


Abstract
Cell morphological phenotypes, including shape, size, intensity, and texture of cellular compartments have been shown to change in response to perturbation with small molecule compounds. Image-based cell profiling or cell morphological profiling has been used to associate changes of cell morphological features with alterations in cellular function and to infer molecular mechanisms of action. Recently, the Library of Integrated Network-based Cellular Signatures (LINCS) Project has measured gene expression and performed image-based cell profiling on cell lines treated with 9515 unique compounds. These data provide an opportunity to study the interdependence between transcription and cell morphology. Previous methods to investigate cell phenotypes have focused on targeting candidate genes as components of known pathways, RNAi morphological profiling, and cataloging morphological defects; however, these methods do not provide an explicit model to link transcriptomic changes with corresponding alterations in morphology. To address this, we propose a cell morphology enrichment analysis to assess the association between transcriptomic alterations and changes in cell morphology. Additionally, for a new transcriptomic query, our approach can be used to predict associated changes in cellular morphology. We demonstrate the utility of our method by applying it to cell morphological changes in a human bone osteosarcoma cell line.
INTRODUCTION
Measurements of the cellular responses to perturbations are crucial to understanding cellular function. Only by altering an aspect of the system and measuring the response can we begin to understand the interdependencies central to cellular systems. Large-scale compendia of perturbation experiments, especially those with multiple complementary readouts, present an opportunity to advance significantly our understanding of cellular function (1,2). Image-based cell profiling quantifies changes in cell morphological features such as shape, size, intensity, and texture of cellular compartments (3). As a result of recent advances in high-throughput cell imaging techniques, comprehensive repositories of morphological and transcriptomic profiles, such as the LINCS Project, have been developed (4,5). These data repositories provide a platform for developing computational methods to integrate multiple sources of cellular information (6).
Several studies have demonstrated that changes in cell morphology can be used to further our understanding of the mechanisms of action of small compound perturbations and to predict the phenotypic impact of novel compounds (7–9). It has also been shown that one can identify drug targets using imaging-based signatures (10). High-throughput image-based profiling has been used to characterize genetic regulators of particular cellular processes such as mitosis and membrane-trafficking (11,12). These findings suggest that it may be possible to classify genes whose expression changes in response to perturbations based on their association with cellular morphological features.
We propose a new approach to identify sets of landmark genes associated with context-dependent morphology alterations from cells exposed to chemical or genetic perturbations, which we term cell morphology enrichment analysis. Specifically, we map a query profile of transcriptomic changes against a LINCS-based reference repository to find similar alteration profiles, use these similar profiles to create a gene set repository for each image-based feature, identify the top features associated with alterations in gene expression, and infer a gene network based on shared association with image-based features. For each query, our approach generates a specific gene set associated with each image-based feature. Providing a clear link between gene expression, cell morphology, and the gene network underlying these changes has the potential to uncover novel interdependences crucial to a cellular response.
MATERIALS AND METHODS
The main goal of this study is the development of a method to associate gene expression changes in response to perturbations with alterations in image-based features. The proposed approach for cell morphology enrichment analysis is composed of five main steps which we briefly describe here, illustrate in Figure 1, and discuss in detail in the rest of the Materials and Methods section.
Figure 1.

Overview of the proposed approach for cell morphology enrichment analysis. The input data, matched transcriptomic and cell morphological profiles, are obtained from the LINCS database in Step I. In Step II, we identify reference transcriptomic profiles that are similar to the gene expression pattern of a query. In Step III, we identify significant associations between alterations in cell morphology and gene expression. This results in query-specific cell morphology associated gene sets, which are used in Step IV to model cell morphology in response to treatment with a compound. Finally, in Step V, cell morphology associated gene sets can be used to performance pathway analysis, rank image-based features based on their predicted change in response to a given perturbation, or infer a gene association network.
Step I: Organization of the LINCS database. We use data from the LINCS project to develop a database of transcriptomic and cell morphological profiles for 9515 drugs and small compound molecules, 978 landmark genes, and 812 image-based features.
Step II: Pattern matching to detect similar gene expression signatures. We compare the query signature of expression changes for landmark genes measured by the L1000 assay to the reference transcriptomic profiles from the database to find drugs and small compound molecules that produce expression changes similar to the query.
Step III: Cell morphology enrichment analysis. We identify genes that are associated with each individual image-based feature within the set of similar profiles from Step II and use these to generate query-specific gene sets for each image-based feature.
Step IV: Validation of the method. We test our central assumption that gene expression changes can be used to identify associated changes in image-based features in two ways. First, we use a LASSO model to predict cell morphology changes in response to compound perturbations using observed transcriptomic changes (Step IVa). Second, we assess a simpler model that examines the correlation between changes of the image-based features for a given sample and the average image-based feature changes of samples with similar transcriptomic profiles to the query (Step IVb).
Step V: Applications of the method. A set of query-specific cell morphological gene sets and related transcriptomic profiles can be used in a variety of applications, such as pathway analysis, identification of the most significant morphological changes in response to treatment with a compound, and gene association network analysis.
LINCS database
The Library of Integrated Network-Based Cellular Signatures (LINCS) is a comprehensive public resource of gene expression signatures and other cellular processes in response to a variety of perturbing agents, including 847 approved drugs and 30 455 small compound molecules (http://www.lincsproject.org/). The LINCS database is divided into 11 categories on the basis of biological processes and 314 subcategories based on the type of experimental assay. The cellular component organization category includes image-based features extracted from the Cell Painting assay, representing changes in shape, texture, and intensity of major cellular components upon treatment with perturbing agents (6). The Cell Painting assay is a fluorescence imaging multiplex cytological profiling assay that uses fluorescent dyes followed by automatic imaging in order to quantify the effects that compound treatments have on cells (6). Transcriptomic measurements in the LINCS project were obtained using either L1000 mRNA profiling or RNA-seq and consist of 1.3 million gene expression profiles. We demonstrate the application of our approach using transcriptomic data from the L1000 mRNA profiling assay and image-based cell-morphology profiling in response to the drugs or small molecule compounds from the LINCS database (Figure 1, Step I). The subset of the LINCS data used in this manuscript can be found at: https://bitbucket.org/isarnassiri/cmeadata
Each cell morphological profile is a vector of numerical values representing changes of 812 image-based features upon treatment with a drug or small compound molecule. Each of these values represents the effect of a given compound treatment on each image-based feature compared to the effect of DMSO (6,13). The CellProfiler software was used to process and transform images into quantitative information (13,14). Gene expression changes in response to each compound perturbation were obtained from lincscloud.org, and the 20 304 compounds that produced the largest gene expression changes were selected (13). Compounds with the strongest signatures are often conserved across contexts, e.g. cell type, dosage, or time point of the compound treatment (13,15). We selected the 9,515 drugs and small compound molecules for which both image-based cell profile and gene expression data were available. This produced gene expression data for 978 genes and cell morphology data for 812 image-based features on which to develop the proposed methods (Figure 1, Step I). We represent the transcriptomic data as T and the cell morphology data as C:
 |
Mapping a query transcriptomic profile against the reference repository
Our proposed method begins by mapping a query of transcriptomic changes, produced by a drug or small compound molecule, to a database of 9515 transcriptomic profiles from the LINCS data, which form the reference database (Figure 1, Step II) (6,13,15). A query profile (Q) is a vector of centered and scaled (as above) expression changes for the 978 landmark genes. First, we convert the query and repository of the transcriptomic profiles to Boolean expression by replacing positive changes with one and negative changes with zero. We generate a confusion matrix for the query with each of the transcriptomic profiles in the reference repository. Next, we use Matthew’s correlation coefficient (MCC) to assess the similarity between the query and each reference profile, where MCC is defined as:
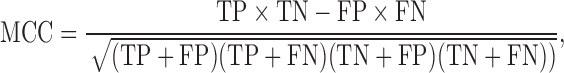 |
where TP is the number of true positives, TN is the number of true negatives, FP is the number of false positives, and FN is the number of false negatives. Here, true positives and true negatives are those genes that are over- or under-expressed in both the reference and query profiles, respectively. Samples with an MCC greater than 0.1 are included in the set of similar transcriptomic profiles and the corresponding cell morphological subset. Specifically, we select the compounds (rows of T) such that:
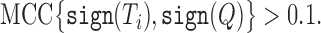 |
We denote the set of similar transcriptomic profiles as Ts.
In assessing similarity between gene expression changes, we focus on the direction of changes in expression, rather than the magnitude to reduce the impact of technical variability between samples. A similar approach has been successfully used as part of the Connectivity Map (15). The magnitude of gene expression changes can be reintroduced in downstream analyses to identify more complex biological interactions.
Similar transcriptomic profiles can be used to reveal shared mechanism of action and phenotypic impact between compounds (15–17) because compounds with similar gene expression signatures tend to interact with similar protein targets (18). We leverage similarities between a query gene signature and those in the LINCS data to infer shared targets in the context of cell morphological phenotypes.
Cell morphology enrichment analysis
We use a stepwise variable selection approach, which we term cell morphology enrichment analysis, to select landmark genes that are associated with a given image-based feature (Figure 1, Step III). This approach uses the least absolute shrinkage and selection operator (LASSO), with cross-validation to select the tuning parameter, and identifies significant associations between alterations in image-based features and gene expression. The LASSO fits a sparse model and consequently focuses on the most significant transcriptomic features (20).
Prior to applying the LASSO, we standardized the transcriptomic (Ts) and cell morphological (Cs) profiles via unitization with zero minimum: (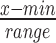 ). To assess the association between each image-based feature and the 978 landmark genes in the subset of samples previously selected (Ts), we use the LASSO method to model each image-based feature as a sparse function of the 978 landmark genes. This produces a set of genes associated with a given image-based feature in a local similarity neighbor of a transcriptomic query, which we call a query-specific cell morphological gene set. Note that a given gene can be assigned to several gene sets if that gene is associated with several image-based features. Each gene set represents a group of genes with similar expression patterns and shared phenotypic impact. Examination of these gene sets can lead to an improved understanding of the biological response to compound perturbations and identification of genetic variations associated with context-specific changes in cell morphological features (1).
). To assess the association between each image-based feature and the 978 landmark genes in the subset of samples previously selected (Ts), we use the LASSO method to model each image-based feature as a sparse function of the 978 landmark genes. This produces a set of genes associated with a given image-based feature in a local similarity neighbor of a transcriptomic query, which we call a query-specific cell morphological gene set. Note that a given gene can be assigned to several gene sets if that gene is associated with several image-based features. Each gene set represents a group of genes with similar expression patterns and shared phenotypic impact. Examination of these gene sets can lead to an improved understanding of the biological response to compound perturbations and identification of genetic variations associated with context-specific changes in cell morphological features (1).
Validation
We leverage the significant cross correlation between the image-based and transcriptomic profiles to predict cell morphological states for a transcriptomic profile of interest (Figure 1, Step IVa) (13). We hypothesize that transcriptomic profiles that are similar to a query gene expression profile can be used to predict changes in image-based features in response to a compound perturbation (15). We applied leave-one-out cross validation (LOOCV) to assess our ability to predict changes in image-based features based on the corresponding transcriptomic alterations (Q). In each LOOCV iteration, we first mapped the transcriptomic response profile of an indicated drug against the reference repository to identify similar profiles. Next, we used the transcriptomic and image-based profiles (minus the query) as training sets for the LASSO. Spearman’s rank correlation coefficient was used to compare the experimental and predicted values for each image-based feature.
Additionally, we assessed the similarity between the image-based features corresponding to a query transcriptomic profile and the image-based features corresponding to transcriptomic profiles that are similar to the query. Specifically, for a query q, we calculated Spearman’s rank correlation coefficient between Cq, 812 and the column means of Cs, 812. We repeated this procedure for each of the 9515 compounds in the data repository (Figure 1, Step IVb).
Applications of the proposed methodology
We envision several potential down-stream applications of the proposed methodology (Figure 1, Step V). To demonstrate these applications, we extracted image-based features from processed images of stained Human Osteosarcoma Cells (U-2 OS) for the nucleus, endoplasmic reticulum, nucleoli, Golgi apparatus, F-actin, mitochondria, and plasma membrane. These images were obtained 48 h after exposure to each of 19 864 compounds (6).
One potential down-stream analysis would be to identify the most significant morphological changes in response to a given perturbation. An estimate of the relative magnitude of image-based feature changes in response to the compound perturbation compared to DMSO has previously been used to quantify the significance of morphological changes (6). By using our proposed method to first identify a query-specific gene set for each image-based feature, we can estimate the cell morphology changes resulting from perturbation with a given compound. In the Results section, we evaluate the ability of this approach to identify the most significant image-based features for situations in which the true cell morphological changes were measured. Specifically, we use the magnitudes of the predicted morphological changes to rank the cell morphological features. We applied Spearman’s rank correlation coefficient to compare the predicted and observed values for each image-based feature.
Our proposed cell morphology enrichment analysis identifies a set of associated genes for each image-based feature. These potentially overlapping sets can be subsequently used to identify associations between genes based on their shared association with cell morphology features (22,23). We use transcriptomic profiles (Ts, 978) to construct a gene-gene interaction network (GN), and weight edges (X⇒Y) based on the proportion of cell morphological-specific gene sets that contain X and also contain Y. The edge weights in the network can be used to assess the strength of the association between two genes and to prune interactions that are not strongly involved in cell morphology phenotypes. We applied a standard association mining algorithm to describe the relationship between genes based on their membership in cell morphology-specific gene sets. Specifically, let l = {l1, l2, ..., lk} be a set of k genes, and C = {c1, c2, ..., cn} be a collection of n subsets of l, with each subset related to an image-based feature. To define an interaction between any two genes (X⇒Y) from the complete digraph of interactions, we use the following thresholds for support and lift:
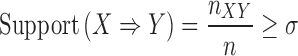 |
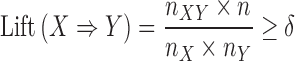 |
where n is the total number of gene sets in C, nXY is the total number of gene sets that contain gene X and Y, and nX is the total number of gene sets that contain gene X. Support is the proportion of gene sets that contain both gene X and gene Y. Lift is the deviation of the support of an indicated rule (X ⇒ Y) from the support expected under independence of X and the Y (24).
To focus on the strongest associations between genes, we select gene pairs that are highly co-expressed and that exceed the Support and Lift thresholds. We identify highly co-expressed gene pairs within the subset of similar transcriptomic profiles (Ts, 978) using the Context Likelihood of Relatedness (CLR) algorithm (25). CLR infers the interactions between genes using a scoring function based on the empirical distribution of mutual information (25).
Finally, one could apply standard pathway analysis methods to examine whether the cell morphology-specific gene sets were associated with certain biological pathways. It is likely that genes associated with a given image-based feature would be involved in biological pathways relevant to that morphological phenotype (27). To demonstrate this application, we use the Funrich package for functional annotation based on predefined gene sets for biological pathways (version 2.1.2) (26).
Implementation
The method is implemented as an R package, called CMEA (Cell Morphology Enrichment Analysis). CMEA is available at: https://github.com/isarnassiri/CMEA. The qgraph package is used for visualization of results (28). All analysis was performed using R (version 3.3.1).
RESULTS
Validation
We began by testing our main assumption that gene expression changes in response to perturbations can be used to identify associated changes in image-based feature. As described in the Materials and Methods section, we used a LOOCV procedure to assess the prediction error of our approach. Specifically, we randomly selected 20 image-based features and assessed the agreement between the predicted and experimental values for 9515 transcriptomic queries (all transcriptomic profiles in the repository). Our approach identified associated genes for each image-based feature for 9442 of the queries. For the remaining 0.8% of the queries, changes in these 20 image-based features were not associated with any gene expression changes. Generally, we observed a significant positive correlation between the predicted and observed image-based feature values based on our proposed approach (Figure 2 and Table 1, column 3).
Figure 2.

Prediction of the image-based feature from transcriptomic data is improved by model learning from similar transcriptomic and image-based profiles. This scatter plot shows the predicted versus experimental values of the Nuclei Area Shape image-based feature in response to treatment with 9515 drugs or small compound molecules. There is a clear improvement in prediction performance when using our proposed subset selection approach (red) versus using all profiles (green).
Table 1.
The results of leave-one-out cross-validation (LOOCV) to compare the performance of different approaches to model the cell morphological changes based on transcriptomic changes. The correlation coefficient (Spearman’s rho) between the experimental and predicted values was used to assess performance. The performance of LASSO is compared with Canonical Correlation Analysis (CCA) and simply using the average of the image-based profiles corresponding to transcriptomic profiles that are similar to the query (MEAN). The All Profiles (AP) column shows the performance using all of the profiles in the repository (minus the query profile). The Set of Similar Profiles (SS) shows the performance using just the set of similar profiles (minus the query profile). Our proposed stepwise subset selection approach substantially improves the prediction of cell morphological alterations from transcriptomic changes. The Set of Similar Permuted Profiles (SSP) shows the performance of LASSO using the image-based profiles with randomly permuted labels. The percentile column shows the 5- and 95-percentiles for the image-based feature changes in response to the perturbation with compound treatments. The distribution of image-based feature changes across major percentiles does not appear to be associated with the ability of the LASSO to predict cell morphological changes based on transcriptomic changes
| LASSO | CCA | MEAN | Percentile All | Percentile SS | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Image-based features | AP | SS | SSP | SS | SS | Q5% | Q95% | Q5% | Q95% | |
| 1 | Cells Area Shape | 0.024 | 0.672 | −0.00235 | 0.547 | 0.672 | 0.666 | 0.774 | 0.140 | 0.792 |
| 2 | Cells Intensity Edge golgi | 0.048 | 0.879 | −0.00003 | 0.804 | 0.853 | 0.502 | 0.574 | 0.133 | 0.853 |
| 3 | Cells Radial Distribution Mean Mit | 0.051 | 0.832 | −0.00004 | 0.798 | 0.863 | 0.195 | 0.247 | 0.059 | 0.642 |
| 4 | Cells Radial Distribution Radial CV Mit | 0.015 | 0.694 | 0.00123 | 0.550 | 0.685 | 0.310 | 0.392 | 0.139 | 0.774 |
| 5 | Cells Texture Angular Second Moment | 0.025 | 0.732 | 0.00218 | 0.560 | 0.693 | 0.651 | 0.774 | 0.215 | 0.831 |
| 6 | Cells Texture Gabor Mitochonderia | 0.004 | 0.876 | −0.00032 | 0.781 | 0.843 | 0.055 | 0.084 | 0.044 | 0.744 |
| 7 | Cells Texture Inverse Difference Moment | 0.002 | 0.833 | 0.00162 | 0.708 | 0.797 | 0.802 | 0.862 | 0.282 | 0.916 |
| 8 | Cytoplasm Intensity Integrated Intensity | 0.006 | 0.883 | −0.00125 | 0.812 | 0.869 | 0.103 | 0.122 | 0.039 | 0.701 |
| 9 | Cytoplasm Intensity vs. Intensity Edge ER | 0.021 | 0.812 | 0.00130 | 0.685 | 0.781 | 0.374 | 0.435 | 0.125 | 0.811 |
| 10 | Cytoplasm Texture Contrast golgi | 0.004 | 0.860 | −0.00177 | 0.745 | 0.827 | 0.107 | 0.149 | 0.056 | 0.716 |
| 11 | Cytoplasm Texture Inverse Difference ER | 0.032 | 0.766 | −0.00004 | 0.686 | 0.783 | 0.713 | 0.796 | 0.365 | 0.919 |
| 12 | Nuclei Area Shape | 0.053 | 0.786 | −0.00264 | 0.659 | 0.754 | 0.184 | 0.284 | 0.141 | 0.779 |
| 13 | Nuclei Texture Contrast | 0.034 | 0.810 | 0.00136 | 0.637 | 0.734 | 0.190 | 0.272 | 0.118 | 0.812 |
| 14 | Nuclei Texture Difference Variance ER | 0.053 | 0.808 | 0.00018 | 0.700 | 0.772 | 0.104 | 0.167 | 0.074 | 0.708 |
| 15 | Nuclei Texture Difference Variance golgi | 0.038 | 0.815 | −0.00086 | 0.634 | 0.736 | 0.118 | 0.183 | 0.055 | 0.747 |
| 16 | Nuclei Texture Entropy | 0.006 | 0.801 | −0.00007 | 0.682 | 0.770 | 0.845 | 0.891 | 0.210 | 0.891 |
| 17 | Nuclei Texture Gabor ER | 0.031 | 0.791 | −0.00184 | 0.614 | 0.718 | 0.150 | 0.247 | 0.081 | 0.731 |
| 18 | Nuclei Texture versus Mitochonderia | 0.038 | 0.614 | −0.00204 | 0.440 | 0.585 | 0.253 | 0.408 | 0.145 | 0.773 |
| 19 | Nuclei Texture versus Average golgi | 0.085 | 0.794 | −0.00268 | 0.723 | 0.801 | 0.323 | 0.449 | 0.252 | 0.835 |
| 20 | Nuclei Texture Variance golgi | 0.026 | 0.749 | −0.00093 | 0.554 | 0.688 | 0.077 | 0.158 | 0.070 | 0.703 |
To test whether our subset selection procedure is necessary, we repeated the LOOCV procedure, omitting the selection of similar transcriptomic profiles (Figure 1, Step II). Omitting the selection of transcriptomic profiles similar to the query profile resulted in a substantial decrease in performance (Table 1 and Figure 2). When using all transcriptomic profiles to identify relationships between gene expression and changes in an indicated image-based feature, the model is dominated by a few extreme gene expression profiles. These findings suggest a link between gene expression and cell morphological changes for a class of compounds that produce similar gene expression changes. This may indicate a similar mechanism of action and/or a degree of context dependence in the association between cell morphology and gene expression.
To test whether our feature selection procedure is necessary, we repeated the LOOCV procedure, omitting the LASSO modeling of the association between cell morphological features and genes (Figure 1, Step III) or replacing it with Canonical Correlation Analysis (CCA). In the former case, after identifying similar transcriptomic profiles to a given query, we simply calculated the arithmetic means of the corresponding image-based profiles. In the latter case, we substituted CCA for the LASSO-based feature selection. The LASSO consistently outperformed CCA and generally performed better than the simple image-based profile average; however, for 3 of the 20 image-based features, the mean actually outperformed the LASSO (Table 1). Scatter-plots for each of the 20 image-based features and each of the methods in Table 1 are included as Supplementary Figures S1–S23.
We assessed the possibility of over-fitting by repeating the LOOCV procedure after randomly permuting the compound labels for the image-based feature profiles. This label shuffling was performed on all 9515 profiles, prior to subset selection based on similarity to the query profile. The label permutation was repeated 100 times, and the correlation between all observed and predicted values is reported. The label permutation resulted in correlations of approximately zero for all image-based features (Table 1).
The 20 image-based features used in the previous assessments are a random sample from the 812 image-based features in the repository. We examined the 5th and 95th percentiles of the image-based morphological changes in response to perturbation with 9515 compounds to assess the range of responses for each image-based feature (Table 1). Neither the percentiles nor the range appear to be associated with prediction performance. To assess whether these 20 image-based features are representative of the full 812 image-based features, we compared the changes for the 20 selected image-based features used above with the distribution of changes across all image-based features (Supplementary Figure S24) and found no evidence of bias in our random sample.
Cell Morphology enrichment analysis
For a given transcriptomic query, we apply our proposed enrichment analysis for all 812 image-based features and create a repository of query-specific cell morphological gene sets. The modeling procedure to link image-based profiles and transcriptomic data involves the selection of genes that are associated with a given image-based feature. We can use these gene sets to investigate the relationship between query-specific cell morphological gene sets and different compound classes. To demonstrate this, we applied our approach to investigate the cell morphological responses to treatment with 3 compounds: NOMILIN, ZARDAVERINE and HYDROCOTARNINE. For each of the compounds, we identified 113, 145, and 143 compounds, respectively, that produced similar gene expression signatures. Our LASSO-based feature selection procedure identified a total of 421, 333, 123 genes in the cell morphological-specific genes sets for NOMILIN, ZARDAVERINE, and HYDROCOTARNINE, respectively (Supplementary Table S2). We did not find a significant similarity between cell morphological gene sets in response to the three compound treatments; instead, we found different gene sets associated with an indicated cell morphological change (Supplementary Figure S25).
Identification of the most significant image-based features in response to treatment with a compound
We leverage the ability of our method to model the interdependence between image-based features and gene expression to estimate the changes in cell morphology that are produced by treatment with a specific compound. To demonstrate this approach, we investigated cell morphological changes in response to treatment with NOMILIN, ZARDAVERINE and HYDROCOTARNINE based on the gene expression alterations those compounds produce. Specifically, we compared the predicted morphological changes with the observed changes using Spearman’s rank correlation. The results showed strong agreement between the predicted and observed morphological changes (Figure 3), and the largest predicted changes in cell morphology were likely to appear among top 10 experimentally observed image-based features (Supplementary Table S1).
Figure 3.

The results of modeling the rank of cell morphological changes in response to treatment with NOMILIN, ZARDAVERINE and HYDROCOTARNINE. For each drug, we identified a group of signature of landmark genes that mimic the query gene expression pattern, and applied the LASSO to model the cell morphological changes upon compound treatment. Spearman’s rank correlation coefficient is used to assess statistical dependence between the ranking based on the predicted and experimental values (A–C). Tables D–G show the first 10 top image-based features based on actual biological effects of drugs, and predicted values based on the gene expression profiles.
Pathway analysis based on cell morphological-specific gene sets
Stimulation of cells with compound treatment leads to changes in cell morphology. A variety of regulatory mechanisms connect changes in gene expression and cell morphology brought about by perturbations, including extracellular matrix proteins, trans-membrane receptors, and cytoskeletal organization (29). In order to identify the cell signaling events that link cell morphology and gene expression, we begin with the transcriptomic and cell morphological changes in response to treatment with NOMILIN, ZARDAVERINE and HYDROCOTARNINE. Next, we construct three repositories of cell morphological gene sets, one for each compound. Then, we infer edges between genes, based on co-expression and shared membership in cell morphological-specific gene sets, as described in the Materials and Methods. Lastly, we use the inferred networks to functionally annotate genes associated with the response to each treatment (Figure 4 and Supplementary Table S5).
Figure 4.

The topology and cellular functions of a gene interaction network (GIN) in response to treatment with NOMILIN (A), HYDROCOTARNINE (B), and ZARDAVERINE (C). On the left are graph-based visualizations of gene interaction networks, including gene-gene interactions selected based on the cell morphological gene sets and transcriptomic profiles in the repository subset (Ts). On the right are bar charts of the top 10 enriched biological pathways in the regulatory networks of osteosarcoma cells in response of the three treatments. We used functional pathway analysis to associate genes in the regulatory networks with predefined gene sets related to biological pathways (26). The sizes of some reference pathways are identical and they share common genes; therefore, the overlap between genes in the GIN and some enriched pathways are similar (e.g. S1P1 pathway and Arf6 downstream pathway).
These functional enrichment analyzes identified genes involved in regulation of cytoskeletal remodeling and growth-activation as two main biological functions of the inferred networks (Figure 4). Activation of the Sphingosine 1-phosphate (S1P) pathway indicates the involvement of the second messenger system in response to the compound treatments by controlling actin binding proteins and the cytoskeleton (30). Enrichment of EGFR receptor (ErbB1) signaling pathway and ATR signaling pathway suggest that morphological changes and cell adhesion can regulate the expression of genes involved in the transformation and growth-activation of fibroblasts (31). The pathways related to Arf6 downstream pathway signaling are involved in the regulation of cytoskeletal remodeling (32). Enrichment of Beta1 integrin pathways indicate a role of Integrin-ECM interactions in the regulation of osteosarcoma cell shape in response to treatment with NOMILIN, ZARDAVERINE and HYDROCOTARNINE (Figure 4) (Supplementary Table S6) (33).
DISCUSSION
In this study, we proposed an approach to link transcriptomic and cell morphological changes in response to perturbations and developed a cell morphology enrichment analysis. We validated the central assumptions of our proposed approach, namely that changes in gene expression can be used to predict corresponding changes in image-based profiles (6,34,35). Application of this approach provides a model to link genes through their shared effect on cell morphology and a clear method to investigate the mechanism of action for therapeutic agents in terms of phenotype impact (15).
The association between transcriptomic and cell morphological changes in response to perturbations appears to exist primarily among compounds that produce similar directional changes in gene expression. Within this local similarity neighborhood, specific changes in image-based features are often associated with changes in the expression of a relatively small number of genes. These associations often differ between similarity neighborhoods, suggesting that there is not a strong global association between transcriptomic and cell morphological responses to perturbations. This could indicate that compounds that produce similar changes in gene expression share similar mechanisms of action which are associated with corresponding morphological alterations.
The link between gene expression and cell morphology may arise via a signal transduction system scheme (35). Upon stimulation by a compound treatment, receptor-mediated activation of adhesion plaques in the cytoskeleton may cause the release of transcription regulators and alter specific gene expression programs (30). Adhesion plaques include Prostaglandin-stimulated second messengers, growth factors, and structural components such as β-integrin (31). Alterations in the cytoskeleton, in addition to its role in determining cell morphology, produce changes in gene expression (36). This could explain the observed association between gene expression and cell morphology.
Enrichment analyses of cellular processes and biological pathways have become a routine part of transcriptomic data analyses (37). Here, we propose an enrichment analysis that connects gene expression profiles to cell morphological phenotypes (3,38). Previous methods to investigate cell phenotypes have focused on targeting candidate genes as components of known pathways, RNAi morphological profiling, and development of databases of morphological defects (3,39–41). Our approach furthers our understanding of the interdependence between gene expression and cell morphology.
The work presented in this manuscript is based on data from the LINCS Project (6,13). A possible weakness of our approach is that the definition and selection of some cell morphological terms reflect technical properties of the image analysis rather than informative biological characteristics of the cell. Additional, there are some highly similar image-based features, for which the same sets of associated genes were identified. This redundancy among morphological features is a limitation of morphological profiling in general, and advances in morphological feature annotation and assessments of biological relevance can be readily incorporated to improve our approach.
We anticipate the results of this study will impact the utility of LINCS L1000 data and provide a blueprint for the integrative analysis of other multi-omics data, such as mass spectrometry-based targeted proteomics (LINCS P100) and epigenomics (NUROLINCS) (42).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the members of the LINCS Consortium for making their data publicly available and easily accessible.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Human Genome Research Institute of the National Institutes of Health [R00-HG006853 to M.N.M.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Funding for open access charge: NIH/NHGRI [R00-HG006853].
Conflict of interest statement. None declared.
REFERENCES
- 1. Caicedo J.C., Cooper S., Heigwer F., Warchal S., Qiu P., Molnar C., Vasilevich A.S., Barry J.D., Bansal H.S., Kraus O. et al. Data-analysis strategies for image-based cell profiling. Nat. Methods. 2017; 14:849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Subramanian A., Narayan R., Corsello S.M., Peck D.D., Natoli T.E., Lu X., Gould J., Davis J.F., Tubelli A.A., Asiedu J.K. et al. A next generation connectivity Map: L1000 platform and the first 1,000,000 profiles. Cell. 2017; 171:1437–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bray M.A., Singh S., Han H., Davis C.T., Borgeson B., Hartland C., Kost-Alimova M., Gustafsdottir S.M., Gibson C.C., Carpenter A.E.. Cell painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc. 2016; 11:1757–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Loo L.H., Wu L.F., Altschuler S.J.. Image-based multivariate profiling of drug responses from single cells. Nat. Methods. 2007; 4:445–453. [DOI] [PubMed] [Google Scholar]
- 5. Breinig M., Klein F.A., Huber W., Boutros M.. A chemical-genetic interaction map of small molecules using high-throughput imaging in cancer cells. Mol. Syst. Biol. 2015; 11:846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wawer M.J., Jaramillo D.E., Dancik V., Fass D.M., Haggarty S.J., Shamji A.F., Wagner B.K., Schreiber S.L., Clemons P.A.. Automated structure-activity relationship mining: connecting chemical structure to biological profiles. J. Biomol. Screen. 2014; 19:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reisen F., Sauty de Chalon A., Pfeifer M., Zhang X., Gabriel D., Selzer P.. Linking phenotypes and modes of action through high-content screen fingerprints. Assay Drug Dev. Technol. 2015; 13:415–427. [DOI] [PubMed] [Google Scholar]
- 8. Tanaka M., Bateman R., Rauh D., Vaisberg E., Ramachandani S., Zhang C., Hansen K.C., Burlingame A.L., Trautman J.K., Shokat K.M. et al. An unbiased cell Morphology-Based screen for new, biologically active small molecules. PLOS Biol. 2005; 3:e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ochoa J.L., Bray W.M., Lokey R.S., Linington R.G.. Phenotype-Guided natural products discovery using cytological profiling. J. Nat. Prod. 2015; 78:2242–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ohnuki S., Oka S., Nogami S., Ohya Y.. High-content, image-based screening for drug targets in yeast. PLos One. 2010; 5:e10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liberali P., Snijder B., Pelkmans L.. A hierarchical map of regulatory genetic interactions in membrane trafficking. Cell. 2014; 157:1473–1487. [DOI] [PubMed] [Google Scholar]
- 12. Neumann B., Walter T., Heriche J.K., Bulkescher J., Erfle H., Conrad C., Rogers P., Poser I., Held M., Liebel U. et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010; 464:721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z., Clark N.R., Ma’ayan A.. Drug-induced adverse events prediction with the LINCS L1000 data. Bioinformatics. 2016; 32:2338–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J. et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006; 7:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamb J., Crawford E.D., Peck D., Modell J.W., Blat I.C., Wrobel M.J., Lerner J., Brunet J.P., Subramanian A., Ross K.N. et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006; 313:1929–1935. [DOI] [PubMed] [Google Scholar]
- 16. Duan Q., Reid S.P., Clark N.R., Wang Z., Fernandez N.F., Rouillard A.D., Readhead B., Tritsch S.R., Hodos R., Hafner M. et al. L1000CDS2: LINCS L1000 characteristic direction signatures search engine. NPJ Syst. Biol. Applic. 2016; 2:16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Caicedo J.C., Singh S., Carpenter A.E.. Applications in image-based profiling of perturbations. Curr. Opin. Biotech. 2016; 39:134–142. [DOI] [PubMed] [Google Scholar]
- 18. Campillos M., Kuhn M., Gavin A.C., Jensen L.J., Bork P.. Drug target identification using side-effect similarity. Science. 2008; 321:263–266. [DOI] [PubMed] [Google Scholar]
- 19. Clark N.R., Hu K.S., Feldmann A.S., Kou Y., Chen E.Y., Duan Q., Ma’ayan A.. The characteristic direction: a geometrical approach to identify differentially expressed genes. BMC Bioinformatics. 2014; 15:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mo Q.X., Wang S.J., Seshan V.E., Olshen A.B., Schultz N., Sander C., Powers R.S., Ladanyi M., Shen R.L.. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:4245–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wawer M.J., Li K., Gustafsdottir S.M., Ljosa V., Bodycombe N.E., Marton M.A., Sokolnicki K.L., Bray M.A., Kemp M.M., Winchester E. et al. Toward performance-diverse small-molecule libraries for cell-based phenotypic screening using multiplexed high-dimensional profiling. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:10911–10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen S.-C., Tsai T.-H., Chung C.-H., Li W.-H.. Dynamic association rules for gene expression data analysis. BMC Genomics. 2015; 16:786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alves R., Rodriguez-Baena D.S., Aguilar-Ruiz J.S.. Gene association analysis: a survey of frequent pattern mining from gene expression data. Brief. Bioinformatics. 2010; 11:210–224. [DOI] [PubMed] [Google Scholar]
- 24. Hahsler M., Grun B., Hornik K.. arules - A computational environment for mining association rules and frequent item sets. J. Stat. Softw. 2005; 14:doi:10.18637/jss.v014.i15. [Google Scholar]
- 25. Faith J.J., Hayete B., Thaden J.T., Mogno I., Wierzbowski J., Cottarel G., Kasif S., Collins J.J., Gardner T.S.. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007; 5:54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pathan M., Keerthikumar S., Ang C.S., Gangoda L., Quek C.Y.J., Williamson N.A., Mouradov D., Sieber O.M., Simpson R.J., Salim A. et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015; 15:2597–2601. [DOI] [PubMed] [Google Scholar]
- 27. Rohban M.H., Singh S., Wu X.Y., Berthet J.B., Bray M.A., Shrestha Y., Varelas X., Boehm J.S., Carpenter A.E.. Systematic morphological profiling of human gene and allele function via cell painting. Elife. 2017; 6:e24060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Epskamp S., Cramer A.O.J., Waldorp L.J., Schmittmann V.D., Borsboom D.. qgraph: Network visualizations of relationships in psychometric data. J. Stat. Softw. 2012; 48:1–18. [Google Scholar]
- 29. Singh S., Wu X.Y., Ljosa V., Bray M.A., Piccioni F., Root D.E., Doench J.G., Boehm J.S., Carpenter A.E.. Morphological profiles of RNAi-Induced gene knockdown are highly reproducible but dominated by seed effects. PLoS One. 2015; 10:e0131370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Donati C., Bruni P.. Sphingosine 1-phosphate regulates cytoskeleton dynamics: Implications in its biological response. BBA-Biomembranes. 2006; 1758:2037–2048. [DOI] [PubMed] [Google Scholar]
- 31. Muthuswamy S.K., Gilman M., Brugge J.S.. Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol. Cell. Biol. 1999; 19:6845–6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bhanot H., Young A.M., Overmeyer J.H., Maltese W.A.. Induction of nonapoptotic cell death by activated Ras requires inverse regulation of Rac1 and Arf6. Mol. Cancer Res. 2010; 8:1358–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wan X., Kim S.Y., Guenther L.M., Mendoza A., Briggs J., Yeung C., Currier D., Zhang H., Mackall C., Li W.J. et al. Beta4 integrin promotes osteosarcoma metastasis and interacts with ezrin. Oncogene. 2009; 28:3401–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ami Y., Shimazui T., Akaza H., Uematsu N., Yano Y., Tsujimoto G., Uchida K.. Gene expression profiles correlate with the morphology and metastasis characteristics of renal cell carcinoma cells. Oncol. Rep. 2005; 13:75–80. [PubMed] [Google Scholar]
- 35. Ben-Ze’ev A. Animal cell shape changes and gene expression. BioEssays. 1991; 13:207–212. [DOI] [PubMed] [Google Scholar]
- 36. Fletcher D.A., Mullins R.D.. Cell mechanics and the cytoskeleton. Nature. 2010; 463:485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nassiri I., Lombardo R., Lauria M., Morine M.J., Moyseos P., Varma V., Nolen G.T., Knox B., Sloper D., Kaput J. et al. Systems view of adipogenesis via novel omics-driven and tissue-specific activity scoring of network functional modules. Sci. Rep. 2016; 6:28851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ljosa V., Caie P.D., ter Horst R., Sokolnicki K.L., Jenkins E.L., Daya S., Roberts M.E., Jones T.R., Singh S., Genovesio A. et al. Comparison of methods for Image-Based profiling of cellular morphological responses to Small-Molecule treatment. J. Biomol. Screen. 2013; 18:1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jin K., Li J.J., Vizeacoumar F.S., Li Z.J., Min R.Q., Zamparo L., Vizeacoumar F.J., Datti A., Andrews B., Boone C. et al. PhenoM: a database of morphological phenotypes caused by mutation of essential genes in Saccharomyces cerevisiae. Nucleic Acids Res. 2012; 40:D687–D694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kiger A.A., Baum B., Jones S., Jones M.R., Coulson A., Echeverri C., Perrimon N.. A functional genomic analysis of cell morphology using RNA interference. J. Biol. 2003; 2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kirsanova C., Brazma A., Rustici G., Sarkans U.. Cellular phenotype database: a repository for systems microscopy data. Bioinformatics. 2015; 31:2736–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cavill R., Jennen D., Kleinjans J., Bried J.J.. Transcriptomic and metabolomic data integration. Brief. Bioinformatics. 2016; 17:891–901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.