cell. Neurosci. 7:229. 10.3389/fncel.2013.00229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel N., Bale T. L. (2009). Examining the intersection of sex and stress in modelling neuropsychiatric disorders. J. Neuroendocrinol. 21, 415–420. 10.1111/j.1365-2826.2009.01843.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldwater D. S., Pavlides C., Hunter R. G., Bloss E. B., Hof P. R., Mcewen B. S., et al. (2009). Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience 164, 798–808. 10.1016/j.neuroscience.2009.08.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez C. R., Boehmer E. D., Kovacs E. J. (2005). The aging innate immune system. Curr. Opin. Immunol. 17, 457–462. 10.1016/j.coi.2005.07.013 [DOI] [PubMed] [Google Scholar]
- Gong Y., Chai Y., Ding J. H., Sun X. L., Hu G. (2011). Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neurosci. Lett. 488, 76–80. 10.1016/j.neulet.2010.11.006 [DOI] [PubMed] [Google Scholar]
- Gorczynski R. M. (2005). CD200 and its receptors as targets for immunoregulation. Curr. Opin. Investig. Drugs 6, 483–488. [PubMed] [Google Scholar]
- Graham J. E., Christian L. M., Kiecolt-Glaser J. K. (2006). Stress, age, and immune function: toward a lifespan approach. J. Behav. Med. 29, 389–400. 10.1007/s10865-006-9057-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray P., Cooney J. (1982). Stress-induced responses and open-field behavior in estrous and nonestrous mice. Physiol. Behav. 29, 287–292. 10.1016/0031-9384(82)90017-8 [DOI] [PubMed] [Google Scholar]
- Greenberg G. D., Laman-Maharg A., Campi K. L., Voigt H., Orr V. N., Schaal L., et al. (2013). Sex differences in stress-induced social withdrawal: role of brain derived neurotrophic factor in the bed nucleus of the stria terminalis. Front. Behav. Neurosci. 7:223. 10.3389/fnbeh.2013.00223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg G. D., Steinman M. Q., Doig I. E., Hao R., Trainor B. C. (2015). Effects of social defeat on dopamine neurons in the ventral tegmental area in male and female California mice. Eur. J. Neurosci. 42, 3081–3094. 10.1111/ejn.13099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller J., Fuchs E., Halasz J., Makara G. B. (1999). Defeat is a major stressor in males while social instability is stressful mainly in females: towards the development of a social stress model in female rats. Brain Res. Bull. 50, 33–39. 10.1016/s0361-9230(99)00087-8 [DOI] [PubMed] [Google Scholar]
- Halliwell B. (1992). Reactive oxygen species and the central nervous system. J. Neurochem. 59, 1609–1623. 10.1007/978-3-642-77609-0_2 [DOI] [PubMed] [Google Scholar]
- Hankin B. L., Abramson L. Y., Moffitt T. E., Silva P. A., Mcgee R., Angell K. E. (1998). Development of depression from preadolescence to young adulthood: emerging gender differences in a 10-year longitudinal study. J. Abnorm. Psychol. 107, 128–140. 10.1037/0021-843x.107.1.128 [DOI] [PubMed] [Google Scholar]
- Hansen I., Klimek L., Mosges R., Hormann K. (2004). Mediators of inflammation in the early and the late phase of allergic rhinitis. Curr. Opin. Allergy. Clin. Immunol. 4, 159–163. 10.1097/00130832-200406000-00004 [DOI] [PubMed] [Google Scholar]
- Harris A. Z., Atsak P., Bretton Z. H., Holt E. S., Alam R., Morton M. P., et al. (2018). A novel method for chronic social defeat stress in female mice. Neuropsychopharmacology 43, 1276–1283. 10.1038/npp.2017.259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry G. J., Kraft A. D. (2012). Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology 33, 191–206. 10.1016/j.neuro.2012.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodes G. E., Pfau M. L., Leboeuf M., Golden S. A., Christoffel D. J., Bregman D., et al. (2014). Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc. Natl. Acad. Sci. U S A 111, 16136–16141. 10.1073/pnas.1415191111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollis F., Van Der Kooij M. A., Zanoletti O., Lozano L., Canto C., Sandi C. (2015). Mitochondrial function in the brain links anxiety with social subordination. Proc. Natl. Acad. Sci. U S A 112, 15486–15491. 10.1073/pnas.1512653112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holly E. N., Shimamoto A., Debold J. F., Miczek K. A. (2012). Sex differences in behavioral and neural cross-sensitization and escalated cocaine taking as a result of episodic social defeat stress in rats. Psychopharmacology 224, 179–188. 10.1007/s00213-012-2846-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert G. W., Li C., Rainnie D. G., Muly E. C. (2014). Effects of stress on AMPA receptor distribution and function in the basolateral amygdala. Brain Struct. Funct. 219, 1169–1179. 10.1007/s00429-013-0557-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner J. E., Bean B. P. (1988). Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc. Natl. Acad. Sci. U S A 85, 1307–1311. 10.1073/pnas.85.4.1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman J. C., Celano C. M., Beach S. R., Motiwala S. R., Januzzi J. L. (2013). Depression and cardiac disease: epidemiology, mechanisms and diagnosis. Cardiovasc. Psychiatry Neurol. 2013:695925. 10.1155/2013/695925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhman K. L., Solomon M. B., Janicki M., Harmon A. C., Lin S. M., Israel J. E., et al. (2003). Conditioned defeat in male and female Syrian hamsters. Horm. Behav. 44, 293–299. 10.1016/j.yhbeh.2003.05.001 [DOI] [PubMed] [Google Scholar]
- Iniguez S. D., Flores-Ramirez F. J., Riggs L. M., Alipio J. B., Garcia-Carachure I., Hernandez M. A., et al. (2018). Vicarious social defeat stress induces depression-related outcomes in female mice. Biol. Psychiatry 83, 9–17. 10.1016/j.biopsych.2017.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin M. R., Miller A. H. (2007). Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav. Immun. 21, 374–383. 10.1016/j.bbi.2007.01.010 [DOI] [PubMed] [Google Scholar]
- Isaac J. T. R., Ashby M. C., Mcbain C. J. (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871. 10.1016/j.neuron.2007.06.001 [DOI] [PubMed] [Google Scholar]
- Jacobson-Pick S., Audet M.-C., Mcquaid R. J., Kalvapalle R., Anisman H. (2013). Social agonistic distress in male and female mice: changes of behavior and brain monoamine functioning in relation to acute and chronic challenges. PLoS One 8:e60133. 10.1371/journal.pone.0060133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway C. A., Jr., Travers P., Walport M., Shlomchik M. J. (2001). Immunobiology: The Immune System in Health and Disease. New York, NY: Garland Science. [Google Scholar]
- Jayatissa M. N., Bisgaard C., Tingström A., Papp M., Wiborg O. (2006). Hippocampal cytogenesis correlates to escitalopram-mediated recovery in a chronic mild stress rat model of depression. Neuropsychopharmacology 31, 2395–2404. 10.1038/sj.npp.1301041 [DOI] [PubMed] [Google Scholar]
- Jiang B., Wang W., Wang F., Hu Z.-L., Xiao J.-L., Yang S., et al. (2013). The stability of NR2B in the nucleus accumbens controls behavioral and synaptic adaptations to chronic stress. Biol. Psychiatry 74, 145–155. 10.1016/j.biopsych.2012.10.031 [DOI] [PubMed] [Google Scholar]
- Joffe R. T., Levitt A. J., Sokolov S. T. (1996). Augmentation strategies: focus on anxiolytics. J. Clin. Psychiatry 57, 25–31; discussion 32–23. [PubMed] [Google Scholar]
- Johnson F. K., Kaffman A. (2018). Early life stress perturbs the function of microglia in the developing rodent brain: new insights and future challenges. Brain Behav. Immun. 69, 18–27. 10.1016/j.bbi.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katon W. J., Richardson L., Lozano P., Mccauley E. (2004). The relationship of asthma and anxiety disorders. Psychosom. Med. 66, 349–355. 10.1097/00006842-200405000-00010 [DOI] [PubMed] [Google Scholar]
- Kendler K. S., Kessler R. C., Heath A. C., Neale M. C., Eaves L. J. (1991). Coping: a genetic epidemiological investigation. Psychol. Med. 21, 337–346. 10.1017/s0033291700020444 [DOI] [PubMed] [Google Scholar]
- Kessler R. C., McGonagle K. A., Swartz M., Blazer D. G., Nelson C. B. (1993). Sex and depression in the National Comorbidity Survey. I: Lifetime prevalence, chronicity and recurrence. J. Affect. Disord. 29, 85–96. 10.1016/0165-0327(93)90026-g [DOI] [PubMed] [Google Scholar]
- Kettenmann H., Hanisch U. K., Noda M., Verkhratsky A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. 10.1152/physrev.00011.2010 [DOI] [PubMed] [Google Scholar]
- Kiecolt-Glaser J. K., Graham J. E., Malarkey W. B., Porter K., Lemeshow S., Glaser R. (2008). Olfactory influences on mood and autonomic, endocrine and immune function. Psychoneuroendocrinology 33, 328–339. 10.1016/j.psyneuen.2007.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecolt-Glaser J. K., Heffner K. L., Glaser R., Malarkey W. B., Porter K., Atkinson C., et al. (2009). How stress and anxiety can alter immediate and late phase skin test responses in allergic rhinitis. Psychoneuroendocrinology 34, 670–680. 10.1016/j.psyneuen.2008.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecolt-Glaser J. K., Preacher K. J., MacCallum R. C., Atkinson C., Malarkey W. B., Glaser R. (2003). Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc. Natl. Acad. Sci. U S A 100, 9090–9095. 10.1073/pnas.1531903100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluver H., Bucy P. C. (1997). Preliminary analysis of functions of the temporal lobes in monkeys. 1939. J. Neuropsychiatry Clin. Neurosci. 9, 606–620. 10.1176/jnp.9.4.606 [DOI] [PubMed] [Google Scholar]
- Kobayashi K., Imagama S., Ohgomori T., Hirano K., Uchimura K., Sakamoto K., et al. (2013). Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 4:e525. 10.1038/cddis.2013.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohman R. A., Crowell B., Kusnecov A. W. (2010). Differential sensitivity to endotoxin exposure in young and middle-age mice. Brain Behav. Immun. 24, 486–492. 10.1016/j.bbi.2009.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollack-Walker S., Watson S. J., Akil H. (1997). Social stress in hamsters: defeat activates specific neurocircuits within the brain. J. Neurosci. 17, 8842–8855. 10.1523/JNEUROSCI.17-22-08842.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koning N., Swaab D. F., Hoek R. M., Huitinga I. (2009). Distribution of the immune inhibitory molecules CD200 and CD200R in the normal central nervous system and multiple sclerosis lesions suggests neuron-glia and glia-glia interactions. J. Neuropathol. Exp. Neurol. 68, 159–167. 10.1097/nen.0b013e3181964113 [DOI] [PubMed] [Google Scholar]
- Koolhaas J. M., de Boer S. F., Buwalda B., van Reenen K. (2007). Individual variation in coping with stress: a multidimensional approach of ultimate and proximate mechanisms. Brain Behav. Evol. 70, 218–226. 10.1159/000105485 [DOI] [PubMed] [Google Scholar]
- Koolhaas J. M., Korte S. M., De Boer S. F., Van Der Vegt B. J., Van Reenen C. G., Hopster H., et al. (1999). Coping styles in animals: current status in behavior and stress-physiology. Neurosci. Biobehav. Rev. 23, 925–935. 10.1016/s0149-7634(99)00026-3 [DOI] [PubMed] [Google Scholar]
- Kopp B. L., Wick D., Herman J. P. (2013). Differential effects of homotypic vs. heterotypic chronic stress regimens on microglial activation in the prefrontal cortex. Physiol. Behav. 122, 246–252. 10.1016/j.physbeh.2013.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisel T., Frank M. G., Licht T., Reshef R., Ben-Menachem-Zidon O., Baratta M. V., et al. (2014). Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 19, 699–709. 10.1038/mp.2013.155 [DOI] [PubMed] [Google Scholar]
- Kumar S., Hultman R., Hughes D., Michel N., Katz B. M., Dzirasa K. (2014). Prefrontal cortex reactivity underlies trait vulnerability to chronic social defeat stress. Nat. Commun. 5:4537. 10.1038/ncomms5537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai S., Wu G., Jiang Z. (2018). Glycyrrhizin treatment facilitates extinction of conditioned fear responses after a single prolonged stress exposure in rats. Cell. Physiol. Biochem. 45, 2529–2539. 10.1159/000488271 [DOI] [PubMed] [Google Scholar]
- LaPlant Q., Chakravarty S., Vialou V., Mukherjee S., Koo J. W., Kalahasti G., et al. (2009). Role of nuclear factor κB in ovarian hormone-mediated stress hypersensitivity in female mice. Biol. Psychiatry 65, 874–880. 10.1016/j.biopsych.2009.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz K. M., Nelson L. H. (2018). Microglia and beyond: innate immune cells as regulators of brain development and behavioral function. Front. Immunol. 9:698. 10.3389/fimmu.2018.00698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levita L., Hoskin R., Champi S. (2012). Avoidance of harm and anxiety: a role for the nucleus accumbens. Neuroimage 62, 189–198. 10.1016/j.neuroimage.2012.04.059 [DOI] [PubMed] [Google Scholar]
- Li Y., Howell E. A., Lagoo A. S., Kuchibhatla M., Pan H., Cohen H. J., et al. (2009). Differential gene expression of interleukin-1 receptor associated kinase-1 and interleukin-1 receptor associated kinase-M in peripheral blood mononuclear cells of young and aged rats following preconditioning with endotoxin. Shock 31, 55–63. 10.1097/shk.0b013e3181778ab2 [DOI] [PubMed] [Google Scholar]
- Li L.-H., Wang Z.-C., Yu J., Zhang Y.-Q. (2014). Ovariectomy results in variable changes in nociception, mood and depression in adult female rats. PLoS One 9:e94312. 10.1371/journal.pone.0094312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist D., Dhabhar F. S., James S. J., Hough C. M., Jain F. A., Bersani F. S., et al. (2017). Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology 76, 197–205. 10.1016/j.psyneuen.2016.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. Y., Coe C. L., Swenson C. A., Kelly E. A., Kita H., Busse W. W. (2002). School examinations enhance airway inflammation to antigen challenge. Am. J. Respir. Crit. Care Med. 165, 1062–1067. 10.1164/ajrccm.165.8.2109065 [DOI] [PubMed] [Google Scholar]
- Liu H.-S., Shi H.-L., Huang F., Peterson K. E., Wu H., Lan Y.-Y., et al. (2016). Astragaloside IV inhibits microglia activation via glucocorticoid receptor mediated signaling pathway. Sci. Rep. 6:19137. 10.1038/srep19137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftis J. M., Huckans M., Morasco B. J. (2010). Neuroimmune mechanisms of cytokine-induced depression: current theories and novel treatment strategies. Neurobiol. Dis. 37, 519–533. 10.1016/j.nbd.2009.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loram L. C., Sholar P. W., Taylor F. R., Wiesler J. L., Babb J. A., Strand K. A., et al. (2012). Sex and estradiol influence glial pro-inflammatory responses to lipopolysaccharide in rats. Psychoneuroendocrinology 37, 1688–1699. 10.1016/j.psyneuen.2012.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord J. M., Butcher S., Killampali V., Lascelles D., Salmon M. (2001). Neutrophil ageing and immunesenescence. Mech. Ageing Dev. 122, 1521–1535. 10.1016/s0047-6374(01)00285-8 [DOI] [PubMed] [Google Scholar]
- Lubach G. R., Coe C. L., Ershler W. B. (1995). Effects of early rearing environment on immune responses of infant rhesus monkeys. Brain Behav. Immun. 9, 31–46. 10.1006/brbi.1995.1004 [DOI] [PubMed] [Google Scholar]
- Lupien S. J., Maheu F., Tu M., Fiocco A., Schramek T. E. (2007). The effects of stress and stress hormones on human cognition: implications for the field of brain and cognition. Brain Cogn. 65, 209–237. 10.1016/j.bandc.2007.02.007 [DOI] [PubMed] [Google Scholar]
- MacDonald J. F., Miljkovic Z., Pennefather P. (1987). Use-dependent block of excitatory amino acid currents in cultured neurons by ketamine. J. Neurophysiol. 58, 251–266. 10.1152/jn.1987.58.2.251 [DOI] [PubMed] [Google Scholar]
- Madrigal J. L., Olivenza R., Moro M. A., Lizasoain I., Lorenzo P., Rodrigo J., et al. (2001). Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology 24, 420–429. 10.1016/s0893-133x(00)00208-6 [DOI] [PubMed] [Google Scholar]
- Maier S. F., Watkins L. R. (2005). Stressor controllability and learned helplessness: the roles of the dorsal raphe nucleus, serotonin and corticotropin-releasing factor. Neurosci. Biobehav. Rev. 29, 829–841. 10.1016/j.neubiorev.2005.03.021 [DOI] [PubMed] [Google Scholar]
- Marrie R. A., Walld R., Bolton J. M., Sareen J., Walker J. R., Patten S. B., et al. (2017). Increased incidence of psychiatric disorders in immune-mediated inflammatory disease. J. Psychosom. Res. 101, 17–23. 10.1016/j.jpsychores.2017.07.015 [DOI] [PubMed] [Google Scholar]
- Martinez M., Phillips P. J., Herbert J. (1998). Adaptation in patterns of c-fos expression in the brain associated with exposure to either single or repeated social stress in male rats. Eur. J. Neurosci. 10, 20–33. 10.1046/j.1460-9568.1998.00011.x [DOI] [PubMed] [Google Scholar]
- Massaad C. A., Klann E. (2011). Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 14, 2013–2054. 10.1089/ars.2010.3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T., Sekiguchi M., Hashimoto A., Tomita U., Nishikawa T., Wada K. (1995). Functional comparison of D-serine and glycine in rodents: the effect on cloned NMDA receptors and the extracellular concentration. J. Neurochem. 65, 454–458. 10.1046/j.1471-4159.1995.65010454.x [DOI] [PubMed] [Google Scholar]
- Mattson M. P., Gleichmann M., Cheng A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. 10.1016/j.neuron.2008.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen B. S., Gianaros P. J. (2010). Central role of the brain in stress and adaptation: links to socioeconomic status, health and disease. Ann. N Y Acad. Sci. 1186, 190–222. 10.1111/j.1749-6632.2009.05331.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim D. B., Weber M. D., Niraula A., Sawicki C. M., Liu X., Jarrett B. L., et al. (2018). Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Mol. Psychiatry 23, 1421–1431. 10.1038/mp.2017.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard C., Pfau M. L., Hodes G. E., Kana V., Wang V. X., Bouchard S., et al. (2017). Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 20, 1752–1760. 10.1038/s41593-017-0010-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaluk P., Wawrzyniak M., Alot P., Szczot M., Wyrembek P., Mercik K., et al. (2011). Influence of matrix metalloproteinase MMP-9 on dendritic spine morphology. J. Cell Sci. 124, 3369–3380. 10.1242/jcs.090852 [DOI] [PubMed] [Google Scholar]
- Miczek K. A. (1979). A new test for aggression in rats without aversive stimulation: differential effects of d-amphetamine and cocaine. Psychopharmacology 60, 253–259. 10.1007/bf00426664 [DOI] [PubMed] [Google Scholar]
- Miguel-Hidalgo J. J., Waltzer R., Whittom A. A., Austin M. C., Rajkowska G., Stockmeier C. A. (2010). Glial and glutamatergic markers in depression, alcoholism, and their comorbidity. J. Affect. Disord. 127, 230–240. 10.1016/j.jad.2010.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A. H., Haroon E., Raison C. L., Felger J. C. (2013). Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress. Anxiety 30, 297–306. 10.1002/da.22084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misaki M., Suzuki H., Savitz J., Drevets W. C., Bodurka J. (2016). Individual variations in nucleus accumbens responses associated with major depressive disorder symptoms. Sci. Rep. 6:21227. 10.1038/srep21227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra R., Sundlass K., Parker K. J., Schatzberg A. F., Lyons D. M. (2006). Social stress-related behavior affects hippocampal cell proliferation in mice. Physiol. Behav. 89, 123–127. 10.1016/j.physbeh.2006.05.047 [DOI] [PubMed] [Google Scholar]
- Moieni M., Irwin M. R., Jevtic I., Olmstead R., Breen E. C., Eisenberger N. I. (2015). Sex differences in depressive and socioemotional responses to an inflammatory challenge: implications for sex differences in depression. Neuropsychopharmacology 40, 1709–1716. 10.1038/npp.2015.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujica-Parodi L. R., Cha J., Gao J. (2017). From anxious to reckless: a control systems approach unifies prefrontal-limbic regulation across the spectrum of threat detection. Front. Syst. Neurosci. 11:18. 10.3389/fnsys.2017.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu R., Takahashi C., Miyake S., Fujimura H., Mochizuki H., Yamashita T. (2012). Angiogenesis induced by CNS inflammation promotes neuronal remodeling through vessel-derived prostacyclin. Nat. Med. 18, 1658–1664. 10.1038/nm.2943 [DOI] [PubMed] [Google Scholar]
- Murrough J. W., Huryk K. M., Mao X., Iacoviello B., Collins K., Nierenberg A. A., et al. (2018). A pilot study of minocycline for the treatment of bipolar depression: effects on cortical glutathione and oxidative stress in vivo. J. Affect. Disord. 230, 56–64. 10.1016/j.jad.2017.12.067 [DOI] [PubMed] [Google Scholar]
- Nair A., Bonneau R. H. (2006). Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J. Neuroimmunol. 171, 72–85. 10.1016/j.jneuroim.2005.09.012 [DOI] [PubMed] [Google Scholar]
- Nasca C., Zelli D., Bigio B., Piccinin S., Scaccianoce S., Nisticò R., et al. (2015). Stress dynamically regulates behavior and glutamatergic gene expression in hippocampus by opening a window of epigenetic plasticity. Proc. Natl. Acad. Sci. U S A 112, 14960–14965. 10.1073/pnas.1516016112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R. J., Trainor B. C. (2007). Neural mechanisms of aggression. Nat. Rev. Neurosci. 8, 536–546. 10.1038/nrn2174 [DOI] [PubMed] [Google Scholar]
- Neylan T. C., Sun B., Rempel H., Ross J., Lenoci M., O’Donovan A., et al. (2011). Suppressed monocyte gene expression profile in men versus women with PTSD. Brain Behav. Immun. 25, 524–531. 10.1016/j.bbi.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai K. A., Laney T., Mezulis A. H. (2013). Different stressors, different strategies, different outcomes: how domain-specific stress responses differentially predict depressive symptoms among adolescents. J. Youth Adolesc. 42, 1183–1193. 10.1007/s10964-012-9866-4 [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A., Kirchhoff F., Helmchen F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- Nolen-Hoeksema S. (2001). Gender differences in depression. Curr. Dir. Psychol. Sci. 10, 173–176. 10.1111/1467-8721.00142 [DOI] [Google Scholar]
- Oliveira L. A., Almeida J., Benini R., Crestani C. C. (2015). CRF1 and CRF2 receptors in the bed nucleus of the stria terminalis modulate the cardiovascular responses to acute restraint stress in rats. Pharmacol. Res. 95–96, 53–62. 10.1016/j.phrs.2015.03.012 [DOI] [PubMed] [Google Scholar]
- Ota K. T., Liu R.-J., Voleti B., Maldonado-Aviles J. G., Duric V., Iwata M., et al. (2014). REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat. Med. 20, 531–535. 10.1038/nm.3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace T. W., Mletzko T. C., Alagbe O., Musselman D. L., Nemeroff C. B., Miller A. H., et al. (2006). Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am. J. Psychiatry 163, 1630–1633. 10.1176/appi.ajp.163.9.1630 [DOI] [PubMed] [Google Scholar]
- Pandey S. P., Rai R., Gaur P., Prasad S. (2015). Development- and age-related alterations in the expression of AMPA receptor subunit GluR2 and its trafficking proteins in the hippocampus of male mouse brain. Biogerontology 16, 317–328. 10.1007/s10522-014-9548-6 [DOI] [PubMed] [Google Scholar]
- Patki G., Solanki N., Atrooz F., Allam F., Salim S. (2013). Depression, anxiety-like behavior and memory impairment are associated with increased oxidative stress and inflammation in a rat model of social stress. Brain Res. 1539, 73–86. 10.1016/j.brainres.2013.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson-Leary J., Eacret D., Chen R., Takano H., Nicholas B., Bhatnagar S. (2017). Inflammation and vascular remodeling in the ventral hippocampus contributes to vulnerability to stress. Transl. Psychiatry 7:e1160. 10.1038/tp.2017.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao H., Li M. X., Xu C., Chen H. B., An S. C., Ma X. M. (2016). Dendritic spines in depression: what we learned from animal models. Neural Plast. 2016:8056370. 10.1155/2016/8056370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J., Xu J., Zheng Y., Wei Y., Zhu X., Lo E. H., et al. (2010). High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke 41, 2077–2082. 10.1161/strokeaha.110.590463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn A. M., Williams A. R., Sivilli T. I., Raison C. L., Pace T. W. W. (2018). The plasma interleukin-6 response to acute psychosocial stress in humans is detected by a magnetic multiplex assay: comparison to high-sensitivity ELISA. Stress 21, 376–381. 10.1080/10253890.2018.1446518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley J. J., Morrison J. H. (2005). Repeated stress and structural plasticity in the brain. Ageing Res. Rev. 4, 271–287. 10.1016/j.arr.2005.03.004 [DOI] [PubMed] [Google Scholar]
- Radley J. J., Rocher A. B., Rodriguez A., Ehlenberger D. B., Dammann M., McEwen B. S., et al. (2008). Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. J. Comp. Neurol. 507, 1141–1150. 10.1002/cne.21588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkowska G., Miguel-Hidalgo J. J., Dubey P., Stockmeier C. A., Krishnan K. R. R. (2005). Prominent reduction in pyramidal neurons density in the orbitofrontal cortex of elderly depressed patients. Biol. Psychiatry 58, 297–306. 10.1016/j.biopsych.2005.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkowska G., Stockmeier C. A. (2013). Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr. Drug. Targets 14, 1225–1236. 10.2174/13894501113149990156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy M. S. (2010). Depression: the disorder and the burden. Indian J. Psychol. Med. 32, 1–2. 10.4103/0253-7176.70510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault P. F., Hoofnagle J. H., Park Y., Mullen K. D., Peters M., Jones D. B., et al. (1987). Psychiatric complications of long-term interferon alfa therapy. Arch. Intern. Med. 147, 1577–1580. 10.1001/archinte.147.9.1577 [DOI] [PubMed] [Google Scholar]
- Rezin G. T., Cardoso M. R., Gonçalves C. L., Scaini G., Fraga D. B., Riegel R. E., et al. (2008). Inhibition of mitochondrial respiratory chain in brain of rats subjected to an experimental model of depression. Neurochem. Int. 53, 395–400. 10.1016/j.neuint.2008.09.012 [DOI] [PubMed] [Google Scholar]
- Riba M., Wulsin L., Rubenfire M. (2011). Psychiatry and Heart Disease: The Mind, Brain, and Heart. Hoboken, NJ: Wiley-Blackwell. [Google Scholar]
- Robinson K. E., Pearson M. M., Cannistraci C. J., Anderson A. W., Kuttesch J. F., Jr., Wymer K., et al. (2015). Functional neuroimaging of working memory in survivors of childhood brain tumors and healthy children: associations with coping and psychosocial outcomes. Child Neuropsychol. 21, 779–802. 10.1080/09297049.2014.924492 [DOI] [PubMed] [Google Scholar]
- Rochfort K. D., Collins L. E., Murphy R. P., Cummins P. M. (2014). Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: consequences for interendothelial adherens and tight junctions. PLoS One 9:e101815. 10.1371/journal.pone.0101815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque S., Mesquita A. R., Palha J. A., Sousa N., Correia-Neves M. (2014). The behavioral and immunological impact of maternal separation: a matter of timing. Front. Behav. Neurosci. 8:192. 10.3389/fnbeh.2014.00192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros-Bernal F., Hunot S., Herrero M. T., Parnadeau S., Corvol J.-C., Lu L., et al. (2011). Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc. Natl. Acad. Sci. U S A 108, 6632–6637. 10.1073/pnas.1017820108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim S., Asghar M., Chugh G., Taneja M., Xia Z., Saha K. (2010). Oxidative stress: a potential recipe for anxiety, hypertension and insulin resistance. Brain Res. 1359, 178–185. 10.1016/j.brainres.2010.08.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim S., Asghar M., Taneja M., Hovatta I., Chugh G., Vollert C., et al. (2011). Potential contribution of oxidative stress and inflammation to anxiety and hypertension. Brain Res. 1404, 63–71. 10.1016/j.brainres.2011.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg S., Paton J. Y., Ahola S., McCann D. C., McGuinness D., Hillary C. R., et al. (2000). The role of acute and chronic stress in asthma attacks in children. Lancet 356, 982–987. 10.1016/s0140-6736(00)02715-x [DOI] [PubMed] [Google Scholar]
- Sarkar S., Malovic E., Harishchandra D. S., Ghaisas S., Panicker N., Charli A., et al. (2017). Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis. 3:30. 10.1038/s41531-017-0032-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasayama D., Hattori K., Wakabayashi C., Teraishi T., Hori H., Ota M., et al. (2013). Increased cerebrospinal fluid interleukin-6 levels in patients with schizophrenia and those with major depressive disorder. J. Psychiatr. Res. 47, 401–406. 10.1016/j.jpsychires.2012.12.001 [DOI] [PubMed] [Google Scholar]
- Satpute A. B., Lieberman M. D. (2006). Integrating automatic and controlled processes into neurocognitive models of social cognition. Brain Res. 1079, 86–97. 10.1016/j.brainres.2006.01.005 [DOI] [PubMed] [Google Scholar]
- Schoenfeld T. J., McCausland H. C., Morris H. D., Padmanaban V., Cameron H. A. (2017). Stress and loss of adult neurogenesis differentially reduce hippocampal volume. Biol. Psychiatry 82, 914–923. 10.1016/j.biopsych.2017.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz J. M., Sholar P. W., Bilbo S. D. (2012). Sex differences in microglial colonization of the developing rat brain. J. Neurochem. 120, 948–963. 10.1111/j.1471-4159.2011.07630.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerstrom S. C., Miller G. E. (2004). Psychological stress and the human immune system: a meta-analytic study of 30 years of inquiry. Psychol. Bull. 130, 601–630. 10.1037/0033-2909.130.4.601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J. H., Miyamoto N., Hayakawa K., Pham L. D., Maki T., Ayata C., et al. (2013). Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J. Clin. Invest. 123, 782–786. 10.1172/JCI65863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiawan E., Wilson A. A., Mizrahi R., Rusjan P. M., Miler L., Rajkowska G., et al. (2015). Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72, 268–275. 10.1001/jamapsychiatry.2014.2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline Y. I., Barch D. M., Donnelly J. M., Ollinger J. M., Snyder A. Z., Mintun M. A. (2001). Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol. Psychiatry 50, 651–658. 10.1016/s0006-3223(01)01263-x [DOI] [PubMed] [Google Scholar]
- Shimamoto A., Debold J. F., Holly E. N., Miczek K. A. (2011). Blunted accumbal dopamine response to cocaine following chronic social stress in female rats: exploring a link between depression and drug abuse. Psychopharmacology 218, 271–279. 10.1007/s00213-011-2364-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama Y., Chaki S. (2006). Neurochemistry of the nucleus accumbens and its relevance to depression and antidepressant action in rodents. Curr. Neuropharmacol. 4, 277–291. 10.2174/157015906778520773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sih A., Bell A. M., Johnson J. C., Ziemba R. E. (2004). Behavioral syndromes: an intergrative overiew. Q. Rev. Biol. 79, 241–277. 10.1086/422893 [DOI] [PubMed] [Google Scholar]
- Silveira Villarroel H., Bompolaki M., Mackay J. P., Miranda Tapia A. P., Michaelson S. D., Leitermann R. J., et al. (2018). NPY induces stress resilience via downregulation of Ih in principal neurons of rat basolateral amygdala. J. Neurosci. 38, 4505–4520. 10.1523/JNEUROSCI.3528-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M., Czéh B., Fuchs E. (2005). Age-dependent susceptibility of adult hippocampal cell proliferation to chronic psychosocial stress. Brain Res. 1049, 244–248. 10.1016/j.brainres.2005.05.006 [DOI] [PubMed] [Google Scholar]
- Slavich G. M., Way B. M., Eisenberger N. I., Taylor S. E. (2010). Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc. Natl. Acad. Sci. U S A 107, 14817–14822. 10.1073/pnas.1009164107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanki N., Salvi A., Patki G., Salim S. (2017). Modulating oxidative stress relieves stress-induced behavioral and cognitive impairments in rats. Int. J. Neuropsychopharmacol. 20, 550–561. 10.1093/ijnp/pyx017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauder A., Kovács M. (2003). Anxiety symptoms in allergic patients: identification and risk factors. Psychosom. Med. 65, 816–823. 10.1097/01.psy.0000088620.66211.b1 [DOI] [PubMed] [Google Scholar]
- Stelzhammer V., Ozcan S., Gottschalk M. G., Steeb H., Hodes G. E., Guest P. C., et al. (2015). Central and peripheral changes underlying susceptibility and resistance to social defeat stress—a proteomic profiling study. Diagn. Neuropsychiatry 1, 1–7. 10.1016/j.dineu.2015.08.001 [DOI] [Google Scholar]
- Straube T., Mentzel H. J., Miltner W. H. (2007). Waiting for spiders: brain activation during anticipatory anxiety in spider phobics. Neuroimage 37, 1427–1436. 10.1016/j.neuroimage.2007.06.023 [DOI] [PubMed] [Google Scholar]
- Strawbridge R., Arnone D., Danese A., Papadopoulos A., Herane Vives A., Cleare A. J. (2015). Inflammation and clinical response to treatment in depression: a meta-analysis. Eur. Neuropsychopharmacol. 25, 1532–1543. 10.1016/j.euroneuro.2015.06.007 [DOI] [PubMed] [Google Scholar]
- Takahashi A., Chung J. R., Zhang S., Zhang H., Grossman Y., Aleyasin H., et al. (2017). Establishment of a repeated social defeat stress model in female mice. Sci. Rep. 7:12838. 10.1038/s41598-017-12811-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki J., Fujimori K., Miura M., Suzuki T., Sekino Y., Sato K. (2012). L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J. Neuroinflammation 9:275. 10.1186/1742-2094-9-275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H., Jin S., Wang J., Zhang G., Kawanokuchi J., Kuno R., et al. (2006). Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 281, 21362–21368. 10.1074/jbc.M600504200 [DOI] [PubMed] [Google Scholar]
- Tang J., Yu W., Chen S., Gao Z., Xiao B. (2018). Microglia polarization and endoplasmic reticulum stress in chronic social defeat stress induced depression mouse. Neurochem. Res. 43, 985–994. 10.1007/s11064-018-2504-0 [DOI] [PubMed] [Google Scholar]
- Trainor B. C., Pride M. C., Villalon Landeros R., Knoblauch N. W., Takahashi E. Y., Silva A. L., et al. (2011). Sex differences in social interaction behavior following social defeat stress in the monogamous California mouse (Peromyscus californicus). PLoS One 6:e17405. 10.1371/journal.pone.0017405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi M. H., Rush A. J., Wisniewski S. R., Nierenberg A. A., Warden D., Ritz L., et al. (2006). Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am. J. Psychiatry 163, 28–40. 10.1176/appi.ajp.163.1.28 [DOI] [PubMed] [Google Scholar]
- Tse Y. C., Montoya I., Wong A. S., Mathieu A., Lissemore J., Lagace D. C., et al. (2014). A longitudinal study of stress-induced hippocampal volume changes in mice that are susceptible or resilient to chronic social defeat. J. Neurosci. 24, 1120–1128. 10.1002/hipo.22296 [DOI] [PubMed] [Google Scholar]
- Tynan R. J., Naicker S., Hinwood M., Nalivaiko E., Buller K. M., Pow D. V., et al. (2010). Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav. Immun. 24, 1058–1068. 10.1016/j.bbi.2010.02.001 [DOI] [PubMed] [Google Scholar]
- Vaváková M., Ďuračková Z., Trebatická J. (2015). Markers of oxidative stress and neuroprogression in depression disorder. Oxid. Med. Cell. Longev. 2015:898393. 10.1155/2015/898393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ver Hoeve E. S., Kelly G., Luz S., Ghanshani S., Bhatnagar S. (2013). Short-term and long-term effects of repeated social defeat during adolescence or adulthood in female rats. Front. Behav. Neurosci. 249, 63–73. 10.1016/j.neuroscience.2013.01.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videbech P. (2000). PET measurements of brain glucose metabolism and blood flow in major depressive disorder: a critical review. Acta. Psychiatr. Scand. 101, 11–20. 10.1034/j.1600-0447.2000.101001011.x [DOI] [PubMed] [Google Scholar]
- Villa A., Vegeto E., Poletti A., Maggi A. (2016). Estrogens, neuroinflammation, and neurodegeneration. Endocr. Rev. 37, 372–402. 10.1210/er.2016-1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees J. L., Tarr A. J., Wohleb E. S., Godbout J. P., Mo X., Sheridan J. F., et al. (2013). Prolonged restraint stress increases IL-6, reduces IL-10 and causes persistent depressive-like behavior that is reversed by recombinant IL-10. PLoS One 8:e58488. 10.1371/journal.pone.0058488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. B., Bozdagi O., Nikitczuk J. S., Zhai Z. W., Zhou Q., Huntley G. W. (2008). Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc. Natl. Acad. Sci. U S A 105, 19520–19525. 10.1073/pnas.0807248105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. L., Han Q. Q., Gong W. Q., Pan D. H., Wang L. Z., Hu W., et al. (2018). Microglial activation mediates chronic mild stress-induced depressive- and anxiety-like behavior in adult rats. J. Neuroinflammation 15:21. 10.1186/s12974-018-1054-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Ye L., Lu H., Chen H., Zhang Y., Huang Y., et al. (2017). TNF-α promotes extracellular vesicle release in mouse astrocytes through glutaminase. J. Neuroinflammation 14:87. 10.1186/s12974-017-0853-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zhang K., Chen X., Liu X., Teng H., Zhao M., et al. (2017). Epigenetic activation of ASCT2 in the hippocampus contributes to depression-like behavior by regulating D-serine in mice. Front. Mol. Neurosci. 10:139. 10.3389/fnmol.2017.00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren B. L., Vialou V. F., Iniguez S. D., Alcantara L. F., Wright K. N., Feng J., et al. (2013). Neurobiological sequelae of witnessing stressful events in adult mice. Biol. Psychiatry 73, 7–14. 10.1016/j.biopsych.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Gould E., Cameron H. A., Daniels D. C., Mcewen B. S. (1992). Phenytoin prevents stress- and corticosterone-induced atrophy of CA3 pyramidal neurons. Hippocampus 2, 431–435. 10.1002/hipo.450020410 [DOI] [PubMed] [Google Scholar]
- Weber M. D., Frank M. G., Tracey K. J., Watkins L. R., Maier S. F. (2015). Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J. Neurosci. 35, 316–324. 10.1523/jneurosci.3561-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger A. H., Gbedemah M., Martinez A. M., Nash D., Galea S., Goodwin R. D. (2017). Trends in depression prevalence in the USA from 2005 to 2015: widening disparities in vulnerable groups. Psychol. Med. 48, 1308–1315. 10.1017/s0033291717002781 [DOI] [PubMed] [Google Scholar]
- Weinhard L., Di Bartolomei G., Bolasco G., Machado P., Schieber N. L., Neniskyte U., et al. (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 9:1228. 10.1038/s41467-018-03566-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman M. M., Klerman G. L. (1992). Depression: current understanding and changing trends. Annu. Rev. Public Health 13, 319–339. 10.1146/annurev.pu.13.050192.001535 [DOI] [PubMed] [Google Scholar]
- Wickens M. M., Bangasser D. A., Briand L. A. (2018). Sex differences in psychiatric disease: a focus on the glutamate system. Front. Mol. Neurosci. 11:197. 10.3389/fnmol.2018.00197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M. A., Grillo C. A., Fadel J. R., Reagan L. P. (2015). Stress as a one-armed bandit: differential effects of stress paradigms on the morphology, neurochemistry and behavior in the rodent amygdala. Neurobiol. Stress 1, 195–208. 10.1016/j.ynstr.2015.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb E. S., Mckim D. B., Sheridan J. F., Godbout J. P. (2014a). Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front. Neurosci. 8:447. 10.3389/fnins.2014.00447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb E. S., Patterson J. M., Sharma V., Quan N., Godbout J. P., Sheridan J. F. (2014b). Knockdown of interleukin-1 receptor type-1 on endothelial cells attenuated stress-induced neuroinflammation and prevented anxiety-like behavior. J. Neurosci. 34, 2583–2591. 10.1523/jneurosci.3723-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb E. S., Powell N. D., Godbout J. P., Sheridan J. F. (2013). Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J. Neurosci. 33, 13820–13833. 10.1523/jneurosci.1671-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S. K. (2014). Individual differences in the neurobiology of social stress: implications for depression-cardiovascular disease comorbidity. Curr. Neuropharmacol. 12, 205–211. 10.2174/1570159x11666131120224413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood C. S., Valentino R. J., Wood S. K. (2017). Individual differences in the locus coeruleus-norepinephrine system: relevance to stress-induced cardiovascular vulnerability. Physiol. Behav. 101, 40–48. 10.1016/j.physbeh.2016.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S. K., Walker H. E., Valentino R. J., Bhatnagar S. (2010). Individual differences in reactivity to social stress predict susceptibility and resilience to a depressive phenotype: role of corticotropin-releasing factor. Endocrinology 151, 1795–1805. 10.1210/en.2009-1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S. K., Wood C. S., Lombard C. M., Lee C. S., Zhang X. Y., Finnell J. E., et al. (2015). Inflammatory factors mediate vulnerability to a social stress-induced depressive-like phenotype in passive coping rats. Biol. Psychiatry 78, 38–48. 10.1016/j.biopsych.2014.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood G. E., Young L. T., Reagan L. P., Chen B., Mcewen B. S. (2004). Stress-induced structural remodeling in hippocampus: prevention by lithium treatment. Proc. Natl. Acad. Sci. U S A 101, 3973–3978. 10.1073/pnas.0400208101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S. K., Zhang X. Y., Reyes B. A., Lee C. S., Van Bockstaele E. J., Valentino R. J. (2013). Cellular adaptations of dorsal raphe serotonin neurons associated with the development of active coping in response to social stress. Biol. Psychiatry 73, 1087–1094. 10.1016/j.biopsych.2013.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.-Z., Bodles A. M., Porter M. M., Griffin W. S. T., Basile A. S., Barger S. W. (2004). Induction of serine racemase expression and D-serine release from microglia by amyloid β-peptide. J. Neuroinflammation 1:2. 10.1186/1742-2094-1-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Liu B. J., Ji S., Wu J. F., Xu C. Q., Du Y. J., et al. (2015). Social defeat stress promotes tumor growth and angiogenesis by upregulating vascular endothelial growth factor/extracellular signal-regulated kinase/matrix metalloproteinase signaling in a mouse model of lung carcinoma. Mol. Med. Rep. 12, 1405–1412. 10.3892/mmr.2015.3559 [DOI] [PubMed] [Google Scholar]
- Yadid G., Overstreet D. H., Zangen A. (2001). Limbic dopaminergic adaptation to a stressful stimulus in a rat model of depression. Brain Res. 896, 43–47. 10.1016/s0006-8993(00)03248-0 [DOI] [PubMed] [Google Scholar]
- Yan W., Chang Y., Liang X., Cardinal J. S., Huang H., Thorne S. H., et al. (2012). High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology 55, 1863–1875. 10.1002/hep.25572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Lundback P., Ottosson L., Erlandsson-Harris H., Venereau E., Bianchi M. E., et al. (2012). Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol. Med. 18, 250–259. 10.2119/molmed.2011.00389 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yassa M. A., Hazlett R. L., Stark C. E., Hoehn-Saric R. (2012). Functional MRI of the amygdala and bed nucleus of the stria terminalis during conditions of uncertainty in generalized anxiety disorder. J. Psychiatr. Res. 46, 1045–1052. 10.1016/j.jpsychires.2012.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L., Huang Y., Zhao L., Li Y., Sun L., Zhou Y., et al. (2013). IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J. Neurochem. 125, 897–908. 10.1111/jnc.12263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate C. A., Jr., Singh J. B., Carlson P. J., Brutsche N. E., Ameli R., Luckenbaugh D. A., et al. (2006). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864. 10.1001/archpsyc.63.8.856 [DOI] [PubMed] [Google Scholar]
- Zhang J., Takahashi H. K., Liu K., Wake H., Liu R., Maruo T., et al. (2011). Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 42, 1420–1428. 10.1161/strokeaha.110.598334 [DOI] [PubMed] [Google Scholar]
- Zhao L., Huang Y., Tian C., Taylor L., Curthoys N., Wang Y., et al. (2012). Interferon-α regulates glutaminase 1 promoter through STAT1 phosphorylation: relevance to HIV-1 associated neurocognitive disorders. PLoS One 7:e32995. 10.1371/journal.pone.0032995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G., Okada M., Yoshida S., Mori F., Ueno S., Wakabayashi K., et al. (2006). Effects of interleukin-1β on hippocampal glutamate and GABA releases associated with Ca2+-induced Ca2+ releasing systems. Epilepsy Res. 71, 107–116. 10.1016/j.eplepsyres.2006.05.017 [DOI] [PubMed] [Google Scholar]