


Abstract
APOBEC3H (A3H) is a mammal-specific cytidine deaminase that potently restricts the replication of retroviruses. Primate A3Hs are known to exert key selective pressures against the cross-species transmission of primate immunodeficiency viruses from chimpanzees to humans. Despite recent advances, the molecular structures underlying the functional mechanisms of primate A3Hs have not been fully understood. Here, we reveal the 2.20-Å crystal structure of the chimpanzee A3H (cpzA3H) dimer bound to a short double-stranded RNA (dsRNA), which appears to be similar to two recently reported structures of pig-tailed macaque A3H and human A3H. In the structure, the dsRNA-binding interface forms a specialized architecture with unique features. The analysis of the dsRNA nucleotides in the cpzA3H complex revealed the GC-rich palindrome-like sequence preference for dsRNA interaction, which is largely determined by arginine residues in loop 1. In cells, alterations of the cpzA3H residues critical for the dsRNA interaction severely reduce intracellular protein stability due to proteasomal degradation. This suggests that cpzA3H stability is regulated by the dsRNA-mediated dimerization as well as by unknown cellular machinery through proteasomal degradation in cells. Taken together, these findings highlight unique structural features of primate A3Hs that are important to further understand their cellular functions and regulation.
INTRODUCTION
The APOBEC3 (A3) proteins are a family of mammal-specific polynucleotide cytidine deaminases, consisting of seven members (A3A, -B, -C, -D, F, -G and -H) in primates. These proteins are antiviral restriction factors that act against retrotransposons and retroviruses such as human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus (SIV) [reviewed in (1,2)]. The antiviral mechanisms are dependent on and/or independent of single-stranded DNA (ssDNA) cytidine deamination (3–13). Moreover, localization of the A3s to the site of nascent viral DNA synthesis is a prerequisite for efficient inhibition (14,15). To successfully replicate, HIV and SIV encode virus infectivity factor (Vif), which antagonizes the antiviral function by targeting the enzymes for ubiquitination–proteasome degradation [reviewed in (1,2)]. The Vif proteins, which form thermodynamically stable heterodimers with a scaffold protein, core-binding factor subunit β (CBF-β) (16–18), specifically interact with the corresponding host-derived A3 proteins and are thereby recruited to the Cullin5-based E3 ubiquitin ligase complex (19).
A3H has a single zinc-binding domain and phylogenetically belongs to the unique Z3 type among primate A3s (20,21). The properties of human A3H (hA3H) have been well documented, especially the existence of seven divergent haplotypes (hap I–VII) circulating in the human population, which drastically differ in stability and phenotype (22–27). hA3H hap II, V and VII are stably expressed, efficiently packaged into virions and potently restrict vif-deficient HIV-1 replication (22,24,25,28–30). In contrast, hA3H hap I has low stability, while the other three haplotypes (hap III, IV and VI) are completely unstable in cells, mainly due to a single amino acid difference of G105 in hap I and an N15 deletion (ΔN15) in hap III, IV and VI (22,25,28–31). Two unstable types of the latter hA3H likely evolved independently under selection (24). The divergent hA3H haplotype distributions are biased in human populations and thus correlate with the global distribution of HIV-1 Vif variants that differ in their hA3H-antagonizing effects (28). This correlation is supported by two important observations by Ooms et al. (32): (i) HIV-1 Vif variants derived from patients encoding unstable hA3H haplotypes largely lack antagonizing ability, while they tend to adapt to the stable hA3H haplotypes; (ii) lower plasma viral loads and higher CD4+ T-cell counts are observed in newly infected treatment-naïve patients with stable hA3H haplotypes than in patients with unstable haplotypes. These results indicate that the polymorphisms of hA3H are tightly associated with individual infection and the global transmission of HIV-1. Recently, Zhang et al. demonstrated that chimpanzee SIV (SIVcpz) and HIV-1 lineage Vifs antagonize chimpanzee A3H (cpzA3H), although no SIVcpz Vifs counteract hA3H hap II, suggesting that hA3H haplotypes may have also played a critical role in the earlier cross-species transmission of SIVcpz/HIV-1 from chimpanzees to humans (33). These accumulating data help to elucidate the critical roles of the primate A3Hs, especially hominoid A3Hs, in the evolutionary conflicts of SIVcpz/HIV-1 and the host restriction factors. However, the molecular structures of primate A3Hs that underlie their functional mechanisms are still not fully understood.
While our manuscript was in preparation, three independent groups have reported the crystal structures of the pig-tailed macaque A3H (pgtA3H) (34), hA3H hapII enzymes (35) and hA3H hap II variant (E56A/W90S/W115A/C116S/C127S/G128Q/S129E/Q130G/L155A) (36). The hA3H variant structure is a monomer in the apo form (36), whereas the other structures are a dimer bridged by a short double-stranded RNA (dsRNA). In the dimer structures of pgtA3H and hA3H hapII, dsRNA with specific sequences have not been observed, suggesting that A3H proteins lack specificity in the recognition of RNA sequences upon dimerization.
In this study, we report the 2.20-Å crystal structure of the cpzA3H dimer bound to dsRNA, which displays an overall structural configuration similar to those of pgtA3H (34) and hA3H hapII (35). Interestingly, our high-resolution structure reveals the unique capacity of cpzA3H to specifically recognize the dsRNA. Extensive analysis of the dsRNA nucleotides in the complex and molecular dynamic (MD) simulations suggest a novel structure-based mechanistic model of cpzA3H’s nucleobase preference for the dsRNA interaction. Furthermore, cell-based cpzA3H expression assays indicate that alterations in the cpzA3H residues critical for dsRNA interaction abolish intracellular protein stability due to proteasomal degradation. This suggests that the stability of cpzA3H is regulated by dsRNA-mediated dimerization as well as by presumptive Vif-like factor-mediated degradation in cells. Moreover, this mechanism is most likely responsible for the intracellular instability of the completely unstable hA3H haplotypes. These findings provide an important basis for the primate A3H structures that underlie their functional mechanisms and may indicate a novel primate A3H regulatory system.
MATERIALS AND METHODS
Plasmids
To construct the pET-41 glutathione S-transferase (GST)-cpzA3H plasmid for expression in bacteria, a DNA fragment of the cpzA3H open reading frame region was amplified by polymerase chain reaction (PCR) using the primer set GST-A3H (SacII_noEK)(+) and A3H (stop) XhoI (–) (Supplementary Table S1). The DNA fragment was cloned into pET-41a(+) (Merck Millipore), which has an N-terminal GST-HIS tag and a thrombin cleavage site. Thrombin cleavage of the GST-cpzA3H protein leaves a dipeptide (glycine-serine) preceding the N-terminus of cpzA3H. The mammalian expression plasmids of pTR600 without an insert (37) or containing a fragment for an amino-terminal FLAG-tagged cpzA3H wild-type (WT) (38) were used. Mutant cpzA3H expression plasmids were constructed by oligonucleotide-directed PCR as previously described (38) using the appropriate primer set listed in Supplementary Table S1. The sequences of both the insert and the boundary regions for the cpzA3H expression plasmids were verified by conventional DNA sequencing methods.
Cells, transfection and immunoblotting
Human embryonic kidney 293T cells (293T) were maintained in Dulbecco’s modified Eagle medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum plus penicillin (100 U/ml) and streptomycin (100 μg/ml) (Thermo Fisher Scientific). For analysis of the intracellular cpzA3H protein levels, the cpzA3H expression plasmid (0.5 μg) was transfected into 12-well plates using FuGENE HD (Promega) according to the manufacturer’s instructions. At 32-h post-transfection, the culture supernatant was gently replaced with fresh medium with or without MG132 (Sigma-Aldrich), chloroquine (Sigma-Aldrich) or bafilomycin A1 (Sigma-Aldrich), followed by an additional 16-h incubation. The cell lysates were prepared with Laemmli buffer (Bio-Rad) containing 2.5% 2-mercaptoethanol (2-ME). The immunoblotting assays were performed as previously described (38). The following antibodies were used: anti-DYKDDDDK tag monoclonal antibody (1:2 × 103 dilution) (Wako) for detecting the FLAG tag; anti-β-tubulin antibody-Loading Control (1:103 dilution) (Abcam); and goat horseradish peroxidase-conjugated secondary antibodies against mouse IgG or rabbit IgG (1:2 × 104 dilution) (Thermo Fisher Scientific).
Protein expression and purification
For recombinant protein production, the Escherichia coli (E. coli) expression system was used as previously described with slight modifications (39). Briefly, pET-41 GST-cpzA3H was transformed into Rosetta 2(DE3)pLysS competent cells (Merck Millipore). The transformed bacteria were grown in Luria-Bertani medium containing kanamycin (50 μg/ml) and chloramphenicol (34 μg/ml) at 37°C to an optical density at 600 nm of ∼0.6. After cooling down to 20°C, the bacterial cultures were induced with 10 mg/ml lactose monohydrate in the presence of 1 μM ZnSO4 at 20°C for 20 h. The bacterial pellets were harvested and resuspended in lysis buffer [5 mM 2-ME, 10% (v/v) glycerol, 1 M NaCl, 100 μg/ml RNase A (Qiagen) and 50 mM Tris–HCl (pH 7.4)]. The suspensions were sonicated, subjected to centrifugation at 30 000 × g for 30 min and then filtered through a 0.8-μm pore-size membrane. The cleared soluble fraction was applied to a glutathione Sepharose 4 Fast Flow (FF) column (GE Healthcare) for affinity purification. The column was washed with five column volumes of lysis buffer and subsequently washed with five column volumes of wash buffer [1 mM 2-ME, 2% glycerol, 500 mM NaCl and 50 mM Tris–HCl (pH 7.4)]. The GST-cpzA3H fraction was obtained using elution buffer [1 mM 2-ME, 2% glycerol, 500 mM NaCl, 40 mM reduced l-glutathione and 50 mM Tris–HCl (pH 7.4)] and was treated with thrombin (∼40 U/ml) (GE Healthcare) and RNase A (1 mg/ml) at 20°C for 16 h. The cpzA3H was further purified using Superdex 75 gel filtration chromatography (GE Healthcare) equilibrated with gel filtration buffer [50 mM NaCl, 300 mM l-arginine HCl, 1 mM Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid sodium salt (pH 7.0)]. To remove the residual GST tag, the peak fraction was passed through a glutathione Sepharose 4 FF column in tandem with a Ni Sepharose High Performance column (GE Healthcare). The flow-through fraction contained the cpzA3H recombinant proteins. To determine the approximate molecular sizes of the proteins, the gel filtration elution profiles of thrombin-treated GST-cpzA3H with or without RNase A were compared with those of standard marker proteins (GE Healthcare).
Protein crystallization
The purified cpzA3H proteins were concentrated to ∼15 mg/ml using an Amicon Ultra-0.5 (Merck Millipore). Crystallization screening of cpzA3H was performed by the hanging-drop vapor-diffusion method using the JCSG-plus screen solution (Molecular Dimensions) at 20°C. The optimized crystallization solution contained 100 mM ammonium formate and 10% (w/v) polyethylene glycol (PEG) 3350.
Data collection and processing
The crystal was soaked in a cryoprotectant solution (100 mM ammonium formate, 10% PEG 3350 and 30% glycerol) for a few seconds and flash cooled in liquid nitrogen. X-ray diffraction data sets were collected at 100 K using synchrotron radiation (λ = 1.00 Å) on the beamline BL41XU at Spring-8, RIKEN, Harima Science Garden City, Japan. The diffraction patterns were processed using XDS (40) and AIMLESS (41).
Analysis of structure data and construction of structure model
The cpzA3H structure was determined using the molecular replacement method starting with the pgtA3H crystal structure (PDB 5W3V) (34) using the program MOLREP (42) implemented in the CCP4 suite (43). The structure of cpzA3H was then refined using the programs REFMAC5 (44) and PHENIX (45) and manually fitted with COOT (46). The electrostatic potential of our structure was visualized with PyMOL 2.0 (Schrödinger, LLC).
In vitro DNA cytidine deamination assays
The deamination assay conditions were adapted from our previous report (47). A total volume of 12 μl for each reaction contained 150 nM of a 40-nt ssDNA substrate labeled at its 5′ end with 6-carboxyfluorescein (Fasmac Co.) in deamination buffer [15 mM Tris–HCl (pH 8.0), 55 mM NaCl, 1 mM TCEP and 3% glycerol]. The ssDNA substrate sequences are listed in Supplementary Table S1. The deamination reactions were performed at 37°C with 5 μM of either the cpzA3H or rhesus macaque A3C recombinant proteins for 1 h. The deamination reaction was terminated by heat denaturation at 94°C for 5 min, followed by incubation at 37°C for 1 h with 0.2 U/μl uracil-DNA glycosylase (UDG) (New England Biolabs) and UDG buffer. The mixture was treated with 0.15 M sodium hydroxide at 37°C for 30 min. Gel Loading Buffer II (Thermo Fisher Scientific) was added to the mixture and heated at 94°C for 5 min. The final products were subjected to 20% denaturing urea-polyacrylamide gel electrophoresis. The gels were scanned with a Typhoon PhosphorImager (GE Healthcare) and analyzed using ImageQuant (GE Healthcare).
Identification of the dsRNA-coding regions in the E. coli genome
The dsRNA sequences identified in our crystal structure were a duplex of 9 and 8 nt (chains C and D, respectively), chain C, 5′-Y1Y2R3CCGGGU and chain D, 5′-Y2′ACCCGGY9′. ‘Y’ and ‘R’ indicate a pyrimidine (C or U) and a purine (G or A), respectively. Of note, chain D starts with Y2′ since the monophosphate observed in the structure at the 5′ end of Y2′ observed in the structure was designated as the first nucleotide. In our refined electron map, the densities of R3 and Y9′ clearly indicated a pyrimidine and a purine, respectively, although the densities were not actually strong enough to clearly determine whether the R3 and the Y9′ were either G or A and either C or U, respectively. To further assess these nucleotide sequences, we analyzed the dsRNA-coding regions in the complete genome of the E. coli BL21(DE3) strain, which was used to produce the cpzA3H–dsRNA complex. First, we generated a PERL script (available at https://github.com/odehir/find-seqmotif) to identify the gene regions encoding the short RNAs in the E. coli complete genomes (the GenBank accession numbers CP001509.3, AM946981.2, NC_012971.2 and NC_012892.2) because the lengths of the nucleotide sequences are too short to search for certain positions using the BLAST program (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Next, sequence combinations paired with the two sequences, 5′-YYRCCGGGU and 5′-YACCCGGY, were selected as plausible origins of the dsRNA sequences using the following criteria: (i) R3 and Y9′ engage in Watson–Crick base pairing and (ii) the two sequences were positioned within a 100-nt distance in the genome. Of note, no sequence motifs were identified in the GST-cpzA3H expression plasmid. RNA secondary structures were predicted with the mfold program (http://unafold.rna.albany.edu/?q=mfold/rna-folding-form) (48).
RESULTS
The crystal structure of the cpzA3H dimer mediated with a short RNA duplex
Despite our exhaustive attempts, we were unable to find methods for successful purification of the hA3H hap II recombinant protein, as it easily precipitates and aggregates. Thus, cpzA3H (∼21.9 kDa), which is orthologous to hA3Hs and has a relatively high solubility, was chosen for bacterial expression. An N-terminal GST-tagged cpzA3H (∼49.6 kDa) was initially expressed using an E. coli expression system and then treated with thrombin to remove the GST tag. The protein mixture was subjected to fractionation using Superdex 75 gel filtration chromatography, as previously described (39). In the absence of RNase A, despite complete thrombin cleavage, a large portion of the cpzA3H eluted in the void fraction (Figure 1A). In contrast, ∼92% of the protein pool treated with thrombin and RNase A was obtained as a fraction containing a protein of ∼40 kDa, which is equivalent to the molecular weight (MW) of a cpzA3H dimer (Figure 1A). We further purified the full-length untagged cpzA3H to >97% purity (Figure 1B) and tested its enzymatic activity in the UDG-based deaminase assay. As shown in Figure 1C, the purified cpzA3H displayed high deaminase activity on the trinucleotide (TTC) motif of 40-nt ssDNA, but less activity on the CCC motif (lanes 2–4). No 5-methylC (5mC) deamination of the cpzA3H was detected (lane 5). The deamination substrate specificities of cpzA3H differ from those of hA3H hap II, which efficiently deaminates 5mC (49).
Figure 1.

Crystal structure of the cpzA3H dimer with an RNA duplex. (A) Gel filtration Superdex 75 elution profile of cpzA3H with (blue) or without (red) RNase A treatment. The standard MW marker proteins conalbumin (75 kDa), ovalbumin (44 kDa), carbonic anhydrase (29 kDa) and RNase A (13.7 kDa) were used. (B) Gel image of purified cpzA3H stained with Coomassie brilliant blue R-250; MWM, MW marker. (C) Deamination activity of cpzA3H. Forty-nucleotide ssDNA substrates containing TTC (lanes 1, 2 and 3), the CCC (lane 4) or the TT5mC (lane 5) motifs were used in the UDG-dependent deamination assay. The recombinant protein rhesus macaque A3C (rheA3C) (lane 1) was used as a control; 5mC, 5-methylcytosine. (D) Ribbon and (E) electrostatic surface representations of cpzA3H seen in four rotated views. The ribbons are colored according to the secondary structures: α-helices (red); β-strands (yellow); loops (gray) and dsRNA (orange/yellow). Coordinated zinc ions are represented as navy spheres. The loops 1 and 7 and the α6 helix in chain A are indicated as L1, L7 and α6, respectively. The accessible surface area of cpzA3H is colored according to the calculated electrostatic potential from –5.0 kT/e (red) to +5.0 kT/e (blue).
We crystalized the untagged cpzA3H and successfully determined the structure at a resolution of 2.20 Å in the space group P21 (Table 1). The overall structure has a dimer architecture with the two monomers connected by a short RNA duplex (Figure 1D). Surprisingly, the cpzA3H monomers have no direct contact in the dimer unit. The cpzA3H protein has the structural fold commonly found in other A3 domain core structures [reviewed in (50,51)], which is composed of six major α helices and five β strands. As reported in our previous analysis of an hA3H structural model, cpzA3H also forms a clustered region of basic residues near the catalytic center (Figure 1E), which with the exception of the A3G N-terminal domain (52,53) differs from the electrostatic surface polarization of other A3 proteins. This basic cpzA3H surface, including loops 1 (L1), 7 (L7) and α6, provides an electrostatic interaction interface for the negatively charged phosphate backbones of the dsRNA. Based on the comparative analysis of primate A3H sequences (Supplementary Figure S1), the residue positions with high posterior probabilities of having evolved under positive selection are located around the dsRNA-binding region, the deaminase catalytic center and the Vif-binding region. Nevertheless, similarities in the residues responsible for the dsRNA interaction are high (Supplementary Figure S1), suggesting that the dsRNA-binding interfaces are highly conserved among primate A3Hs.
Table 1.
Data collection and refinement statistics
| cpzAPOBEC3H | |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 38.34, 86.97, 77.36 |
| α, β, γ (°) | 90.00, 102.63, 90.00 |
| Resolution (Å) | 43.49–2.20 (2.27–2.20)a |
| R merge | 0.073 (0.471) |
| I / σI | 8.5 (2.1) |
| Completeness (%) | 97.3 (94.7) |
| Redundancy | 3.1 (2.8) |
| Refinement | |
| Resolution (Å) | 43.49–2.20 |
| No. of reflections | 24454 |
| R work/Rfree (%) | 25.11/32.02 |
| No. of atoms | 3477 |
| Protein | 3028 |
| RNA | 370 |
| Ligand/ion | 2 |
| Water | 77 |
| B-factors(Å2) | 51.36 |
| Protein | 51.59 |
| RNA | 50.02 |
| Ligand/ion | 49.48 |
| Water | 48.81 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.010 |
aValues in parentheses are for the highest resolution shell.
Structural features of the dsRNA that bridges the cpzA3H molecules within the dimer
The dsRNA contains a 9-mer strand composed of ribonucleotides (nt1–9) with a 3′-monophosphate (chain C) as well as a second strand having eight ribonucleotides (nt2′–9′) with 5′- and 3′-monophosphates (chain D). The structure forms seven Watson–Crick base pairs (nt3–9 and nt3′–9′), a 5′-overhang of two bases at one end (nt1 and nt2) and a one-base 5′-overhang at the other end (nt2′) (Figure 2A). The double-stranded portion of the RNA is an A-form duplex in which the sugar geometries have C3′-endo puckers. Interestingly, in chain C, a kinked strand conformation is observed at the 5′-overhang portion, where the nt1 ribose displays sterically unfavored C3′-exo puckering. A comparison of the dsRNA phosphate backbones in the crystal with that of expected A- or B-form duplexes simulated in silico also supports the possibility that the cpzA3H dimer adopts an A-form RNA duplex (Supplementary Figure S2). These results suggest that the double-stranded duplex form is one of the critical determinants mediating cpzA3H dimerization. In our refined crystal structure, the dsRNA density was consistent with two ribonucleotide strands of 5′-Y1Y2R3CCGGGU (chain C) and 5′-Y2′ACCCGGY9′ (chain D), whereas ‘Y’ and ‘R’ indicate a pyrimidine (C or U) and a purine (G or A), respectively. The R3 and Y9′ densities were too weak to be assigned as either G or A and either C or U, respectively, because of a mixture of bases or insufficient data acquisition. However, they clearly engage in Watson–Crick base pairing. To further assess these nucleotide sequences, we searched the RNA-coding regions in the complete genome of the E. coli BL21(DE3) strain, which we used to express our cpzA3H–dsRNA production and based our analysis on two criteria: (i) the R3-Y9′ is either a G-C or A-U base pair; (ii) the two strands are located within a 100-base distance in the genome. The results showed that seven sequence motifs with G-C base pairing at the R3-Y9′ were matched, although no motifs with A-U at the position were detected (Figure 2B). In addition, no motifs were identified in the GST-cpzA3H expression plasmid sequence. Therefore, the dsRNA structure in the crystal structure was modeled using one of the sequence motifs containing 5′-CUGCCGGGU (chain C) and 5′-UACCCGGC (chain D) (Figure 2A).
Figure 2.

Identification of Escherichia coli-derived nucleotide sequences analogous to the dsRNA detected in the cpzA3H crystal structure. (A) Diagram of the dsRNA secondary structure modeled in the crystal structure. (B) Seven of our identified nucleotide sequences and their putative RNA secondary structures are shown. The dsRNA sequences identified in the crystal structure are shown as the ‘dsRNA motif’, consisting of 5′-YYRCCGGGU and 5′-YACCCGGY (Y, pyrimidine—C or U; R, purine—G or A). Using an ‘in-house’ program, the locations of these two nucleotide sequences were determined in the complete genome of E. coli BL21(DE3), which was used for cpzA3H protein expression. Based on the genome sequences around the identified positions, the RNA secondary structures were predicted with the mfold program. The nucleotides analogous to the dsRNA motif are highlighted in red. No dsRNA motifs were identified in the GST-cpzA3H expression plasmid.
The dsRNA-interaction interfaces of cpzA3H
The surface of the dsRNA-interaction within the cpzA3H protein surface consists largely of basic or aromatic residues in L1, L5, L7 and α6 (Figure 3A). Two aromatic residues, Y23 and W115, provide critical driving forces through π–π stacking interactions with the C1 and G3 nucleobases, respectively. In addition, the W115 indole ring is underpinned by the T-shaped ‘π–π networks’ of the aromatic clusters W82–Y112–Y113–W115 and H114–W115 (Figure 3B). In contrast to the dsRNA nucleobase–cpzA3H interactions, the Y113 hydroxyl moiety (Oη) solely interacts with one ribose, U2 (or U2′), via a hydrogen bond to the 2′-hydroxyl group (O2′) (Supplementary Figure S3A); no other cpzA3H residues directly contacting riboses can be observed. An MD simulation for the dsRNA–cpzA3H complex suggests that the Y113 Oη-U2 O2′ bond is unlikely to be steady and it is occasionally broken (Supplementary Figure S3B). Thus, these results imply that Y113 plays no major role in the specificity of the RNA interaction and that cpzA3H discriminates dsRNA from dsDNA without recognizing the nucleotide sugar moieties. Electrostatic surface potential analysis revealed that the two basic cpzA3H surfaces face each other across the negatively charged dsRNA (Figure 3C). In addition, as previously predicted in our hA3H structural model, there is a positively charged channel starting from the RNA 3′-end, which can stretch far enough to accommodate a presumed extension of the negatively charged RNA strand (Figure 3C). These results indicate that electrostatic interaction mediated by dsRNA is also a key feature of cpzA3H dimerization.
Figure 3.

Interactions between cpzA3H proteins and the RNA duplex. (A) Overall structure of cpzA3H–dsRNA complex. The cpzA3H and the dsRNA are represented with gray ribbons and orange/yellow sticks, respectively. cpzA3H residues responsible for the RNA interaction are highlighted with cyan sticks. The loop 5 is indicated as L5. L1′ and α6′ are in chain B. (B) The electron density map (2FO-FC, contoured at the 1.5σ level) around the termini of the RNA duplex. The aromatic residues involved in the nucleobase interactions are highlighted with cyan sticks. (C) A hypothetical path for the extended ssRNA portion (beige dashed line). The electrostatic surface potential is colored between –5.0 kT/e (red) and +5.0 kT/e (blue).
In the crystal structure, L1, L7 and α6 constitute the basic surface for the dsRNA interaction. The arginine cluster in L1 fits in the dsRNA major groove (Figure 4A and B); R16, R20, R21 and R26 in both chains clamp the phosphate backbone. In addition, the embedded R17′ and R18′ in chain B potentially form hydrogen bonds with the G6 N7 and the G7 N7, while R17 and R18 in chain A do not create hydrogen bonds with any nucleobases in the groove (Supplementary Figure S3C). As the side chains of R17′ and R18′ were refined based on the relatively low densities of their atoms, these hydrogen bonds did not appear to be fully fixed at the same positions. Furthermore, a 20-ns MD simulation of the cpzA3H–dsRNA complex showed that the guanidino group atoms of R17, R18, R17′ and R18′ are mapped in the groove. In addition, the R17 side chain frequently recognizes the Watson–Crick faces of G8′, but the G7′ face less often (frequencies of 83% and 22%, respectively). Similarly, R17′ forms hydrogen bonds mainly with G7 and transiently with G8 (58% and 10%, respectively) (Supplementary Figure S3D and E). In contrast, R18 and R18′ form hydrogen bonds infrequently with G7′ and G6, respectively (4% and 8%). These observations suggest that the basic L1 residues, positioned symmetrically in the dimer unit, are flexible in recognizing the Watson–Crick faces of the guanines within their accessible distances. Furthermore, the four basic residues on α6 (K168, R175, R176 and R179) also participate in the RNA backbone recognition via their electrostatic interactions (Figure 4C). In addition, these basic residue clusters on L1 as well as α6 are aligned along the helical curvature of the dsRNA phosphate backbones (Figure 4B and C), which also appear to contribute to the specificity of the dsRNA interaction of cpzA3H.
Figure 4.

Roles of A3H loop 1 (L1) and the α6 helix (α6) in the interaction with dsRNA. (A) RNA–duplex interaction interfaces formed by positively charged cpzA3H residues. The overall complex structure is represented with gray ribbons (cpzA3H) and an electrostatic surface (RNA). The basic residues involved in the RNA interaction are represented with cyan sticks. (B) Enlarged view of the interaction site between L1 and dsRNA. Basic residues R17, R18, R20 and R21 of L1 protrude into the negatively charged major groove of dsRNA. (C) Basic residues of L1 and α6 are aligned along the curvature of the RNA phosphate backbone. (D) The α6 position is fixed via the formation of a hydrogen bond between the L1 stem N15 (gray stick) and the α6 L166. The structure is shown without dsRNA is represented.
Interestingly, the helical structure axis of α6 is distorted in the middle (Figure 4D and Supplementary Figure S4A), partly due to the fit of the curvature architecture for the helical dsRNA recognition. The distortion located between K168 and A172 is attributed to a hydrogen bond between the L166 main chain and the N15 side chain. This hydrogen bond holds not only the distortion, but also the α6–L1 configuration. This interpretation is supported by our MD simulation data showing that the apo form of cpzA3H ΔN15 displays an unbent α6 helical structure compared with the WT protein (Supplementary Figure S4B). The N15–L166 hydrogen bond formed at the bottom position of L1 is also observed in the structures of other A3s and activation-induced cytidine deaminase (AID) structures that have been reported previously [(54,55), and reviewed in (50,51)]. Although the glutamine residue equivalent to N15 is highly conserved in APOBEC2, A3s, APOBEC4 and AID, hA3H hap III, IV and VI are missing N15 (ΔN15) (22,24). Furthermore, the MD simulation data suggest that the N15 mutation decreases accessibility of L1, especially R17, in the dsRNA major groove (Supplementary Figure S3D and E), which likely reduces the binding affinity for the dsRNA-interaction with A3H. These observations suggest that the N15–L166 hydrogen bond plays a fundamental role in maintaining the integrity of the dsRNA-interaction interface of primate A3Hs.
Intracellular instability of cpzA3H mutants
In our previous report, we demonstrated that mutations at the putative nucleotide-binding residues of hA3H hap II—R17D/R18D, Y113D/H114D and W115A—resulted in significantly lower intracellular protein levels (52). To investigate whether such intracellular instability can also be observed in the case of cpzA3H, we transiently expressed each cpzA3H mutant, which contained single or double mutations at the critical residues, including the N15, and analyzed the amounts of protein by immunoblotting. As shown in Figure 5 (no inhibitor), the expression of cpzA3H mutants ΔN15, N15S, R17D/R18D, H114A, W115A and R175D/R176D was drastically reduced; in one case, R20D/R21D expression was detected, but it was lower than that of the WT. In contrast, the Y113A and Y113F mutants were expressed at similar levels to WT. Interestingly, these reductions in the intracellular cpzA3H mutant levels were sharply rescued by the addition of MG132. In contrast, neither of the lysosome inhibitors chloroquine and bafilomycin A1 blocked the instability of the mutants. These findings demonstrated that the cpzA3H mutations critical for dsRNA binding cause intracellular instability facilitated by the proteasomal degradation pathway. In addition, the results suggest that the cpzA3H dimerization mediated by dsRNA increases its protein stability in cells. Data indicating that the Y113 residue might not be essential for cpzA3H to interact with dsRNA (Supplementary Figure S3B), appear to reflect the observation that the Y113A and the Y113F mutants are as stable as WT in cells.
Figure 5.
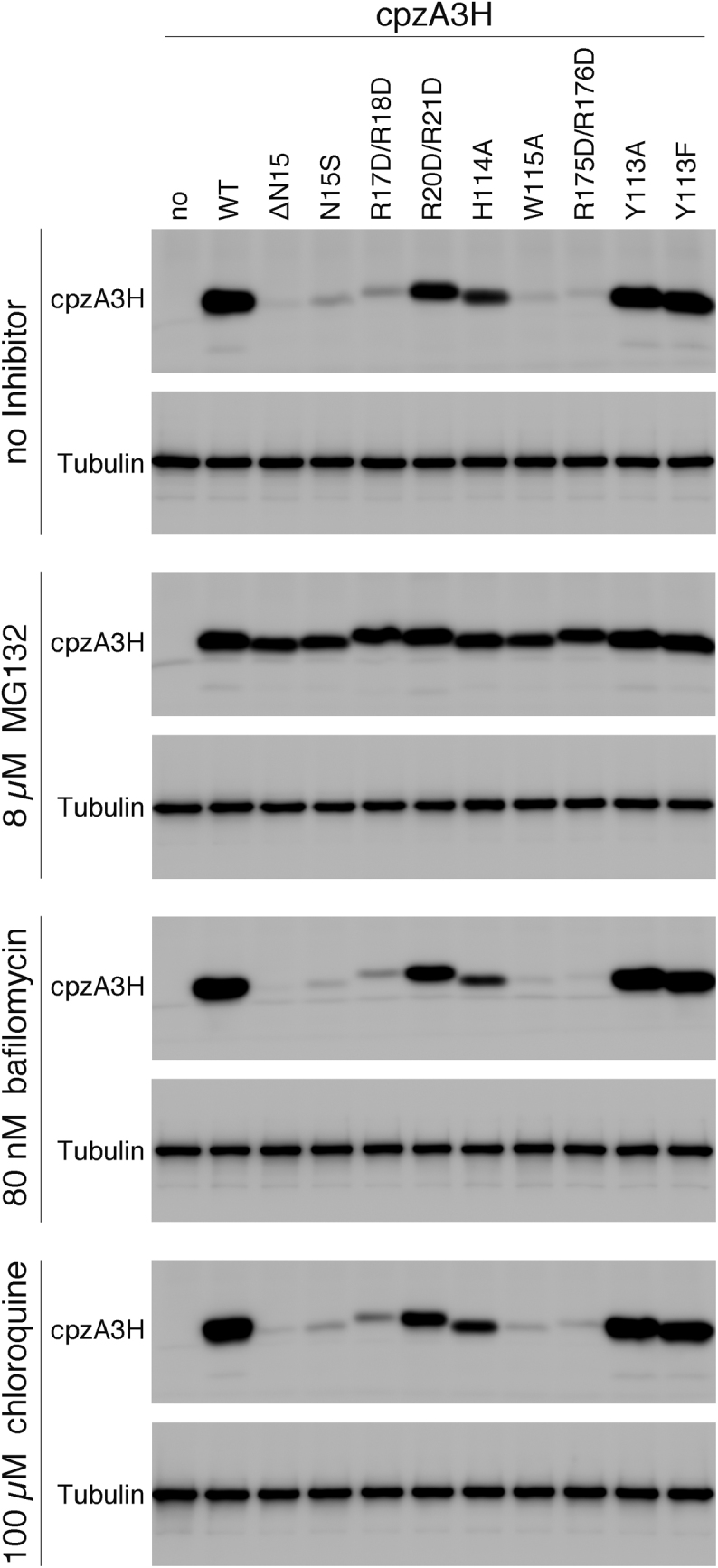
Effect of cpzA3H mutations in the RNA recognition interface on the intracellular levels of cpzA3H. WT cpzA3H and mutants were transiently expressed in 293T cells in the absence of an inhibitor or in the presence of MG132 (8 μM), chloroquine (100 μM) or bafilomycin A1 (80 nM). The proteins were detected by western blotting with the anti-DYKDDDDK tag monoclonal antibody (Anti-FLAG) or anti-β-tubulin antibody (Anti-β-Tubulin).
The putative Vif-interaction interface in cpzA3H dimer
Previous studies by our group and by Ooms et al. identified 11 hA3H residues critical for the HIV-1–Vif interaction and mapped the positively charged region using homology models of monomeric hA3H or cpzA3H structures rather than their dimeric forms (38,56). To verify whether the two regions in the dimer together form one Vif-binding site upon their dimerization, we mapped the critical residues onto our cpzA3H dimer complex and analyzed the interfaces. As shown in Figure 6A, the two putative Vif-binding regions are separated and positioned at the opposite end in the cpzA3H–dsRNA dimer complex. These interface configurations indicate that the Vif-interaction interface is composed of the monomeric A3H unit and that the two interfaces are in a dimeric unit. In addition, our in silico docking simulation of the interaction between cpzA3H and the Vif–CBF-β complex suggests that the A3H Vif-binding surface has sufficient space for access of the Vif complex (Supplementary Figure S5A) and a reasonable contact area, ∼877.9 Å2, for a single unit of the Vif–CBF-β complex (Supplementary Figure S5B). Furthermore, the Vif-interaction surface is not overlapped with the positively charged path required to accommodate the putatively extended RNA strand (Figure 6B). These results suggest that the putative A3H Vif-interaction interface is composed of one A3H molecule unit and is accessible by Vif without potential steric hindrances, even though cpzA3H forms a dimer through dsRNA bridging. Therefore, it is conceivable that two units of the Vif–CBF-β complex can bind to one A3H dimer simultaneously. This may be one of the reasons why the Vif interaction with A3H is more complex than that with A3G, as observed in an in-depth biochemical analysis (57).
Figure 6.

A view of the Vif-interaction interface in the cpzA3H dimer. (A) Two separate A3H sites are responsible for Vif binding. cpzA3H and dsRNA are represented in gray ribbons with a transparent surface and orange/yellow ribbons with nucleobases, respectively. The residues (magenta sticks) critical for HIV-1–Vif interaction are mapped on the dimer structure. (B) Highlighted view of the Vif-binding interface and its unique residue Q97 in the chimpanzee-bonobo lineage. Residue Q97 is one of the most important determinants for the differential sensitivity of HIV-1/SIVcpz Vifs. A putative path for the extended ssRNA portion is also shown (orange dashed line).
DISCUSSION
In the present study, we determined the high-resolution crystal structure of the chimpanzee A3H dimer bound to a short dsRNA. The structure reveals that cpzA3H interacts with an RNA duplex through its unique interface, which overlaps with the ssDNA substrate recognition surface for deamination. The cpzA3H dsRNA-binding interface consists of three key elements: (i) two aromatic-residue networks in L1 and L7 that form π–π stacking interactions with the dsRNA nucleobases; (ii) the protrusion of L1 into the duplex major groove and (iii) alignment of the basic L1 and α6 residues that fit the helical curvature of the A-form dsRNA phosphate backbones. Analysis of the dsRNA nucleotides in the complex revealed the Guanine-Cytosine(GC)-rich sequence preference for dsRNA interaction, which is largely determined by one L1 residue R17. Interestingly, the mutagenesis of cpzA3H demonstrated that the alteration of the residues critical for the dsRNA interaction reduces intracellular protein stability through the action of the cellular proteasomal degradation pathway. This result suggests that the dsRNA-mediated dimerization stabilizes and protects cpzA3H from such degradation.
Recently, while our manuscript was in preparation, the crystal structures of pgtA3H (34), hA3H hap II (35) and hA3H hap II variant (36) were determined by three independent groups. The hA3H variant structure is a monomer in the apo form, presumably due to introduction of mutations in the expression clone (36). In contrast, the other structures, including our cpzA3H, appear to be similar in that they are dimers bound to dsRNA. This suggests that structural dimerization principles are highly conserved among primate A3Hs. However, more detailed structural comparisons show that the cpzA3H side chain configurations, particularly the aromatic residues, which are responsible for the dsRNA interaction differ from those of hA3H, but are almost identical to those of pgtA3H (Supplementary Figure S6). A tryptophan is often observed in π–π interactions between proteins and RNA, especially with G or C bases (58), and here too, the W115 indole interacts with the G3 base in cpzA3H as well as in pgtA3H and hA3H. However, W115 is underpinned by the multiple T-stacking interactions of W82–Y112–Y113–W115 and H114–W115 in cpzA3H and pgtA3H, but not in hA3H. These aromatic clusters likely stabilize the position of the W115 indole ring so that it can recognize the nucleobase. The highly conserved nature of these aromatic amino acids is probably responsible for preservation of this function among the primate A3Hs (Supplementary Figure S1). Additionally, the π–π stacking interaction between Y23 and C1, which results in the kinked RNA strand with the C3′-exo-puckered C1 ribose, can be seen in cpzA3H as well as in pgtA3H. Currently, it is unclear what biological functions are linked to such unique distortions in the RNA bound to the primate A3Hs.
One principal finding in this study is the first determination of the nucleobases in the primate A3H-dsRNA crystal structure in combination with database searches for short RNAs. Importantly, the dsRNA density in our crystal structure was consistent with two specific ribonucleotide strands: 5′-Y1Y2R3CCGGGU (chain C) and 5′-Y2′ACCCGGY9′(chain D) (Figures 2 and 3). This finding suggests that there is a sequence preference for the dsRNA recognition upon cpzA3H dimerization. In the crystal structure, there are three direct interactions between the duplexed nucleobases and the cpzA3H residues, although the electron densities of the L1 residues are weakly displayed. Additionally, our MD simulations suggest that the R17 and R17′ of L1′/L1 embedded in the dsRNA major groove are key interaction residues that function to form hydrogen bonds with the Watson–Crick face of G8′ (or G7′) and G7 (or G8). The second interaction of π-stacking between W115 and G3 is also involved, although it is likely to enhance the strength of the interaction rather than the specificity. Thus, residues R17/R17′ significantly contribute to the base preference recognition for the dsRNA. Because they are symmetrically positioned in the dimer unit, the nucleobase preference of GC-rich palindrome-like sequences in the 7-base-paired dsRNA may be rationalized as the preferred sequence motif in the interaction. This interpretation is supported in part by the previous observation that hA3H preferentially targets G-rich RNA sequences in cells (59). The interaction preference for guanines in the Watson–Crick face is primarily due to the fact that arginine–guanine hydrogen bonds are more energetically favorable than bonds formed between arginine and the other nucleobases (60,61).
Importantly, we demonstrated that substitutions of the cpzA3H residues important for dsRNA recognition drastically reduce protein stability in cells, which depends on the proteasomal degradation pathway (Figure 5). This finding suggests that the dsRNA-mediated cpzA3H dimerization may be linked to a particular system that regulates the intracellular stability of the protein. One possible mechanistic model can be proposed: the dsRNA-mediated dimerization may mask a cpzA3H site recognized by an unknown cellular Vif-like factor, which promotes ubiquitination–proteasome degradation. Indeed, analogous hA3H mutants are also present at significantly lower levels than WT in 293T cells (52). In conjunction with the high conservation of these A3H residues among primates, the A3H regulatory systems might be common to primate cells. Recently, Shaban et al. demonstrated that dsRNA-binding-deficient hA3H mutants are enzymatically hyperactive and that RNA interaction is required for hA3H cytoplasmic localization (35). These findings might be explained by a mechanism by which dsRNA-mediated dimerization/oligomerization prevents the large A3H enzyme complex from migrating into the nucleus, where it can access genomic DNA and induce DNA damage. Interestingly, it has been reported that one of the hA3H haplotypes, hap I, may cause mutations that drive cancer (62). Taken together, our findings suggest that dsRNA-mediated dimerization of primate A3Hs tightly regulates their antiviral activity in the cytoplasm and prevents harmful mutagenesis in the nucleus via an as yet unidentified cellular system, thereby eliminating dsRNA-binding deficient A3Hs in cells.
Among the seven hA3H haplotypes, hap III, IV and V are completely unstable, largely due to the absence of N15 (ΔN15) at the normally conserved position (22,30,31). In the structure of cpzA3H, we found that the N15–L166 hydrogen bond plays a crucial role, as it stabilizes the position of L1 and α6 for the dsRNA interaction (Figure 4D and Supplementary Figure S4A and B). In addition, the MD simulation suggests that because the L1 element of ΔN15 is one residue shorter than WT L1, the basic mutant L1 tip has poor access into the dsRNA major groove as required for the WT interaction (Supplementary Figure S3D and E). Furthermore, our data indicate that the N15 mutations ΔN15 and N15S significantly decrease intracellular protein stability through proteasomal degradation, as is the case for dsRNA-binding deficient mutants (Figure 5). Indeed, a previous report showed that hA3H hap III and the hA3H ΔN15 mutant have diminished cellular-RNA-binding activity, packaging into virions and antiviral activity (31). Therefore, we propose that hA3H hap III, IV and V may be degraded in a similar manner as cpzA3H mutants displaying an unstable phenotype in cells. This instability may be largely attributed to their defect in dsRNA-mediated dimerization and high sensitivity to the unknown Vif-like factor. Of note, unstable hA3H hap I has a key substitution, R105G, in β4 that is distal from the dsRNA interaction interface, suggesting that another mechanistic model may exist for hA3H hap I instability in cells.
Our determination of the high-resolution cpzA3H structure has revealed structural mechanisms that define cpzA3H dimerization mediated by an RNA duplex and its nucleobase-binding preference. In addition, our cell-based assays demonstrate that dsRNA-mediated A3H dimerization regulates its intracellular stability through proteasome degradation. These novel findings will provide important insights to understand new structure–function mechanisms of A3 cytidine deaminases and offer a possible breakthrough for exploration of ancestral factors related to primate lentiviral Vif proteins.
DATA AVAILABILITY
Crystallography, atomic coordinates and structure factors have been deposited in the Protein Data Bank with code 5Z98. The PERL scripts to search for short sequence motifs are available at https://github.com/odehir/find-seqmotif.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Judith G. Levin for valuable discussions. The synchrotron radiation experiments were performed at the BL41XU of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2017B2720).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Japan Society for the Promotion of Science (JSPS) KAKENHI [JP15H04740 to Y.I., JP17K08872 to H.O.]; Japan Agency for Medical Research and Development (AMED) [JP17fk0410304 to Y.I.]. Funding for open access charge: Nagoya Medical Center, Japan.
Conflict of interest statement. None declared.
REFERENCES
- 1. Desimmie B.A., Delviks-Frankenberrry K.A., Burdick R.C., Qi D., Izumi T., Pathak V.K.. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 2014; 426:1220–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harris R.S., Dudley J.P.. APOBECs and virus restriction. Virology. 2015; 479–480:131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iwatani Y., Chan D.S., Wang F., Maynard K.S., Sugiura W., Gronenborn A.M., Rouzina I., Williams M.C., Musier-Forsyth K., Levin J.G.. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007; 35:7096–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chaurasiya K.R., McCauley M.J., Wang W., Qualley D.F., Wu T., Kitamura S., Geertsema H., Chan D.S., Hertz A., Iwatani Y. et al. . Oligomerization transforms human APOBEC3G from an efficient enzyme to a slowly dissociating nucleic acid-binding protein. Nat. Chem. 2014; 6:28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Newman E.N., Holmes R.K., Craig H.M., Klein K.C., Lingappa J.R., Malim M.H., Sheehy A.M.. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005; 15:166–170. [DOI] [PubMed] [Google Scholar]
- 6. Morse M., Huo R., Feng Y., Rouzina I., Chelico L., Williams M.C.. Dimerization regulates both deaminase-dependent and deaminase-independent HIV-1 restriction by APOBEC3G. Nat. Commun. 2017; 8:597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pollpeter D., Parsons M., Sobala A.E., Coxhead S., Lang R.D., Bruns A.M., Papaioannou S., McDonnell J.M., Apolonia L., Chowdhury J.A. et al. . Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat. Microbiol. 2018; 3:220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bishop K.N., Holmes R.K., Sheehy A.M., Davidson N.O., Cho S.J., Malim M.H.. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004; 14:1392–1396. [DOI] [PubMed] [Google Scholar]
- 9. Harris R.S., Bishop K.N., Sheehy A.M., Craig H.M., Petersen-Mahrt S.K., Watt I.N., Neuberger M.S., Malim M.H.. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003; 113:803–809. [DOI] [PubMed] [Google Scholar]
- 10. Iwatani Y., Takeuchi H., Strebel K., Levin J.G.. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 2006; 80:5992–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mangeat B., Turelli P., Caron G., Friedli M., Perrin L., Trono D.. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003; 424:99–103. [DOI] [PubMed] [Google Scholar]
- 12. Zhang H., Yang B., Pomerantz R.J., Zhang C., Arunachalam S.C., Gao L.. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003; 424:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mbisa J.L., Barr R., Thomas J.A., Vandegraaff N., Dorweiler I.J., Svarovskaia E.S., Brown W.L., Mansky L.M., Gorelick R.J., Harris R.S. et al. . Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 2007; 81:7099–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goila-Gaur R., Khan M.A., Miyagi E., Kao S., Strebel K.. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology. 2007; 4:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J., Chen Y., Li M., Carpenter M.A., McDougle R.M., Luengas E.M., Macdonald P.J., Harris R.S., Mueller J.D.. APOBEC3 multimerization correlates with HIV-1 packaging and restriction activity in living cells. J. Mol. Biol. 2014; 426:1296–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang W., Du J., Evans S.L., Yu Y., Yu X.F.. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature. 2012; 481:376–379. [DOI] [PubMed] [Google Scholar]
- 17. Jager S., Kim D.Y., Hultquist J.F., Shindo K., LaRue R.S., Kwon E., Li M., Anderson B.D., Yen L., Stanley D. et al. . Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature. 2012; 481:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo Y., Dong L., Qiu X., Wang Y., Zhang B., Liu H., Yu Y., Zang Y., Yang M., Huang Z.. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature. 2014; 505:229–233. [DOI] [PubMed] [Google Scholar]
- 19. Yu X., Yu Y., Liu B., Luo K., Kong W., Mao P., Yu X.F.. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003; 302:1056–1060. [DOI] [PubMed] [Google Scholar]
- 20. LaRue R.S., Andrésdóttir V., Blanchard Y., Conticello S.G., Derse D., Emerman M., Greene W.C., Jonsson S.R., Landau N.R., Löchelt M. et al. . Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 2009; 83:494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. OhAinle M., Kerns J.A., Malik H.S., Emerman M.. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J. Virol. 2006; 80:3853–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang X., Abudu A., Son S., Dang Y., Venta P.J., Zheng Y.H.. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J. Virol. 2011; 85:3142–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ooms M., Majdak S., Seibert C.W., Harari A., Simon V.. The localization of APOBEC3H variants in HIV-1 virions determines their antiviral activity. J. Virol. 2010; 84:7961–7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. OhAinle M., Kerns J.A., Li M.M., Malik H.S., Emerman M.. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe. 2008; 4:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harari A., Ooms M., Mulder L.C., Simon V.. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J. Virol. 2009; 83:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duggal N.K., Fu W., Akey J.M., Emerman M.. Identification and antiviral activity of common polymorphisms in the APOBEC3 locus in human populations. Virology. 2013; 443:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dang Y., Siew L.M., Wang X., Han Y., Lampen R., Zheng Y.H.. Human cytidine deaminase APOBEC3H restricts HIV-1 replication. J. Biol. Chem. 2008; 283:11606–11614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Refsland E.W., Hultquist J.F., Luengas E.M., Ikeda T., Shaban N.M., Law E.K., Brown W.L., Reilly C., Emerman M., Harris R.S.. Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PLoS Genet. 2014; 10:e1004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ooms M., Krikoni A., Kress A.K., Simon V., Munk C.. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J. Virol. 2012; 86:6097–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li M.M., Emerman M.. Polymorphism in human APOBEC3H affects a phenotype dominant for subcellular localization and antiviral activity. J. Virol. 2011; 85:8197–8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhen A., Du J., Zhou X., Xiong Y., Yu X.F.. Reduced APOBEC3H variant anti-viral activities are associated with altered RNA binding activities. PLoS One. 2012; 7:e38771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ooms M., Brayton B., Letko M., Maio S.M., Pilcher C.D., Hecht F.M., Barbour J.D., Simon V.. HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe. 2013; 14:411–421. [DOI] [PubMed] [Google Scholar]
- 33. Zhang Z., Gu Q., de Manuel Montero M., Bravo I.G., Marques-Bonet T., Haussinger D., Munk C.. Stably expressed APOBEC3H forms a barrier for cross-species transmission of simian immunodeficiency virus of chimpanzee to humans. PLoS Pathog. 2017; 13:e1006746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bohn J.A., Thummar K., York A., Raymond A., Brown W.C., Bieniasz P.D., Hatziioannou T., Smith J.L.. APOBEC3H structure reveals an unusual mechanism of interaction with duplex RNA. Nat. Commun. 2017; 8:1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shaban N.M., Shi K., Lauer K.V., Carpenter M.A., Richards C.M., Salamango D., Wang J., Lopresti M.W., Banerjee S., Levin-Klein R. et al. . The antiviral and cancer genomic DNA deaminase APOBEC3H is regulated by an RNA-Mediated dimerization mechanism. Mol. Cell. 2018; 69:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ito F., Yang H., Xiao X., Li S.-X., Wolfe A., Zirkle B., Arutiunian V., Chen X.S.. Understanding the structure, multimerization, subcellular localization and mC selectivity of a genomic mutator and Anti-HIV factor APOBEC3H. Sci. Rep. 2018; 8:3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Green T.D., Newton B.R., Rota P.A., Xu Y., Robinson H.L., Ross T.M.. C3d enhancement of neutralizing antibodies to measles hemagglutinin. Vaccine. 2001; 20:242–248. [DOI] [PubMed] [Google Scholar]
- 38. Nakashima M., Tsuzuki S., Awazu H., Hamano A., Okada A., Ode H., Maejima M., Hachiya A., Yokomaku Y., Watanabe N. et al. . Mapping region of human restriction factor APOBEC3H critical for interaction with HIV-1 Vif. J. Mol. Biol. 2017; 429:1262–1276. [DOI] [PubMed] [Google Scholar]
- 39. Kitamura S., Ode H., Nakashima M., Imahashi M., Naganawa Y., Kurosawa T., Yokomaku Y., Yamane T., Watanabe N., Suzuki A. et al. . The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 2012; 19:1005–1010. [DOI] [PubMed] [Google Scholar]
- 40. Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Evans P.R., Murshudov G.N.. How good are my data and what is the resolution. Acta Crystallogr. D Biol. Crystallogr. 2013; 69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vagin A., Teplyakov A.. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:22–25. [DOI] [PubMed] [Google Scholar]
- 43. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al. . Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vagin A.A., Steiner R.A., Lebedev A.A., Potterton L., McNicholas S., Long F., Murshudov G.N.. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2184–2195. [DOI] [PubMed] [Google Scholar]
- 45. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. . PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 47. Nakashima M., Ode H., Kawamura T., Kitamura S., Naganawa Y., Awazu H., Tsuzuki S., Matsuoka K., Nemoto M., Hachiya A. et al. . Structural insights into HIV-1 Vif-APOBEC3F interaction. J. Virol. 2015; 90:1034–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gu J., Chen Q., Xiao X., Ito F., Wolfe A., Chen X.S.. Biochemical characterization of APOBEC3H Variants: Implications for their HIV-1 restriction activity and mC modification. J. Mol. Biol. 2016; 428:4626–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aydin H., Taylor M.W., Lee J.E.. Structure-Guided analysis of the human APOBEC3-HIV restrictome. Structure. 2014; 22:668–684. [DOI] [PubMed] [Google Scholar]
- 51. Vasudevan A.A., Smits S.H., Hoppner A., Haussinger D., Koenig B.W., Munk C.. Structural features of antiviral DNA cytidine deaminases. Biol. Chem. 2013; 394:1357–1370. [DOI] [PubMed] [Google Scholar]
- 52. Mitra M., Singer D., Mano Y., Hritz J., Nam G., Gorelick R.J., Byeon I.J., Gronenborn A.M., Iwatani Y., Levin J.G.. Sequence and structural determinants of human APOBEC3H deaminase and anti-HIV-1 activities. Retrovirology. 2015; 12:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xiao X., Li S.X., Yang H., Chen X.S.. Crystal structures of APOBEC3G N-domain alone and its complex with DNA. Nat. Commun. 2016; 7:12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pham P., Afif S.A., Shimoda M., Maeda K., Sakaguchi N., Pedersen L.C., Goodman M.F.. Structural analysis of the activation-induced deoxycytidine deaminase required in immunoglobulin diversification. DNA Repair. 2016; 43:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Qiao Q., Wang L., Meng F.L., Hwang J.K., Alt F.W., Wu H.. AID recognizes structured DNA for class switch recombination. Mol. Cell. 2017; 67:361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ooms M., Letko M., Simon V.. The structural interface between HIV-1 Vif and human APOBEC3H. J. Virol. 2017; 91:e02289-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baig T.T., Feng Y., Chelico L.. Determinants of efficient degradation of APOBEC3 restriction factors by HIV-1 Vif. J. Virol. 2014; 88:14380–14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Baker C.M., Grant G.H.. Role of aromatic amino acids in protein-nucleic acid recognition. Biopolymers. 2007; 85:456–470. [DOI] [PubMed] [Google Scholar]
- 59. York A., Kutluay S.B., Errando M., Bieniasz P.D.. The RNA binding specificity of human APOBEC3 Proteins resembles that of HIV-1 nucleocapsid. PLoS Pathog. 2016; 12:e1005833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Allers J., Shamoo Y.. Structure-based analysis of protein-RNA interactions using the program ENTANGLE. J. Mol. Biol. 2001; 311:75–86. [DOI] [PubMed] [Google Scholar]
- 61. Lustig B., Arora S., Jernigan R.L.. RNA base-amino acid interaction strengths derived from structures and sequences. Nucleic Acids Res. 1997; 25:2562–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Starrett G.J., Luengas E.M., McCann J.L., Ebrahimi D., Temiz N.A., Love R.P., Feng Y., Adolph M.B., Chelico L., Law E.K. et al. . The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat. Commun. 2016; 7:12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallography, atomic coordinates and structure factors have been deposited in the Protein Data Bank with code 5Z98. The PERL scripts to search for short sequence motifs are available at https://github.com/odehir/find-seqmotif.