


Abstract
Lamins are the main components of the nucleoskeleton. Whereas their 3D organization was recently described using cryoelectron tomography, no structural data highlights how they interact with their partners at the interface between the inner nuclear envelope and chromatin. A large number of mutations causing rare genetic disorders called laminopathies were identified in the C-terminal globular Igfold domain of lamins A and C. We here present a first structural description of the interaction between the lamin A/C immunoglobulin-like domain and emerin, a nuclear envelope protein. We reveal that this lamin A/C domain both directly binds self-assembled emerin and interacts with monomeric emerin LEM domain through the dimeric chromatin-associated Barrier-to-Autointegration Factor (BAF) protein. Mutations causing autosomal recessive progeroid syndromes specifically impair proper binding of lamin A/C domain to BAF, thus destabilizing the link between lamin A/C and BAF in cells. Recent data revealed that, during nuclear assembly, BAF’s ability to bridge distant DNA sites is essential for guiding membranes to form a single nucleus around the mitotic chromosome ensemble. Our results suggest that BAF interaction with lamin A/C also plays an essential role, and that mutations associated with progeroid syndromes leads to a dysregulation of BAF-mediated chromatin organization and gene expression.
INTRODUCTION
Laminopathies are rare genetic disorders caused by mutations in genes encoding lamins or by abnormalities in the processing of lamin A (1). They display a large variety of clinical symptoms including cardiomyopathy, muscular dystrophy, lipodystrophy, mandibuloacral dysplasia, restrictive dermopathy, peripheral neuropathy, and premature ageing. A major fraction of the disease-causing mutations comprises single point mutations with poorly understood functional implications. Most of them are distributed all along the LMNA gene coding for both prelamin A and lamin C. These two proteins are, together with lamins B1 and B2, essential components of the nucleoskeleton. They share an N-terminal region of 566 residues, and whereas lamin C only possesses six additional specific C-terminal residues, prelamin A presents 98 additional residues, is farnesylated, carboxymethylated and then cleaved to become mature lamin A. Disease-causing mutations are observed in the large region common to lamins A and C (lamin A/C) as well as in the lamin A specific C-terminal region (2). At the cellular level, lamins A and C form filaments that are mainly present at the nuclear periphery but are also observed in the nucleoplasm (3). Laminopathies are characterized by nuclear morphological abnormalities and an altered pattern of heterochromatin distribution that is more severe in progeroid syndromes, including Hutchinson-Gilford progeria syndrome (HGPS), mandibuloacral dysplasia (MAD), atypical-Werner syndrome (WS) and restrictive dermopathy (RD) (4,5). The lamin filament network interacts with chromatin at different stages of the cell cycle: in ana-telophase, lamin A and C dimers are recruited at the core regions of sister chromosomes; in interphase cells, they relocalize both at the nuclear envelope and within the nucleoplasm, where lamins A and C provide anchor points for chromatin (6–9). The complex molecular organization of the nuclear envelope, nucleoskeleton and their interface with chromatin was recently described using cryo-electron tomography (10,11). Within the densely packed environment observed close to the nuclear envelope, only nuclear pores, lamin filaments and chromatin could be recognized. Lack of high resolution 3D structures for proteins anchored at the inner nuclear membrane as well as complexes between these proteins, the lamina and chromatin still precludes any further description of the nuclear periphery architecture and of the structural defects caused by mutations in the LMNA gene coding for lamins A and C.
We set out to understand how the C-terminal immunoglobulin-like (Igfold) domain of lamin A/C interacts with the inner nuclear membrane protein emerin and the chromatin-associated protein Barrier-to-Autointegration Factor (BAF). Indeed, a large number of disease-causing mutations were identified in this lamin Igfold domain (12,13). Mutations causing muscle diseases affect residues of the hydrophobic core, suggesting that destabilization of the lamin A/C Igfold domain is responsible for these diseases (13). Mutations causing either lipodystrophies or progeroid syndromes involve residues localized on two different solvent-exposed sites of the lamin A/C Igfold domain, suggesting that these accessible regions are binding sites for unknown lamin partners and that disruption of these binding events cause the diseases (12–14). Lamin A/C binds to the inner nuclear membrane protein emerin, as shown by blot overlay, coimmunoprecipitation and yeast 2-hybrid assays (15–17). Moreover, the tail region of lamin A/C (residues 384–566, comprising the Igfold domain) is responsible for emerin binding (17,18). The central region of emerin (residues 70–178) is essential for lamin binding (15,18), and its N-terminal domain (residues 1–45, the LEM domain) interacts with BAF, as shown by a panel of techniques going from blot overlay and coimmunoprecipitation assays to X-ray crystallography analyses (15,19).
The structural inter-dependence of lamins, emerin and BAF was revealed by downregulating either lamins A and C or BAF or the LEM domain proteins emerin and LEM2 in Caenorhabditis elegans embryos. If any one component was missing, the other two failed to co-assemble, with severe consequences for mitotic spindle assembly and positioning (20), mitotic chromosome segregation and postmitotic nuclear assembly (21,22). In vitro experiments revealed that human lamin A binds weakly (Kd of 1 μM) but directly to BAF and that lamin A and BAF simultaneously bind to emerin (23). Moreover, BAF enhances binding of prelamin A tails to emerin (24). Finally, during nuclear assembly, BAF enriches around the mitotic chromosome ensemble, and its ability to bridge distant DNA sites is essential for guiding membranes to form a single nucleus (25).
We now demonstrate that emerin oligomerization regulates direct lamin A/C binding, and that emerin monomer also interacts with lamin A/C through BAF. We present the 3D structure of a complex involving lamin A/C, emerin and BAF. We propose a model in which BAF dimer bound to two DNA molecules interacts on one side with emerin and on the other side with lamin A/C, and we analyse the impact of mutations causing autosomal recessive progeroid syndromes of the assembly of this complex.
MATERIALS AND METHODS
Protein expression and purification
Human WT emerin fragments from amino acid 1 to amino acid 187 (EmN), from amino acid 67 to 170 (EmC170), from amino acid 67 to 187 (EmC187) and from amino acid 67 to amino acid 221 (EmC), human BAF with all cysteines mutated in alanine and human lamin B1 tail from amino acid 409 to amino acid 586 were expressed using a pETM13 vector as N-terminal octa-histidine fusions in Escherichia coli BL21 DE3 Star (Novagen). The emerin and BAF cDNAs were optimized for expression in Escherichia coli (Genscript). Human wild-type lamin A/C fragment from amino acid 411 to amino acid 566 (LamIgF) was expressed using a pGEX vector as an N-terminal GST fusion in E. coli BL21 DE3 Star (Novagen). All LamIgF mutant expression vectors (R435C, R453W, R471C, R482W, R527H, A529V and K542N) were obtained by mutagenesis using the Quikchange (Agilent) kit from the LamIgF expression vector. Bacteria were cultured in rich medium (lysogeny broth, LB) or 15N-labeled minimum medium, induced at an optimal density of 1 with 0.5 mM isopropyl β-d-1-thiogalactopyranoside, grown overnight at 293 K, and lysed in 50 mM Tris–HCl pH 8.0, 300 mM NaCl, 40 mM imidazole, 5% glycerol, 1% Triton X-100 and 1 mM phenylmethanesulfonylfluoride.
For EmN, EmC170, EmC187, EmC and BAF, after sonication at room temperature and centrifugation at 20 000 g, for 20 min, at 277 K, the pellet was resuspended in buffer C8 (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 20 mM imidazole, 8 M urea). A second centrifugation step was performed at 20 000 g, for 20 min, at 293 K. The soluble extract was then loaded onto Ni-NTA beads (GE-Healthcare) equilibrated with buffer C8. Proteins were eluted directly with buffer E8 (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 M imidazole, 8 M urea). Then, proteins were refolded by dialysis in buffer D1 (20 mM Tris–HCl pH 8.0, 30 mm NaCl, 2 mM DTT) for EmN, EmC170, EmC187 and EmC and buffer D2 (50 mm Tris–HCl pH 8.0, 150 mm NaCl, 2 mM DTT) for BAF. After refolding, they were incubated with the TEV protease during 3 h at room temperature and finally dialyzed into the selected buffer. The final yield was typically 10 mg of purified protein per liter of bacterial culture (26). Self-assembly was initiated using proteins that were dialyzed in a buffer containing 20 mM Tris–HCl pH 8.0 and 30 mM NaCl. After dialysis, proteins were concentrated until 600 μM, reduced with 5 mM DTT and stored at room temperature during one week.
For lamin B1 tail, after sonication at 277 K, benzonase (SigmaAldrich) addition and centrifugation at 20 000 g, for 30 min, at 277 K, the soluble extract was loaded onto Ni-NTA beads (GE-Healthcare) equilibrated with buffer C (50 mM Tris–HCl pH 8.0, 150 mM NaCl and 20 mM imidazole). The protein was then eluted directly with buffer E (50 mM Tris–HCl pH 8.0, 150 mM NaCl and 1 M imidazole) and dialyzed in buffer C. It was incubated with the TEV protease during 3 h at room temperature, loaded again onto Ni-NTA beads and finally, after flow-through recovery, dialyzed into the selected buffer. The final yield was typically 20 mg of purified protein per liter of bacterial culture.
For LamIgF (WT and mutants), after sonication at 277 K, benzonase (SigmaAldrich) addition and centrifugation at 20 000 g, for 30 min, at 277 K, the soluble extract was loaded onto GST-beads (GE-Healthcare) equilibrated with buffer T2 (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 2 mM DTT). After 2 h of incubation at 277 K, GST-beads were washed with buffer T2 and 1 M NaCl, and then thrombin was added and incubated with the beads overnight. The protein was recovered in the flow-through and finally dialyzed into the selected buffer. The final yield was typically 2 mg of purified protein per liter of bacterial culture.
Liquid-state NMR spectroscopy
NMR experiments were performed on a 700 MHz Bruker Avance spectrometer equipped with a triple resonance cryogenic probe. To study interaction between the lamin fragments and BAF, two-dimensional 1H–15N HSQC spectra were acquired at 303 K, on a 3-mm-diameter NMR sample tube containing 100–200 μM uniformly 15N-labeled lamin peptides and non-labeled BAF, in 20 mM sodium phosphate pH 7.0, 100 mM NaCl and 80:20% H2O/D2O. To study the interaction between lamin fragments and EmN or EmC, two-dimensional 1H–15N HSQC spectra were acquired at 303 K, in a 3-mm-diameter NMR sample tube containing 150 μM uniformly 15N-labeled lamin peptides and non-labeled EmN or EmC, in 20 mM Tris–HCl pH 8.0, 30 mM NaCl and 80:20% H2O/D2O. The 1H, 15N, 13C NMR signals of lamin A/C domain from aa 411 to aa 553 were already assigned previously in a phosphate buffer at pH 6.3 and 303 K (27). In order to assign the NMR signals of lamin A/C region from aa 411 to aa 566 (LamIgF) in our conditions (20 mM sodium phosphate pH 7.0, 100 mM NaCl, 303 K), we took advantage of these previous assignments, but also recorded additional 3D 1H–15N–13C correlation spectra using 3D HNCACB, CBCA(CO)NH, HNCO, HN(CA)CO and HN(CO)(CA)NH pulse sequences on a 3-mm-diameter NMR sample tube containing 500 μM uniformly 15N/13C-labeled LamIgF. The data were processed using Topspin3.1 (Bruker) and analyzed using CCPNMR (28).
Size-exclusion chromatography
Size-exclusion chromatography experiments aiming at identifying interactions between emerin, BAF and lamin fragments (LamIgF WT and mutants, lamin B1 tail) were performed using a Superdex-75 10/300 GL column (GE Healthcare) pre-equilibrated with buffer G (20 mM Tris–HCl pH 8.0, 30 mM NaCl, 2 mM DTT). 500 μl of proteins concentrated at 150 μM were injected for each experiment at a flow rate of 0.5 ml/min at 277 K.
Isothermal titration calorimetry
ITC was performed using a high-precision VP-ITC calorimetry system. To characterize interactions between the BAF dimer and LamIgF (WT and mutants), all proteins were dialyzed against 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 10 mM β-mercaptoethanol and protease inhibitors (Roche). BAF (20 μM) in the calorimetric cell at 288 K was titrated with LamIgF WT (at a concentration of 100 μM in the injection syringe) or BAF in the calorimetric cell at 283 K was titrated with LamIgF mutants (at a concentration of 100 μM in the injection syringe). To characterize interactions between the BAF dimer and EmN, all proteins were dialyzed against 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 10 mM β-mercaptoethanol and protease inhibitors (Roche). BAF (30 μM) in the calorimetric cell at 288 K was titrated with EmN (at a concentration of 150 μM in the injection syringe). Analyses of the data were performed using the Origin software provided with the instrument.
Negative-staining electron microscopy
To observe self-assembled EmN and EmC proteins, with or without LamIgF WT or lamin B1 tail, protein samples containing 2% uranyl acetate were deposited on glow-discharged carbon-coated copper grids. Data collection was performed using a Tecnai Spirit transmission electron microscope (FEI) equipped with a LaB6 filament, operating at 100 keV. Images were recorded using a K2 Base camera (Gatan, 4k × 4k) at 15 000 magnification (at the level of the microscope) with a pixel size of 0.25 nm at the specimen level.
X-ray crystallography
Crystallization and data collection
The EmN-Baf-LamIgF complex was purified by gel filtration (Superdex 75, 10/300 GL) in 50 mM Tris–HCl pH 8.0, 100 mM NaCl and concentrated to 3 mg/ml. Crystallization was initiated one week after the gel filtration and in these conditions, EmN was proteolyzed and the final complex was composed of LEM-BAF-LamIgF. For crystallization, 1 μl of the complex was mixed with 1 μl of the reservoir solution and equilibrated against a 500-μl reservoir by hanging drop at 277 K. Crystals were grown in 25% (w/v) polyethylene glycol (PEG) 3.350, 20 mM Bis-Tris pH 5.5, 30 mM NaCl and 0.2 M NH4SO4. They were flash-cooled in liquid nitrogen, using a cryo-protection solution of 25% PEG 3.350, 20 mM Bis-Tris pH 5.5 and 0.2 M NH4SO4, supplemented with 27% (V/V) ethylene glycol.
Structure determination and refinement
The 3D structure of the complex was solved by molecular replacement using Molrep in CCP4 (29,30). The coordinates of the BAF dimer with DNA (PDB entry: 2BZF), the coordinates of lamin A/C globular domain (PDB entry 1IFR) and the coordinates of the emerin LEM domain (PDB entry: 2ODC) were used as templates. The resulting model was rebuilt using PHENIX suite (31), manual correction was performed with Coot (32) according to |Fo| – |Fc| and 2|Fo| – |Fc| electron density maps, and further refinement was carried out with phenix.refine. All structure figures were prepared using PyMOL (Schrödinger, LLC). Structure coordinates were deposited to the PDB, with entry 6GHD.
Fluorescence-based thermal shift assay
The thermal stability of LamIgF proteins was monitored by a fluorescence-based thermal shift assay performed with a QuantStudio 12K Flex instrument (LifeTechnologies). 10 μg of purified protein was mixed with the SYPRO Orange dye (diluted 800-fold from a 5000-fold stock solution, Invitrogen) in 50 mM Tris pH 8.0, 150 mM NaCl, 10% glycerol. Reactions were carried out in duplicate in a 96-well fast PCR plate at a final volume of 20 μl (33 μM) and each experiment was repeated at least twice independently. The samples were submitted to a denaturation kinetic from 283 to 368 K at a rate of 3 K/min and fluorescence of Sypro Orange dye was recorded in real time. The protein denaturation temperature (Td ± s.e.m.) was calculated using the Protein Thermal Shift software v1.3 (LifeTechnologies) as the maximum of the derivative of the resulting fluorescence curves.
Proximity ligation assays
Transfection
HeLa cells were obtained from American Type Culture Collection and cultured in Minimum Essential Medium containing Glutamax (Gibco), 1% non-essential amino acids and 10% fetal bovine serum. HeLa cells were transfected using XtremeGene 9 (Roche) with the plasmids pEGFC3-BAF encoding full length BAF and pcmvTAG2A-FLAG-Lamin A encoding mature lamin A (aa 1-646). After 24 h, cells were processed for either immunoblotting, immunofluorescence or Proximity ligation assay.
Immunoblotting
Whole cell protein extracts were suspended in Laemmli sample buffer, separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked for 1.5 h in TBST (10 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween-20) containing 5% dry milk, incubated with mouse anti FLAG antibody (Sigma; 1:600) and rabbit anti-BAF (Santacruz, 1:200) for 1 h in TBST/1% milk, washed 4 times and incubated with HRP-conjugated secondary antibodies. After four washes in TBST, proteins were detected by enhanced chemiluminescence.
Immunofluorescence microscopy
Cells were fixed with 3% paraformaldehyde for 12 min at R.T., permeabilized with phosphate buffered saline containing 0.5% Triton for 5 min at R.T., and quenched with 2% bovine serum albumin diluted in phosphate buffered saline containing 0.1% Triton. Primary antibodies were rabbit anti-FLAG (Sigma, 1:300) and mouse anti-GFP (Roche, 1:200). Fluorescent labeled secondary antibodies (donkey anti-mouse Cy2 1:60 and donkey anti-rabbit 594 1:200) were from Jackson ImmunoResearch. DNA was stained with Hoechst 33258 (1 μg/ml).
Proximity ligation assay
PLA was used to detect interactions between GFP-BAF and FLAG-lamin A, either WT or mutant (R435C, R527H, A529V, K542N, R453W, R482W), based on proximity (<40 nm) of two secondary antibodies directed against these tagged proteins. After cell fixation, cell permeabilisation and quenching (as above), pairs of primary antibodies, rabbit anti-FLAG 1:300 and mouse anti-GFP 1:200, were added on HeLa cells expressing GFP-BAF together with FLAG-lamin A, for 40 min at R.T. Next, Duolink PLA probe anti-rabbit plus, Duolink PLA probe anti-mouse minus and Duolink detection reagents orange (detected with a Cy3 filter) were used according to manufacturer's instructions (Olink, Bioscience). At last, cells were mounted in Duolink mounting medium with Dapi (Olink, Bioscience). Confocal microscopy image acquisition was performed using a LSM 700 Laser scanning microscope (Zeiss) at the imaging facility of the BFA institute. Quantitative analysis of PLA signals was done on images using ImageJ, as reported in (33). Data were then analyzed by comparing median values for the total number of pixels showing a signal (Cy3) per nucleus. Data of the three independent experiments were normalized to 1 (100%) for cells expressing FLAG-lamin A WT. Statistical analysis were performed using Kruskal–Wallis tests.
RESULTS
The lamin A/C Igfold domain interacts with the self-assembled emerin fragment EmN
To determine how the lamin A/C tail interacts with emerin, we generated two fragments of the emerin nucleoplasmic region (Figure 1A). We tested binding between the tail region common to lamins A and C (LamIgF comprising aa 411 to aa 566) and these emerin fragments. As previously published, the whole emerin nucleoplasmic region from aa 1 to aa 221 is poorly soluble (34). We thus produced two overlapping emerin peptides, which we called EmN (26) and EmC (Figure 1A). EmN (aa 1 to aa 187) comprises a LEM domain and a region that is intrinsically disordered (34,35). We previously showed that, in vitro, it can be observed either as a monomer or as an oligomer (26) (Figure 1B, left panel). EmC (aa 67 to aa 221) is entirely unstructured as observed by NMR (Supplementary Figure S1A), and after purification immediately oligomerizes as observed by fluorescence (Supplementary Figure S1B) and negative-staining electron microscopy (EM; Figure 1B, right panel). In order to identify an interaction between lamin A/C and emerin, we produced a 15N labeled LamIgF peptide, and mixed the labeled peptide with either monomeric EmN, oligomeric EmN or oligomeric EmC (Figure 1C). The oligomeric state of emerin fragments in the NMR samples was systematically checked by EM (Supplementary Figure S2). Nuclear Magnetic Resonance (NMR) analysis of these samples revealed that LamIgF only interacts with oligomeric EmN in these conditions. This interaction is specific to lamin A/C as NMR analysis of a sample containing 15N labeled lamin B1 tail and oligomeric EmN did not demonstrate any binding (Figure 1C, lower and right panel; the lamin A/C and B1 Igfold domains share 52% of sequence identity).
Figure 1.

The lamin A/C Igfold domain directly binds to emerin self-assembled nucleoplasmic region. (A) Architecture of the emerin protein, highlighting its LEM domain (in orange) and its transmembrane domain (in dark grey), as well as the 2 fragments of the nucleoplasmic region that could be obtained as soluble peptides (EmN and EmC). (B) Negative staining EM images of EmN and EmC fragments obtained after purification, concentration and incubation at 293 K (white bar: 100 nm). EmN filaments were observed at 600 μM after 1 week (26), whereas EmC filaments were already visible at 300 μM after 1 day. The fragment corresponding to the region common to EmN and EmC did not self-assemble in these conditions (Supplementary Figure S1B). (C) Superimposition of the solution-state NMR 1H–15N HSQC spectra of the lamin Igfold domain (lamin A/C in gray or lamin B1 in green) recorded in the absence or presence of emerin fragments EmN or EmC (EmN in red, EmC in purple; 1:1 ratio). The emerin fragments were EmN filaments (upper left and lower right views), EmN monomers (lower left view) and EmC filaments (upper right view). All these spectra were recorded at 303 K on a 700 MHz spectrometer.
The Igfold domain of lamin A/C forms a ternary complex with the LEM domain of emerin and BAF
The chromatin-associated protein BAF binds to both lamin A/C and emerin (36,37). We examined the role of BAF in mediating an interaction between LamIgF, EmN and BAF by Size-Exclusion Chromatography (SEC). Consistent with our previous NMR results, we observed that LamIgF and monomeric EmN did not co-elute under the conditions used (Figure 2A). However, they co-eluted in the presence of BAF. Similarly, we tested the binding between the lamin B1 tail fragment, EmN and BAF. In this case, no binding could be observed between lamin B1 and BAF, precluding the formation of a ternary complex with EmN. NMR further confirmed that the lamin B1 tail fragment did not interact with BAF (Supplementary Figure S3). Isothermal Titration Calorimetry (ITC) experiments showed that LamIgF and EmN bind to BAF with an affinity of 3.2 ± 1.2 μM and 0.7 ± 0.2 μM, respectively (Figure 2B). The LamIgF/BAF interaction is enthalpy driven, whereas the EmN / BAF interaction is both enthalpy and entropy driven (Supplementary Table S1). Crystal structure of the ternary complex was solved at 1.9 Å resolution (Figure 3A; Supplementary Table S2). One BAF dimer interacts on one side with the LEM domain of emerin and on the other side with the Igfold domain of lamin A/C. The emerin fragment outside of the LEM domain is not visible in the electron density map. SDS-PAGE revealed that it was proteolyzed between protein purification and crystallization.
Figure 2.

The lamin A/C Igfold domain binds to emerin monomeric LEM domain through BAF. (A) Detection of a ternary complex including LamIgF, EmN and BAF by size exclusion chromatography using a Superdex 75 10/300GL column. Each gel corresponds to the elution of a different set of proteins, using the same column, the same protein concentrations and the same buffer: from the bottom EmN, LamIgF, BAF, lamin B1 tail (LB1; the lower band corresponds to a degradation product), BAF and lamin B1 tail, EmN and LamIgF, EmN and BAF, LamIgF and BAF and finally EmN, LamIgF and BAF. The bands corresponding to the different complexes are boxed: EmN and BAF in orange, LamIgF and BAF in green and the ternary complex in blue. (B) ITC titration of BAF onto LamIgF (upper panel) and BAF onto EmN (lower panel). 200 μM LamIgF or EmN were injected in a cell containing 40 μM of BAF at a temperature of 288 K. The affinities deduced from the fitting are 3.2 ± 1.2 μM and 0.7 ± 0.2 μM, respectively. The corresponding thermodynamics parameters (in kcal/mol) are plotted in the low right corner of the ITC figures.
Figure 3.

Three-dimensional structure of the complex between LamIgF, emerin LEM domain and BAF. (A) Cartoon representation of the complex, with emerin LEM domain (residues 3–44) in orange, BAF dimer in yellow-green and green (residues 4–89), and lamin A/C Igfold domain (residues 432–544) in gray. The interfaces corresponding to the emerin / BAF and BAF / lamin interactions are indicated by red circles and numbered as 1 and 2, respectively. (B) Superimposition of the 3D structure of the BAF dimer bound to the emerin LEM domain, as determined in this work (same colors as in (A)), and as revealed using NMR by Clore et al. (PDB reference 2ODG (19); in cyan). (C) Zoom on the EmN/BAF interface, with residues >30% buried within the interface displayed in sticks. On the emerin side, the interface is mainly formed by residues Gly24 to Lys37 (labeled residues on loop α1α2 and helix α2). On the BAF side, it is mainly formed by helices α3 and α4 of one monomer and loop α2α3 and helix α3 of the other monomer.
Dimerization of BAF is essential for both lamin and emerin binding
The 3D structure of the ternary complex highlights that BAF dimerization is essential for emerin and lamin A/C binding. First, BAF monomers 1 and 2 both interact with the LEM domain of emerin (interfaces of 395 and 211 Å2, respectively) (Figure 3B). BAF monomer 1 loop α2α3 (Phe39), helix α3 (Gln48, Val51, Leu52) and helix α4 (Leu58, Glu61, Trp62) contact a LEM surface comprising the β-strand (Gly24, Pro25), loop β1α2 (Ser29, Thr30) and helix α2 (Leu33, Tyr34, Lys36, Lys37). BAF monomer 2 loop α2α3 (Arg37, Phe39) and helix α3 (Glu48) contact an overlapping LEM surface comprising helix α1 (Thr13), the β-strand (Pro25) and loop β1α2 (Val27). A similar 3D structure between BAF dimer and emerin LEM domain was previously modeled based on NMR data (Figure 3C; PDB reference 2ODG (19)). When we superimposed the Cα atoms of the 2 structures, we obtained root mean square deviation values of 1.3, 1.4 and 0.9 Å for the LEM domain, BAF monomer 1 and BAF monomer 2, respectively, demonstrating that the NMR and X-ray structures are highly similar. If we superimposed only the Cα atoms of dimeric BAF, we measured a root mean square deviation of 2.8 Å between the LEM domains of the 2 structures. Consistently, the surface buried at the LEM/BAF interface yielded 510 Å2 in the NMR structure compared to 606 Å2 in the X-ray structure. Altogether, we observed a slightly different positioning of the LEM domain onto the BAF dimer and a consistent larger surface complementarity in the X-ray structure.
Both BAF monomers also contribute to lamin A/C binding, through interfaces of 270 and 192 Å2, respectively (Figure 4A). BAF monomer 1 helix α1 (Val11, Ala12), loop α1α2 (Pro14) and helix α5 (Glu83, Ala87, Phe88) contact lamin A/C strand β1 (His433, Ala434, Arg435) and strand β9 (Met540, Lys542), whereas BAF monomer 2 helix α1 (Ala12), loop α1α2 (Pro14) and helix α5 (Ala87, Phe88) contact lamin A/C strand β1 (Arg435) and loop β8β9 (Glu537). It was reported that BAF mutant G47E lost its ability to bind to lamin A as well as prelamin A in cells (5). Gly47 is located at the center of the dimerization interface (in magenta on Figure 4A). Mutation G47E might destabilize the BAF dimer, therefore hindering BAF interaction with both emerin LEM domain and lamin A/C Igfold.
Figure 4.
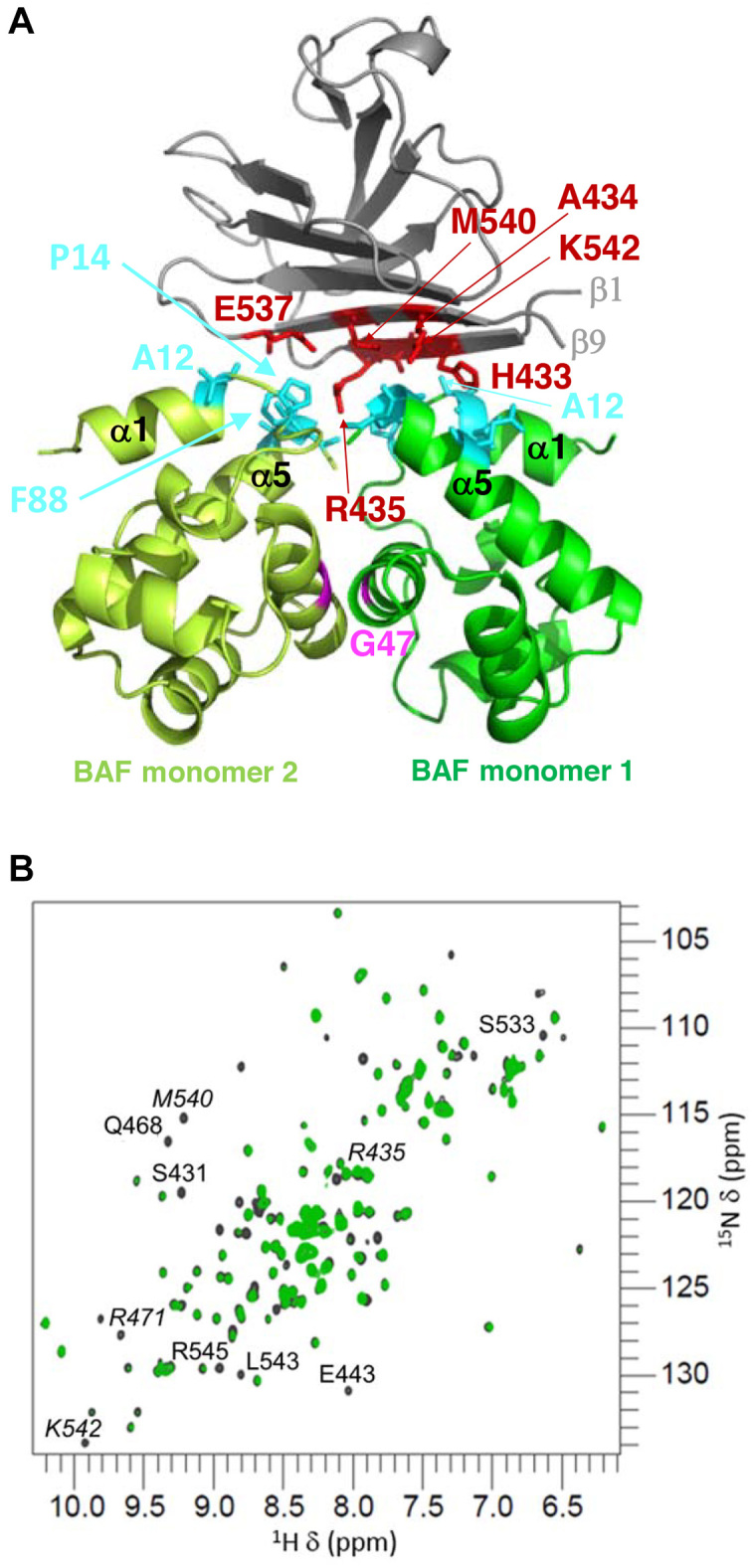
Lamin A/C Igfold domain binds BAF through a β-sheet including residues mutated in recessive progeroid syndromes. (A) Cartoon representation of the complex between the lamin A/C Igfold domain and the BAF dimer (main chain colors as in Figure 3). On the lamin side, the interface is mainly polar: it is formed by residues His433, Ala434, Arg435, Glu537, Met540 and Lys542 (in red sticks). On the BAF side, it is formed by residues from helix α1, loop α1α2 and helix α5 from both monomers. At the BAF monomer-monomer interface, Gly47 is displayed in magenta. (B) 1H–15N HSQC spectrum of LamIgF (150 μM) in the absence (grey) and presence (green) of BAF (150 μM) (ratio: 1 LamIgF for 0.5 BAF dimer). The labelled peaks exhibit a significant intensity decrease after addition of BAF. The peaks labelled in italics correspond to residues mutated in patients with progeroid diseases (R435(39,40),(42), R471C (44), M540 (43),(14), K542 (41)).
The interface between lamin A/C and BAF involves residues mutated in progeroid syndromes
By solving the 3D structure of a ternary complex formed by BAF, EmN and LamIgF, we revealed a yet undescribed interface between the BAF dimer and the lamin A/C Igfold domain. On the BAF side, Ala12, Pro14 and Phe88 of both monomers are more than 50% buried in the complex (Figure 4A). An Ala12 to Thr amino acid substitution occurs in patients with a Nestor-Guillermo progeria syndrome (38). Our 3D structure predicts that this substitution causes a defect in BAF/lamin binding. Consistently, in cells, it significantly decreases BAF binding to lamin A and prelamin A (5).
On the lamin side, Arg435, Met540 and K542 are more than 50% buried at the lamin/BAF monomer 2 interface (Figure 4A). Arg435 side-chain is hydrogen-bonded to Phe88 backbone oxygens of both BAF monomers, while Lys542 side-chain is bonded to BAF monomer 2 Asp86 backbone oxygen. These lamin residues are mutated in patients with severe autosomal recessive progeroid syndromes (RD and HGPS (14,39–43)). They belong to a hot spot for mutations causing progeroid diseases (14). In order to verify that in solution, this hot spot surface is contacting BAF, we followed the NMR 1H and 15N signals of LamIgF while adding BAF to the NMR sample. We observed that the intensities of the NMR signals corresponding to several residues of strand β1 (as Arg435), loop β1β2, strand β4 (as Arg471), loop β8β9 and strand β9 (as Met540 and Lys542) were significantly decreased (Figure 4B). This demonstrated that the β-sheet formed by strands β1 and β9 is indeed involved in BAF binding in solution.
Lamin A/C Igfold mutations causing progeroid syndromes decrease binding affinity for BAF
To identify the consequences of the LamIgF mutations causing progeroid diseases on the formation of the lamin-BAF complex, we produced a set of LamIgF peptides mutated on sites responsible for severe diseases. We focused our interest on five homozygous LamIgF mutations, R435C (strand β1) (39,40,42), R471C (strand β4) (44), R527H (45–48), A529V (49) (strand β8) and K542N (strand β9) (41), which were identified in patients with atypical progeroid syndromes. Two of these mutations cause syndromes generally appearing during early childhood as RD (R435C) and HGPS (K542N), characterized by severe premature aging features, whereas mutations R471C, R527H and A529V cause a disease generally appearing later, called MAD, characterized by growth retardation and skeletal abnormalities. In the case of R471C, the disease was particularly severe and the phenotype combined MAD, progeria, and rigid spine muscular dystrophy (44). We also produced as control variants the mutants R453W and R482W that cause Emery-Dreifuss Muscular Dystrophy (EDMD) and Dunnigan-type Familial Partial Lipodystrophy (FPLD), respectively. We first verified that the LamIgF mutated peptides were well folded and had a thermal stability close to that of WT LamIgF (i.e. ±5 K relatively to WT; Supplementary Figure S4; see also the X-ray structure of mutant R482W in (50)). We then measured their affinity for BAF by ITC (Figure 5A; Supplementary Table S1). We observed that the 2 control mutants R453W and R482W bind BAF as LamIgF WT, whereas mutant R435C does not bind BAF in our conditions. Mutants R471C and A529V exhibit a 5-fold decreased affinity for BAF. In the case of K542N and R527H, the heat release due to BAF binding was so weak that no affinity could be measured. We also tested the binding of LamIgF mutants to BAF by SEC (Figure 5B). We observed that mutants R453W and R482W coeluted together with BAF as WT LamIgF. However, mutant R435C did not induce any elution volume shift of BAF, and mutants R471C, A529V, R527H and K542N only weakly shifted BAF elution volume. We concluded that the five tested mutations causing atypical progeroid syndromes decrease the affinity of lamin A/C for BAF. Moreover, the binding defect seems to globally correlate with the disease severity (Figures 5A and B).
Figure 5.

Impact of mutants causing recessive progeroid syndromes on BAF binding depends on the disease severity. (A) ITC titration of BAF onto LamIgF, as measured at 283K. Whereas variants R453W and R482W show WT binding, variants A529V and R471C exhibit a significantly lower binding enthalpy and affinity as compared to LamIgF WT. No affinity could be measured for R527H and K542N and no binding could be detected for R435C. In the lower right panel, the spatial distribution of residues mutated in (A) is displayed on the lamin A/C Igfold structure (PDB reference 1IFR (12)). The two residues mutated in control variants, causing muscle and adipose tissue diseases respectively, are colored in green, whereas the 5 residues mutated in variants with a lower affinity for BAF are colored in cyan, purple and red (for weak binding, no measurable affinity and no detected binding, respectively). (B) Size exclusion chromatography of LamIgF mutants mixed to BAF (Superdex 75 10/300GL). Each gel corresponds to the elution of a different set of proteins, using the same column, the same concentrations and the same buffer: from bottom BAF, BAF and R435C, BAF and K542N, BAF and R527H, BAF and R471C, BAF and A529V, BAF and R482W, BAF and R453W and BAF and WT LamIgF. The bands corresponding to the BAF-LamIgF complex are boxed in red.
Finally, we transfected HeLa cells with plasmids coding for GFP-BAF and FLAG-mature lamin A (aa 1 to aa 646) WT or variants and detected using an in situ Proximity Ligation Assay the proximities between BAF and lamin A in these cells. We thus tested the impact of mutations related to RD (R435C), HGPS (K542N) and MAD (R527H, A529V), as well as EDMD (R453W) and FPLD (R482W), on these proximities. FLAG-lamin variants were expressed at similar protein levels as FLAG-lamin WT; they localized both at the nuclear periphery and in the nucleoplasm (Figure 6A and B). We observed that all 4 lamin A mutations associated with atypical progeroid syndromes similarly reduced the frequency of the proximity events between BAF and lamin A (Figures 6C and 7). Lamin A mutations associated with muscle and adipose tissue diseases had an intermediate impact: they also reduced the number of proximity events but their impact was significantly lower than the impact of mutations associated with progeroid syndromes in cells (Figures 6C and 7).
Figure 6.

Mature lamin mutants with mutations causing recessive progeroid syndromes impair BAF binding in cells. (A) HeLa cells expressing GFP-BAF (WT) together with either WT, R435C, R527H, A529V, K542N, R453W or R482W FLAG-LAm were fixed, and processed for immunofluorescence using as primary antibodies, a mix of anti GFP and anti-FLAG antibodies. Observation was done at the confocal microscope. Are shown in the left panels, merges for GFP-BAF (green), FLAG-LAm (red) and DNA (blue), and in the right panels, the FLAG-LAm alone. Scale bar: 20 μm. (B) Whole cell protein extracts prepared from HeLa cells control (ctrl) or expressing GFP-BAF (WT) together with either WT, R435C, R527H, A529V, K542N, R453W or R482W FLAG-LAm were analyzed by western blot using anti FLAG and anti BAF antibodies. (C) HeLa cells expressing GFP-BAF (WT) together with either WT, R435C, R527H, A529V, K542N, R453W or R482W FLAG-LAm were fixed, labeled with a mix of anti GFP and anti-FLAG antibodies and further processed for PLA before observation at the confocal microscope. The PLA signals (red) are shown merged with DNA (blue) and GFP-BAF (green) or alone (PLAssay). Signals for GFP-BAF and DNA are shown alone in the two right panels, respectively. Scale bar: 15 μm.
Figure 7.

Quantification of the decrease in proximity events between mature lamin A and BAF due to mutations causing either recessive progeroid syndromes, EDMD or FPLD. Quantification of PLA signals per nucleus was performed as reported in (33). (A) Proximity events were compared between either WT lamin A and BAF, or lamin A variants associated with progeroid syndromes and BAF. (B) The impact of lamin mutations causing either a progeroid syndrome (R435C), EDMD (R453W) or FPLD (R482W) on the proximity between mature lamin A and BAF was measured. The upper graphs show the results of three independent experiments carried out with each mutant. The lower graphs represent the mean values ± stdevpa (n = 3 experiments; *P < 0.05 with Kruskal and Wallis test).
DISCUSSION
The mechanical roles of lamins and their functions in gene regulation are often viewed as independent activities, but recent findings suggest a highly cross-linked and interdependent regulation of these different activities (51–53). Lamins interact with inner nuclear membrane proteins to mediate mechanical signaling but also contact chromatin-associated proteins that contribute to gene transcription regulation. Most of these interactions depend on the lamin ability to assemble into higher-order structures (54,55). Therefore, lamin assembly has been studied intensively in vitro and the resulting filaments have been observed in cells (11). In contrast, little is known about the contacts of lamins with their partners in vivo. Lamins are extensively modified and interact with a large number of proteins (56,57), the inner nuclear membrane (58) and chromatin (59). Their tail region is responsible for association with partners. It contains a globular Igfold domain that is mutated in several laminopathies (13). Until this study, it was not known how this domain interacted with partners anchored at the inner nuclear membrane and established contacts with chromatin.
The lamin A/C Igfold domain directly binds to self-assembled emerin but also indirectly interacts with monomeric emerin through BAF
We showed that the lamin A/C Igfold domain interacts with the inner nuclear membrane protein emerin through two mechanisms: it either directly binds to self-assembled EmN or interacts indirectly with monomeric EmN through the chromatin-associated protein BAF. It was previously published that direct lamin A tail interaction with emerin is disrupted not by mutations in the LEM domain, but by mutations in emerin region from aa 70 to aa 164: mutations around positions 70, 76, 95, 112, as well as 141 and 164, decreased binding of lamin A to emerin (15,37). These results are consistent with our analysis. Indeed, the disordered region of emerin between aa 50 and aa 132 is necessary for emerin self-assembly (26) and LamIgF only binds directly to self-assembled emerin. Similarly, GST fusions of an emerin fragment from aa 1 to aa 132 is sufficient to bind mature lamin A tails (18), and we showed that emerin aa 1 to aa 132 is sufficient for self-assembly (26). Finally, the EDMD-associated emerin deletion mutation del95-99, which impairs EmN self-assembly in vitro, causes a decrease of lamin A/C binding in cells (26). Altogether these results strongly suggest that emerin self-assembly is necessary for direct binding to lamin A/C in cells. On the other hand, we showed that the monomeric LEM domain (emerin aa 1 to aa 45) indirectly interacts with the lamin A/C Igfold domain through a dimer of BAF, consistent with previous data showing that BAF enhanced binding of prelamin A tails to emerin (24).
A defect at the interface between lamin A/C and the chromatin-associated BAF protein is observed in atypical progeroid syndromes
By analyzing the 3D structure of our ternary complex comprising domains of lamin A/C, BAF and emerin, we noticed that the interface between lamin A/C and BAF coincides with a hot spot of lamin A/C residues mutated in progeroid syndromes (14). Proximities between lamin A/C and BAF were detected in situ using a proximity assay; they were particularly enriched at the nuclear periphery in HeLa cells (Supplementary Figure S5). The four tested mutations associated with progeroid syndromes reduced the frequency of proximity events between lamin A and BAF. This decrease was significantly larger than the decrease observed in the case of control mutations R453W and R482W associated with muscle and adipose tissue diseases, respectively: mutations associated with progeroid syndromes reduced the number of PLA signals to ∼50% of the signals measured for WT lamin A and BAF. Consistently, in vitro, SEC experiments revealed that LamIgF mutants R471C, A529V, R527H and K542N weakly interact with BAF, and mutant R435C does not bind BAF. ITC experiments provided only estimations for affinity decreases, due to the weak affinities of the mutants. However, an affinity decrease of ∼5-fold could be calculated for mutants R471C and A529T, whereas essentially no binding could be detected for R527H, K542N and R435C. Interestingly, this affinity scale matches with the severity of the associated diseases: R471C and A529V cause MAD that generally appears during late childhood (44,49), whereas K542N causes a HGPS with an onset between 18 and 24 months (41) and R435C causes the most severe disease, RD, which appears during the first months after birth (39,40,42); only mutant R527H, which causes a MAD (45–48), also shows a strong BAF binding defect under the conditions we used. We conclude that a destabilization of the interface between lamin A/C and BAF might mediate the disease-mechanism in atypical progeroid syndromes due to homozygous mutations in LamIgF.
Another progeroid syndrome called the Nestor-Guillermo Progeria Syndrome occurs in patients older than the average lifespan of progeroid patients. This syndrome is due to the homozygous BAF mutation A12T (38). The mutated protein is expressed and stable (60). In cells, it still co-immunoprecipitates with endogeneous emerin, lamin A and histone H3. Our preliminary data show that, in vitro, mutation A12T does not compromise BAF 3D structure (Supplementary Figure S6A), and BAF mutant A12T interacts with LamIgF, but with an affinity lower than BAF WT and BAF S4E (Supplementary Figure S6B). Immunoprecipitation experiments performed in co-transfected cells also showed a reduced interaction between BAF mutant A12T and lamin A (5). Finally, BAF mutant A12T exhibited a marked defect in double-stranded DNA binding compared to WT BAF (60). Altogether, these data strongly suggest that the complex between BAF, lamin A/C, DNA and emerin is destabilized by the mutation A12T, highlighting the similarity between the mechanisms of autosomal recessive progeroid syndromes due to mutations in LamIgF and BAF.
Complexes including lamin A/C and emerin are assembled at different stages of the cell cycle
In cells, formation and localization of complexes including lamin A/C, BAF and emerin is highly regulated and depend on the cell cycle, in accord with the disassembly/reformation of the nuclear envelope that delimits the interphase nuclear compartment. We have identified two types of complexes including lamin A/C and emerin, however the biological relevance of these two modes of interaction is still unclear. It is known that at the end of mitosis, in ana-telophase, first BAF, and then emerin and lamin A/C accumulate at the core region of chromosomes (61). On the BAF side, mutation G25E, at the interface with DNA, impairs localization of BAF at the core region of chromosomes, emerin localization during telophase and further emerin and lamin A accumulation at reforming nuclear envelopes. On the emerin side, the LEM domain is necessary and sufficient for the core localization of emerin. Superimposition of our 3D structure with that of a complex between BAF dimer and DNA (PDB reference 2BZF (62)) revealed that the BAF dimer can simultaneously bind to emerin, lamin and DNA (Figure 8, left panel). We propose that BAF mediates the interaction between emerin and lamin A at the core region.
Figure 8.

A first model of the interface between the LEM-domain proteins, the nucleoskeleton and the chromatin-associated protein BAF. (A) During the ana/telophase, BAF mediates an interaction between the lamin A/C Igfold domain and emerin anchored at the endoplasmic reticulum (ER) or at the inner nuclear membrane (INM). Superimposition of the X-ray structure of the LamIgF-BAF-LEM complex onto the X-ray structure of the BAF protein complexed to DNA (PDB reference 2BZF (62)) suggests that BAF can simultaneously interact with lamin A/C, emerin and DNA. The interface between lamin A/C and BAF is disrupted by mutations causing autosomal recessive progeroid syndromes, as symbolized by the red arrow. (B) In interphase cells, the lamin A/C Igfold domain binds self-assembled emerin anchored at the inner nuclear membrane (INM), as symbolized by the black arrow. It is also able to interact with BAF and histones, thus creating an interface between the lamina and chromatin. This interface is disrupted by mutations causing autosomal recessive progeroid syndromes, as symbolized by the red arrow.
In interphase cells, lamin A/C is assembled into polymers that bind to emerin (37). This interaction might not require a well-folded LEM domain. Indeed, an emerin protein mutated in its LEM domain (residues 24–27 being changed into alanines), which does not interact with BAF (15), localizes normally at the nuclear envelope (63), consistent with its ability to directly bind lamin A/C in vitro (15). We suggest that lamin A/C directly interacts with emerin that is self-assembled at the nuclear envelope (18,64) (Figure 8, right panel). During in vitro self-assembly, emerin exhibits a large LEM domain conformational change, strongly suggesting that self-assembled emerin does not bind BAF (34). However, we still detect proximities between lamin A/C and BAF at the nuclear periphery of interphase cells (Supplementary Figure S5).
In conclusion, the Igfold domain of lamin A/C can both directly interact with self-assembled emerin and bind to monomeric emerin through the chromatin-associated protein BAF. A defect in the interaction between lamin A/C and BAF might lead to a less efficient targeting of emerin at the reforming nuclear envelope in telophase. However, diseases associated with mutations disrupting the lamin A/C–BAF interaction are much more severe than diseases associated with emerin loss of function. This suggests that the impact of these lamin A/C and BAF mutations goes beyond LEM-domain protein mislocalization and loss of function. We propose that the autosomal recessive progeroid syndrome-causing mutations perturb the BAF-mediated interaction between lamin A/C and chromatin, thus leading to a dysregulation of chromatin organization and gene expression. Consistently, an abnormal nuclear envelope–chromatin interface was previously described using electron microscopy in the case of progeroid syndromes caused by lamin A or BAF mutations (5,65–67). More work is needed to understand how the lamin–BAF complex interacts with DNA and histones, and how a defect in this complex compromises the interactions between lamina and chromatin at the nuclear periphery in autosomal recessive progeroid syndromes.
DATA AVAILABILITY
Structure coordinates were deposited to the PDB, with entry 6GHD.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. H.J. Worman for providing the plasmid coding for GST-LamIgF and reviewing the manuscript and Prof. G. Lattanzi for providing the plasmid coding for GFP-BAF. We also thank N. Vadrot for her help with cell cultures.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Commissariat à l’énergie atomique et aux énergies alternatives, Centre National de la Recherche Scientifique, University Paris South and University Paris Diderot; French Infrastructure for Integrated Structural Biology (https://www.structuralbiology.eu/networks/frisbi) [ANR-10-INSB-05-01, Acronym FRISBI]; French Association against Myopathies (AFM) [17243 and 20018 to S.Z.-J. and PhD fellowship number 18159 to C.S.]. Funding for open access charge: Grant 20018 (French Association against Myopathies).
Conflict of interest statement. None declared.
REFERENCES
- 1. Worman H.J. Nuclear lamins and laminopathies. J. Pathol. 2012; 226:316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Worman H.J., Bonne G.. “Laminopathies”: a wide spectrum of human diseases. Exp. Cell Res. 2007; 313:2121–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gruenbaum Y., Medalia O.. Lamins: the structure and protein complexes. Curr. Opin. Cell Biol. 2015; 32:7–12. [DOI] [PubMed] [Google Scholar]
- 4. Robin J.D., Magdinier F.. Physiological and pathological aging affects chromatin dynamics, structure and function at the nuclear edge. Front. Genet. 2016; 7:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Loi M., Cenni V., Duchi S., Squarzoni S., Lopez-Otin C., Foisner R., Lattanzi G., Capanni C.. Barrier-to-autointegration factor (BAF) involvement in prelamin A-related chromatin organization changes. Oncotarget. 2016; 7:15662–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burke B., Gerace L.. A cell free system to study reassembly of the nuclear envelope at the end of mitosis. Cell. 1986; 44:639–652. [DOI] [PubMed] [Google Scholar]
- 7. Glass J.R., Gerace L.. Lamins A and C bind and assemble at the surface of mitotic chromosomes. J. Cell Biol. 1990; 111:1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taniura H., Glass C., Gerace L.. A chromatin binding site in the tail domain of nuclear lamins that interacts with core histones. J. Cell Biol. 1995; 131:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lopez-Soler R.I., Moir R.D., Spann T.P., Stick R., Goldman R.D.. A role for nuclear lamins in nuclear envelope assembly. J. Cell Biol. 2001; 154:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mahamid J., Pfeffer S., Schaffer M., Villa E., Danev R., Cuellar L.K., Forster F., Hyman A.A., Plitzko J.M., Baumeister W.. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science. 2016; 351:969–972. [DOI] [PubMed] [Google Scholar]
- 11. Turgay Y., Eibauer M., Goldman A.E., Shimi T., Khayat M., Ben-Harush K., Dubrovsky-Gaupp A., Sapra K.T., Goldman R.D., Medalia O.. The molecular architecture of lamins in somatic cells. Nature. 2017; 543:261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dhe-Paganon S., Werner E.D., Chi Y.I., Shoelson S.E.. Structure of the globular tail of nuclear lamin. J. Biol. Chem. 2002; 277:17381–17384. [DOI] [PubMed] [Google Scholar]
- 13. Krimm I., Ostlund C., Gilquin B., Couprie J., Hossenlopp P., Mornon J.P., Bonne G., Courvalin J.C., Worman H.J., Zinn-Justin S.. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure. 2002; 10:811–823. [DOI] [PubMed] [Google Scholar]
- 14. Verstraeten V.L., Broers J.L., van Steensel M.A., Zinn-Justin S., Ramaekers F.C., Steijlen P.M., Kamps M., Kuijpers H.J., Merckx D., Smeets H.J. et al. Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin A accumulation. Hum. Mol. Genet. 2006; 15:2509–2522. [DOI] [PubMed] [Google Scholar]
- 15. Lee K.K., Haraguchi T., Lee R.S., Koujin T., Hiraoka Y., Wilson K.L.. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001; 114:4567–4573. [DOI] [PubMed] [Google Scholar]
- 16. Vaughan A., Alvarez-Reyes M., Bridger J.M., Broers J.L., Ramaekers F.C., Wehnert M., Morris G.E., Whitfield W.G.F., Hutchison C.J.. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J. Cell Sci. 2001; 114:2577–2590. [DOI] [PubMed] [Google Scholar]
- 17. Sakaki M., Koike H., Takahashi N., Sasagawa N., Tomioka S., Arahata K., Ishiura S.. Interaction between emerin and nuclear lamins. J. Biochem. (Tokyo). 2001; 129:321–327. [DOI] [PubMed] [Google Scholar]
- 18. Berk J.M., Simon D.N., Jenkins-Houk C.R., Westerbeck J.W., Gronning-Wang L.M., Carlson C.R., Wilson K.L.. The molecular basis of emerin-emerin and emerin-BAF interactions. J. Cell Sci. 2014; 127:3956–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cai M., Huang Y., Suh J.Y., Louis J.M., Ghirlando R., Craigie R., Clore G.M.. Solution NMR structure of the barrier-to-autointegration factor-Emerin complex. J. Biol. Chem. 2007; 282:14525–14535. [DOI] [PubMed] [Google Scholar]
- 20. Qi R., Xu N., Wang G., Ren H., Li S., Lei J., Lin Q., Wang L., Gu X., Zhang H. et al. The lamin-A/C-LAP2alpha-BAF1 protein complex regulates mitotic spindle assembly and positioning. J. Cell Sci. 2015; 128:2830–2841. [DOI] [PubMed] [Google Scholar]
- 21. Liu J., Lee K.K., Segura-Totten M., Neufeld E., Wilson K.L., Gruenbaum Y.. MAN1 and emerin have overlapping function(s) essential for chromosome segregation and cell division in Caenorhabditis elegans. PNAS. 2003; 100:4598–4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Margalit A., Segura-Totten M., Gruenbaum Y., Wilson K.L.. Barrier-to-autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. PNAS. 2005; 102:3290–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holaska J.M., Lee K.K., Kowalski A.K., Wilson K.L.. Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. J. Biol. Chem. 2003; 278:6969–6975. [DOI] [PubMed] [Google Scholar]
- 24. Bengtsson L., Wilson K.L.. Barrier-to-autointegration factor phosphorylation on Ser-4 regulates emerin binding to lamin A in vitro and emerin localization in vivo. Mol. Biol. Cell. 2006; 17:1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Samwer M., Schneider M.W.G., Hoefler R., Schmalhorst P.S., Jude J.G., Zuber J., Gerlich D.W.. DNA Cross-Bridging shapes a single nucleus from a set of mitotic chromosomes. Cell. 2017; 170:956–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herrada I., Samson C., Velours C., Renault L., Ostlund C., Chervy P., Puchkov D., Worman H.J., Buendia B., Zinn-Justin S.. Muscular dystrophy mutations impair the nuclear envelope emerin Self-assembly properties. ACS Chem. Biol. 2015; 10:2733–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krimm I., Couprie J., Ostlund C., Worman H.J., Zinn-Justin S.. 1H, 13C and 15N resonance assignments of the C-terminal domain of human lamin A/C. J. Biomol. NMR. 2002; 22:371–372. [DOI] [PubMed] [Google Scholar]
- 28. Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., Pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D.. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005; 59:687–696. [DOI] [PubMed] [Google Scholar]
- 29. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D, Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vagin A., Teplyakov A.. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997; 30:1022–1025. [Google Scholar]
- 31. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D, Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D, Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vadrot N., Duband-Goulet I., Cabet E., Attanda W., Barateau A., Vicart P., Gerbal F., Briand N., Vigouroux C., Oldenburg A.R. et al. The p.R482W substitution in A-type lamins deregulates SREBP1 activity in Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2015; 24:2096–2109. [DOI] [PubMed] [Google Scholar]
- 34. Samson C., Celli F., Hendriks K., Zinke M., Essawy N., Herrada I., Arteni A.A., Theillet F.X., Alpha-Bazin B., Armengaud J. et al. Emerin self-assembly mechanism: role of the LEM domain. FEBS J. 2017; 284:338–352. [DOI] [PubMed] [Google Scholar]
- 35. Samson C., Herrada I., Celli F., Theillet F.X., Zinn-Justin S.. 1H, 13C and 15N backbone resonance assignment of the intrinsically disordered region of the nuclear envelope protein emerin. Biomol. NMR Assign. 2016; 10:179–182. [DOI] [PubMed] [Google Scholar]
- 36. Margalit A., Brachner A., Gotzmann J., Foisner R., Gruenbaum Y.. Barrier-to-autointegration factor—a BAFfling little protein. Trends Cell Biol. 2007; 17:202–208. [DOI] [PubMed] [Google Scholar]
- 37. Berk J.M., Tifft K.E., Wilson K.L.. The nuclear envelope LEM-domain protein emerin. Nucleus. 2013; 4:298–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Puente X.S., Quesada V., Osorio F.G., Cabanillas R., Cadinanos J., Fraile J.M., Ordonez G.R., Puente D.A., Gutierrez-Fernandez A., Fanjul-Fernandez M. et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am. J. Hum. Genet. 2011; 88:650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Madej-Pilarczyk A., Rosinska-Borkowska D., Rekawek J., Marchel M., Szalus E., Jablonska S., Hausmanowa-Petrusewicz I.. Progeroid syndrome with scleroderma-like skin changes associated with homozygous R435C LMNA mutation. Am. J. Med. Genet. A. 2009; 149A:2387–2392. [DOI] [PubMed] [Google Scholar]
- 40. Youn G.J., Uzunyan M., Vachon L., Johnson J., Winder T.L., Yano S.. Autosomal recessive LMNA mutation causing restrictive dermopathy. Clin. Genet. 2010; 78:199–200. [DOI] [PubMed] [Google Scholar]
- 41. Plasilova M., Chattopadhyay C., Pal P., Schaub N.A., Buechner S.A., Mueller H., Miny P., Ghosh A., Heinimann K.. Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilford progeria syndrome. J. Med. Genet. 2004; 41:609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Starke S., Meinke P., Camozzi D., Mattioli E., Pfaeffle R., Siekmeyer M., Hirsch W., Horn L.C., Paasch U., Mitter D. et al. Progeroid laminopathy with restrictive dermopathy-like features caused by an isodisomic LMNA mutation p.R435C. Aging (Albany NY). 2013; 5:445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bai S., Lozada A., Jones M.C., Dietz H.C., Dempsey M., Das S.. Mandibuloacral dysplasia caused by LMNA mutations and uniparental disomy. Case Rep. Genet. 2014; 2014:508231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zirn B., Kress W., Grimm T., Berthold L.D., Neubauer B., Kuchelmeister K., Muller U., Hahn A.. Association of homozygous LMNA mutation R471C with new phenotype: mandibuloacral dysplasia, progeria, and rigid spine muscular dystrophy. Am. J. Med. Genet. A. 2008; 146A:1049–1054. [DOI] [PubMed] [Google Scholar]
- 45. Novelli G., Muchir A., Sangiuolo F., Helbling-Leclerc A., D’Apice M.R., Massart C., Capon F., Sbraccia P., Federici M., Lauro R. et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 2002; 71:426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Simha V., Agarwal A.K., Oral E.A., Fryns J.P., Garg A.. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J. Clin. Endocrinol. Metab. 2003; 88:2821–2824. [DOI] [PubMed] [Google Scholar]
- 47. Shen J.J., Brown C.A., Lupski J.R., Potocki L.. Mandibuloacral dysplasia caused by homozygosity for the R527H mutation in lamin A/C. J. Med. Genet. 2003; 40:854–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Garavelli L., D’Apice M.R., Rivieri F., Bertoli M., Wischmeijer A., Gelmini C., De Nigris V., Albertini E., Rosato S., Virdis R. et al. Mandibuloacral dysplasia type A in childhood. Am. J. Med. Genet. A. 2009; 149A:2258–2264. [DOI] [PubMed] [Google Scholar]
- 49. Garg A., Cogulu O., Ozkinay F., Onay H., Agarwal A.K.. A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia. J. Clin. Endocrinol. Metab. 2005; 90:5259–5264. [DOI] [PubMed] [Google Scholar]
- 50. Magracheva E., Kozlov S., Stewart C.L., Wlodawer A., Zdanov A.. Structure of the lamin A/C R482W mutant responsible for dominant familial partial lipodystrophy (FPLD). Acta Crystallogr. F, Struct. Biol. Cryst. Commun. 2009; 65:665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ho C.Y., Jaalouk D.E., Vartiainen M.K., Lammerding J.. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature. 2013; 497:507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cho S., Irianto J., Discher D.E.. Mechanosensing by the nucleus: From pathways to scaling relationships. J. Cell Biol. 2017; 216:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Osmanagic-Myers S., Dechat T., Foisner R.. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015; 29:225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Strelkov S.V., Herrmann H., Geisler N., Lustig A., Ivaninskii S., Zimbelmann R., Burkhard P., Aebi U.. Divide-and-conquer crystallographic approach towards an atomic structure of intermediate filaments. J. Mol. Biol. 2001; 306:773–781. [DOI] [PubMed] [Google Scholar]
- 55. Herrmann H., Strelkov S.V., Burkhard P., Aebi U.. Intermediate filaments: primary determinants of cell architecture and plasticity. J. Clin. Invest. 2009; 119:1772–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dorner D., Gotzmann J., Foisner R.. Nucleoplasmic lamins and their interaction partners, LAP2alpha, Rb, and BAF, in transcriptional regulation. FEBS J. 2007; 274:1362–1373. [DOI] [PubMed] [Google Scholar]
- 57. Simon D.N., Wilson K.L.. Partners and post-translational modifications of nuclear lamins. Chromosoma. 2013; 122:13–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. King M.C., Lusk C.P.. A model for coordinating nuclear mechanics and membrane remodeling to support nuclear integrity. Curr. Opin. Cell Biol. 2016; 41:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Camozzi D., Capanni C., Cenni V., Mattioli E., Columbaro M., Squarzoni S., Lattanzi G.. Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies. Nucleus. 2014; 5:427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Paquet N., Box J.K., Ashton N.W., Suraweera A., Croft L.V., Urquhart A.J., Bolderson E., Zhang S.D., O’Byrne K.J., Richard D.J.. Nestor-Guillermo Progeria Syndrome: a biochemical insight into Barrier-to-Autointegration Factor 1, alanine 12 threonine mutation. BMC Mol. Biol. 2014; 15:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Haraguchi T., Kojidani T., Koujin T., Shimi T., Osakada H., Mori C., Yamamoto A., Hiraoka Y.. Live cell imaging and electron microscopy reveal dynamic processes of BAF-directed nuclear envelope assembly. J. Cell Sci. 2008; 121:2540–2554. [DOI] [PubMed] [Google Scholar]
- 62. Bradley C.M., Ronning D.R., Ghirlando R., Craigie R., Dyda F.. Structural basis for DNA bridging by barrier-to-autointegration factor. Nat. Struct. Mol. Biol. 2005; 12:935–936. [DOI] [PubMed] [Google Scholar]
- 63. Haraguchi T., Koujin T., Segura-Totten M., Lee K.K., Matsuoka Y., Yoneda Y., Wilson K.L., Hiraoka Y.. BAF is required for emerin assembly into the reforming nuclear envelope. J. Cell Sci. 2001; 114:4575–4585. [DOI] [PubMed] [Google Scholar]
- 64. Barton L.J., Soshnev A.A., Geyer P.K.. Networking in the nucleus: a spotlight on LEM-domain proteins. Curr. Opin. Cell Biol. 2015; 34:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Columbaro M., Capanni C., Mattioli E., Novelli G., Parnaik V.K., Squarzoni S., Maraldi N.M., Lattanzi G.. Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell. Mol. Life Sci. 2005; 62:2669–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shumaker D.K., Dechat T., Kohlmaier A., Adam S.A., Bozovsky M.R., Erdos M.R., Eriksson M., Goldman A.E., Khuon S., Collins F.S. et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. PNAS. 2006; 103:8703–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Scaffidi P., Misteli T.. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005; 11:440–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structure coordinates were deposited to the PDB, with entry 6GHD.