

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Uncoupling of G6b-B from Shp1 and Shp2 results in severe macrothrombocytopenia and aberrant platelet function.
G6b-B inhibits CLEC-2 signaling primarily through the protein-tyrosine phosphatase Shp2.
Abstract
The immunoreceptor tyrosine-based inhibitory motif (ITIM)–containing receptor G6b-B has emerged as a key regulator of platelet homeostasis. However, it remains unclear how it mediates its effects. Tyrosine phosphorylation of ITIM and immunoreceptor tyrosine-based switch motif (ITSM) within the cytoplasmic tail of G6b-B provides a docking site for Src homology 2 domain–containing protein-tyrosine phosphatases Shp1 and Shp2, which are also critical regulators of platelet production and function. In this study, we investigate the physiological consequences of uncoupling G6b-B from Shp1 and Shp2. To address this, we generated a transgenic mouse model expressing a mutant form of G6b-B in which tyrosine residues 212 and 238 within ITIM and ITSM were mutated to phenylalanine. Mice homozygous for the mutation (G6b-B diY/F) were macrothrombocytopenic, as a result of the reduction in platelet production, and had large clusters of megakaryocytes and myelofibrosis at sites of hematopoiesis, similar to those observed in G6b-deficient mice and patients. Platelets from G6b-B diY/F mice were hyporesponsive to collagen, as a result of the significant reduction in the expression of the immunoreceptor tyrosine-based activation motif (ITAM)–containing collagen receptor complex GPVI–FcR γ-chain, as well as thrombin, which could be partially rescued by costimulating the platelets with adenosine diphosphate. In contrast, platelets from G6b-B diY/F, G6b KO, and megakaryocyte-specific Shp2 KO mice were hyperresponsive to antibody-mediated cross-linking of the hemi-ITAM–containing podoplanin receptor CLEC-2, suggesting that G6b-B inhibits CLEC-2–mediated platelet activation through Shp2. Findings from this study demonstrate that G6b-B must engage with Shp1 and Shp2 to mediate its regulatory effects on platelet homeostasis.
Visual Abstract
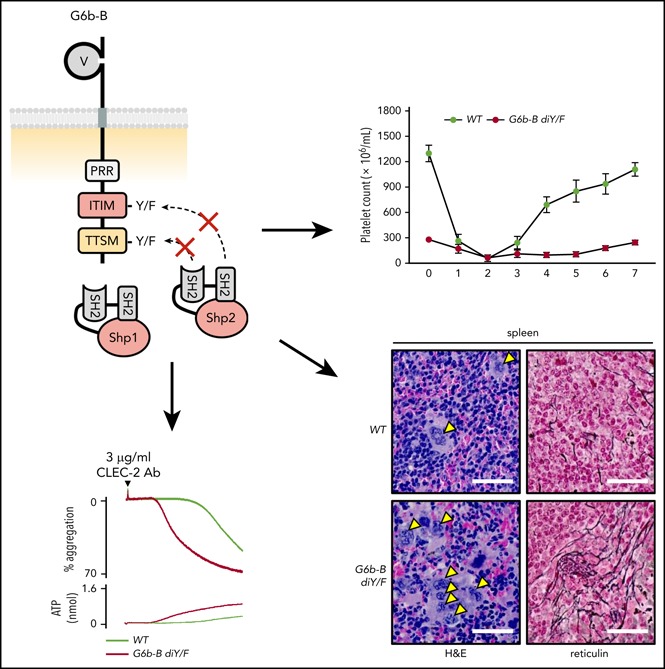
Introduction
Platelets are small fragments of megakaryocytes (MKs) that play a critical role in thrombosis, hemostasis, and the maintenance of vascular integrity.1,2 They do so by adhering to exposed extracellular matrix proteins at sites of vascular injury, where they become activated and form a hemostatic plug, preventing excessive blood loss and stimulating wound repair. The mechanisms required to maintain hemostasis also facilitate the formation of occlusive thrombi, leading to ischemia in acute coronary heart disease and stroke, 2 of the leading causes of death worldwide. Therefore, it is critical to understand the molecular mechanisms controlling platelet production and function to devise new and improved ways of regulating these processes.
Immunoreceptor tyrosine-based inhibition motif (ITIM)–containing receptors are important inhibitors of platelet activation.3 They function through a conserved intracellular ITIM (consensus sequence: I/V/LxYxxL/V),4 usually in tandem with another ITIM or an immunoreceptor tyrosine-based switch motif (ITSM; consensus sequence: TxYxxV/I).5 Tyrosine residues within ITIM and ITSM are phosphorylated by Src family kinases (SFKs), providing docking sites for the structurally related Src homology 2 (SH2) domain-containing protein-tyrosine phosphatases (PTPs) Shp1 and Shp2.4 This interaction predominantly inhibits signaling from immunoreceptor tyrosine-based activation motif (ITAM)–containing receptors (consensus sequence: YxxI/Lx6-12YxxI/L). Characterization of knockout (KO) mouse models has also revealed ITIM-containing receptors as regulators of platelet number.6-8
G6b-B is a type I transmembrane glycoprotein consisting of a single extracellular immunoglobulin-like variable-type domain, a transmembrane region, and a cytoplasmic tail. The cytoplasmic tail contains a juxtamembrane proline-rich region, an ITIM, and a C-terminal ITSM. An inhibitory role for G6b-B in regulating ITAM-mediated platelet activation was shown using G6b-B–deficient mice (G6b KO) and G6b-B–GPVI double-heterozygous mice (G6b+/−Gp6+/−).6 Unexpectedly, G6b KO mice also exhibited severe macrothrombocytopenia and aberrant proplatelet formation. Concomitant deletion of GPVI and CLEC-2 failed to fully rescue the G6b KO phenotype, suggesting other physiological functions of G6b-B that go beyond inhibiting ITAM receptor signaling.6 Intriguingly, patients lacking G6b-B exhibit a phenotype similar to that of G6b KO mice, including macrothrombocytopenia, MK clusters in the bone marrow, and myelofibrosis.9,10 Indeed, recent findings using a humanized G6b-B mouse model confirmed that human and mouse G6b-B perform analogous physiological functions.9
G6b-B is thought to mediate its functions through association with Shp1 and Shp2.6,11 Activation of Shp phosphatases depends upon phosphopeptide binding of the N-terminal SH2 (N-SH2) domain, which normally blocks access of substrates to the PTP catalytic site.12 MK-specific Shp2-KO and, to a lesser extent, Shp1-KO mouse models (Ptpn11fl/fl;PF4-Cre+ and Ptpn6fl/fl;Pf4-Cre+, referred to as Shp2 KO and Shp1 KO, respectively) phenocopy multiple features of G6b KO mice.13 However, Shp1/Shp2 conditional double-KO (DKO) mice (Ptpn6fl/fl;Ptpn11fl/fl;Pf4-Cre+, referred to as Shp1/Shp2 DKO) exhibited a more severe phenotype than G6b KO mice,13,14 demonstrating additional roles of Shp1 and Shp2 in MKs and platelets.
The aim of this study was to determine the role of G6b-B–mediated compartmentalization of Shp1 and Shp2 in regulating platelet production and function. To address this, we generated and characterized a novel knock-in (KI) mouse model, in which tyrosine (Y) residues 212 and 238 within the ITIM and ITSM of G6b-B were mutated to phenylalanine (F). Mice homozygous for these loss-of-function mutations (G6bdiYF/diYF, referred to as G6b-B diY/F) were severely macrothrombocytopenic and showed a significant downregulation of the ITAM-containing GPVI–FcRγ-chain complex, highlighting that the biological function of G6b-B is dependent upon recruitment of Shp1 and Shp2. We also demonstrate that the inhibitory effect of G6b-B on CLEC-2 signaling is primarily mediated by Shp2 and provide structural insights into the interaction between phosphorylated G6b-B and Shp2.
Materials and methods
Antibodies and reagents
Fluorescein isothiocyanate (FITC)-conjugated P-selectin, GPVI, GPIbα, and α2 and R-phycoerythrin (PE)–conjugated JON/A antibodies were from Emfret Analytics (Eibelstadt, Germany). PE-conjugated anti-αIIb antibody was from BD Biosciences (Oxford, United Kingdom), fibrinogen-488, phalloidin-488, and Alexa Fluor 488–conjugated goat anti-rat and Src phosphotyrosine-418 (Src p-Tyr418) antibodies were from Thermo Fisher (Loughborough, United Kingdom). Rat anti-mouse CLEC-2 antibody and IgG2B isotype-control antibody were from Serotec (Oxford, United Kingdom). G6b-B–specific antibodies were raised as previously described.6 For immunoblotting, anti-Shp1 and anti-Shp2 antibodies were from Santa Cruz Biotechnology (Dallas, TX), anti-phosphotyrosine (p-Tyr), FcRγ-chain, and PAR4 antibodies were from Merck Millipore (Nottingham, United Kingdom), and Syk phosphotyrosine-525/26 (Syk p-Tyr525/526) was from Cell Signaling Technology (Hitchin, United Kingdom). All other reagents were from Sigma-Aldrich (Poole, United Kingdom).
Mice
The G6b-B diY/F KI mouse model was generated, as outlined in supplemental Methods (available on the Blood Web site), by Taconic Biosciences (Cologne, Germany). G6b constitutive KO and conditional Shp1- and Shp2-KO mouse models, referred to as G6b KO, Shp1 KO, and Shp2 KO mice, respectively, were generated as previously described.6,13 Genotypes of litter-matched control mice (wild-type [WT]) were G6b+/+, Ptpn6fl/fl;Pf4-Cre−, and Ptpn11fl/fl;Pf4-Cre−. All mouse models used in this study were on a C57BL/6 background. Procedures were undertaken with UK Home Office approval, in accordance with the Animals (Scientific Procedures) Act of 1986.
Statistical analysis
All data are presented as mean ± standard error of the mean (SEM), unless stated otherwise. Unless stated otherwise, statistical significance was analyzed by a Student t test or 2-way ANOVA, followed by the indicated post hoc test, using GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
Detailed descriptions of all other methods can be found in supplemental Methods.
Results
Verification of G6b-B diY/F mouse model
To determine the physiological importance of the interaction among G6b-B–Shp1–Shp2 in platelet production and function, we generated a G6b-B diY/F mouse model (supplemental Figure 1) expressing a mutant form of G6b-B that is unable to engage with Shp1 or Shp2. The specific abolition of Shp1 and Shp2 docking sites allowed for delineation of the importance of this binding event and downstream signaling, independent of other potential signaling events. G6b-B diY/F mice were born at Mendelian frequencies (supplemental Table 1), were fertile, and survived for >25 weeks without any overt developmental or growth defects. Surface and total protein expression of G6b-B diY/F in platelets from KI mice was comparable to that of G6b-B in platelets from WT mice (Figure 1A-B). As expected, neither Shp1 nor Shp2 coimmunoprecipitated with G6b-B diY/F from resting or collagen-stimulated platelets (Figure 1C), demonstrating that interactions among G6b-B–Shp1–Shp2 were indeed lost. Immunoprecipitation of Shp2 also confirmed absence of the interaction (supplemental Figure 2).
Figure 1.

Loss of ITIM/ITSM phosphorylation prevents G6b-B–Shp1–Shp2 interaction. Flow cytometric (A) and western blot (B) analysis of G6b-B expression in platelets from WT and G6b-B diY/F mice. Mean ± SEM (n = 6) of G6b-FITC median fluorescence intensity (MFI), with rat IgG2A subtracted (Ai) and representative histogram (Aii). Representative blots (Bi) and quantification, normalized to tubulin reblots (mean ± SEM, n = 3) (Bii). (C) Lysates were prepared using washed platelets (5 × 108/mL) under basal conditions and 30 μg/mL collagen–stimulated conditions (90 seconds, 37°C, stirring at 1200 rpm) from WT and G6b-B diY/F mice. Coimmunoprecipitation of Shp1 and Shp2 was investigated by western blotting, following immunoprecipitation using anti–G6b-B or nonimmune rabbit polyclonal antibodies.
Reduced platelet production in G6b-B diY/F mice
G6b-B diY/F mice were severely macrothrombocytopenic, as observed in G6b KO mice. Platelet count was reduced by 76% and platelet volume was increased by 36% in G6b-B diY/F mice compared with WT mice (supplemental Table 2). These changes were independent of age (supplemental Figure 3). Increased platelet volume is frequently associated with immature platelets, which was confirmed by an increased proportion of reticulated platelets in G6b-B diY/F mice (Figure 2A). Thrombocytopenia in these mice was not a consequence of reduced platelet lifespan, measured as the proportional slope to give the rate of clearance of biotinylated platelets from the circulation (Figure 2B).
Figure 2.

Uncoupling of G6b-B–Shp1–Shp2 disrupts platelet production. (A) Percentage of reticulated platelets following staining with Retic-Count (mean ± SEM, n = 6). (B) Clearance of platelets in WT and G6b-B diY/F mice, following labeling with IV NHS-biotin. (Bi) Biotin-labeled platelets measured by streptavidin-PE binding in tail vein–sampled whole blood (mean ± SEM, n = 5 or 6 per data point). (Bii) Rate of platelet elimination calculated from slope of loss of biotinylated platelets (mean ± SEM, n = 6). (Ci) Platelet recovery following anti-GPIbα antibody–mediated platelet depletion in WT and G6b-B diY/F mice (n = 8-20 per time point). (Cii) Platelet-recovery rate calculated from recovery data for WT and G6b-B diY/F mice between days 3 and 7 (mean ± SEM, n = 8-11). Representative images (Di) and quantification of the number of MKs (Dii) in hematoxylin and eosin (H&E)–stained spleen and femur sections from WT and G6b-B diY/F mice (mean ± SEM, n = 6 mice, 5 images per mouse). Arrowheads indicate MKs. Representative images of reticulin staining showing myelofibrosis of WT and G6b-B diY/F spleens and femurs (scale bars, 50 μm). (Diii) Spleen/body weight ratio in WT, G6b-B diY/F, and G6b KO mice (mean ± SEM, n = 22). ***P < .001.
Because clearance was normal, reduced platelet counts had to be due to defects in production. Therefore, we investigated platelet recovery following anti-GPIbα antibody-mediated depletion (Figure 2Ci). Proportional slopes for days 3-7 were calculated and compared for WT and G6b-B diY/F mice. A fourfold reduction in the number of platelets produced per day was identified in G6b-B diY/F mice (Figure 2Cii), suggesting aberrant platelet production by MKs. This was supported by increased MK clusters and myelofibrosis in spleens and femurs and an 86% increase in spleen/body weight ratio (Figure 2D). MKs tended to be localized in distinct large clusters throughout the bone marrow and red pulp of spleens, similar to that observed in G6b-B–deficient patients.9 Interestingly, lymphocyte counts were also increased in G6b-B diY/F mice, as observed in patients and G6b KO mice, likely due to inflammation caused by the severe myelofibrosis.
Increased bleeding is caused by aberrant platelet function in G6b-B diY/F mice
A primary function of platelets is to prevent blood loss following injury. Therefore, we investigated the effect of the loss of G6b-B–Shp1–Shp2 interactions on hemostasis. Overall, blood loss increased in G6b-B diY/F and G6b KO mice, although for both genotypes there were mice with apparently normal bleeding (Figure 3A). The probability of abnormal bleeding in genetically modified mice was determined by modeling blood loss to a bimodal distribution. The mean probability of abnormal bleeding was 0.47 (95% confidence interval, 0.23-0.72) in G6b-B diY/F mice and 0.79 (95% confidence interval, 0.49-0.94) in G6b KO mice. Pairwise testing among the 3 groups (likelihood ratio test with no adjustment for multiple testing) indicated a clear difference between WT (probability of increased bleeding = 0) and both G6b-B diY/F (P = .0006) and G6b KO (P = 1.0 × 10−6) mice. Although G6b-B diY/F mice had a lower probability of bleeding than G6b KO mice, the difference was less clear-cut (P = .071). Because platelet counts must decrease by >90% for defective hemostasis to be observed,15 aberrant platelet function is the most likely underlying explanation of this bleeding phenotype, warranting further investigation.
Figure 3.

Aberrant platelet activation in G6b-B diY/F mice. (A) Blood loss/body weight ratio following excision of 5 mm of tail tip in isoflurane-anesthetized mice of the indicated genotypes. Data were analyzed using a bimodal function, comprising a gamma and normal distribution for low- and high-bleeding tendency, respectively. Likelihood ratio tests were performed to determine whether the probability of bleeding (Pbleed) differed between genotypes. (B-D) Flow cytometric measurement of P-selectin exposure and fibrinogen binding of WT and G6b-B diY/F platelets in whole blood using ITAM-activating (Bi-ii) and G protein–coupled receptor–activating (Ci-ii) agonists (mean ± SEM, n = 5 or 6 per stimulation). Thrombin stimulation was in the presence of 10 μM glycine-proline-arginine-proline to prevent blood clotting. (Di-ii) Rescue of activating PAR4 peptide (PAR4p)-stimulated fibrinogen binding in the presence of 10 μM ADP (mean ± SEM, n = 5 or 6). Platelets were gated using forward and side scatter, and 10 000 events were collected. *P < .05; **P < .01; ***P < .001.
To determine the cause of platelet functional defects, we measured surface receptor expression levels in G6b-B diY/F mice by flow cytometry (supplemental Figure 4). Expression of GPVI, the main receptor facilitating signaling in response to collagen, was reduced by 81%. Only minor changes were observed in other platelet receptors: a 22% reduction in the collagen integrin α2 subunit, a 7% increase in the fibrinogen integrin αIIb subunit, and a 20% reduction in CLEC-2 expression. GPIbα surface expression was unaltered. Western blotting showed comparable expression levels of the receptors investigated by flow cytometry and a small increase in the thrombin receptor PAR4 (supplemental Figure 5). Interestingly, alterations in receptor levels did not fully correlate between G6b-B diY/F and G6b KO mice; GPIbα levels were significantly reduced in G6b KO platelets,6 suggesting residual functional roles of G6b-B diY/F. This may explain differences in the bleeding distributions in G6b-B diY/F and G6b KO mice described above.
We next set out to identify how these changes in receptor expression affect platelet function. Due to the severe thrombocytopenia of G6b-B diY/F mice, a flow cytometry–based assay was first used to assess platelet activation in whole blood. P-selectin exposure and fibrinogen binding were used as markers of α-granule release and αIIbβ3 integrin activation, respectively. Not surprisingly, responses to the GPVI-specific agonist collagen-related peptide (CRP) were significantly reduced in G6b-B diY/F platelets (Figure 3B). Neither was a consequence of reduced P-selectin or αIIbβ3 expression (supplemental Figures 4 and 5). Convulxin, a more potent GPVI agonist than CRP, induced higher levels of P-selectin exposure and fibrinogen binding in G6b-B diY/F platelets but still failed to reach the levels observed in WT platelets (Figure 3B). P-selectin exposure was marginally reduced in G6b-B diY/F platelets in response to antibody-mediated cross-linking of CLEC-2, whereas fibrinogen binding was normal (Figure 3B).
No difference was observed between G6b-B diY/F and WT mice in response to adenosine diphosphate (ADP) or the thromboxane A2 analog U46619, both of which elicited only weak responses when used alone or in combination (Figure 3C). However, P-selectin surface exposure was significantly reduced in G6b-B diY/F platelets in response to thrombin and PAR4 peptide (Figure 3Ci). Fibrinogen-binding was normal in response to thrombin and reduced in response to PAR4 peptide (Figure 3Cii), presumably because thrombin also binds to GPIbα and is a more potent agonist than PAR4 peptide. Interestingly, P-selectin surface exposure was not rescued in G6b-B diY/F platelets costimulated with 10 μM ADP and PAR4 peptide (Figure 3Di). In contrast, αIIbβ3 activation was increased to levels observed in WT mouse platelets (Figure 3Dii), suggesting a more severe impairment in α-granule release than integrin activation. Comparable results were obtained for most agonists using washed platelets at a normalized count (2 × 107/mL) (supplemental Figure 6). Interestingly G6b-B diY/F platelets showed increased CLEC-2 antibody–mediated P-selectin exposure and unaltered PAR4 peptide–stimulated fibrinogen binding compared with WT platelets, both of which were significantly reduced in whole blood activations. Increased relative levels of secondary activators, such as ADP and thromboxane A2, which are known to be essential for CLEC-2 signaling16 and contributed to the PAR4-mediated defect (Figure 3D), are likely responsible for these differences, although an effect of the platelet-washing procedure cannot be excluded.
To further investigate platelet functional defects, we measured platelet aggregation and adenosine triphosphate (ATP) secretion from dense granules in response to select agonists by lumi-aggregometry. Moderate reductions in aggregation and ATP secretion were observed in G6b-B diY/F platelets compared with WT platelets in response to 30 μg/mL collagen, whereas platelets from G6b-B diY/F mice failed to respond to 30 μg/mL CRP (Figure 4A; supplemental Figure 7). In contrast, G6b-B diY/F platelets, which exhibited a minor reduction in CLEC-2 expression, responded more rapidly to 3 μg/mL anti–CLEC-2 antibody than did WT platelets (Figure 4A; supplemental Figure 7). No difference was seen when platelets were stimulated with 3 nM of the tetravalent, CLEC-2 binding snake toxin rhodocytin (Figure 4A; supplemental Figure 7), which is a more robust agonist than the divalent anti–CLEC-2 antibody. The amplitude of platelet aggregation was marginally reduced in G6b-B diY/F platelets in response to 0.06 U/mL thrombin and 10 μM U46619, correlating with decreased ATP secretion (Figure 4A; supplemental Figure 7). Aggregation was also significantly reduced in response to 10 μM ADP (Figure 4A; supplemental Figure 7). Taken together, these findings demonstrate moderate to severe impairments in platelet aggregation and ATP secretion to all agonists tested, with the exception of anti–CLEC-2 antibody, which was enhanced.
Figure 4.

Aberrant platelet function in G6b-B diY/F mice. (A) Mean aggregation and ATP release traces in washed platelets (2 × 108/mL). For ADP aggregations, platelets were washed in the presence of 0.02 U/mL apyrase and supplemented with 1 μM CaCl2 and 50 μg/mL fibrinogen (mean ± SEM, n = 4 or 5). (Bi) Representative images of WT and G6b-B diY/F washed platelet (2 × 107/mL) spreading on fibrinogen under basal conditions and 0.1 U/mL thrombin–preactivated conditions (scale bars, 5 μm). Quantification of surface area coverage (Bii), platelet perimeter (Biii), total platelets per image (Biv), and 4 stages of spreading in the presence of thrombin (Bv) (mean ± SEM, n = 5 or 6). ***P < .001.
Integrin αIIbβ3 activation and outside-in signaling were next interrogated by analyzing platelet adhesion and spreading on fibrinogen (Figure 4B). The number of adhered platelets, platelet perimeter, and surface area coverage of G6b-B diY/F platelets were normal compared with WT platelets, under basal conditions. However, G6b-B diY/F platelets failed to maximally spread following preactivation with 0.1 U/mL thrombin (Figure 4Bi-ii), correlating with the reduced P-selectin exposure to thrombin described above (Figure 3Ci). Morphological analysis of thrombin-treated platelets revealed a significant increase in G6b-B diY/F platelets with filopodia and a concomitant decrease in those forming lamellipodia compared with WT platelets (Figure 4Bv), likely due to the defective thrombin-mediated platelet activation identified in aggregation studies.
In light of these defects in platelet activation and function in response to specific agonists, a more physiologically relevant flow adhesion–based assay was performed to assess platelet recruitment to 3 surfaces: collagen; von Willebrand factor binding peptide (vWF-BP) and laminin; and vWF-BP, laminin, and rhodocytin (Figure 5). Platelet adhesion to vWF-BP and laminin, via GPIbα and α6β1, respectively, are important for initial platelet rolling and adhesion to exposed extracellular matrix. A shear rate of 1000 s−1 was chosen as a standard for phenotyping mice, to allow investigation of hemostasis and thrombosis. Under almost all conditions tested, G6b-B diY/F and G6b KO platelet adherence and thrombus formation were significantly reduced when quantifying morphological parameters and surface area coverage (Figure 5A,C). Thrombus contraction and multilayer thrombus formation were also significantly reduced upon adhesion to collagen (Figure 5C), as were formation of procoagulant platelets, α-granule release, and integrin activation, as measured by annexin V–647, anti–P-selectin–FITC, and JON/A-PE antibody binding, respectively (Figure 5B-C). Interestingly, G6b-B diY/F platelet αIIbβ3 activation on vWF-BP–laminin–rhodocytin was unaltered, whereas it was reduced in G6b KO samples. This could indicate a residual function of G6b-B, independent of Shp1 and Shp2 interactions. Previous studies using human blood showed an effect of low platelet counts on adhesion and activation in this assay, particularly on collagen-coated surfaces.17 Therefore, the observed reductions are due, at least in part, to thrombocytopenia in the loss-of-function and KO models.
Figure 5.

Reduced platelet adhesion and activation under flow conditions. PPACK, heparin, and fragmin anticoagulated whole blood from WT, G6b-B diY/F and G6b KO mice was flowed for 3.5 minutes (1000 s−1 shear) over coverslips coated with collagen, vWF-BP + laminin, and vWF-BP + laminin + rhodocytin. Representative brightfield images (A) and representative fluorescence images (B) following staining with Alexa Fluor 647–conjugated annexin V, P-selectin–FITC, and JON/A-PE antibodies to measure phosphatidylserine-positive platelets, α-granule release, and αIIbβ3 integrin activation (αIIbβ3act), respectively (scale bars, 10 μm). (C) Heat map showing the effect size of morphology scores and surface area coverage (SAC) of the indicated parameters, normalized to WT (n = 5 or 6).
Collectively, evidence from the in vitro and ex vivo assays demonstrates that a combination of reduced platelet counts and defective activation responses produces the hemostatic defect observed in G6b-B diY/F mice (Figure 3A). The combination of platelet defects is likely also responsible for the bleeding symptoms identified in G6b loss-of-function patients.9
G6b-B inhibits CLEC-2 signaling via Shp2
Because ITIM-containing receptors are most commonly characterized as negative regulators of ITAM-containing receptor signaling, we set out to determine at which stage G6b-B targets this pathway in platelets. The increased aggregation response to anti–CLEC-2 antibody in G6b-B diY/F mice (Figure 4A) made this hemi-ITAM–containing receptor ideal for further investigation. Washed platelets from G6b KO, G6b-B diY/F, Shp1 KO, Shp2 KO, and WT mice were activated with 10 μg/mL anti–CLEC-2 antibody for 3 minutes, and key proximal tyrosine phosphorylation signaling events were assessed by western blotting (Figure 6). As expected from aggregation studies (Figure 4A),6,13 platelets from G6b KO, G6b-B diY/F, and Shp2 KO mice exhibited increased whole-cell tyrosine phosphorylation (p-Tyr) following antibody-mediated CLEC-2 stimulation compared with WT platelets (Figure 6A-C). Platelets from Shp1 KO mice did not (Figure 6D),13 suggesting that G6b-B mediates an inhibitory effect on CLEC-2 signaling through Shp2 specifically. Rhodocytin-mediated tyrosine phosphorylation in G6b-B diY/F platelets was normal (supplemental Figure 8), correlating with the normal aggregation and ATP secretion responses (Figure 4A).
Figure 6.

G6b-B–Shp2 regulates CLEC-2 signaling. Washed platelets (4 × 108/mL) from G6b KO (A), G6b-B diY/F (B), Shp1 KO (C), and Shp2 KO (D) mice and litter-matched WT controls were activated with 10 μg/mL activating CLEC-2 antibody in the presence of 10 μM lotrafiban, followed by lysing and investigation of named phospho-tyrosine sites by western blotting. Representative western blots from 3 independent experiments. *P < .05; **P < .01.
Using phospho-specific antibodies, we investigated the site of action of G6b-B–Shp2 on CLEC-2 signaling. We focused on SFKs, which phosphorylate the tyrosine residue within the hemi-ITAM of the CLEC-2 receptor, and spleen tyrosine kinase (Syk), which binds the phosphorylated hemi-ITAMs of 2 adjacent CLEC-2 receptors and mediates downstream effects. SFK activation was indirectly measured as trans-autophosphorylation of the highly conserved tyrosine residue 418 in Src (Src p-Tyr418) and Syk activation was measured as trans-autophosphorylation of tyrosine residues 519/520 (p-Tyr519/20), both of which directly correlate with activity. Following quantification, Src p-Tyr418 was not significantly altered in CLEC-2–stimulated platelets from all mouse models, whereas Syk p-Tyr519/520 was significantly higher in CLEC-2–stimulated G6b KO, G6b-B diY/F, and Shp2 KO platelets compared with WT and Shp1 KO platelets (Figure 6). It should be noted that comigrating plasma proteins present in G6b KO and G6b-B diY/F samples occasionally interfered with the migration and appearance of Syk blots. This was controlled for when quantifying by normalizing Syk p-Tyr519/20 band intensities to total Syk band intensities for each sample. These findings suggest that G6b-B–Shp2 inhibits CLEC-2 signaling by directly dephosphorylating CLEC-2, to abolish Syk association, and/or Syk, attenuating its activity.
Shp2 interacts directly with phosphorylated G6b-B in an extended orientation
Because Shp2 appears to be the dominant mediator of G6b-B function in mouse platelets,13 and the proteins are known to associate by coimmunoprecipitation and direct phosphopeptide-binding experiments,6,11,18 we investigated the structural basis of their interaction in solution using heteronuclear magnetic resonance (NMR) spectroscopy. Recombinant N-SH2 and C-terminal SH2 (C-SH2) domains of Shp2 were incubated with phosphopeptides corresponding to the ITIM (p-ITIM) and ITSM (p-ITSM) of G6b-B. This revealed that the N-SH2 and C-SH2 domains of Shp2 preferentially interact with p-ITIM and p-ITSM, respectively, of G6b-B (Figure 7A-B). The chemical shift perturbations between the apo and bound forms of C-SH2 and p-ITSM exhibited slow exchange, indicating strong interactions. This accounts for the increased affinity of the tandem SH2 domain for the dual p-ITIM and p-ITSM peptide over that seen with isolated N-SH2.18 The tandem SH2–phosphopeptide interaction showed comparable chemical shift perturbations, indicating synergistic binding of the doubly phosphorylated peptide to both SH2 domains (Figure 7C-D). The direct interaction of Shp2 SH2 domains with G6b-B cognate binding motifs explains the critical role of the 2 residues mutated in the G6b-B diY/F mice reported here. Moreover, the structural model of the complex is fully supported by earlier studies reporting the binding affinity to be 0.5 nM.18 The structural studies show that the Y/F substitutions would eliminate critical contacts that allow the deep simultaneous insertion of G6b-B’s ITIM and ITSM phosphotyrosine residues into Shp2 N-SH2 and C-SH2 domains, respectively, in a manner that is consistent with canonical SH2 ligand binding modes.19 Moreover, the interaction with Shp2 tandem SH2 domains would also be compromised by G6b-B deletions and point mutations in Shp2, including L210I, R223H/C, and A238V, which have been found in human tumors,20-22 suggesting wider roles for Shp2 uncoupling in cancer.
Figure 7.

Structural basis of G6b-B recognition by SHP2. The specific interactions are shown by comparison of N-SH2 apo (black) and N-SH2 + 1 equivalent p-ITIM (sky blue) (A), C-SH2 apo (black) and C-SH2 + 1 equivalent p-ITSM (red) (B), and tandem SH2 apo (teal) and tandem SH2 + 1 equivalent p-ITIM+p-ITSM (red) (C). Selected chemical shift perturbations in SOFAST-HMQC NMR spectra are labeled. (D) Lowest-energy model of tandem SH2 bound to p-ITIM+p-ITSM generated by HADDOCK based on restraints generated from the NMR data. Residues showing large chemical shift perturbations are shown in blue and are defined as active residues directly involved in interaction with the peptide. Residues showing small, yet significant, chemical shift perturbations are shown in cyan and are defined as passively involved residues.
Discussion
This study establishes the physiological significance of tyrosine phosphorylation of the ITIM and ITSM domains of G6b-B in regulating platelet number and function. Uncoupling of G6b-B from Shp1 and Shp2 leads to a severe platelet phenotype, closely resembling that observed in G6b-B–deficient mice and patients.6,9 The primary features of the G6b-B loss-of-function and KO phenotypes included severe macrothrombocytopenia due to reduced platelet production, large MK clusters and myelofibrosis at sites of hematopoiesis, downregulation of the GPVI–FcRγ-chain complex, and increased signaling through CLEC-2 in platelets. Increased platelet activation via the CLEC-2 pathway was specifically due to Shp2 recruitment to G6b-B, which acted to inhibit the hemi-ITAM–Syk–dependent pathway.
Minor differences in the phenotypes of G6b diY/F KI and KO mouse models may be due to residual functions of G6b-B diY/F, a dominant-negative effect of this mutant form of the receptor, or functional roles of splice isoforms of G6b-B, which have been hypothesized to exist. Despite the apparent difference in bleeding between G6b-B diY/F KI and KO mice, this did not reach significance. Potential incomplete penetrance of the G6b-B diY/F phenotype correlates with an identified null patient presenting with no clinical symptoms.9 However, variability in humans beyond G6b mutations could explain the observed differences among patients.
We previously reported a strong overlap between the phenotype of Shp1/2 DKO mice and G6b conditional-KO (G6bfl/fl; Pf4-Cre+) mice.13 However, the phenotype of Shp1/2 DKO mice was more severe than that of G6b conditional-KO mice, including severe developmental defects in MKs that were not observed in G6b-B–deficient mice, reflecting other functional roles of Shp1 and Shp2. Thus, the G6b-B diY/F KI mouse model provides a more refined approach to demonstrate that G6b-B signals exclusively through Shp1 and Shp2. This G6b-B loss-of-function mouse model also allows for expression of other splice isoforms of G6b-B. This includes G6b-A–like, which is predicted to share the same extracellular, transmembrane, and juxtamembrane regions as G6b-B but has an entirely different cytoplasmic tail, lacking ITIM and ITSM. Although we have demonstrated expression of the G6b-A splice variant in human platelets using a custom anti–G6b-A antibody,9 generation of an antibody specific to the mouse G6b-A–like protein has been unsuccessful. This is due to lack of antigenic sites in the predicted mouse G6b-A–like protein. The function of human G6b-A also remains unknown, but advances can now be made through the use of the custom antibody and humanized G6b mouse model described by Hofmann et al.9
G6b-B appears to signal primarily through Shp2 in mice, because Shp2 KO mice more closely phenocopied G6b KO and G6b-B diY/F mice than Shp1 KO mice.13 This may be accounted for by the affinity of Shp2 for G6b-B being 100-fold greater than Shp118 and/or the relative stoichiometry of the 2 PTPs. Shp2 is expressed at levels sixfold greater than Shp1 in mouse platelets,23 meaning that, in the absence of Shp1, the highly expressed Shp2 may provide better compensation than Shp1 in the absence of Shp2. Structural characterization of Shp2 modular interaction with phospho-G6b-B–ITIM–ITSM peptides revealed simultaneous binding of its N-SH2 domain with the phospho-ITIM and its C-SH2 domain with the phospho-ITSM, resulting in a high-affinity interaction that is dependent on the phosphotyrosines that were substituted in KI mice. This double mutation would compromise the complexed orientation that alleviates the intramolecular interaction between the backside of the N-SH2 domain of Shp2 and the PTP catalytic site that normally allows activation of the phosphatase.12 Moreover, the double mutation would have prevented the high-affinity stable interaction with G6b-B that provides localization of Shp2 to the plasma membrane, with its PTP domain freely available for engagement with substrates, including phosphorylated CLEC-2 and/or Syk.
One of the main phenotypes of the G6b-B diY/F mouse is the severe macrothrombocytopenia, which was found to be due to reduced platelet production and not increased clearance. Comparison of the proportionate slopes of reduction in biotinylated platelets in circulation showed no significant difference compared with WT mice, which does not correlate with the increased clearance of G6b KO platelets reported previously.6 However, this most likely reflects the method of analysis, because the total platelet counts are not significantly different. Therefore, reduced platelet counts are due to decreased platelet production, which is supported by the reduced platelet recovery following depletion and observed extramedullary hematopoiesis.
Analysis of the regulation of ITAM-containing receptor signaling by G6b-B in the G6b-B diY/F mouse model is complicated by the reduced platelet counts and severe downregulation of the collagen receptor complex GPVI–FcRγ. This downregulation of GPVI–FcRγ is thought to be due to loss of inhibition of Syk by G6b-B recruited Shp1 and Shp2, activating negative-feedback mechanisms, such as shedding.6 Both flow cytometric– and lumi-aggregometry–based assays showed reduced G6b-B diY/F response to GPVI-specific agonists. Therefore, the predicted increase in signaling in the absence of G6b-B–Shp1–Shp2 interaction is not sufficient to overcome the downregulation of GPVI.
In contrast, flow cytometry, aggregation, and biochemical studies showed the expected hyperreactivity of G6b-B diY/F platelets to CLEC-2–mediated hemi-ITAM signaling. Similar increases in signaling were observed in G6b KO and Shp2 KO mice but not in Shp1 KO mice. Syk, but not SFK, activation was increased in these mice, indicating that Syk and/or the CLEC-2 receptor itself are the targets of G6b-B–Shp2 phosphatase activity. The pathophysiological consequences of increased CLEC-2 activity is not known and cannot be ascertained using the G6b KO or G6b diY/F mouse models due to the concomitant reductions in platelet counts and accompanying platelet anomalies. Thus, other means of investigating the effects of increased CLEC-2 signaling on vessel development, permeability, and other physiological processes will have to be implemented.
Other than in washed platelets stimulated with CLEC-2 antibody, P-selectin exposure was significantly reduced in response to all agonists. The absence of α-granule release rescue upon PAR4p and ADP costimulation indicates that defects are not due to loss of secondary signaling.24 Together with the reduced spreading, this suggests a positive-regulatory role for G6b-B in regulating α-granule release. Interestingly, agonists that activate G protein–coupled receptors (GPCRs) also showed attenuated aggregation responses in G6b-B diY/F mouse platelets. This may indicate direct involvement of the G6b-B–Shp1–Shp2 signaling complex in regulation of GPCR-mediated tyrosine kinase signaling,25,26 or it may be the indirect consequence of other platelet defects, including aberrant secretion or receptor turnover. The regulation of GPCR signaling by ITIM-containing receptors is highly provocative and warrants further investigation.
Despite that Shp2 KO mice most closely resemble G6b KO and G6b-B diY/F phenotypes, some features do not overlap.13,14 Shp1 KO mice show normal platelet production, clearance, and reactivity to most agonists. However, the absence of Shp1 in platelets reduces GPVI surface expression, as observed in G6b-B diY/F platelets. Therefore, platelets from both models are hyporesponsive to GPVI-specific activation. GPVI is known to be shed from the platelet surface once activated,27 indicating that loss of G6b-B–Shp1–mediated inhibition causes activation of this negative-feedback mechanism.
These findings confirm the hypothesis that the regulation of platelet homeostasis by G6b-B is dependent upon recruitment and signal transduction by the PTPs Shp1 and Shp2. Loss of this interaction severely impairs the formation of functional platelets that are essential for the maintenance of hemostasis. Because GPVI and CLEC-2 share common downstream signaling proteins, it is of note that they are differentially regulated by Shp1 and Shp2.13 Whether this is due to differences in compartmentalized recruitment of Shp1 and Shp2 into proximity with GPVI and CLEC-2, which may be localized differentially in the plasma membrane, or to differences in targeting of proteins specific to each pathway remains to be determined.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Inga Hofmann (University of Wisconsin) and Mark Fleming (Boston Children’s Hospital) for interpretation of mouse pathology and constructive feedback on the manuscript.
M.J.G. is funded by a Medical Research Council PhD studentship (GBT1564), A.M. is a British Heart Foundation Intermediate Basic Science Research Fellow (FS/15/58/31784), and Y.A.S. is a British Heart Foundation Senior Basic Science Research Fellow (FS/13/1/29894). P.G. was funded by Cancer Research UK, M.J. was supported by the Biotechnology and Biological Sciences Research Council, and M.O. received Bloodwise and Cancer Research UK support. Henry Wellcome Building–NMR provided NMR facility access through Wellcome Trust support. Rhodocytin was purified as part of the project financed by Deutsche Forschungsgemeinschaft Grant EB177/13-1.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contributions: M.J.G. performed experiments, analyzed data, and wrote and revised the manuscript; J.P.v.G., P.G., T.V., C.W.S., M.J., and J.W.M.H. performed experiments, analyzed data, and revised the manuscript; S.H. and M.J.E.K. performed experiments and analyzed data; B.M.E.T. analyzed data; G.E.J. analyzed data and revised the manuscript; J.A.E. supplied essential reagents; M.O. revised the manuscript and contributed intellectually; A.M. designed experiments, interpreted data, and wrote and revised the manuscript; and Y.A.S. conceptualized the study, analyzed data, and wrote and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for M.O. is Department of Biochemistry, University of Alberta, Edmonton, AB, Canada.
Correspondence: Yotis A. Senis, Institute of Cardiovacular Sciences, College of Medical and Dental Sciences, University of Birmingham, Birmingham B15 2TT, United Kingdom; e-mail: y.senis@bham.ac.uk.
REFERENCES
- 1.George JN. Platelets. Lancet. 2000;355(9214):1531-1539. [DOI] [PubMed] [Google Scholar]
- 2.Hartwig JH. The platelet: form and function. Semin Hematol. 2006;43(1 suppl 1):S94-S100. [DOI] [PubMed] [Google Scholar]
- 3.Coxon CH, Geer MJ, Senis YA. ITIM receptors: more than just inhibitors of platelet activation. Blood. 2017;129(26):3407-3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daëron M, Jaeger S, Du Pasquier L, Vivier E. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol Rev. 2008;224(1):11-43. [DOI] [PubMed] [Google Scholar]
- 5.Cannons JL, Tangye SG, Schwartzberg PL. SLAM family receptors and SAP adaptors in immunity. Annu Rev Immunol. 2011;29(1):665-705. [DOI] [PubMed] [Google Scholar]
- 6.Mazharian A, Wang YJ, Mori J, et al. . Mice lacking the ITIM-containing receptor G6b-B exhibit macrothrombocytopenia and aberrant platelet function. Sci Signal. 2012;5(248):ra78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Washington AV, Gibot S, Acevedo I, et al. . TREM-like transcript-1 protects against inflammation-associated hemorrhage by facilitating platelet aggregation in mice and humans. J Clin Invest. 2009;119(6):1489-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan X, Shi P, Dai J, et al. . Paired immunoglobulin-like receptor B regulates platelet activation. Blood. 2014;124(15):2421-2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann I, Geer MJ, Vögtle T, et al. . Congenital macrothrombocytopenia with focal myelofibrosis due to mutations in human G6b-B is rescued in humanized mice. Blood. 2018;132(13):1399-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melhem M, Abu-Farha M, Antony D, et al. . Novel G6B gene variant causes familial autosomal recessive thrombocytopenia and anemia. Eur J Haematol. 2017;98(3):218-227. [DOI] [PubMed] [Google Scholar]
- 11.Senis YA, Tomlinson MG, García A, et al. . A comprehensive proteomics and genomics analysis reveals novel transmembrane proteins in human platelets and mouse megakaryocytes including G6b-B, a novel immunoreceptor tyrosine-based inhibitory motif protein. Mol Cell Proteomics. 2007;6(3):548-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92(4):441-450. [DOI] [PubMed] [Google Scholar]
- 13.Mazharian A, Mori J, Wang YJ, et al. . Megakaryocyte-specific deletion of the protein-tyrosine phosphatases Shp1 and Shp2 causes abnormal megakaryocyte development, platelet production, and function. Blood. 2013;121(20):4205-4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Paola J. SHPing in different directions in platelet production. Blood. 2013;121(20):4018-4019. [DOI] [PubMed] [Google Scholar]
- 15.Morowski M, Vögtle T, Kraft P, Kleinschnitz C, Stoll G, Nieswandt B. Only severe thrombocytopenia results in bleeding and defective thrombus formation in mice. Blood. 2013;121(24):4938-4947. [DOI] [PubMed] [Google Scholar]
- 16.Pollitt AY, Grygielska B, Leblond B, Désiré L, Eble JA, Watson SP. Phosphorylation of CLEC-2 is dependent on lipid rafts, actin polymerization, secondary mediators, and Rac. Blood. 2010;115(14):2938-2946. [DOI] [PubMed] [Google Scholar]
- 17.Nagy M, Mastenbroek TG, Mattheij NJA, et al. . Variable impairment of platelet functions in patients with severe, genetically linked immune deficiencies. Haematologica. 2018;103(3):540-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coxon CH, Sadler AJ, Huo J, Campbell RD. An investigation of hierachical protein recruitment to the inhibitory platelet receptor, G6B-b. PLoS One. 2012;7(11):e49543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gopalasingam P, Quill L, Jeeves M, Overduin M. SH2 domain structures and interactions. In: Kurochkina N, ed. SH Domains: Structure, Mechanisms and Applications. Cham, Switzerland: Springer International Publishing; 2015:159-185. [Google Scholar]
- 20.Giannakis M, Mu XJ, Shukla SA, et al. . Genomic correlates of immune-cell infiltrates in colorectal carcinoma [published correction appears in Cell Rep. 2016;17(4):1206]. Cell Rep. 2016;15(4):857-865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers [published correction appears in Nature 2012;491(7423):288]. Nature. 2012;489(7417):519-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cerami E, Gao J, Dogrusoz U, et al. . The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeiler M, Moser M, Mann M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol Cell Proteomics. 2014;13(12):3435-3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harper MT, van den Bosch MT, Hers I, Poole AW. Platelet dense granule secretion defects may obscure α-granule secretion mechanisms: evidence from Munc13-4-deficient platelets. Blood. 2015;125(19):3034-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dorsam RT, Kim S, Murugappan S, et al. . Differential requirements for calcium and Src family kinases in platelet GPIIb/IIIa activation and thromboxane generation downstream of different G-protein pathways. Blood. 2005;105(7):2749-2756. [DOI] [PubMed] [Google Scholar]
- 26.Canobbio I, Cipolla L, Guidetti GF, et al. . The focal adhesion kinase Pyk2 links Ca2+ signalling to Src family kinase activation and protein tyrosine phosphorylation in thrombin-stimulated platelets. Biochem J. 2015;469(2):199-210. [DOI] [PubMed] [Google Scholar]
- 27.Gardiner EE, Andrews RK. Platelet receptor expression and shedding: glycoprotein Ib-IX-V and glycoprotein VI. Transfus Med Rev. 2014;28(2):56-60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.