


Abstract
Recently, Type III-A CRISPR-Cas systems were found to catalyze the synthesis of cyclic oligoadenylates (cOAs), a second messenger that specifically activates Csm6, a Cas accessory RNase and confers antiviral defense in bacteria. To test if III-B CRISPR-Cas systems could mediate a similar CRISPR signaling pathway, the Sulfolobus islandicus Cmr-α ribonucleoprotein complex (Cmr-α–RNP) was purified from the native host and tested for cOA synthesis. We found that the system showed a robust production of cyclic tetra-adenylate (c-A4), and that c-A4 functions as a second messenger to activate the III-B-associated RNase Csx1 by binding to its CRISPR-associated Rossmann Fold domain. Investigation of the kinetics of cOA synthesis revealed that Cmr-α–RNP displayed positively cooperative binding to the adenosine triphosphate (ATP) substrate. Furthermore, mutagenesis of conserved domains in Cmr2α confirmed that, while Palm 2 hosts the active site of cOA synthesis, Palm 1 domain serves as the primary site in the enzyme–substrate interaction. Together, our data suggest that the two Palm domains cooperatively interact with ATP molecules to achieve a robust cOA synthesis by the III-B CRISPR-Cas system.
INTRODUCTION
CRISPR-Cas systems provide a heritable adaptive immunity against the invasion of viruses and plasmids in prokaryotes. The antiviral immunity is achieved in three steps: first, a short fragment (protospacer) of an invading nucleic acid is recognized and acquired as a new spacer in a CRISPR locus (1–3); second, CRISPR loci are transcribed, yielding precursor CRISPR RNAs (crRNAs) that are further processed into mature crRNAs (4) and finally, crRNAs generated from the new spacer form ribonucleoprotein complexes (RNPs) with Cas proteins, and the resulting RNPs recognize invading nucleic acids by sequence complementarity to the corresponding protospacer on recurring invasive genetic elements and target them for destruction (5). CRISPR-Cas systems are classified into six different types (Type I–VI) based on their gene synteny and mechanisms of interference (6,7). These antiviral systems have evolved distinct mechanisms of recognition and destruction of invading nucleic acids. Among them, Type I, II and V systems target double-stranded DNA, and the interference activity is dependent on a short motif flanking the protospacer (known as protospacer adjacent motif ), while Type VI systems bind to the target RNA that complements the crRNA and are activated for indiscriminate RNA degradation (8–12).
Investigation of CRISPR-Cas10 (Type III) systems has revealed that mechanisms of their nucleic acid interference are very different from those of all other known CRISPR-Cas systems. Two subtypes have been characterized in detail, including III-A Csm and III-B Cmr systems. Early studies showed that the Staphylococcus epidermidis Csm (SeCsm) system mediates DNA interference (13,14), while the Pyrococcus furiosus Cmr (PfCmr) effector complex exhibits RNA cleavage activity (15). Further studies show that Type III effector complexes cleave target RNA in 6-nt intervals (16–21), and the active site is located in Cmr4 or Csm3, the large backbone subunit of each subtype system (18,21,22). Strikingly, investigation of the Sulfolobus islandicus Cmr-α (Cmr-α) system by an invader plasmid assay revealed that it mediates transcription-dependent DNA interference (23), and the system shows dual DNA/RNA interference (23,24). Subsequently, the same activities were found for other Type III systems (25–27). Furthermore, in vitro characterization of both Csm and Cmr effector complexes showed that they all possess target RNA-activated DNA cleavage activity (28–32). In addition, the Sulfolobus solfataricus III-D (SsCsm) system was shown to cleave plasmid DNA targets in vitro (33). As recently reviewed (34–36), all investigated Type III systems exhibit multiple nucleic acid interference activities, including the backbone RNA cleavage, the target RNA-activated DNA cleavage, in which the former activity relies on the nuclease domain of the large backbone subunit, while the latter depends on the HD nuclease domain of the Cas10 subunit (Csm1 or Cmr2).
More recently, it was reported that Csm–RNPs of Streptococcus thermophilus (StCsm) and Enterococcus italicus (EiCsm) catalyze the synthesis of cyclic oligoadenylates (cOAs) (37,38). Upon the binding of the cognate target RNA to a binary Csm–RNP complex, the Palm 2 (or cyclase) domain of the Csm1 subunit in these RNPs is activated for cOA synthesis. The resulting cOAs function as a second messenger that binds the CRISPR-associated Rossmann Fold (CARF) domain of Csm6, a Cas accessory ribonuclease, and activates its RNase activity from the higher eukaryotes and prokaryotes nucleotide-binding (HEPN) domain (37,38). Consistent with these results, csm6 is essential for the prevention of plasmid transfer and virus multiplication by SeCsm in S. epidermidis and Staphylococcus aureus, respectively (39,40). To date, the synthesis of cOA second messenger and the CRISPR signaling remain to be investigated for Type III-B CRISPR-Cas systems.
We have employed S. islandicus Rey15A (41) as the model to investigate molecular mechanisms of III-B CRISPR-Cas systems. The organism contains a complete I-A system including an adaptation module, an interference module and two CRISPR arrays as well as two disparate III-B Cmr modules (Cmr-α and Cmr-β), among which the last two systems are not located in a close neighborhood to any CRISPR array (42). Nevertheless, both Cmr systems can utilize crRNAs produced from the I-A CRISPR arrays and mediate nucleic acid interference (23,24,43). Furthermore, the S. islandicus Cmr-α ribonucleoprotein complex (Cmr-α–RNP) cleaves target RNAs by the large backbone subunit Cmr4 and performs single-stranded DNA (ssDNA) degradation upon binding to the cognate target RNA (31), and Cmr1α facilitates the dual DNA/RNA cleavage by interacting with the target RNA (44). On the other hand, cmr-α genes are clustered with csx1 (23) encoding a Cmr-associated CARF domain RNase (45). The CARF domain of the enzyme was found to bind to 3′-tetradenylates and the interaction strongly activates the Csx1 RNase activity (46).
In this study, we demonstrated that the CRISPR signaling pathway is evolutionarily conserved in Type III-B CRISPR-Cas systems since Cmr-α–RNP converts adenosine triphosphate (ATP) to cyclic tetraadenylate (c-A4) that also functions as a second messenger to activate the RNase activity of Csx1. Characterization of the catalysis of cOA synthesis by Cmr-α–RNP further revealed that the enzyme displays robust cOA synthesis, probably resulting from the cooperative substrate binding in enzyme–substrate interaction.
MATERIALS AND METHODS
Construction of pAC-cmr2α and its derivatives and purification of Cmr-α–RNPs from S. islandicus
The cmr2α gene (SiRe_0894) was amplified from S. islandicus REY15A (42) by polymerase chain reaction (PCR) using FastPfu DNA polymerase (TransGene, Beijing China) with the primer set of Cmr2-up-NheI and Cmr2-dw-SalI (Supplementary Table S1). The resulting PCR fragment was digested with NheI and SalI and inserted into the same sites on pSeSD1 (47), giving pCmr2α plasmid (Supplementary Table S3). Then, the cmr2α expression cassette on pCmr2α was amplified by PCR, using the primer pair of MRS-up and MRS-dw (Supplementary Table S1), including the artificial arabinose-inducible promoter araS-SD, the coding sequence of 6× His-tag, and that of cmr-2α plus the transcriptional terminator. The resulting PCR product was digested with SmalI and XhoI and insertion of the expression cassette into the SmaI and SalI sites on pAC-MS1 (31) yielded pAC-cmr2α (Supplementary Figure S1).
Mutated cmr2α genes carrying substitutions in the HD, Palm 1 or Palm 2 domain were generated by splicing, overlapping and extension PCR (SOE-PCR) following the published procedure (48), using the primers listed in Supplementary Table S1. Specifically, the first DNA fragment (Cmr2HD-L, Cmr2P2-L or Cmr2P1-L) was generated by PCR using MRS-up and one of the following primers: Cmr2HD-L-dw, Cmr2P2-L-dw or Cmr2P1-L-dw, whereas the second DNA fragment (Cmr2HD-R, Cmr2P2-R or Cmr2P1-R) was obtained by PCR using the primer combination of MRS-dw with one of the following: Cmr2HD-R-up, Cmr2P2-R-up or Cmr2P1-R-up. PCR fragments containing mutated HD, P1 or P2 domain were obtained by PCR using the primer set of MRS-up and MRS-dw, with the corresponding PCR fragment as DNA template (i.e. Cmr2HD-L+R, Cmr2P2-L+R or Cmr2P1-L+R). The resulting SOE-PCR fragments were digested with SmalI and XhoI, and ligation of each restricted SOE-PCR fragment with SmaI+SalI-digested pAC-MS1 yielded pAC-cmr2α-HD, pAC-cmr2α-P1 and pAC-cmr2α-P2 individually. All mutations were confirmed by determination of DNA sequences of the constructed plasmids. Each plasmid was then electroporated into S. islandicus MF1 (31) as previously described, giving transformants that were suitable for Cmr-α–RNP purification.
To purify Cmr-α–RNPs carrying substitution mutations in HD, Palm 1 or Palm 2 domain, these transformants were grown in SCV media (Basal media supplemented with 0.2% sucrose, 0.2% Casamino acids and 1% vitamin solution) at 78°C (41). When cultures were grown to A600 = 0.7, cell mass was harvested and used for Cmr-α–RNP purification as previously described (31,49). Briefly, cell pellets were resuspended in Buffer A (20 mM hydroxyethyl-piperazineethane-sulfonic acid (HEPES) pH 7.5, 30 mM Imidazole, 500 mM NaCl), giving cell suspensions that were treated by French press to disrupt cells. Each resulting cell extract was loaded onto a 1 ml HisTrap HP column (GE Healthcare, Waukesha, WI, USA). The Cmr-α effector complex was purified by stepwise elution with buffers containing 30, 70 and 200 mM imidazole successively. Fractions containing the target protein were concentrated and further purified by size exclusion chromatography (SEC) in Buffer C (20 mM Tris–HCl (pH 7.5), 250 mM NaCl), using a Superdex 200 Hiload column (GE Healthcare, Waukesha, WI, USA). Sample fractions collected during SEC were analyzed by sodium dodecylsulphate-polyacrylamide gel electrophoresis and those containing the complete set of Cmr-α components were pooled together and used for further analysis.
Conversion of ATP to cyclic oligoadenylates (cOAs) by Cmr-α–RNP
Biochemical assay for testing ATP conversion by Cmr-α–RNP consisted of the following components, including 5 mM MgCl2, 5 mM DTT, ∼1 nM α32P-ATP (PerkinElmer, Waltham, MA, USA), 80 nM SS1-46, 100 μM ATP and 40 nM Cmr-α–RNP in 20 mM Mes (pH 6.0). The assay was conducted at 70°C for 20 min, and the reaction was stopped by addition of 10 μl 2× RNA loading dye. After denaturing by heating to 95°C for 2 min and cooling on ice for 5 min, all samples were analyzed by denaturing 24% polyacrylamide gel electrophoresis (PAGE). Labeled nucleotides were detected by exposing the gel to a phosphor screen and scanned with a Typhoon FLA 7000 (GE Healthcare, Waukesha, WI, USA).
Liquid chromatography and mass spectrometry (LC-MS) analysis
A 100 μl ATP reaction was set up as described above except for the omission of α32P-ATP. The reaction mixture was incubated at 70°C for 20 min. After inactivation of the enzyme by incubation at 95°C for 5 min and subsequently cooling down on ice, the reaction mixture was loaded onto a Kinetex EVO C18 column (100 × 2.1 mm, 1.7 μm) (Phenomenex, Torrance, CA, USA), which was pre-equilibrated with buffer A (5 mM ammonium acetate) at 30°C at 0.3 ml/min. The products were purified by stepwise elution with different concentrations of buffer B (100% acetonitrile): 0–2 min, 0% buffer B; 2–22 min, 20% buffer B; 22–25 min, 50% buffer B, 25–29 min 100% buffer B.
Electrospray ionization mass spectrometry data were acquired using a micrOTOF-QII (Bruker, Billerica, MA, USA) in MS Scan mode from 100 to 2000 m/z in the negative ionization mode. The employed ESI parameters were as following: Cappilary voltage: 4.5 kV; End plate offset: -500, Nebulizer: 1.2 Bar, Dry Heater: 200°C, Dry Gas: 9 L/min, collision cell RF: 800 Vpp.
Nuclease S1 degradation analysis
To visualize the degradation of ATP reaction product, ∼50 nM labeled ATP reaction product was incubated with 0.2 U/μl Nuclease S1 (Thermo Fisher Scientific, Waltham, MA, USA) in a 10-μl mixture containing 2 μl 5× reaction buffer at 37°C. The reactions were stopped by supplementing 2× RNA loading dye at indicated time points and stored on ice. Then, all samples were analyzed denaturing PAGE in a 24% gel with results recorded by phosphor imaging as described above.
Enzymatic probing of nucleotide products produced by Cmr-α–RNP
T4 polynucleotide kinase (PNK; New England Biolabs, Ipswich, MA, USA), Fast alkaline phosphatase (FastAP; Thermo Fisher Scientific, Waltham, MA, USA) and poly(A) polymerase (PAP; New England Biolabs, Ipswich, MA, USA) were employed for the characterization of ATP reaction products. Aliquots of the labeled cOAs were treated with each enzyme by following the instruction from their manufacturers. All reactions were performed in a 10-μl mixture containing ∼50 nM labeled ATP reaction product and 1 μl of each enzyme. The enzyme treatments were conducted at 37°C for 60 min and analyzed by denaturing PAGE in a 24% PA gel with results recorded by phosphor imaging.
To further investigate whether the ATP reaction products could be modified, reaction mixtures (in 10 μl) were set up in Buffer A (50 mM Tris–HCl, pH 7.6, 10 mM MgCl2, 5 mM Dithiothreitol (DTT), 0.1 mM spermidine) with the following components: (i) ∼100 nM ATP reaction product from each liquid chromatography fraction collected from the two predominant peaks (Figure 2A), (ii) 1 U/μl T4 PNK and (iii) 1 nM γ32P-ATP. After incubation at 37°C for 60 min, all treated samples were analyzed by denaturing PAGE in a 24% gel, with the ATP reaction product generated with α32P-ATP as a reference. The results were recorded by phosphor imaging.
Figure 2.

Identification of the Cmr-α-mediated ATP reaction products. (A) Liquid chromatography (upper panel) and mass spectrometry (two lower panels) analysis of the ATP reaction products by Cmr-α; *: the peak showing a MW of 657.10 in the MS analysis was also present in the reference sample; **: peak 3 with a MW of 1315.22 could be the tail of the main peak. (B) The ATP reaction product (∼40 nM) was treated with 1 U/μl PNK, 0.05 U/μl poly(A) polymerase (PAP), 0.1 U/μl alkaline phosphatase (FastAP) and 0.2 U/μl Nuclease S1, respectively, followed by analysis of denaturing gel electrophoresis. The reactions with PNK, PAP and FastAP were incubated at 37°C for 60 min and the S1 nuclease incubation time was indicated above the gel. If applicable, 1 mM ATP was supplemented into the reaction mixture. (C) The fractions of the peaks from Figure 2A (1: peak 1; 2: peak 2) were labeled with γ32P-ATP by PNK. The ATP reaction product generated with α32P-ATP was also loaded as size marker (the first lane).
Nucleic acid cleavage by the wild-type and mutated Cmr-α effector complexes
Radio-labeled SS1-40 RNA and S10 ssDNA (Supplementary Table S2) were substrate for the backbone RNA cleavage and the target RNA-activated ssDNA cleavage, respectively. These nucleic acids were synthesized from Integrated DNA Technologies (IDT, Coralville, IA, USA.), gel-purified and 5′ labeled with γ32P-ATP using T4 PNK (New England Biolabs, Ipswich, MA, USA) as described previously (31).
Backbone RNA cleavage was assayed in a 10-μl reaction mixture containing 50 mM Tris-Cl (pH 7.5), 10 mM MgCl2, 5 mM DTT, 20 nM of labeled SS1-40 and 20 nM of the wild-type or one of the mutated Cmr-α–RNP as specified in each experiment. The cleavage was conducted at 70°C for 20 min, and the reaction was stopped by addition of 10 μl of 2× RNA loading dye. After heating to 95°C for 2 min and cooling down on ice for 5 min, RNA cleavage was analyzed by denaturing PAGE in a 18% gel with the results recorded by phosphor imaging.
The DNA cleavage assay was conducted in a 10-μl reaction mixture contains 50 mM Tris-Cl (pH 7.5), 10 mM MgCl2, 5 mM DTT, 200 nM SS1-46, 20 nM labeled S10-60 ssDNA and 20 nM of the wild-type or a mutated Cmr-α–RNP. The mixtures were incubated at 70°C for 60 min and analyzed as described for the backbone RNA cleavage assay.
Csx1 RNA cleavage assay
Csx1 RNA cleavage was assayed in a 10-μl reaction mixture containing 20 mM Mes (pH 6.0), 5 mM DTT, 500 nM unlabeled SS1-40, 20 nM labeled SS1-40, 100 nM Csx1 or one of its mutants in the presence of c-A4, the circular ligand, or CAAAA, the linear ligand. Reaction mixtures were incubated at 70°C for 20 min and analyzed as described for the backbone RNA cleavage assay.
Csx1 ligand binding assay
The affinity of Csx1 to two ligands, c-A4 and CAAAA, was analyzed using electrophoretic mobility shift assay (EMSA). The assay was set up in a 10-μl reaction mixture containing 20 mM Mes (pH 6.0), 8% glycerine, 50 mM DTT, 50 nM labeled c-A4 or CAAAA, and a series of different concentrations of Csx1 or one of its mutants as indicated in each experiment. Reaction mixtures were incubated at 70°C for 10 min and analyzed by non-denaturing PAGE in a 10% gel, and the results were recorded by phosphor imaging.
Cmr-α substrate-binding assay
Interaction between ATP and Cmr-α–RNP, or one of the mutated effector complexes was analyzed by EMSA. The basic binding reaction contained 200 nM Cmr-α (or a mutant effector complex) and 1 nM γ32P-ATP to which 1 mM ethylenediaminetetraacetic acid (EDTA) or 5 mM MgCl2 was added. The resulting mixtures were incubated at 70°C for 10 min. The effect of target RNA on Cmr-α ATP binding was evaluated by addition of 300 nM SS1-46 into the mixtures and further incubated for 3 min at 70°C. Samples were then analyzed by non-denaturing PAGE on 10% gels in 0.5× TB electrophoresis buffer (45 mM Tris-borate). ATP–Cmr-α–RNP complexes were detected as for the analysis of Csx1–ligand interaction described above.
Kinetic study of ATP incorporation by Cmr-α–RNP
Kinetics of the ATP reaction by Cmr-α was determined in a mixture containing 20 mM Mes 9 (pH 6.00), 5 mM MgCl2, 5 mM DTT, 200 nM SS1-46 RNA and ∼1 nM α32P-ATP (PerkinElmer), with the concentrations of Cmr-α and ATP specified in the experiments. After incubation at 70°C for 5 min, the reactions were analyzed by denaturing PAGE in a 24% gel and ATP reaction products were quantified by ImageQuant TL 8.1 (GE Healthcare, Waukesha, WI, USA). The ATP incorporation rate was calculated as the amount of incorporated ATP (μM) per min. To ensure the experiments could reflect the initial reaction rate, different amounts of the Cmr-α effector complex were supplemented into the reactions so that the percentage of incorporated ATP was always <30% of the total substrate. Four nanomolar of Cmr-α was assayed for the ATP reaction with 5–1500 μM substrate, with the resulting data used for simulation to fit the Michaelis–Menten model (Equation 1). Hill plot analysis was conducted with two datasets using the simplified Hill plot model (Equation 2): (i) ATP reaction products produced by 20 nM Cmr-α in the presence of 1–10 μM substrate and (ii) ATP reaction products produced by 80 nM Cmr-α in the presence of 0.02–1 μM of substrate. The simulation was performed by using OriginPro 2017 software (OriginLab Co., Northampton, MA, USA).
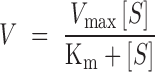 |
(1) |
 |
(2) |
Equation (2) represents a simplified Hill plot model (Equation 3) (50), in which it is assumed that, when the V value is very low in reference to Vmax, ‘Vmax V’ can be regarded as equal to ‘Vmax’.
V’ can be regarded as equal to ‘Vmax’.
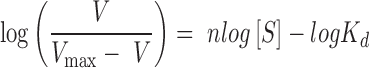 |
(3) |
In Equations (2) and (3), ‘n’ represents the Hill coefficient, reflecting the extent of cooperativity in substrate binding by an enzyme with multiple binding sites (51).
RESULTS
Activation of cyclic tetra-adenylate (c-A4) synthesis by binding of the cognate target RNA to Cmr-α–RNP
Recently, two III-A Csm–RNPs were shown to catalyze cOA synthesis with ATP as the substrate, and the ATP conversion reaction requires the formation of a tertiary Csm complex between Csm–RNP and the cognate target RNA, and mismatches between the 5′-repeat handle of the crRNA and the corresponding 3′ region of the cognate target RNA (37,38). To test if the cOA synthesis could be evolutionarily conserved in III-B Cmr–RNPs, we purified the native Cmr-α–RNP that only contained crRNAs derived from the lacS SS1 spacer. The purified Cmr-α–RNP was then tested for ATP conversion (see ‘Materials and Methods’ section) in presence of one of the following RNAs: (i) SS1-46 (the cognate target RNA), (ii) SS1-50 (target RNA fully complementary to SS1 crRNA), (iii) SS1-40 (a target RNA lacking the 3′-flanking sequence corresponding to the crRNA repeat handle) and (iv) S10 RNA, a non-target RNA to the Cmr-α–RNP complex. All four reaction mixtures were incubated at 70°C for 20 min and subjected to denaturing PAGE to detect ATP reaction products. A single predominant product was observed only in the presence of SS1-46, which was absent from the remaining three reactions (Figure 1A). These results indicated that only the cognate RNA target that carries a non-complementary 3′-flanking region has the capability to activate the binary Cmr-α–RNP for the generation of the ATP reaction product.
Figure 1.

Cmr-α-mediated ATP conversion and its regulation. (A) Cmr-α converts ATP to a dominant product upon binding to target RNA. The Cmr-α complex (40 nM) was incubated with 1 nM α32P-ATP and 100 μM ATP in the presence of different RNA oligos (80 nM) and then the samples were analyzed with denaturing gel electrophoresis. The interactions of the Cmr-α complex with different RNA oligos are depicted above the gel. The 5′-handle of crRNA, the spacer sequence of crRNA and the supplemented RNA oligos are shown in red, black and blue, respectively. (B) Pre-incubation of Cmr-α and target RNA abolishes the ATP reaction. The Cmr-α complex (40 nM) and SS1-46 RNA (40 nM) were pre-incubated until 1 nM α32P-ATP and 100 μM ATP were supplemented into the reaction mixture at indicated delay time points. Then, the samples were further incubated for 20 min, followed by analysis of denaturing gel electrophoresis.
The metal ion dependence of the ATP reaction was then investigated by replacement of Mg2+ with one of the following divalent metal ions in the reaction, including Mn2+, Zn2+, Co2+, Ni2+, Cu2+ and Ca2+. We found that Mg2+, Mn2+, Zn2+ and Co2+ strongly facilitated the ATP reaction, Ni2+ showed a lower efficiency, but Cu2+ and Ca2+ failed to facilitate the ATP reaction (Supplementary Figure S2).
Early studies also showed that the cleavage of the cognate target RNA inactivates the DNase activity of CRISPR-Cas10 systems (28–30), and more recently, the cOA synthesis by Csm–RNPs (37). To investigate if the target RNA regulation could also apply to the ATP reaction by Type III-B systems, the Cmr-α–RNP was incubated at 70°C with SS1-46 for 5–40 min (pre-incubation). Then, ATP was added to the mixtures to initiate the ATP reaction. After incubation at the same temperature for additional 20 min, ATP reaction products were analyzed by denaturing PAGE and quantified. As shown in Figures 1B, 5 min pre-incubation reduced the production of the main product by the Cmr-α complex (indicated by an arrowhead) by 87%, i.e. exhibiting 13% whereas 10 min pre-incubation basically abolished the ATP reaction. Since these results are in agreement with the target RNA cleavage profile by the same effector complex, which reached the equilibrium within 10 min (Supplementary Figure S3) (31), we concluded that the mechanism of the inactivation of the ATP reaction by target RNA cleavage is conserved in III-A and III-B CRISPR-Cas systems.
LC-MS was then used to determine the molecular weight (MW) of the ATP reaction products, and this revealed a predominant product (>70%) with a MW of 1315.2 Da, corresponding either to cyclic tetra-adenylate (c-A4) or to linear A4 containing the terminal 2′, 3′-cyclic phosphate and 5′-hydroxyl group (A4>p) and a minor product of a MW of 986.2 Da matching to either cyclic tri-adenylate (c-A3) or the corresponding linear A3 (A3>p) (Figure 2A). Estimation of MW of these products by denaturing PAGE revealed that the product migrated more slowly than a 5-nt RNA marker in the gel (Figure 2B), a characteristic that is compatible with the circular form of oligoadenylates (c-A3 and c-A4) but incompatible with their corresponding linear form (A4>p or A3>p), in analogy to the results reported for the ATP reaction products of StCsm-RNP (37).
To further demonstrate that the ATP reaction products should be circular, the products were treated with a polynucleotide kinase, a poly(A) RNA polymerase or an alkaline phosphatase to probe reactive chemical groups both at the 5′- and the 3′-position. Aliquots of the reaction products were mixed with each of the enzymes individually and incubated as described in ‘Materials and Methods’ section, and the treated samples were analyzed by denaturing PAGE. As shown in Figure 2B, none of the three enzymes could modify the tri- or tetra-adenylate, indicating that these products do not have any free hydroxyl or phosphate groups at the 5′- and 3′-position. Moreover, labeling of purified products (obtained from Peak 1 and 2 in Figure 2A) by T4 polynucleotide kinase with γ32P-ATP failed to yield any labeled products (Figure 2C), further confirming the absence of any 5′-hydroxy group from the reaction products. In addition, testing the presence of the 3′-5′ phosphodiester bond in the OAs product by S1 nuclease digestion (52) revealed that the products were readily degraded by the enzyme (Figure 2B). Altogether these data indicated that the oligonucleotides synthesized by Cmr-α–RNP are c-A3 and c-A4 containing 3′-5′ phosphodiester bonds.
Cyclic tetra-adenylate activates the Csx1 RNase by interaction with the CARF domain
In III-A CRISPR-Cas10 systems, c-A6 functions as a second messenger to activate Csm6 (37,38), a Cas accessory protein belonging to the Csm6/Csx1 superfamily of Cas accessory proteins (53). Proteins of this superfamily carry an N-terminal CARF domain predicted for ligand binding (45) and a C-terminal HEPN nuclease domain (54). The corresponding member in the S. islandicus Cmr-α system is Csx1 (45,55). The S. islandicus Csx1 was found to play a role in invader DNA silencing by the III-B DNA interference in vivo (23). In addition, the Csx1–CARF domain was found to bind to tetraadenylate, and the interaction strongly activates the nuclease activity from its HEPN domain (46). To test if c-A4 synthesized by Cmr-α could also activate Csx1 in the same fashion, 520 nM of SS1-40 RNA substrate was incubated with 100 nM Csx1 with increasing concentrations of c-A4 for 20 min. All samples were then analyzed by denaturing PAGE. As shown in Figure 3A, c-A4 strongly activated the RNA cleavage activity of Csx1. The extent of Csx1 RNase activation by c-A4 and CAAAA was estimated by quantification of residual substrates, and comparison of the resulting data revealed a ∼3.7-fold difference (Figure 3A and Supplementary Figure S4A). These results indicated that the circular ligand is a better activator to Csx1 RNase than the linear one.
Figure 3.

c-A4 activates the RNA cleavage activity of SisCsx1 by binding to its CARF domain. (A) Activation of SisCsx1 RNA cleavage by c-A4 and a linear poly(A) oligo (CAAAA). The non-cleaved RNA substrate was quantified after incubation with SiCsx1 in the presence of increasing concentrations of c-A4 or CAAAA. Error bar represents S.D. of three independent experiments. The EC50 value determined for c-A4 and CAAAA is 7.4 and 27.3 nM, respectively. (B) Comparison of the binding affinity of SisCsx1 to c-A4 and CAAAA. The two ligands were incubation with 0.16, 0.5, 1.5 and 4.5 μM SiCsx1 and analyzed by non-denaturing PAGE. The percentage of bound ligand was calculated. Error bar represents S.D. of three independent experiments. (C) Schematic of SisCsx1 domain structure. Conserved amino acid residues subjected to mutagenesis are indicated, including one HEPN domain mutant (M1: R399A-H404A) and three CARF domain mutants (M2: G95L-A97L-A99L, M3: D50L-S51L and M6: H154L). (D) RNA substrate was incubated with the wild-type SisCsx1 or each mutant in the presence of 100 nM c-A4, followed by analysis of denaturing PAGE. (E) Labeled c-A4 was incubated with the wild-type and mutated SisCsx1 proteins, followed by analysis of non-denaturing PAGE.
To gain an insight into the enhanced Csx1 activation by c-A4, the binding of each ligand to Csx1 was investigated by EMSA. We found that both ligands formed a complex of large molecular weight (Supplementary Figure S4B). These complexes (Csx1–c-A4 and Csx1–CAAAA) were then quantified, and the resulting data were then plotted against Csx1 concentrations. This revealed that, although the interaction between Csx1 and each ligand was weak, the enzyme showed ∼4–10-fold higher affinity to c-A4 than to CAAAA (Figure 3B). Therefore, the better interaction between c-A4 and Csx1 could be accounted for the elevated level of Csx1 RNase activity with the circular ligand (Figure 3A).
Then, the Csx1 mutants of the CARF or HEPN domain previously constructed (M1, M2, M3 and M6, Figure 3C) (46) were tested for RNA cleavage and their interaction with the c-A4 ligand. This showed that: (i) none of the CARF mutants were active in RNA degradation in the presence of 100 nM c-A4 (Figure 3D), neither were they capable of binding to the c-A4 ligand (Figure 3E), and (ii) M1, the HEPN domain mutant retained the c-A4 binding activity (Figure 3E). Therefore, albeit c-A4 is a more efficient ligand for Csx1 activation, both the circular and the linear ligands interact with the CARF domain of the enzyme and allosterically regulate the enzyme activity from the HEPN domain.
Cmr-α–RNP-mediated ATP reaction is characterized by cooperative substrate binding
Kinetics of the ATP reaction was then investigated by incubation of 4 nM Cmr-α–RNP with a large range of ATP concentrations (5–1500 μM), and the resulting products were analyzed by denaturing PAGE and quantified (Supplementary Figure S5A). The ATP incorporation rates (V) were calculated for all tested substrate concentrations [S] and plotting of the former against the latter yielded the enzyme kinetic data that were fitted into the Michaelis–Menten equation as described in ‘Materials and Methods’ section. The simulation revealed a Vmax of 5.21 ± 0.22 μM/min with a Km value of 233.40 ± 33.04 μM, suggesting a robust ATP incorporation by Cmr-α–RNP (Figure 4A). Nevertheless, changing both V and [S] into a logarithmic scale revealed that the relationship of V and [S] did not fit into the Michaelis–Menten equation in the low substrate range (Figure 4B).
Figure 4.

Michaelis–Menten modeling of the ATP reaction by Cmr-α–RNP. (A) Four nanomolar Cmr–α was incubated with increasing concentrations of ATP and the ATP incorporation rate (V) was plotted to the substrate concentration [S] (blue line). Error bar represents S.D. of three independent experiments. The V versus [S] relationship was further fitted into the Michaelis–Menten model (red line). F-test shows the P value <0.001 for the simulation. (B) The curves from (A) are shown in a logarithmic scale.
To yield a further insight into how Cmr-α–RNP catalyzes the ATP incorporation at low substrate concentrations, we analyzed the ATP incorporation rate of 20 nM Cmr-α–RNP with 1∼10 μM ATP and that of 80 nM Cmr-α–RNP with 0.02∼1 μM ATP, respectively. Plotting of V versus [S] revealed that the two factors exhibit a polynomial relationship in both experiments (Figure 5A and B; Supplementary Figure S5B and C). To test if the data could meet a cooperative binding model, we fitted the data to a simplified Hill plot model as described in ‘Materials and Methods’ section and estimated their Hill coefficient, a value that reflects the extent of cooperativity in substrate binding by a multiple substrate-binding enzyme (51). The analyses revealed that the Hill plot simulation was almost perfect, with an ‘n’ value of 2.33 ± 0.027 for the 80 nM Cmr-α data, and 2.42 ± 0.056 for the 20 nM Cmr-α data (Figure 5C). Considering that the Hill coefficient never exceeds 2.1 in the absence of positive cooperativity in previous simulations (51), these results indicated that Cmr-α-mediated c-A4 synthesis exhibits the feature of cooperative substrate binding.
Figure 5.

Cmr-α-mediated ATP incorporation exhibits cooperative substrate binding. Cmr-α of 80 nM (A) or 20 nM (B) was incubated with different concentration ranges of ATP for which ATP incorporation rates (V) were determined and plotted against substrate concentrations [S] (blue line). Error bar represents S.D. of three independent experiments. (C) The logV versus log[S] relationship for the 80 nM Cmr-α experiements (blue dots) and 20 nM Cmr-α experiments (red dots) was fitted into a linear model: 80 nM Cmr-α (blue dashed line) and 20 nM Cmr-α (red dashed line). F-test indicates the P value is <0.001 for both simulations.
Cmr-α only binds to ATP in the presence of divalent metal ions
Since both target RNA and divalent metal ions are essential for the Cmr-α-mediated ATP reaction (Figure 1A and Supplementary Figure S2), we set to analyze whether both factors could have a role in the enzyme–substrate interaction. The experiments were conducted as described in ‘Materials and Methods’ section. This revealed that, whereas Mg2+ facilitated the binding of ATP to Cmr-α, the target RNA did not show any influence to the enzyme–substrate interaction (Figure 6A). We also found that binding of metal ions to Cmr-α is required, but not sufficient, for cOA synthesis, since Ca2+ and Ni2+ facilitated the enzyme–substrate interaction (Supplementary Figure S6) but failed to activate cOA synthesis by Cmr-α (Supplementary Figure S2). Nevertheless, even at the highest concentration tested here for this enzyme (250 nM), only ∼1.0% of ATP was associated with Cmr-α, suggesting that the effector complex only has a low affinity to ATP (Figure 6B).
Figure 6.

Cmr-α exhibits a weak and divalent metal ion-dependent ATP binding activity. (A) About 1 nM γ32P-ATP was mixed with 200 nM Cmr-α in the presence of 1 mM EDTA or 5 mM MgCl2 and incubated at 70°C for 10 min. One set of these samples was supplemented with 300 nM SS1-46 and further incubated for 3 min at 70°C. All samples were analyzed by non-denaturing PAGE. (B) About 1 nM γ32P-ATP was incubated with increasing concentrations of Cmr-α (50, 100, 200 and 250 nM) in the presence of 5 mM MgCl2 and analyzed by non-denaturing PAGE.
Both Palm domains functioned in generation of the second messenger c-A4
Cas10 proteins of Type III CRISPR-Cas systems contain one HD nuclease domain and two DNA polymerase/cyclase-like (Palm) domains (56). It has been shown that the HD domain is responsible for the target-RNA activated ssDNA cleavage (28–32) and that the conserved GGDD motif in the cyclase domain (Palm2) is the active site for cOA synthesis in two III-A Csm–RNPs (37,38). Furthermore, the structural analysis reveals that both Palm domains are involved in coordinating nucleotide binding (57,58). However, how the two Palm domains could function in cOA synthesis remain to be clarified.
To test how these Cmr2α domains could affect the cOA production, conserved amino acids in each domain were mutated by alanine substitution (Figure 7A, Supplementary Figure S7), and the resulting cmr2α mutants were employed for purification of the corresponding mutated Cmr-α effector complexes including Cmr-α-Cmr2αHD (H14A-D15A), Cmr-α-Cmr2αP2 (D667A-D668A) and Cmr-α-Cmr2αP1 (S214A-S218A) (Supplementary Figure S8A). These Cmr-α–RNPs were analyzed for target RNA cleavage, ssDNA cleavage and c-A4 generation activities as well as for ATP affinity. We found that: (i) none of these mutations impaired the backbone RNA cleavage (Supplementary Figure S8B); (ii) the HD mutation abolished ssDNA cleavage but showed little effect on cOA synthesis (Supplementary Figure S8C); (iii) while the substitutions in Palm 2 essentially abolished cOA synthesis, the Palm 1 domain substitutions strongly impaired the cOA production (40% residual activity, Figure 7B and Supplementary Figure S9A) and (iv) while Cmr-α-Cmr2αP1 exhibited 33% residual ATP binding, Cmr-α-Cmr2αP2 showed less influence (possessing 62% residual ATP affinity) (Figure 7C and Supplementary Figure S9B). Therefore, our work not only confirmed that the DD motif in Palm 2 is the catalytic site of cA4 synthesis, in analogy to other studied Type III CRISPR-Cas systems (37,38), but also suggested that Palm 1 is very important in promoting cooperative substrate binding for robust cOA production.
Figure 7.

Both Palm1 and Palm2 domains of Cmr2α function in c-A4 generation. (A) Schematic of Cmr–2α domain structure. The conserved amino acids in HD, Palm1 and Palm2 domains (subjected to mutagenesis) are indicated. (B) Relative cOA synthesis activity of the wild-type Cmr-α–RNP (WT) and its mutant derivatives. (C) ATP binding affinity of the wild-type Cmr-α–RNP (WT) and its mutant derivatives. Cmr-αHD, Cmr-αP1 and Cmr-αP2: effector complexes carrying substitution mutations in the HD, Palm 1 or Palm 2 domain of Cmr2α. The amount of synthesized cOA/bound ATP by the wild-type Cmr-α complex was arbitrarily set to 1.
DISCUSSION
Here, we show that the S. islandicus Cmr-α catalyzes the synthesis of cOA second messengers with ATP and that the ATP reaction is subjected to the regulation of target RNA binding and cleavage, in analogy to the cOA synthesis by S. thermophilus and E. italicus Csm–RNPs (37,38). Since all tested Type III CRISPR-Cas systems possess the target RNA-activated indiscriminate DNase activity (28–32), altogether these data demonstrate that the multiple nucleic acid interference activities and the cOA signaling are evolutionarily conserved at least in Type III-A and III-B CRISPR-Cas10 systems of both bacterial and archaeal origin.
Our research has also gained important insights into the cOA synthesis mechanism by Type III CRISPR-Cas systems. First, we show that both Palm domains of Cmr2α, a Cas10 protein function in cOA synthesis. This is because mutagenesis of Cmr2α functional domains has revealed that, whereas the Palm 1 domain is more important in substrate binding than Palm 2 (Figure 7C), the latter hosts the active site of cOA synthesis (Figure 7B). Structural analysis of the P. furiosus Cmr2 (PfCmr2), either alone or in complex with PfCmr3 (57,58), has revealed that both Palm domains interact with nucleotides: first, the two α-helices in PfCmr2 Palm 1 interact with the adenine ring of one nucleotide in all three structures, occupying the same position; second, one of the PfuCmr2-Cmr3 subcomplexes contained two ATP molecules, in which the second ATP is present in the Palm 2 domain (58). The higher substrate affinity and more frequent occurrence of ATP-bound forms suggest that Palm 1 could bind ATP first and then Palm 2, but this assumption remains to be tested experimentally. Nevertheless, these findings indicate that both Palm 1 and Palm 2 functions in substrate binding and the polymerization reaction. In addition, the conservation of the S-X-X-X-S motif in the Palm 1 domain in a number of Cmr2 and Csm1 proteins suggests a functional conservation in Cas10 proteins (Supplementary Figure S10).
Investigation of the kinetics of Cmr-α-mediated cOA synthesis has revealed that the ATP reaction involves cooperative substrate binding (Figure 5). It has been reported that cooperative substrate binding can facilitate enzymatic reaction since the binding of the first substrate molecule to an enzyme induces a conformational change that makes the second substrate-binding site more accessible to substrate molecule (59) such as diguanylate cyclases (60). We propose that Type III CRISPR-Cas could have adopted the same mechanism in cOA synthesis as shown in the model shown in Figure 8. Upon the binding of the cognate target RNA to a Type III effector complex, the Palm 1 site in Cas10 proteins serves as the primary binding site of the enzyme–substrate interaction, and the Palm 1–ATP interaction induces a conformation change that allows the Palm 2 also effectively interacts with ATP to start the cOA synthesis, forming the first phosphodiester bond. Then, the effector complex translocates on the 2-nt intermediate, allowing the enzyme to interact with new incoming ATP. The process is repeated for three times to yield the chain elongation in the nucleotide synthesis. Finally, the complex recaptures the first nucleotide of the oligoadenylate RNA at the last step of the synthesis, and condensation between the 3′-hydroxyl group of the last adenylate of the oligonucleotides and the 5′-triphosphate group of the first adenylate circularizes the tetraadenylate, giving c-A4 (Figure 8).
Figure 8.

A model for Cmr-α-mediated c-A4 synthesis. Cmr-α is represented with Palm1 (P1), Palm2 (P2) and D2 (D2) domains of the Cmr–2α, three of the five conserved domains identified in the P. furiosus Cmr2 (57). Binding of the cognate target RNA to Cmr-α yields a ternary Cmr-α complex (A). Upon the binding of the first ATP molecule to the ternary Cmr-α, the substrate-enzyme intermediate adopts a conformational change and becomes more accessible to a second ATP molecule (B and C). Nucleophilic attack from the 3′-hydroxyl group (3′-OH) of the first ATP molecule to the 5′-triphosphate group (5′-P) of the second ATP molecule yields a phosphodiester bond between the two nucleotides (D). Cmr-α–RNP translocates on the 2-nt intermediate, freeing one of the ATP-binding sites (E). The process is repeated, leading to the formation of the third and the fourth phosphodiester bond (E and F). Finally, the substrate-free active site in Cmr-α recaptures the first nucleotide of the poly-A4 RNA (F and G) and circularizes the tetraadenylate in a condensation reaction.
Nevertheless, it remains to be investigated if other Type III CRISPR-Cas systems have also adopted the cooperative substrate binding mechanism in their cOA synthesis. This is because functional diversification has already been observed for Cmr and Csm systems. For example, while the DD motif of Palm 2 only plays a minor role in ATP binding in the Cmr-α–RNP system, suggesting that ATP molecules bind to the effector complex even when the polymerase domain is in the inactive form. By contrast, the DD motif in StCsm1 is essential for ATP binding by StCsm-RNP, the corresponding effector complex (37). Therefore, it is interesting to study if the diversification of Cmr and Csm systems could have influenced the conservation of cooperative substrate binding in cOA synthesis.
In analogy to c-A6 synthesized by Type III-A Csm systems (37,38), the Sis-Cmr-α-synthesized c-A4 functions as the second messenger to activate Csx1, a Cas accessory protein that has been implicated in the Cmr-α DNA interference (23). In the process of data analysis, we noticed some striking differences in cOA synthesis and Csm6/Csx1 activation. First, Cmr-α–RNP exhibits a robust c-A4 synthetic activity, which should be stronger than that of c-A6 synthesis by StCsm-RNP and EiCsm-RNP (37,38). Second, SisCsx1 and TtCsm6 that interact with c-A4 show a relatively weak RNase activity relative to StCsm6, StCsm6′ and EiCsm6, which bind to c-A6 ((37,38) and this work). Those data suggest a reverse relation between the rate of cOA synthesis by a Type III RNP and the activation efficiency of CARF-domain RNase by the corresponding cOA ligand. The reverse relation might be attributed to the coevolution of the two enzymes in the CRISPR signaling pathway such that the intrinsic drawback of the weakened ligand–enzyme interaction is complemented by an elevated level of ligand synthesis, and vice versa. Such a coevolution would ensure a proper function of CRISPR signaling in antiviral defense.
In a previous work, we showed that RNAs carrying a tetraadenylate tail are capable of strongly activating the indiscriminate RNase of SisCsx1. Here, we further show that the efficiency of the SisCsx1 activation by c-A4 is a few folds higher than that by the linear one. Since it has been reported that linear RNAs carrying a poly-A tail are intermediates of the RNA decay pathway present in many prokaryotes (61), including S. solfataricus (62,63). The finding that both linear and circular tetraadenylate can serve as a second messenger to activate Csx1 has important implication in the biology of this organism. We hypothesize that the dynamic interaction between poly-A-tailed RNAs and SisCsx1 could reflect that some microorganisms have explored the cOA–CARF domain pathway for other stress regulation such that cellular metabolic activity is to be greatly reduced under stressful conditions. For example, a programmed cell death pathway can be induced by DNA damage treatment (64), and the induction of the programmed cell death could be coupled to a novel type of DNA damage response regulation recently been discovered in S. islandicus since lack of DNA damage response regulation greatly facilitates cell death (65,66). To this end, it is very tempting to investigate whether linear and/or circular tetraadenylate could play a role in the stress-responsive regulation in this archaeon.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of both research groups of for stimulating discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation of China [31771380]; Danish Council for Independent Research [DFF-4181-00274]; Novo Nordisk Foundation Center for Protein Research, Novo Nordisk Foundation [NNF14CC0001]. Funding for open access charge: National Science Foundation of China [31771380].
Conflict of interest statement. None declared.
REFERENCES
- 1. Amitai G., Sorek R.. CRISPR-Cas adaptation: insights into the mechanism of action. Nat. Rev. Microbiol. 2016; 14:67–76. [DOI] [PubMed] [Google Scholar]
- 2. Sternberg S.H., Richter H., Charpentier E., Qimron U.. Adaptation in CRISPR-Cas systems. Mol. Cell. 2016; 61:797–808. [DOI] [PubMed] [Google Scholar]
- 3. Jackson S.A., McKenzie R.E., Fagerlund R.D., Kieper S.N., Fineran P.C., Brouns S.J.. CRISPR-Cas: adapting to change. Science. 2017; 356:aal5056. [DOI] [PubMed] [Google Scholar]
- 4. Charpentier E., Richter H., van der Oost J., White M.F.. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol. Rev. 2015; 39:428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Plagens A., Richter H., Charpentier E., Randau L.. DNA and RNA interference mechanisms by CRISPR-Cas surveillance complexes. FEMS Microbiol. Rev. 2015; 39:442–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J., Charpentier E., Haft D.H. et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koonin E.V., Makarova K.S., Zhang F.. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017; 37:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marraffini L.A. CRISPR-Cas immunity in prokaryotes. Nature. 2015; 526:55–61. [DOI] [PubMed] [Google Scholar]
- 9. Mohanraju P., Makarova K.S., Zetsche B., Zhang F., Koonin E.V., van der Oost J.. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016; 353:aad5147. [DOI] [PubMed] [Google Scholar]
- 10. Wright A.V., Nunez J.K., Doudna J.A.. Biology and applications of CRISPR systems: harnessing nature's toolbox for genome engineering. Cell. 2016; 164:29–44. [DOI] [PubMed] [Google Scholar]
- 11. Barrangou R., Horvath P.. A decade of discovery: CRISPR functions and applications. Nat. Microbiol. 2017; 2:17092. [DOI] [PubMed] [Google Scholar]
- 12. Han W., She Q.. CRISPR history: discovery, characterization, and prosperity. Prog. Mol. Biol. Transl. Sci. 2017; 152:1–21. [DOI] [PubMed] [Google Scholar]
- 13. Marraffini L.A., Sontheimer E.J.. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008; 322:1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marraffini L.A., Sontheimer E.J.. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010; 463:568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M., Terns M.P.. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009; 139:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Staals R.H., Agari Y., Maki-Yonekura S., Zhu Y., Taylor D.W., van Duijn E., Barendregt A., Vlot M., Koehorst J.J. et al. Structure and activity of the RNA-targeting Type III-B CRISPR-Cas complex of Thermus thermophilus. Mol. Cell. 2013; 52:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hale C.R., Cocozaki A., Li H., Terns R.M., Terns M.P.. Target RNA capture and cleavage by the Cmr type III-B CRISPR-Cas effector complex. Genes Dev. 2014; 28:2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benda C., Ebert J., Scheltema R.A., Schiller H.B., Baumgartner M., Bonneau F., Mann M., Conti E.. Structural model of a CRISPR RNA-silencing complex reveals the RNA-target cleavage activity in Cmr4. Mol. Cell. 2014; 56:43–54. [DOI] [PubMed] [Google Scholar]
- 19. Staals R.H., Zhu Y., Taylor D.W., Kornfeld J.E., Sharma K., Barendregt A., Koehorst J.J., Vlot M., Neupane N., Varossieau K. et al. RNA targeting by the type III-A CRISPR-Cas Csm complex of Thermus thermophilus. Mol. Cell. 2014; 56:518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tamulaitis G., Kazlauskiene M., Manakova E., Venclovas C., Nwokeoji A.O., Dickman M.J., Horvath P., Siksnys V.. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol. Cell. 2014; 56:506–517. [DOI] [PubMed] [Google Scholar]
- 21. Osawa T., Inanaga H., Sato C., Numata T.. Crystal structure of the CRISPR-Cas RNA silencing Cmr complex bound to a target analog. Mol. Cell. 2015; 58:418–430. [DOI] [PubMed] [Google Scholar]
- 22. Zhu X., Ye K.. Cmr4 is the slicer in the RNA-targeting Cmr CRISPR complex. Nucleic Acids Res. 2015; 43:1257–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deng L., Garrett R.A., Shah S.A., Peng X., She Q.. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol. Microbiol. 2013; 87:1088–1099. [DOI] [PubMed] [Google Scholar]
- 24. Peng W., Feng M., Feng X., Liang Y.X., She Q.. An archaeal CRISPR type III-B system exhibiting distinctive RNA targeting features and mediating dual RNA and DNA interference. Nucleic Acids Res. 2015; 43:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldberg G.W., Jiang W., Bikard D., Marraffini L.A.. Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature. 2014; 514:633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A.. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell. 2015; 161:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ichikawa H.T., Cooper J.C., Lo L., Potter J., Terns R.M., Terns M.P.. Programmable type III-A CRISPR-Cas DNA targeting modules. PLoS One. 2017; 12:e0176221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elmore J.R., Sheppard N.F., Ramia N., Deighan T., Li H., Terns R.M., Terns M.P.. Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR-Cas system. Genes Dev. 2016; 30:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Estrella M.A., Kuo F.T., Bailey S.. RNA-activated DNA cleavage by the Type III-B CRISPR-Cas effector complex. Genes Dev. 2016; 30:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kazlauskiene M., Tamulaitis G., Kostiuk G., Venclovas C., Siksnys V.. Spatiotemporal control of Type III-A CRISPR-Cas immunity: coupling DNA degradation with the target RNA recognition. Mol. Cell. 2016; 62:295–306. [DOI] [PubMed] [Google Scholar]
- 31. Han W., Li Y., Deng L., Feng M., Peng W., Hallstrom S., Zhang J., Peng N., Liang Y.X., White M.F. et al. A type III-B CRISPR-Cas effector complex mediating massive target DNA destruction. Nucleic Acids Res. 2017; 45:1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu T.Y., Iavarone A.T., Doudna J.A.. RNA and DNA targeting by a reconstituted thermus thermophilus Type III-A CRISPR-Cas system. PLoS One. 2017; 12:e0170552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang J., Graham S., Tello A., Liu H., White M.F.. Multiple nucleic acid cleavage modes in divergent type III CRISPR systems. Nucleic Acids Res. 2016; 44:1789–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pyenson N.C., Marraffini L.A.. Type III CRISPR-Cas systems: when DNA cleavage just isn’t enough. Curr. Opin. Microbiol. 2017; 37:150–154. [DOI] [PubMed] [Google Scholar]
- 35. Tamulaitis G., Venclovas C., Siksnys V.. Type III CRISPR-Cas immunity: major differences brushed aside. Trends Microbiol. 2017; 25:49–61. [DOI] [PubMed] [Google Scholar]
- 36. Liu T., Pan S., Li Y., Peng N., She Q.. Type III CRISPR-Cas system: introduction and its application for genetic manipulations. Curr. Issues Mol. Biol. 2018; 26:1–14. [DOI] [PubMed] [Google Scholar]
- 37. Kazlauskiene M., Kostiuk G., Venclovas C., Tamulaitis G., Siksnys V.. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017; 357:605–609. [DOI] [PubMed] [Google Scholar]
- 38. Niewoehner O., Garcia-Doval C., Rostol J.T., Berk C., Schwede F., Bigler L., Hall J., Marraffini L.A., Jinek M.. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017; 548:542–548. [DOI] [PubMed] [Google Scholar]
- 39. Hatoum-Aslan A., Maniv I., Samai P., Marraffini L.A.. Genetic characterization of antiplasmid immunity through a type III-A CRISPR-Cas system. J. Bacteriol. 2014; 196:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jiang W., Samai P., Marraffini L.A.. Degradation of phage transcripts by CRISPR-associated RNases enables Type III CRISPR-Cas immunity. Cell. 2016; 164:710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peng N., Han W., Li Y., Liang Y., She Q.. Genetic technologies for extremely thermophilic microorganisms of Sulfolobus, the only genetically tractable genus of crenarchaea. Sci. China Life Sci. 2017; 60:370–385. [DOI] [PubMed] [Google Scholar]
- 42. Guo L., Brugger K., Liu C., Shah S.A., Zheng H., Zhu Y., Wang S., Lillestol R.K., Chen L., Frank J. et al. Genome analyses of Icelandic strains of Sulfolobus islandicus, model organisms for genetic and virus-host interaction studies. J. Bacteriol. 2011; 193:1672–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peng W., Li H., Hallstrom S., Peng N., Liang Y.X., She Q.. Genetic determinants of PAM-dependent DNA targeting and pre-crRNA processing in Sulfolobus islandicus. RNA Biol. 2013; 10:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Y., Zhang Y., Lin J., Pan S., Han W., Peng N., Liang Y.X., She Q.. Cmr1 enables efficient RNA and DNA interference of a III-B CRISPR-Cas system by binding to target RNA and crRNA. Nucleic Acids Res. 2017; 45:11305–11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chylinski K., Makarova K.S., Charpentier E., Koonin E.V.. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014; 42:6091–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Han W., Pan S., Lopez-Mendez B., Montoya G., She Q.. Allosteric regulation of Csx1, a type IIIB-associated CARF domain ribonuclease by RNAs carrying a tetraadenylate tail. Nucleic Acids Res. 2017; 45:10740–10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peng N., Deng L., Mei Y., Jiang D., Hu Y., Awayez M., Liang Y., She Q.. A synthetic arabinose-inducible promoter confers high levels of recombinant protein expression in hyperthermophilic archaeon Sulfolobus islandicus. Appl. Environ. Microbiol. 2012; 78:5630–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Warrens A.N., Jones M.D., Lechler R.I.. Splicing by overlap extension by PCR using asymmetric amplification: an improved technique for the generation of hybrid proteins of immunological interest. Gene. 1997; 186:29–35. [DOI] [PubMed] [Google Scholar]
- 49. Zhang J., White M.F.. Expression and purification of the CMR (Type III-B) complex in Sulfolobus solfataricus. Methods Mol. Biol. 2015; 1311:185–194. [DOI] [PubMed] [Google Scholar]
- 50. Stefan M.I., Le Novere N.. Cooperative binding. PLoS Comput. Biol. 2013; 9:e1003106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weiss J.N. The Hill equation revisited: uses and misuses. FASEB J. 1997; 11:835–841. [PubMed] [Google Scholar]
- 52. Oleson A.E., Sasakuma M.. S1 nuclease of Aspergillus oryzae: a glycoprotein with an associated nucleotidase activity. Arch. Biochem. Biophys. 1980; 204:361–370. [DOI] [PubMed] [Google Scholar]
- 53. Makarova K.S., Anantharaman V., Grishin N.V., Koonin E.V., Aravind L.. CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front. Genet. 2014; 5:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Anantharaman V., Makarova K.S., Burroughs A.M., Koonin E.V., Aravind L.. Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol. Direct. 2013; 8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vestergaard G., Garrett R.A., Shah S.A.. CRISPR adaptive immune systems of Archaea. RNA Biol. 2014; 11:156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Makarova K.S., Aravind L., Wolf Y.I., Koonin E.V.. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct. 2011; 6:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cocozaki A.I., Ramia N.F., Shao Y., Hale C.R., Terns R.M., Terns M.P., Li H.. Structure of the Cmr2 subunit of the CRISPR-Cas RNA silencing complex. Structure. 2012; 20:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Osawa T., Inanaga H., Numata T.. Crystal structure of the Cmr2-Cmr3 subcomplex in the CRISPR-Cas RNA silencing effector complex. J. Mol. Biol. 2013; 425:3811–3823. [DOI] [PubMed] [Google Scholar]
- 59. Ricard J., Cornish-Bowden A.. Co-operative and allosteric enzymes: 20 years on. Eur. J. Biochem. 1987; 166:255–272. [DOI] [PubMed] [Google Scholar]
- 60. Oliveira M.C., Teixeira R.D., Andrade M.O., Pinheiro G.M., Ramos C.H., Farah C.S.. Cooperative substrate binding by a diguanylate cyclase. J. Mol. Biol. 2015; 427:415–432. [DOI] [PubMed] [Google Scholar]
- 61. Dreyfus M., Regnier P.. The poly(A) tail of mRNAs: bodyguard in eukaryotes, scavenger in bacteria. Cell. 2002; 111:611–613. [DOI] [PubMed] [Google Scholar]
- 62. Hou L., Klug G., Evguenieva-Hackenberg E.. Archaeal DnaG contains a conserved N-terminal RNA-binding domain and enables tailing of rRNA by the exosome. Nucleic Acids Res. 2014; 42:12691–12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martens B., Hou L., Amman F., Wolfinger M.T., Evguenieva-Hackenberg E., Blasi U.. The SmAP1/2 proteins of the crenarchaeon Sulfolobus solfataricus interact with the exosome and stimulate A-rich tailing of transcripts. Nucleic Acids Res. 2017; 45:7938–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Han W., Xu Y., Feng X., Liang Y.X., Huang L., Shen Y., She Q.. NQO-Induced DNA-Less cell formation is associated with chromatin protein degradation and dependent on A0A1-ATPase in sulfolobus. Front. Microbiol. 2017; 8:1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sun M., Feng X., Liu Z., Han W., Liang Y.X., She Q.. An Orc1/Cdc6 ortholog functions as a key regulator in the DNA damage response in Archaea. Nucleic Acids Res. 2018; 46:6697–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Feng X., Sun M., Han W., Liang Y.X., She Q.. A transcriptional factor B paralog functions as an activator to DNA damage-responsive expression in archaea. Nucleic Acids Res. 2018; 46:7085–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.