


Abstract
Protein synthesis requires both high speed and accuracy to ensure a healthy cellular environment. Estimates of errors during protein synthesis in Saccharomyces cerevisiae have varied from 10−3 to 10−4 errors per codon. Here, we show that errors made by 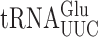 in yeast can vary 100-fold, from 10−6 to 10−4 errors per codon. The most frequent errors require a G•U mismatch at the second position for the near cognate codon GGA (Gly). We also show, contrary to our previous results, that yeast tRNAs can make errors involving mismatches at the wobble position but with low efficiency. We have also assessed the effect on misreading frequency of post-transcriptional modifications of tRNAs, which are known to regulate cognate codon decoding in yeast. We tested the roles of mcm5s2U34 and t6A37 and show that their effects depend on details of the codon anticodon interaction including the position of the modification with respect to the base mismatch and the nature of that mismatch. Both mcm5 and s2 modification of wobble uridine strongly stabilizes G2•U35 mismatches when
in yeast can vary 100-fold, from 10−6 to 10−4 errors per codon. The most frequent errors require a G•U mismatch at the second position for the near cognate codon GGA (Gly). We also show, contrary to our previous results, that yeast tRNAs can make errors involving mismatches at the wobble position but with low efficiency. We have also assessed the effect on misreading frequency of post-transcriptional modifications of tRNAs, which are known to regulate cognate codon decoding in yeast. We tested the roles of mcm5s2U34 and t6A37 and show that their effects depend on details of the codon anticodon interaction including the position of the modification with respect to the base mismatch and the nature of that mismatch. Both mcm5 and s2 modification of wobble uridine strongly stabilizes G2•U35 mismatches when 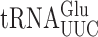 misreads the GGA Gly codon but has weaker effects on other mismatches. By contrast, t6A37 destabilizes U1•U36 mismatches when
misreads the GGA Gly codon but has weaker effects on other mismatches. By contrast, t6A37 destabilizes U1•U36 mismatches when 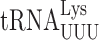 misreads UAA or UAG but stabilizes mismatches at the second and wobble positions.
misreads UAA or UAG but stabilizes mismatches at the second and wobble positions.
INTRODUCTION
Ribosomes decode the information in mRNAs using tRNAs to produce a polypeptide product. The efficiency and fidelity of this process are critical to the health of the cell and systems have evolved both to optimize speed and accuracy (1). A critical step in terms of accuracy and the cause of the most frequent errors is the recruitment of aminoacyl-tRNAs (aa-tRNAs). Recruitment is governed by a suite of interactions between the ribosome and the codon–anticodon complex (2). The occasional ‘misreading’ errors result from the acceptance of an incorrect aa-tRNA resulting in the substitution of one amino acid by another in the protein product. We have shown for Escherichia coli that these errors occur in vivo at frequencies up to 3.5 × 10−3 per decoding event (3) but some errors are no more frequent than 2 × 10−6 (4), which is orders of magnitude less frequent than has been supposed (5).
The understanding of how the ribosome discriminates between correct (cognate) and incorrect (near and non-cognate) aa-tRNAs has advanced recently. Aa-tRNAs bind to the A site in a ternary complex with an elongation factor (EF-Tu in bacteria or its cognate EF-1A in eukaryotes) and guanosine triphosphate (GTP). The ribosome controls acceptance in a two-stage process before (selection) or after (proofreading) hydrolysis of the GTP. The two stages are composed of several distinct kinetic steps. Cognate tRNAs are known to accelerate activation of the intrinsic GTPase activity of EF-Tu/EF-1A and accommodation of the aminoacyl-tRNA into the ribosomal P site after GTP is hydrolyzed (6–8). Ogle et al. (9,10) proposed that cognate but not near-cognate ternary complex can efficiently bind the A site and induce a large-scale rearrangement of the ribosome called domain closure that both prevents dissociation of the ternary complex and activates the EF-Tu GTPase. This large-scale rearrangement involves induced fit in which conformational changes in constituents of the A site allows them to contact all three base pairs of the codon–anticodon complex (11) suggesting that the inability of near-cognate complexes to induce these changes explained the preference for cognate complexes. More recently, X-ray crystallography of near-cognate bacterial complexes showed that near-cognate tRNAs can form G•U mismatches that adopt a geometry indistinguishable from canonical Watson–Crick pairs, interact with the A site equivalently and induce domain closure (12,13). Rozov et al. (14) proposed that closure of the small subunit generates a rigid geometrical mold that constrains some mismatched pairs, but not others (15), to adopt Watson–Crick geometry. A recent ensemble cryoEM study of recruitment of cognate and near-cognate ternary complexes binding to 70S ribosomes argues strongly for induced fit (16). Ternary complex recruitment was shown to involve three distinct pre-accommodation structures with the final structure of both cognate and near-cognate complexes resembling a previously characterized structure (A/T). In the A/T structure, a cognate ternary complex inserts aa-tRNA into the decoding center such that the paired codon–anticodon complex is fully engaged with three rRNA nucleotides of the decoding site: G530, A1492 and A1493 (17). The interaction of these nucleotides is now known to occur step-wise through intermediate states and is consistent with induced fit. The conformational flexibility of step-wise tRNA recruitment seen in the cryoEM study (16) contradicts a model in which a rigid decoding center forces the mismatched pair into Watson–Crick geometry (the mold model). The fact that the A site interacts equivalently with cognate and certain near-cognate codon–anticodon complexes suggests that some mismatches are indistinguishable from canonical Watson–Crick pairs (molecular mimicry model) (12–15).
Despite advances in understanding the steps leading to aa-tRNA selection in vitro, in vivo analysis of misreading error remains important to understand fully how ribosomes maintain translational accuracy. The higher and lower-frequency errors that we have observed appear to be fundamentally different with the higher frequency events depending on acceptance of tRNAs making a small subset of nucleotide mismatches (4). The nature of these mismatches confirms some predictions based on structural analysis. The most frequent errors predominately involve the same G•U mismatched base pairs shown to mimic cognate Watson–Crick pairs during A site binding (14). Other highly frequent errors require U•U or U•C mismatches, which may also mimic Watson–Crick pairs (14); the frequency of acceptance of aa-tRNAs forming these mismatches contradicts the prediction of Rozov et al. (14) that the lack of hydrogen bonding in U-U pairs would reduce their ability to induce errors. An experiment involving unbiased assays of nearly all possible errors using a mass spectrometry approach produced essentially the same conclusion (18). Two studies measuring misreading of nonsense codons also found similar mismatches at the first two codon positions but identified other mismatches associated with significant frequency of selection including A•C, G•A and A•A wobble mismatches in the third or wobble position (19,20); the structures of these mismatches in the A site are not available.
Our studies of misreading errors by 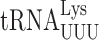 in E. coli (21) and S. cerevisiae (22) identified some differences in the phenomenology of these errors. Overall, the frequency of misreading errors in S. cerevisiae is less than in E. coli (22,23). In addition, errors involving mismatches at the third, or wobble position of the codon predominate in E. coli but were not detected in yeast suggesting that S. cerevisiae might differ from E. coli fundamentally in its ability to discriminate against this type of error (22). The absolute frequency of misreading errors depends on several variables. One source of variation is the effect of competition by cognate tRNAs for the mutant codons; higher misreading error frequencies result from lower competition by low-abundance cognate tRNAs (3). Post-transcriptional modifications can further modulate misreading errors by stabilizing or destabilizing reading by either the misreading tRNA or its competing cognate. The highest diversity of modifications is within the anticodon loop, particularly positions 34 and 37. These modifications increase the efficiency of cognate decoding (24,25) by increasing codon–anticodon stacking energy (26) and they have been proposed to ‘preorder’ the anticodons into a conformation appropriate for cognate recognition (27). By optimizing decoding rates these modifications are thought to help maintain proteome integrity by reducing co-translational protein misfolding caused by sporadic pausing during elongation (28). Modifications of wobble nucleotide U34 (xm5U, xm5s2U and xm5Um) are thought to restrict decoding to A and G ending codons (29,30). U34 modifications also have important roles in regulating translational errors (31). We have shown that in E. coli the mnm5 modification destabilizes misreading by
in E. coli (21) and S. cerevisiae (22) identified some differences in the phenomenology of these errors. Overall, the frequency of misreading errors in S. cerevisiae is less than in E. coli (22,23). In addition, errors involving mismatches at the third, or wobble position of the codon predominate in E. coli but were not detected in yeast suggesting that S. cerevisiae might differ from E. coli fundamentally in its ability to discriminate against this type of error (22). The absolute frequency of misreading errors depends on several variables. One source of variation is the effect of competition by cognate tRNAs for the mutant codons; higher misreading error frequencies result from lower competition by low-abundance cognate tRNAs (3). Post-transcriptional modifications can further modulate misreading errors by stabilizing or destabilizing reading by either the misreading tRNA or its competing cognate. The highest diversity of modifications is within the anticodon loop, particularly positions 34 and 37. These modifications increase the efficiency of cognate decoding (24,25) by increasing codon–anticodon stacking energy (26) and they have been proposed to ‘preorder’ the anticodons into a conformation appropriate for cognate recognition (27). By optimizing decoding rates these modifications are thought to help maintain proteome integrity by reducing co-translational protein misfolding caused by sporadic pausing during elongation (28). Modifications of wobble nucleotide U34 (xm5U, xm5s2U and xm5Um) are thought to restrict decoding to A and G ending codons (29,30). U34 modifications also have important roles in regulating translational errors (31). We have shown that in E. coli the mnm5 modification destabilizes misreading by 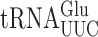 but actually stabilizes errors by
but actually stabilizes errors by 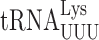 . In the former case, the modification appears to increase discrimination against near-cognate decoding by the tRNA but in the latter the modification appears to generally support decoding by stabilizing a functional conformation of the very weakly structured tRNA anticodon. Comparable divergent effects of wobble queuosine (Q) on
. In the former case, the modification appears to increase discrimination against near-cognate decoding by the tRNA but in the latter the modification appears to generally support decoding by stabilizing a functional conformation of the very weakly structured tRNA anticodon. Comparable divergent effects of wobble queuosine (Q) on 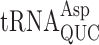 and
and 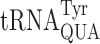 can be explained similarly. Modifications on base 37, adjacent to the anticodon, appear to have a different function of increasing codon–anticodon stacking energy (32). The t6A37 modification decreases frameshifting in yeast (33) while ms2i6A37 increases misreading errors in bacteria (31,34) and Schizosaccharomyces pombe (35).
can be explained similarly. Modifications on base 37, adjacent to the anticodon, appear to have a different function of increasing codon–anticodon stacking energy (32). The t6A37 modification decreases frameshifting in yeast (33) while ms2i6A37 increases misreading errors in bacteria (31,34) and Schizosaccharomyces pombe (35).
Here we validate a reporter-based system to measure misreading errors by t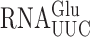 in yeast and use it, and a second reporter of errors by
in yeast and use it, and a second reporter of errors by 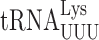 , to determine the effect of anticodon loop modifications of the two tRNAs on misreading frequencies. We demonstrate that wobble position misreading events do occur in yeast but with much reduced frequency compared to in bacteria. This difference is not caused by the eukaryotic-specific wobble modifications of these two tRNAs. Rather, the bacterial and yeast systems appear to differ in their intrinsic abilities to reduce these errors. We do find a difference in the effect of anticodon loop modifications in yeast compared to bacteria. In bacteria these modifications affect the decoding activity or stability of tRNA and the lack of modification had similar effects on all misread codons for each tRNA. In yeast, by contrast, the modifications regulate misreading in a codon-specific manner by altering the selection of the tRNA differently on various misread codons.
, to determine the effect of anticodon loop modifications of the two tRNAs on misreading frequencies. We demonstrate that wobble position misreading events do occur in yeast but with much reduced frequency compared to in bacteria. This difference is not caused by the eukaryotic-specific wobble modifications of these two tRNAs. Rather, the bacterial and yeast systems appear to differ in their intrinsic abilities to reduce these errors. We do find a difference in the effect of anticodon loop modifications in yeast compared to bacteria. In bacteria these modifications affect the decoding activity or stability of tRNA and the lack of modification had similar effects on all misread codons for each tRNA. In yeast, by contrast, the modifications regulate misreading in a codon-specific manner by altering the selection of the tRNA differently on various misread codons.
MATERIALS AND METHODS
Strains and growth conditions
The E. coli strain used in this study for cloning and plasmid propagation is in DH5α (F– Φ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17 phoA supE44 λ– thi-1 gyrA96 relA1) (36). All bacterial strains are cultured at 37°C in Luria-Bertani (LB) media (10 g NaCl, 10 g tryptone and 5 g yeast extract per liter) supplemented with ampicillin (100 μg/ml) or chloramphenicol (25 μg/ml) as required.
The S. cerevisiae used in this study is in the BY4742 background (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) (37). Yeast strains were grown either in Yeast peptone dextrose (YPD) media (Difco) or Synthetic Complete media lacking uracil (SC-Ura) (1.7 g Yeast Nitrogen Base w/o amino acids, 5 g ammonium sulphate supplemented with 2% glucose, amino acids and adenine but lacking uracil for selective purposes). Single mutants (elp3Δ, ncs6Δ, sua5Δ) were created by sporulating a corresponding heterozygous diploid strain in the BY4743 background (MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 LYS2/lys2Δ0 met15Δ0/MET15 ura3Δ0/ura3Δ0) followed by selection of a G418 resistant ascospore on YPD + G418 (200 μg/ml). The double mutants (elp3Δ ncs2Δ, elp3Δ ncs6Δ and elp3Δ sua5Δ) were generated by one-step polymerase chain reaction (PCR)-based gene replacement (38), using the NATMX marker for deletion and positive selection on YPD plates supplemented with nourseothricin (100 μg/ml) (39). Successful deletion was confirmed by PCR. Yeast transformation was carried out as described before (40). To create yeast strains with the hyper accurate and error prone ribosomes, we started with RPS23BΔ yeast strain in a BY4742 background and subsequently introduce a vector carrying RPS23A as either the wild-type or mutant copies (RPS23A-K62R and RPS23A A113V) (41) Translational errors were induced by addition of a sublethal concentration (200 μg/ml) of the antibiotic paromomycin (42).
Plasmids
The construction of the K529 dual luciferase reporter system used in this study, based on the plasmid pDB688 (42) (Supplementary Figure S1), has been described (22). To construct the E537 β-galactosidase reporter plasmids we introduced active site (E537) mutants of β-galactosidase into pANU7 (Supplementary Figure S2), a yeast-based vector (43) that provides bla (ampicillin resistance in bacteria) and URA3 (uracil auxotrophy in yeast) as selection markers. A BamHI–SacI fragment of the pJC27 vector (44) encompassing the mutant lacZ was ligated into the pANU7 vector between unique BamHI and SacI sites and identified by screening using X-gal containing plates to create plasmids carrying 14 codons near-cognate for the glutamic acid (Glu) codons GAA/GAG and 7 synonymous non-cognate codons. All plasmids were confirmed by sequencing (Genewiz).
Preparation of cell extracts and enzyme assays
β-galactosidase protein assays were performed on yeast strains transformed with reporter plasmids and grown in selective medium to an OD600 of 0.8−1.0. Transformant cells expressing wild-type β-galactosidase were diluted for assay 1000-fold compared to mutants and assayed to quantify β-galactosidase activity using the Promega β-Glo system according to manufacturer’s specification, using 96-well LUMITRAC plates (Greiner Bio One). Activities in Relative Light Units were measured using a in a Modulus II Microplate Multimode Reader (Turner BioSystems) according to manufacturer’s directions. Assays of our dual luciferase reporters were performed using the Dual-Luciferase Reporter Assay System (Promega) essentially as described (3) and quantified similarly. For both assays, three to six replicate biological samples were assayed each in three technical repeats. Statistical significance of results was determined using a two-tailed, homoscedastic Student’s t-test.
RESULTS
Misreading errors by 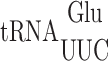 occur at the same codons but are less frequent than in bacteria, especially wobble position errors
occur at the same codons but are less frequent than in bacteria, especially wobble position errors
In previous work we have reported frequencies of misreading errors for several tRNAs in E. coli (3,4,31) but only for 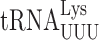 in S. cerevisiae (22). Comparison of the frequency of errors by
in S. cerevisiae (22). Comparison of the frequency of errors by 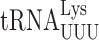 in E. coli and S. cerevisiae revealed two significant differences. Errors at individual codons in S. cerevisiae were 3- to 5-fold less frequent than in E. coli and wobble position errors (on the Asn codons AAU and AAC) appeared to be absent in S. cerevisiae whereas they were quite frequent in E. coli. The differences suggest that S. cerevisiae, and perhaps eukaryotes in general, might have evolved mechanisms to reduce misreading errors, especially with respect to wobble position errors. To test this conclusion, we exploited a set of misreading error reporters based on active site mutants altering glutamic acid 537 (E537) of E. coli β-galactosidase, encoded by the lacZ gene (4). Two isoaccepting tRNAs decode Glu codons,
in E. coli and S. cerevisiae revealed two significant differences. Errors at individual codons in S. cerevisiae were 3- to 5-fold less frequent than in E. coli and wobble position errors (on the Asn codons AAU and AAC) appeared to be absent in S. cerevisiae whereas they were quite frequent in E. coli. The differences suggest that S. cerevisiae, and perhaps eukaryotes in general, might have evolved mechanisms to reduce misreading errors, especially with respect to wobble position errors. To test this conclusion, we exploited a set of misreading error reporters based on active site mutants altering glutamic acid 537 (E537) of E. coli β-galactosidase, encoded by the lacZ gene (4). Two isoaccepting tRNAs decode Glu codons, 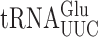 and
and 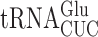 . The former is much more abundant (45) so errors in this reporter system probably nearly exclusively reflect errors by that tRNA. As discussed below, mutants that alter wobble U modification of
. The former is much more abundant (45) so errors in this reporter system probably nearly exclusively reflect errors by that tRNA. As discussed below, mutants that alter wobble U modification of 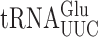 alter the frequency of all misreading events, which validates this conclusion. To quantify all possible misreading errors by
alter the frequency of all misreading events, which validates this conclusion. To quantify all possible misreading errors by 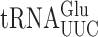 , we measured the activities of 14 near-cognate and 7 synonymous non-cognate mutants (Figure 1A). As in E. coli (4), a majority of the mutants produced very little activity, averaging 2 × 10−6 times wild-type; these include 10 of the near-cognate and all of the synonymous non-cognate mutants (Figure 1B). The remaining four mutants, the Gly codons GGA/GGG and the Asp codons GAU/GAC, produced 46- to 150-fold more activity. GGA and GGG misreading requires middle position G2•U35 mismatches (we refer throughout to base mismatches in codon–anticodon order with subscripts to indicate the positions of the nucleotides in the mRNA codon and tRNA) and GAU/GAC requires U3•U34 or C3•U34 third position or wobble error. Comparing the results to E. coli shows that these four mutants produced on average 3.5-fold fewer errors in S. cerevisiae. Previously we showed that errors by
, we measured the activities of 14 near-cognate and 7 synonymous non-cognate mutants (Figure 1A). As in E. coli (4), a majority of the mutants produced very little activity, averaging 2 × 10−6 times wild-type; these include 10 of the near-cognate and all of the synonymous non-cognate mutants (Figure 1B). The remaining four mutants, the Gly codons GGA/GGG and the Asp codons GAU/GAC, produced 46- to 150-fold more activity. GGA and GGG misreading requires middle position G2•U35 mismatches (we refer throughout to base mismatches in codon–anticodon order with subscripts to indicate the positions of the nucleotides in the mRNA codon and tRNA) and GAU/GAC requires U3•U34 or C3•U34 third position or wobble error. Comparing the results to E. coli shows that these four mutants produced on average 3.5-fold fewer errors in S. cerevisiae. Previously we showed that errors by 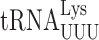 are also lower in S. cerevisiae than in E. coli (3).
are also lower in S. cerevisiae than in E. coli (3).
Figure 1.

Error frequencies in yeast can vary by 100-fold as measured by 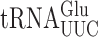 on various near-cognate codons. (A) Genetic code and identity of cytoplasmic tRNAs in Saccharomyces cerevisiae. The codons recognized by each tRNA are indicated by black circles connected by bars; the tRNAs are identified by anticodon and position 37 nt and the encoded amino acid. The Glu codons decoded by
on various near-cognate codons. (A) Genetic code and identity of cytoplasmic tRNAs in Saccharomyces cerevisiae. The codons recognized by each tRNA are indicated by black circles connected by bars; the tRNAs are identified by anticodon and position 37 nt and the encoded amino acid. The Glu codons decoded by 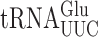 are in italics; near-cognate codons for
are in italics; near-cognate codons for 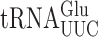 are highlighted in black and synonymous non-cognates in gray. (B) The activity of E537 mutants of β-galactosidase expressed with or without treatment with the antibiotic paromomycin. Statistical significance of the effect of paromomycin is shown (*, P-value < 0.05; **, P-value < 0.01; ***, P-value < 0.001). For mutants showing a significant change the codon–anticodon complexes predicted for corresponding misreading events are shown (the upper line represents the codon, the lower the anticodon). Vertical lines represent Watson–Crick pairs, filled circles canonical wobble pairs and open circles non-Watson–Crick pairs that have been shown to mimic Watson–Crick geometry.
are highlighted in black and synonymous non-cognates in gray. (B) The activity of E537 mutants of β-galactosidase expressed with or without treatment with the antibiotic paromomycin. Statistical significance of the effect of paromomycin is shown (*, P-value < 0.05; **, P-value < 0.01; ***, P-value < 0.001). For mutants showing a significant change the codon–anticodon complexes predicted for corresponding misreading events are shown (the upper line represents the codon, the lower the anticodon). Vertical lines represent Watson–Crick pairs, filled circles canonical wobble pairs and open circles non-Watson–Crick pairs that have been shown to mimic Watson–Crick geometry.
One of the indications that the activity expressed by a reporter gene results from misreading is that the activity is greater than that of synonymous mutants. This is clear for the GGA and GGG Gly mutants, which have distinctly different activities that are also far greater than that of the synonymous non-cognate mutants (GGU/GGC). The activities of the two wobble position mutants (GAU/GAC) were nearly identical and these codons lack synonymous non-cognates. Thus, the GAU/GAC activity could result not from misreading but from the substitution of the wild-type Glu by the mutant Asp, both acidic amino acids. To distinguish this type of functional replacement from misreading we tested the effect of error-modulating treatments on the activity of these two mutants. Sub-lethal concentrations of error-inducing aminoglycoside antibiotic paromomycin caused a significant increase in activity for all four high activity mutants (Figure 1B and Supplementary Table S1). The low activity Gln CAA/CAG mutants showed a small but significant increase, which reflects a C1•C36 first position mismatch error, but the frequency of these errors was about 100-fold lower than those due to G2•U35 or Y3•U34 mismatches. As a second test, we tested the effect of error-modulating mutants of ribosomal protein uS12, encoded by the S. cerevisiae RPS23A gene, that confer either hyperaccurate (rps23A-A113V) or error-prone (rps23A-K62R) phenotypes (46). The activity of the four high-activity E537 mutants was decreased by rps23A-A113V an average of 2.0-fold and increased by rps23A-K62R an average of 3.0-fold (Supplementary Table S2). We conclude that the activity of the four high activity mutants is due to misreading.
We previously failed to demonstrated errors by 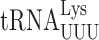 in S. cerevisiae involving wobble position mismatches on the Asn codons AAU and AAC and suggested that yeast might lack wobble position errors in general (22). Using the same RPS23A error-modulating mutations we found no decrease in the activity of the AAU and AAC mutants in the presence of RPS23A-A113V but a significant increase in the presence of rps23A-K62R (Supplementary Table S2). These data show that under error-inducing conditions
in S. cerevisiae involving wobble position mismatches on the Asn codons AAU and AAC and suggested that yeast might lack wobble position errors in general (22). Using the same RPS23A error-modulating mutations we found no decrease in the activity of the AAU and AAC mutants in the presence of RPS23A-A113V but a significant increase in the presence of rps23A-K62R (Supplementary Table S2). These data show that under error-inducing conditions 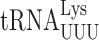 can misread by wobble misreading although the activity of the AAU/AAC mutants in non-error inducing conditions is that of the mutant protein and any activity due to misreading is obscured by this background.
can misread by wobble misreading although the activity of the AAU/AAC mutants in non-error inducing conditions is that of the mutant protein and any activity due to misreading is obscured by this background.
Wobble uridine modification of 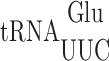 and
and 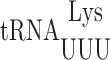 regulates misreading in a codon context-dependent manner
regulates misreading in a codon context-dependent manner
Both 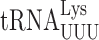 and
and 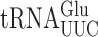 recognize A or G-ending codons by third position pairing with the modified nucleotide 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U34). This modification has been thought to block misreading by U3•U34 or C3•U34 mismatching (47). In S. cerevisiae the mcm5 modification is added to the unmodified wobble U34 by the Elongator complex (Elp1-Elp6) (48) and s2 by the Ncs6•Ncs2 complex (49). The hypermodified mcm5s2U nucleotide is present on three tRNAs that decode pairs of synonymous codons from the third column of the genetic code (Figure 1A), which includes all codons with A in the middle position. The mcm5 and s2 are introduced independently (25,48,50). In an elp3Δ strain the wobble nucleotide of
recognize A or G-ending codons by third position pairing with the modified nucleotide 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U34). This modification has been thought to block misreading by U3•U34 or C3•U34 mismatching (47). In S. cerevisiae the mcm5 modification is added to the unmodified wobble U34 by the Elongator complex (Elp1-Elp6) (48) and s2 by the Ncs6•Ncs2 complex (49). The hypermodified mcm5s2U nucleotide is present on three tRNAs that decode pairs of synonymous codons from the third column of the genetic code (Figure 1A), which includes all codons with A in the middle position. The mcm5 and s2 are introduced independently (25,48,50). In an elp3Δ strain the wobble nucleotide of 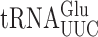 and
and 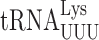 is s2U34 rather than the mcm5s2U34 found in the ELP3+ wild-type (48,51). In the elp3Δ strain there is little or no change in any other post-transcriptional modification of
is s2U34 rather than the mcm5s2U34 found in the ELP3+ wild-type (48,51). In the elp3Δ strain there is little or no change in any other post-transcriptional modification of 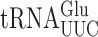 indicating that any elp3Δ phenotype must result from lack of mcm5 (48). In either an ncs2Δ or ncs6Δ strain, mcm5s2U34 in
indicating that any elp3Δ phenotype must result from lack of mcm5 (48). In either an ncs2Δ or ncs6Δ strain, mcm5s2U34 in 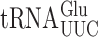 is replaced by mcm5U34 (52). A detailed analysis of the effect on tRNA modification of ncs2Δ or ncs6Δ has not been performed but lack of the s2 modification is known to have no significant effect on either aminoacylation or the concentration of the modified tRNAs (25,52,53). Eliminating both modification systems is lethal in a W303 genetic background but overexpressing
is replaced by mcm5U34 (52). A detailed analysis of the effect on tRNA modification of ncs2Δ or ncs6Δ has not been performed but lack of the s2 modification is known to have no significant effect on either aminoacylation or the concentration of the modified tRNAs (25,52,53). Eliminating both modification systems is lethal in a W303 genetic background but overexpressing 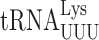 suppresses the lethality, suggesting that the lack of the modification reduces the efficiency of codon recognition (25). In the S288c genetic background lack of both modifications is not lethal (28,51).
suppresses the lethality, suggesting that the lack of the modification reduces the efficiency of codon recognition (25). In the S288c genetic background lack of both modifications is not lethal (28,51).
The effect on misreading of the mcm5 and s2 moieties of mcm5s2U34 can be determined by comparing the activities of the misreading reporters in a strain with U34 (elp3Δ ncs6Δ) to those with mcm5U34 (ncs6Δ) or s2U34 (elp3Δ) or the effect of both by comparing with the activities in the wild-type parental strain. We determined the effect of these modifications by quantifying errors by 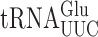 using the four error-prone mutants of E537 of β-galactosidase, and those by
using the four error-prone mutants of E537 of β-galactosidase, and those by 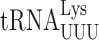 using mutants of K529 of firefly luciferase. The ratio of enzyme activity in these strains varied widely according to the codon being misread. For reporters of first and second position misreading, the presence of mcm5 modification significantly increased errors at UAA, UAG and GGA (an average of 1.8-fold), decreased those at GGG (1.4-fold) and had no significant effect on errors at AGG strain (Table 1). The presence of the s2 modification significantly increased errors at all codons except GGG (an average of 2-fold) but the increases were significantly greater for the A-ending than the G-ending codons (UAA and GAA versus UAG and AGG.) The presence of both modifications increased errors at UAA (7.7-fold), UAG (1.6-fold) and GGA (42-fold) and decreased errors at AGG (1.2-fold) and GGG (1.5-fold). A combination the two modifications showed strong positive synergism for errors at UAA and GGA suggesting that the two modifications cooperatively increase the frequency of misreading errors at these two codons. For AGG and GGG, the combination showed weak negative synergism and for UAG no synergism. In general, the greatest individual or combined effects of these modifications were on A-ending codons, UAA and GGA, and the effects on G-ending codons were either significantly less or actually negative.
using mutants of K529 of firefly luciferase. The ratio of enzyme activity in these strains varied widely according to the codon being misread. For reporters of first and second position misreading, the presence of mcm5 modification significantly increased errors at UAA, UAG and GGA (an average of 1.8-fold), decreased those at GGG (1.4-fold) and had no significant effect on errors at AGG strain (Table 1). The presence of the s2 modification significantly increased errors at all codons except GGG (an average of 2-fold) but the increases were significantly greater for the A-ending than the G-ending codons (UAA and GAA versus UAG and AGG.) The presence of both modifications increased errors at UAA (7.7-fold), UAG (1.6-fold) and GGA (42-fold) and decreased errors at AGG (1.2-fold) and GGG (1.5-fold). A combination the two modifications showed strong positive synergism for errors at UAA and GGA suggesting that the two modifications cooperatively increase the frequency of misreading errors at these two codons. For AGG and GGG, the combination showed weak negative synergism and for UAG no synergism. In general, the greatest individual or combined effects of these modifications were on A-ending codons, UAA and GGA, and the effects on G-ending codons were either significantly less or actually negative.
Table 1.
Effect on misreading errors of addition to U34 of mcm5, s2 or mcm5s2 modifications
| elp3Δncs6Δ | ncs6Δ | elp3Δ | Wild-type | |||
|---|---|---|---|---|---|---|
| U34 | mcm5U34 | s2U34 | mcm5s2U34 | |||
| Misreading tRNA | Codon misread | Mismatch | Activity relative to wild-type reporter (× 10−4) | |||
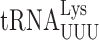
|
UAA | U1•U36 | 0.22 ± 0.01 | 0.45 ± 0.02*** (2.0×) | 0.52 ± 0.09** (2.4×) | 1.7 ± 0.17*** (7.7×) |
| UAG | 3.0 ± 0.2 | 5.3 ± 0.40*** (1.8×) | 4.4 ± 0.18*** (1.5×) | 4.8 ± 0.35** (1.6×) | ||
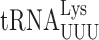
|
AGG | G2•U35 | 9.1 ± 0.2 | 9.2 ± 0.11 (1.0×) | 12 ± 0.29*** (1.3×) | 7.9 ± 0.22** (0.86×) |
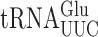
|
GGA | G2•U35 | 0.05 ± 0.003 | 0.08 ± 0.008* (1.6×) | 0.14 ± 0.002*** (2.8×) | 2.1 ± 0.1*** (42×) |
| GGG | 0.35 ± 0.03 | 0.26 ± 0.02* (0.74×) | 0.34 ± 0.01 (0.97×) | 0.23 ± 0.02*** (0.66×) | ||
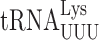
|
AAU | Y3•U34 | 0.85 ± 0.05 | 1.3 ± 0.11** (1.5×) | 1.9 ± 0.16*** (2.2×) | 1.3 ± 0.08** (1.5×) |
| AAC | 0.75 ± 0.05 | 1.3 ± 0.11** (1.7×) | 1.3 ± 0.11*** (1.7×) | 1.3 ± 0.10*** (1.7×) | ||
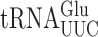
|
GAU | Y3•U34 | 1.8 ± 0.27 | 0.87 ± 0.07** (0.48×) | 1.9 ± 0.13 (1.1×) | 0.95 ± 0.06*** (0.53×) |
| GAC | 0.65 ± 0.06 | 0.80 ± 0.06 (1.2×) | 0.76 ± 0.08 (1.2×) | 0.94 ± 0.06* (1.4×) | ||
Standard errors: *, P-value < 0.05; **, P-value < 0.01; ***, P-value < 0.001.
The effect of U modification on near-cognate decoding is generally similar to their effect on cognate decoding. Introducing s2 at U34 increases the affinity of cognate binding to A but not G-ending codons both in vitro and in vivo (54,55) and recent kinetic analysis shows that s2 slows dissociation of 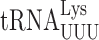 from its cognate codon AAA during both initial selection and proofreading and accelerates acceptance further in two other ways (56). The mcm5 modification also promotes decoding of both A and G-ending codons (57) though the preference for A-ending codons is weaker than for s2 (58); details of the kinetic basis of this effect are not available for mcm5. The synergism we observed is consistent with in vitro data suggesting that the maximum effect of mcm5 on cognate decoding requires s2 (26). The negative synergism on two G-ending codons suggests that at least for near-cognate decoding, the combination of the two modifications interact to limit misreading; the mechanism of this synergism is unclear.
from its cognate codon AAA during both initial selection and proofreading and accelerates acceptance further in two other ways (56). The mcm5 modification also promotes decoding of both A and G-ending codons (57) though the preference for A-ending codons is weaker than for s2 (58); details of the kinetic basis of this effect are not available for mcm5. The synergism we observed is consistent with in vitro data suggesting that the maximum effect of mcm5 on cognate decoding requires s2 (26). The negative synergism on two G-ending codons suggests that at least for near-cognate decoding, the combination of the two modifications interact to limit misreading; the mechanism of this synergism is unclear.
Misreading errors involving wobble position mismatches (U3•U34 or C3•U34) largely were increased by mcm5 and s2 modifications (Table 1). The presence of either or both modification increased all wobble misreading errors by 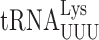 but with no synergism. The effect on wobble errors by
but with no synergism. The effect on wobble errors by 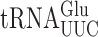 was less consistent. The presence of either mcm5 or s2 had no significant effect on errors involving U3•U34 or C3•U34 matches with the exception of errors at GAU, which were significantly decreased by mcm5 modification. These data are generally inconsistent with the proposal that these wobble U modifications restrict wobble mismatch errors although the negative effect of mcm5 on some errors by
was less consistent. The presence of either mcm5 or s2 had no significant effect on errors involving U3•U34 or C3•U34 matches with the exception of errors at GAU, which were significantly decreased by mcm5 modification. These data are generally inconsistent with the proposal that these wobble U modifications restrict wobble mismatch errors although the negative effect of mcm5 on some errors by 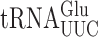 suggests that they can have that effect depending on the codon sequence context.
suggests that they can have that effect depending on the codon sequence context.
It had been thought that xm5 modifications block misreading of pyrimidine-ending codons by restricting nucleotide conformation (29) but structural results challenged that proposal for mnm5U in bacteria (27). In vivo analysis in bacteria, however, shows that mnm5 modification does limit recognition of pyrimidine ending codons by 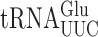 and
and 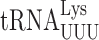 (31,59). Based on these results, we suspected that the extremely low level of wobble errors in S. cerevisiae might result from mcm5 modification more severely limiting pyrimidine•pyrimidine mismatches. Our data show the opposite, that these errors are extremely low for tRNAs with unmodified wobble U and that the presence of either modification generally increases them. The direct comparison of errors involving a s2U wobble nucleotide pairing with pyrimidines shows that they are much more frequent in E. coli than in S. cerevisiae, which suggests that some other aspect of translation in yeast must limit these errors.
(31,59). Based on these results, we suspected that the extremely low level of wobble errors in S. cerevisiae might result from mcm5 modification more severely limiting pyrimidine•pyrimidine mismatches. Our data show the opposite, that these errors are extremely low for tRNAs with unmodified wobble U and that the presence of either modification generally increases them. The direct comparison of errors involving a s2U wobble nucleotide pairing with pyrimidines shows that they are much more frequent in E. coli than in S. cerevisiae, which suggests that some other aspect of translation in yeast must limit these errors.
N6 -Threonylcarbamoyladenosine modification at position 37 regulates misreading errors in 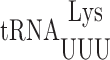
We previously demonstrated that nucleotide 37 modifications can modulate translation accuracy in E. coli in the case of 2-methylthio-N6-isopentenyladenosine (ms2i6A37) in 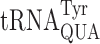 (31). We extended this analysis of the role of modifications in this position in yeast but of the two yeast tRNAs studied here only
(31). We extended this analysis of the role of modifications in this position in yeast but of the two yeast tRNAs studied here only 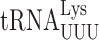 has a modified nucleotide 37, N6 -threonylcarbamoyladenosine (t6A) (34,60). The t6A modification is present in all tRNAs that decode codons with a first position A, which corresponds to the codons of the third row of the standard genetic code (Figure 1A). The purpose of this modification appears to be to compensate for the weakness of the A1•U36 pair formed when these tRNAs read their cognate codon (61) by the t6A37 in the anticodon stacking on the first base of the codon (27). The enzyme responsible for modifying tRNAs with t6A in E. coli is essential although the essentiality of the modification itself has not been demonstrated (62). In yeast, however, the modification is not essential, which allows us to test genetically the modification's role in modulating misreading errors in yeast.
has a modified nucleotide 37, N6 -threonylcarbamoyladenosine (t6A) (34,60). The t6A modification is present in all tRNAs that decode codons with a first position A, which corresponds to the codons of the third row of the standard genetic code (Figure 1A). The purpose of this modification appears to be to compensate for the weakness of the A1•U36 pair formed when these tRNAs read their cognate codon (61) by the t6A37 in the anticodon stacking on the first base of the codon (27). The enzyme responsible for modifying tRNAs with t6A in E. coli is essential although the essentiality of the modification itself has not been demonstrated (62). In yeast, however, the modification is not essential, which allows us to test genetically the modification's role in modulating misreading errors in yeast.
Biosynthesis of t6A is a complex process involving the Sua5 protein and the KEOPS complex (Kae1, Bud32, Gon7, Pcc1 and Cgi121) (34,62). Sua5 is responsible for synthesizing the intermediate threonyl-carbamoyl-AMP (TC-AMP) and the KEOPS complex transfers the threonyl-carbamoyl moiety to tRNAs. To study the effect of t6A modification at position 37 on misreading by 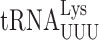 we introduced the K529 reporter plasmids into a sua5Δ strain; this analysis was repeated with mutants lacking the Bud32 and Kae1 subunits of the KEOPS complex with similar results (Supplementary Table S3). We compared activities of our firefly luciferase misreading reporters in strains lacking Sua5 (sua5Δ and elp3Δ sua5Δ) and those in which it is present (the wild-type parent and elp3Δ). In each case, the presence of t6A reduced the activity of UAA (4.5-fold) and UAG (9-fold) termination codon mutants (Table 2). Misreading these codons requires a U1•U36 first position mismatch. By contrast, the presence of t6A significantly increased misreading of the other three error-prone codons, AGG (G2•U35 mismatch), AAU and AAC (U3•U34 and C3•U34 mismatches). All of these effects were similar in the presence or absence of mcm5 modification, suggesting that the effect of t6A is independent of the effect of wobble modifications. In the case of the UAA codon, the frequency of misreading is much greater in the presence of mcm5 than in its absence, consistent with the stabilizing effect of mcm5 on A3•U34 pairing. In the absence of t6A, misreading of UAG is increased only 1.4-fold by addition of mcm5; we suspect that in the absence of t6A about 72% of UAG misreading is by
we introduced the K529 reporter plasmids into a sua5Δ strain; this analysis was repeated with mutants lacking the Bud32 and Kae1 subunits of the KEOPS complex with similar results (Supplementary Table S3). We compared activities of our firefly luciferase misreading reporters in strains lacking Sua5 (sua5Δ and elp3Δ sua5Δ) and those in which it is present (the wild-type parent and elp3Δ). In each case, the presence of t6A reduced the activity of UAA (4.5-fold) and UAG (9-fold) termination codon mutants (Table 2). Misreading these codons requires a U1•U36 first position mismatch. By contrast, the presence of t6A significantly increased misreading of the other three error-prone codons, AGG (G2•U35 mismatch), AAU and AAC (U3•U34 and C3•U34 mismatches). All of these effects were similar in the presence or absence of mcm5 modification, suggesting that the effect of t6A is independent of the effect of wobble modifications. In the case of the UAA codon, the frequency of misreading is much greater in the presence of mcm5 than in its absence, consistent with the stabilizing effect of mcm5 on A3•U34 pairing. In the absence of t6A, misreading of UAG is increased only 1.4-fold by addition of mcm5; we suspect that in the absence of t6A about 72% of UAG misreading is by 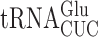 , which forms a G3•C34 pair in the wobble position and therefore is insensitive to the lack of mcm5. The opposite effects of the presence of t6A modification on misreading shows that it affects near-cognate decoding differently based on the position of the mismatch. Its stabilization of second and wobble position errors may result from increased stacking energy stabilizing the conformation of the anticodon loop to promote decoding, as with cognate decoding (44). A conservative model for destabilizing a U3•U34 mismatch would be that t6A stacking on U3 alters the geometry of the pair required to increase acceptance. These opposite effects of the presence of t6A modification on misreading of sense and nonsense mutations mirror its opposite effects indicated by the two phenotypes associated with mutants of SUA5 in reducing the efficiency of initiation codon selection (63) but increasing the efficiency of nonsense codon readthrough (33).
, which forms a G3•C34 pair in the wobble position and therefore is insensitive to the lack of mcm5. The opposite effects of the presence of t6A modification on misreading shows that it affects near-cognate decoding differently based on the position of the mismatch. Its stabilization of second and wobble position errors may result from increased stacking energy stabilizing the conformation of the anticodon loop to promote decoding, as with cognate decoding (44). A conservative model for destabilizing a U3•U34 mismatch would be that t6A stacking on U3 alters the geometry of the pair required to increase acceptance. These opposite effects of the presence of t6A modification on misreading of sense and nonsense mutations mirror its opposite effects indicated by the two phenotypes associated with mutants of SUA5 in reducing the efficiency of initiation codon selection (63) but increasing the efficiency of nonsense codon readthrough (33).
Table 2.
Effect on misreading of addition of the t6A37 modification
| sua5Δ | Wild-type | elp3Δsua5Δ | elp3Δ | |||
|---|---|---|---|---|---|---|
| mcm5s2U34 A37 | mcm5s2U34 t6A37 | s2U34 A37 | s2U34 t6A37 | |||
| Activity relative to wild-type reporter (× 10−4) | ||||||
| Misreading tRNA | Codon misread | Mismatch | (Change from sua5Δ) | (Change from elp3Δ sua5Δ) | ||
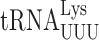
|
UAA | U1•U36 | 7.8 ± 0.37 | 1.7 ± 0.17*** (0.22×) | 0.92 ± 0.02 | 0.52 ± 0.09 (0.57×) |
| UAG | 43 ± 1.6 | 4.8 ± 0.35*** (0.11×) | 31 ± 1.3 | 4.4 ± 0.18*** (0.14×) | ||
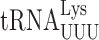
|
AGG | G2•U35 | 5.8 ± 0.2 | 7.9 ± 0.22*** (1.4×) | 6.6 ± 0.03 | 12 ± 0.29*** (1.8×) |
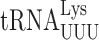
|
AAU | Y3•U34 | 0.75 ± 0.03 | 1.3 ± 0.11*** (1.7×) | 0.82 ± 0.02 | 1.9 ± 0.16** (2.3×) |
| AAC | 0.72 ± 0.04 | 1.3 ± 0.10*** (1.8×) | 0.64 ± 0.02 | 1.3 ± 0.11** (2.0×) | ||
DISCUSSION
Protein synthesis is a kinetically regulated process with tRNA selection in the ribosomal decoding site consisting of many discrete steps. Several of these steps distinguish kinetically between correct (cognate) and incorrect (near or non-cognate) tRNAs with the discrimination resulting from structural dynamics of the ribosome and induced fit (64). Recent X-ray crystallographic results suggest that some near-cognate tRNAs can induce ribosomal structural rearrangements identical to those during cognate tRNA binding including rearrangement of the decoding site to allow non-sequence specific contacts between the codon–anticodon complex and elements of the A site (65). An important question is whether these interactions occur during initial selection since the solved crystal structures are of complexes post initial selection (64). The result of our in vivo misreading analysis affords an important commentary on this question because it demonstrates that acceptance of near-cognate tRNAs is largely restricted to those that involve specific nucleotide mismatches including G•U, U•U or C•U. The G•U and U•U mismatches interact with the A site nucleotides G530, A1492 and A1493 equivalently with Watson–Crick pairs; C•U has not been investigated (65). A comparison of our results reported here with previous studies of misreading errors in E. coli (3,4,31) and S. cerevisiae (22) demonstrate that these errors predominate and that other near-cognate errors are either much less frequent or undetectable by our system, with errors no higher than 2 × 10−6 per codon. Clearly, then, there is congruence between those mismatches that can induce cognate-like A site interactions and those that result in substantial misreading errors. It is very attractive to conclude that their ability to interact with the A site similarly to a cognate tRNA explains their propensity to misread and, correspondingly, the infrequency or lack of errors involving other mismatches predicts their inability to interact as stably. Rozov et al. (14,65) show that the distance between the paired U•U nucleotides is too great to allow hydrogen bonding and suggested that tRNAs with this mismatch should dissociate more readily than those with a G•U Watson–Crick mimic mismatch. Our results show that errors using this mismatch are often as frequent or more frequent than G•U mismatch errors, suggesting that lack of U•U hydrogen bonding per se does not disqualify near-cognates from inducing errors.
Recently, Blanchet et al. (19) and Roy et al. (20) using nonsense codon readthrough assays demonstrated misreading involving the same G1•U36, U1•U36 mismatches, but also A3•G36, G3•G36 and C3•A36 mismatches. We have found that errors dependent on purine–purine wobble mismatches were extremely infrequent but could be increased to high levels in error-prone conditions (4). Misreading requiring C3•A36 mismatches between 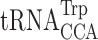 and UGA has long been known (66) but we lack any in vivo reporter for that misreading error. Significantly, Blanchet et al. (19) identified these errors using error-inducing conditions involving a PSI+ background deficient in eukaryotic release factor 3 (eRF3); prolonged pausing at the nonsense codon could drastically increase the opportunity for misreading. Roy et al. (20) showed that although errors were elevated in the PSI+ background, the distribution of misreading errors was relatively unchanged in normal PSI– cells. It is significant that these errors are confined to the wobble position where base pair geometry is less constrained. The proposed Watson–Crick mimicry model is limited to the more strictly monitored first and second positions. Wobble position errors of this type, however, could also be explained by the purine–purine pairs adopting Hoogsteen pairing and the C•A pairing through tautomerism (65). The fact that we fail to find these errors at other codon positions suggests that selection of tRNAs making these errors can occur only at the less monitored wobble position presumably because of their steric clashes in the other positions.
and UGA has long been known (66) but we lack any in vivo reporter for that misreading error. Significantly, Blanchet et al. (19) identified these errors using error-inducing conditions involving a PSI+ background deficient in eukaryotic release factor 3 (eRF3); prolonged pausing at the nonsense codon could drastically increase the opportunity for misreading. Roy et al. (20) showed that although errors were elevated in the PSI+ background, the distribution of misreading errors was relatively unchanged in normal PSI– cells. It is significant that these errors are confined to the wobble position where base pair geometry is less constrained. The proposed Watson–Crick mimicry model is limited to the more strictly monitored first and second positions. Wobble position errors of this type, however, could also be explained by the purine–purine pairs adopting Hoogsteen pairing and the C•A pairing through tautomerism (65). The fact that we fail to find these errors at other codon positions suggests that selection of tRNAs making these errors can occur only at the less monitored wobble position presumably because of their steric clashes in the other positions.
The details of the structure of individual tRNAs resulting from post-transcriptional modification is known to modulate translational error frequency (reviewed in 67). Here, we show that modifications modulate misreading in distinct ways in S. cerevisiae and E. coli. Our study in E. coli showed that the presence of anticodon loop modifications either increased or decreased errors by each targeted tRNA largely independent of the codon being misread (31). By contrast, in S. cerevisiae the effect of the anticodon loop modifications differed for a particular tRNA depending on the codon being read. The presence of a particular modification on misreading was frequently opposite on various of its near-cognate codons. The presence of the mcm5U34 modification increased misreading frequency on most codons, but it had no significant effect or actually decreased misreading on several codons. The s2 modification similarly increased misreading on most codons but had little or no effect on several others. Generally, misreading was increased for first and second position mismatches on A-ending codons, especially in the case of the s2 modification, but in several cases the modification actually reduced errors, especially for G-ending codons. The effect of the two modifications, mcm5 and s2, was greatest and highly synergistic for two A-ending codons—for 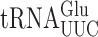 misreading GGA and for
misreading GGA and for 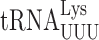 misreading UAA. The strong synergism suggests that the modifications alter near-cognate codon recognition in distinct ways. The effect on third or wobble position mismatches was more similar for A and G-ending codons and showed no evidence of synergism, which suggests that in this case, where the modified base is mismatched, the two modifications do not play distinct roles in supporting decoding.
misreading UAA. The strong synergism suggests that the modifications alter near-cognate codon recognition in distinct ways. The effect on third or wobble position mismatches was more similar for A and G-ending codons and showed no evidence of synergism, which suggests that in this case, where the modified base is mismatched, the two modifications do not play distinct roles in supporting decoding.
A full understanding of how modifications modulate errors will require structural analysis of modified near-cognate tRNAs engaged at the A site. Previous work has demonstrated unexpected cognate A site interactions. The mnm5s2U34 base in bacterial 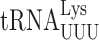 stacks on the adjacent U35 and the amino group of the mnm5 group apparently hydrogen bonds with the 2′OH of U33. These interactions stabilize the cognate codon–anticodon helix and influence its conformation (14). Similarly, t6A37 of
stacks on the adjacent U35 and the amino group of the mnm5 group apparently hydrogen bonds with the 2′OH of U33. These interactions stabilize the cognate codon–anticodon helix and influence its conformation (14). Similarly, t6A37 of 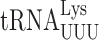 forms a cross-strand stack with codon nucleotide A1 to stabilize the weak A1•U36 base pair (14). The question is whether during near-cognate decoding they might have different or even opposite effects derived from their interacting differently with a mismatched codon–anticodon complex. We know, for example, that mnm5 destabilizes all error-prone near-cognate decoding in E. coli (31), which suggests that it has a different role than increasing stacking energy for these complexes. The stabilization effect of t6A37 on the weak A1•U36 base pair becomes a destabilizing effect on U1•U36 mispairing when
forms a cross-strand stack with codon nucleotide A1 to stabilize the weak A1•U36 base pair (14). The question is whether during near-cognate decoding they might have different or even opposite effects derived from their interacting differently with a mismatched codon–anticodon complex. We know, for example, that mnm5 destabilizes all error-prone near-cognate decoding in E. coli (31), which suggests that it has a different role than increasing stacking energy for these complexes. The stabilization effect of t6A37 on the weak A1•U36 base pair becomes a destabilizing effect on U1•U36 mispairing when 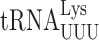 decodes UAA or UAG. Clearly the same stabilizing stacking interaction by t6A37 on A1•U36 is missing for U1•U36, perhaps replaced by an interaction that displaces U1 from pairing with U36. Rozov et al. (15) showed that when
decodes UAA or UAG. Clearly the same stabilizing stacking interaction by t6A37 on A1•U36 is missing for U1•U36, perhaps replaced by an interaction that displaces U1 from pairing with U36. Rozov et al. (15) showed that when 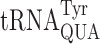 misreads the His codon CAC the hypermodified base queuosine (Q34) stabilizes a conformation of codon nucleotide of C1 away from pairing with anticodon nucleotide A36, blocking formation of the C•A mismatch. An unusual interaction like this may explain effects of modifications like t6A on near-cognate decoding that are opposite to their effects on cognates.
misreads the His codon CAC the hypermodified base queuosine (Q34) stabilizes a conformation of codon nucleotide of C1 away from pairing with anticodon nucleotide A36, blocking formation of the C•A mismatch. An unusual interaction like this may explain effects of modifications like t6A on near-cognate decoding that are opposite to their effects on cognates.
Recent cryo-electron microscopy results provide a detailed view of the process of tRNA assembly involving stepwise assembly of the final A/T complex in a bacterial ribosome (16). These data identify three steps for recruitment of a cognate or near-cognate EF-Tu ternary complex to the ribosomal A site. In these three steps the tRNA increasingly approaches complete pairing with the mRNA codon. With the third step the codon–anticodon complex fully engages with the ribosomal A site and the ribosome shifts to the ‘closed’ conformation. Importantly, for the near-cognate tRNA the G2•U35 pair only adopts Watson–Crick geometry in this third complex; in the second step the pair is in a non-Watson–Crick conformation. Also, from the first to the third step the elements that recognize a cognate codon–anticodon complex of increasingly move into position to interact. This result is inconsistent with the ‘mold’ hypothesis of Rozov et al., which proposed that the A site adopts a rigid structure that forces the codon–anticodon complex into Watson–Crick geometry (14). The adjustment of the positions of the interacting nucleotides between the first and third step is consistent with the induced fit model (64). However, the concept proposed by Demeshkina et al. (12) that acceptance of the near-cognate tRNA does involve Watson–Crick mimicry by the mismatched base pair was confirmed by the new data.
The issue of the reduced occurrence of wobble misreading errors by 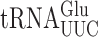 and
and 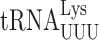 in S. cerevisiae relative to E. coli might be explained by differences in the process of tRNA recruitment. The three-step process in bacteria involves a stage before formation of the final cognate A/T complex in which the wobble bases are paired and stabilized by rRNA base C1054 stacking on the anticodon wobble nucleotide and partially stabilized by hydrogen bonding to G530 in a ‘semi-on’ conformation; in the near-cognate complex C1054 stacking is disrupted and G530 is in an ‘off’ conformation (16). The wobble bases pair in the near-cognate case only in the third structure in which the A/T codon–anticodon complex interacts fully with the A site. The weak wobble interaction with the A site in structure 2 no doubt contributes to the instability of near-cognate complexes implied in the relative rarity of structure 3 for the non-cognate complex, which implies a higher degree of EF-Tu ternary complex dissociation consistent with kinetic data. The fact that structure 2 does not monitor the wobble pair for the near-cognate case may explain why complexes with G2•U35 and Y3•U34 mismatches are accepted at approximately equal frequencies despite the presumably much lower stability of the latter pair. Formation of the wobble pair only in concert with latching of the A site may reduce the destabilizing effect of Y3•U34 versus R3•U34, leading to more frequent wobble misreading in bacteria. The much lower frequency of these errors in S. cerevisiae may result from eukaryotic ribosomes transitioning through an unlatched open complex in which wobble pairing is required and the presence of Y3•U34 may destabilize tRNAs with that mismatch. Cryoelectron microscopy of similar pre-A/T structures for yeast ribosomes could resolve this issue.
in S. cerevisiae relative to E. coli might be explained by differences in the process of tRNA recruitment. The three-step process in bacteria involves a stage before formation of the final cognate A/T complex in which the wobble bases are paired and stabilized by rRNA base C1054 stacking on the anticodon wobble nucleotide and partially stabilized by hydrogen bonding to G530 in a ‘semi-on’ conformation; in the near-cognate complex C1054 stacking is disrupted and G530 is in an ‘off’ conformation (16). The wobble bases pair in the near-cognate case only in the third structure in which the A/T codon–anticodon complex interacts fully with the A site. The weak wobble interaction with the A site in structure 2 no doubt contributes to the instability of near-cognate complexes implied in the relative rarity of structure 3 for the non-cognate complex, which implies a higher degree of EF-Tu ternary complex dissociation consistent with kinetic data. The fact that structure 2 does not monitor the wobble pair for the near-cognate case may explain why complexes with G2•U35 and Y3•U34 mismatches are accepted at approximately equal frequencies despite the presumably much lower stability of the latter pair. Formation of the wobble pair only in concert with latching of the A site may reduce the destabilizing effect of Y3•U34 versus R3•U34, leading to more frequent wobble misreading in bacteria. The much lower frequency of these errors in S. cerevisiae may result from eukaryotic ribosomes transitioning through an unlatched open complex in which wobble pairing is required and the presence of Y3•U34 may destabilize tRNAs with that mismatch. Cryoelectron microscopy of similar pre-A/T structures for yeast ribosomes could resolve this issue.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Eric Westhof for insightful discussions of the relationship between translational error and base mismatching.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM 29480, in part]; National Science Foundation [1645795]. Funding for open access charge: National Science Foundation [1645795].
Conflict of interest statement. None declared.
REFERENCES
- 1. Wohlgemuth I., Pohl C., Mittelstaet J., Konevega A.L., Rodnina M.V.. Evolutionary optimization of speed and accuracy of decoding on the ribosome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011; 366:2979–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ogle J.M., Carter A.P., Ramakrishnan V.. Insights into the decoding mechanism from recent ribosome structures. Trends Biochem. Sci. 2003; 28:259–266. [DOI] [PubMed] [Google Scholar]
- 3. Kramer E.B., Farabaugh P.J.. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA. 2007; 13:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Manickam N., Nag N., Abbasi A., Patel K., Farabaugh P.J.. Studies of translational misreading in vivo show that the ribosome very efficiently discriminates against most potential errors. RNA. 2014; 20:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parker J. Errors and alternatives in reading the universal genetic code. Microbiol. Rev. 1989; 53:273–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pape T., Wintermeyer W., Rodnina M.. Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. EMBO J. 1999; 18:3800–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gromadski K.B., Rodnina M.V.. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol. Cell. 2004; 13:191–200. [DOI] [PubMed] [Google Scholar]
- 8. Rodnina M.V., Wintermeyer W.. Fidelity of aminoacyl-tRNA selection on the ribosome: kinetic and structural mechanisms. Annu. Rev. Biochem. 2001; 70:415–435. [DOI] [PubMed] [Google Scholar]
- 9. Ogle J.M., Brodersen D.E., Clemons W.M. Jr, Tarry M.J., Carter A.P., Ramakrishnan V.. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001; 292:897–902. [DOI] [PubMed] [Google Scholar]
- 10. Ogle J.M., Ramakrishnan V.. Structural insights into translational fidelity. Annu. Rev. Biochem. 2005; 74:129–177. [DOI] [PubMed] [Google Scholar]
- 11. Ogle J.M., Murphy F.V., Tarry M.J., Ramakrishnan V.. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002; 111:721–732. [DOI] [PubMed] [Google Scholar]
- 12. Demeshkina N., Jenner L., Westhof E., Yusupov M., Yusupova G.. New structural insights into the decoding mechanism: translation infidelity via a G·U pair with Watson-Crick geometry. FEBS Lett. 2013; 587:1848–1857. [DOI] [PubMed] [Google Scholar]
- 13. Demeshkina N., Jenner L., Westhof E., Yusupov M., Yusupova G.. A new understanding of the decoding principle on the ribosome. Nature. 2012; 484:256–259. [DOI] [PubMed] [Google Scholar]
- 14. Rozov A., Demeshkina N., Khusainov I., Westhof E., Yusupov M., Yusupova G.. Novel base-pairing interactions at the tRNA wobble position crucial for accurate reading of the genetic code. Nat. Commun. 2016; 7:10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rozov A., Demeshkina N., Westhof E., Yusupov M., Yusupova G.. Structural insights into the translational infidelity mechanism. Nat. Commun. 2015; 6:7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Loveland A.B., Demo G., Grigorieff N., Korostelev A.A.. Ensemble cryo-EM elucidates the mechanism of translation fidelity. Nature. 2017; 546:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schmeing T.M., Voorhees R.M., Kelley A.C., Gao Y.G., Murphy F.V.T., Weir J.R., Ramakrishnan V.. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009; 326:688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Z., Shah B., Bondarenko P.V.. G/U and certain wobble position mismatches as possible main causes of amino acid misincorporations. Biochemistry. 2013; 52:8165–8176. [DOI] [PubMed] [Google Scholar]
- 19. Blanchet S., Cornu D., Argentini M., Namy O.. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res. 2014; 42:10061–10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roy B., Leszyk J.D., Mangus D.A., Jacobson A.. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:3038–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kramer E., Farabaugh P.. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA. 2007; 13:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kramer E.B., Vallabhaneni H., Mayer L.M., Farabaugh P.J.. A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA. 2010; 16:1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kramer E.B., Farabaugh P.J.. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA. 2007; 13:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Björk G., Hagervall T.. Transfer RNA Modification: Presence, synthesis, and function. EcoSal Plus 2014. 2014; 6:doi:10.1128/ecosalplus.ESP-0007-2013. [DOI] [PubMed] [Google Scholar]
- 25. Bjork G.R., Huang B., Persson O.P., Bystrom A.S.. A conserved modified wobble nucleoside (mcm5s2U) in lysyl-tRNA is required for viability in yeast. RNA. 2007; 13:1245–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Durant P.C., Bajji A.C., Sundaram M., Kumar R.K., Davis D.R.. Structural effects of hypermodified nucleosides in the Escherichia coli and human tRNALys anticodon loop: the effect of nucleosides s2U, mcm5U, mcm5s2U, mnm5s2U, t6A, and ms2t6A. Biochemistry. 2005; 44:8078–8089. [DOI] [PubMed] [Google Scholar]
- 27. Murphy F.V., Ramakrishnan V., Malkiewicz A., Agris P.F.. The role of modifications in codon discrimination by tRNA(Lys)UUU. Nat. Struct. Mol. Biol. 2004; 11:1186–1191. [DOI] [PubMed] [Google Scholar]
- 28. Nedialkova D.D., Leidel S.A.. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell. 2015; 161:1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yokoyama S., Watanabe T., Murao K., Ishikura H., Yamaizumi Z., Nishimura S., Miyazawa T.. Molecular mechanism of codon recognition by tRNA species with modified uridine in the first position of the anticodon. Proc. Natl. Acad. Sci. U.S.A. 1985; 82:4905–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Agris P.F. Decoding the genome: a modified view. Nucleic Acids Res. 2004; 32:223–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manickam N., Joshi K., Bhatt M.J., Farabaugh P.J.. Effects of tRNA modification on translational accuracy depend on intrinsic codon-anticodon strength. Nucleic Acids Res. 2016; 44:1871–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weixlbaumer A., Murphy F.V., Dziergowska A., Malkiewicz A., Vendeix F.A., Agris P.F., Ramakrishnan V.. Mechanism for expanding the decoding capacity of transfer RNAs by modification of uridines. Nat. Struct. Mol. Biol. 2007; 14:498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin C.A., Ellis S.R., True H.L.. The Sua5 protein is essential for normal translational regulation in yeast. Mol. Cell. Biol. 2010; 30:354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. El Yacoubi B., Hatin I., Deutsch C., Kahveci T., Rousset J.P., Iwata-Reuyl D., Murzin A.G., de Crecy-Lagard V.. A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J. 2011; 30:882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lamichhane T.N., Blewett N.H., Crawford A.K., Cherkasova V.A., Iben J.R., Begley T.J., Farabaugh P.J., Maraia R.J.. Lack of tRNA modification isopentenyl-A37 alters mRNA decoding and causes metabolic deficiencies in fission yeast. Mol. Cell. Biol. 2013; 33:2918–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taylor R.G., Walker D.C., McInnes R.R.. E. coli host strains significantly affect the quality of small scale plasmid DNA preparations used for sequencing. Nucleic Acids Res. 1993; 21:1677–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D.. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998; 14:115–132. [DOI] [PubMed] [Google Scholar]
- 38. Reid R.J., Sunjevaric I., Keddache M., Rothstein R., Kedacche M.. Efficient PCR-based gene disruption in Saccharomyces strains using intergenic primers. Yeast. 2002; 19:319–328. [DOI] [PubMed] [Google Scholar]
- 39. Goldstein A.L., McCusker J.H.. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999; 15:1541–1553. [DOI] [PubMed] [Google Scholar]
- 40. Gietz R.D., Schiestl R.H.. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007; 2:31–34. [DOI] [PubMed] [Google Scholar]
- 41. Alksne L.E., Anthony R.A., Liebman S.W., Warner J.R.. An accuracy center in the ribosome conserved over 2 billion years. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:9538–9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Salas-Marco J., Bedwell D.M.. Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J. Mol. Biol. 2005; 348:801–815. [DOI] [PubMed] [Google Scholar]
- 43. Belcourt M.F., Farabaugh P.J.. Ribosomal frameshifting in the yeast retrotransposon Ty: tRNAs induce slippage on a 7 nucleotide minimal site. Cell. 1990; 62:339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Curran J.F., Yarus M.. Use of tRNA suppressors to probe regulation of Escherichia coli release factor 2. J. Mol. Biol. 1988; 203:75–83. [DOI] [PubMed] [Google Scholar]
- 45. Percudani R., Pavesi A., Ottonello S.. Transfer RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J. Mol. Biol. 1997; 268:322–330. [DOI] [PubMed] [Google Scholar]
- 46. Anthony R.A., Liebman S.W.. Alterations in ribosomal protein RPS28 can diversely affect translational accuracy in Saccharomyces cerevisiae. Genetics. 1995; 140:1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Agris P.F., Vendeix F.A., Graham W.D.. tRNA’s wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 2007; 366:1–13. [DOI] [PubMed] [Google Scholar]
- 48. Huang B., Johansson M.J., Bystrom A.S.. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA. 2005; 11:424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cavuzic M., Liu Y.. Biosynthesis of sulfur-containing tRNA modifications: a comparison of bacterial, archaeal, and eukaryotic pathways. Biomolecules. 2017; 7:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Esberg A., Huang B., Johansson M.J., Bystrom A.S.. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol. Cell. 2006; 24:139–148. [DOI] [PubMed] [Google Scholar]
- 51. Klassen R., Grunewald P., Thuring K.L., Eichler C., Helm M., Schaffrath R.. Loss of anticodon wobble uridine modifications affects tRNA(Lys) function and protein levels in Saccharomyces cerevisiae. PLoS One. 2015; 10:e0119261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang B., Lu J., Bystrom A.S.. A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. RNA. 2008; 14:2183–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deng W., Babu I.R., Su D., Yin S., Begley T.J., Dedon P.C.. Trm9-Catalyzed tRNA modifications regulate global protein expression by Codon-Biased translation. PLoS Genet. 2015; 11:e1005706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Testa S.M., Disney M.D., Turner D.H., Kierzek R.. Thermodynamics of RNA-RNA duplexes with 2- or 4-thiouridines: implications for antisense design and targeting a group I intron. Biochemistry. 1999; 38:16655–16662. [DOI] [PubMed] [Google Scholar]
- 55. Kumar R.K., Davis D.R.. Synthesis and studies on the effect of 2-thiouridine and 4-thiouridine on sugar conformation and RNA duplex stability. Nucleic Acids Res. 1997; 25:1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ranjan N., Rodnina M.V.. Thio-Modification of tRNA at the wobble position as regulator of the kinetics of decoding and translocation on the ribosome. J. Am. Chem. Soc. 2017; 139:5857–5864. [DOI] [PubMed] [Google Scholar]
- 57. Johansson M.J., Esberg A., Huang B., Bjork G.R., Bystrom A.S.. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol. Cell. Biol. 2008; 28:3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yarian C., Marszalek M., Sochacka E., Malkiewicz A., Guenther R., Miskiewicz A., Agris P.F.. Modified nucleoside dependent Watson-Crick and wobble codon binding by tRNALysUUU species. Biochemistry. 2000; 39:13390–13395. [DOI] [PubMed] [Google Scholar]
- 59. Hagervall T.G., Pomerantz S.C., McCloskey J.A.. Reduced misreading of asparagine codons by Escherichia coli tRNALys with hypomodified derivatives of 5-methylaminomethyl-2-thiouridine in the wobble position. J. Mol. Biol. 1998; 284:33–42. [DOI] [PubMed] [Google Scholar]
- 60. Srinivasan M., Mehta P., Yu Y., Prugar E., Koonin E.V., Karzai A.W., Sternglanz R.. The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J. 2011; 30:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grosjean H., Westhof E.. An integrated, structure- and energy-based view of the genetic code. Nucleic Acids Res. 2016; 44:8020–8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. El Yacoubi B., Lyons B., Cruz Y., Reddy R., Nordin B., Agnelli F., Williamson J.R., Schimmel P., Swairjo M.A., de Crecy-Lagard V.. The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res. 2009; 37:2894–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Na J.G., Pinto I., Hampsey M.. Isolation and characterization of SUA5, a novel gene required for normal growth in Saccharomyces cerevisiae. Genetics. 1992; 131:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rodnina M.V., Fischer N., Maracci C., Stark H.. Ribosome dynamics during decoding. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017; 372:20160182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rozov A., Demeshkina N., Westhof E., Yusupov M., Yusupova G.. New structural insights into translational miscoding. Trends Biochem. Sci. 2016; 41:798–814. [DOI] [PubMed] [Google Scholar]
- 66. Hirsh D. Tryptophan transfer RNA as the UGA suppressor. J. Mol. Biol. 1971; 58:439–458. [DOI] [PubMed] [Google Scholar]
- 67. Atkins J.F., Bjork G.R.. A gripping tale of ribosomal frameshifting: extragenic suppressors of frameshift mutations spotlight P-site realignment. Microbiol. Mol. Biol. Rev. 2009; 73:178–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.