


Abstract
DNA methylation has been associated with transcriptional repression and detection of differential methylation is important in understanding the underlying causes of differential gene expression. Bisulfite-converted genomic DNA sequencing is the current gold standard in the field for building genome-wide maps at a base pair resolution of DNA methylation. Here we systematically investigate the underlying features of detecting differential DNA methylation in CpG and non-CpG contexts, considering both the case of mammalian systems and plants. In particular, we introduce DMRcaller, a highly efficient R/Bioconductor package, which implements several methods to detect differentially methylated regions (DMRs) between two samples. Most importantly, we show that different algorithms are required to compute DMRs and the most appropriate algorithm in each case depends on the sequence context and levels of methylation. Furthermore, we show that DMRcaller outperforms other available packages and we propose a new method to select the parameters for this tool and for other available tools. DMRcaller is a comprehensive tool for differential methylation analysis which displays high sensitivity and specificity for the detection of DMRs and performs entire genome wide analysis within a few hours.
INTRODUCTION
DNA methylation is one of the most common epigenetic modifications that is stably inherited and affects gene regulation (1,2). Predominantly, it involves the addition of a methyl group to the carbon-5 position of cytosines and is associated with transcriptional repression. Detection of the methylated cytosines is usually accomplished by bisulfite treatment of the DNA, which leads to unmethylated cytosines being converted to uracil, whilst the 5-methylcytosines remaining unaffected. Given the reduction in cost of DNA sequencing, genome wide bisulfite converted DNA sequencing (BS-seq) has become the method of choice to determine methylation distribution at genomic scale. This approach, despite generating methylation information at single base resolution for theoretically every cytosine in the genome, frequently requires further analysis to efficiently extract methylation information for entire loci, or to highlight methylation differences in regions across different conditions, cell types or genotypes.
There are several tools available to detect differentially methylated regions (DMRs) from BS-seq datasets and whilst some of them are implemented in different programming languages (e.g. (3)) or through a web interface, the majority are provided as R packages. Due to its statistical power and the available libraries and packages for bioinformatics analysis, R is the programming language of choice for analysing genomic datasets (4). In particular, Bioconductor (5) represents a collection of R packages aimed to bioinformatics analysis and currently contains more than 1500 packages. The most popular R packages used for detecting differential methylated regions include: methylKit (6), bsseq (7), BiSeq (8), methylSig (9), DSS (10), RnBeads (11), methylPipe (12), BEAT (13) and MD3 (14).
Most of these tools were developed for mammalian systems, where DNA is predominantly methylated in CpG context (15–18), and, consequently, they were designed to detect DMRs only in CpG context; e.g. bsseq (7), BiSeq (8) and RnBeads (11). Nevertheless, in plants, non-CpG methylation (in CpHpG and CpHpH contexts, where H can be A, C or T) is also present, playing an important role in epigenetic regulation of transcription (19–23). In addition, recent work has shown the existence of non-CpG methylation in mammalian cell lines (16–18). Whilst there are some tools that can detect differential methylation in a context dependent manner (such as methylKit, methylSig and methylPipe), they mainly consist of partitioning the genome in tilling bins and pooling together the methylation levels of all cytosines in each bin.
Here, we present a new R package DMRcaller, which can compute DMRs between two samples in a methylation context dependent manner. This tool takes as input already pre-processed BS-seq data in the form of methylation calls at each cytosine in the genome. DMRcaller implements several methods to detect DMRs and is highly configurable by allowing a wide range of parameters to be controlled. We provide evidence that DMRcaller displays the highest accuracy when computing DMRs compared to other available tools, outperforming other methods. Most importantly, we show that the best method to detect DMRs depends on the methylation context (CpG, CpHpG or CpHpH) and DMRcaller implements several methods, which makes this package a comprehensive tool for differential methylation analysis. In addition, our results confirm that the detected DMRs are highly reproducible in biological replicates, thus, further providing evidence on the accuracy of the method. Finally, we show that the computing time of DMRs is fast, allowing computing DMRs on whole genome BS-seq datasets generated from human cells/tissues within a few hours.
MATERIALS AND METHODS
Description of DMRcaller
We developed an R/Bioconductor (4,5) package called DMRcaller (available at http://bioconductor.org/packages/DMRcaller/), which computes DMRs between two conditions. DMRcaller uses as input a tab delimited text file with the following columns: chromosome, position, strand, number of reads from methylated DNA, total number of reads, Cytosine context (CG, CHG or CHH) and trinucleotide context (were, instead of H, the exact nucleotide is included). This format is the same of the CX report generated by the popular aligner Bismark (24), however the analysis performed by DMRcaller is independent of the aligners that were used as long as the methylation data is formatted accordingly. Figure 1 presents the workflow that one can use to call DMRs using CX report files from two conditions.
Figure 1.

BS-seq analysis workflow using DMRcaller.
Data preprocessing
The package computes DMRs with three methods: (i) neighbourhood (DMRcaller-N), (ii) bins (DMRcaller-B) and (iii) noise filter (DMRcaller-NF). The main difference between these methods is the way the methylation data is preprocessed. In the case of the neighbourhood method, the algorithm considers each cytosine independently and, using the raw number of reads and reads from methylated DNA, it calls differentially methylated cytosines (DMCs), which can be extended to DMRs. The second method (DMRcaller-B) splits the genome into tilling bins and then pools all reads and reads from methylated DNA in each bin before calling differentially methylated bins. Finally, the noise filter method (DMRcaller-NF), preprocess the methylation data by applying a smoothing kernel on the total number of reads and the number of reads from methylated DNA before computing the differential methylation. The noise filter method uses the same assumption of BSmooth (7), in particular, that neighbouring cytosines display correlated methylation. DMRcaller implements four kernels for noise filtering: (i) uniform, (ii) triangular, (iii) Gaussian and (iv) Epanechnicov; see Supplementary Section S1. For example, the smoothed number of methylated or total reads at a position in the genome is the weighted average over a window (of specified size), where, in the case of a triangular, Gaussian and Epanechnicov kernels, the contribution of distant Cytosines is lower than the contribution of closer ones. As consequence of the smoothing, each position in the genome acquired a smoothed value for both the total number of reads and a number of reads from methylated DNA and, using these corrected values, the function calls the differentially methylated positions (DMPs) using Algorithm 1.
Calling DMRs
To detect DMRs, in the case of all methods, the algorithm performs the statistical test for each position, cytosine or bin and then marks as DMRs all positions, cytosines or bins that satisfy the following three conditions:
Algorithm 1. Select DMPs, cytosines, bins or regions.
the difference in methylation levels between the two conditions is statistically significant according to the statistical test
the difference in methylation proportion between the two conditions is higher than a threshold value
the mean number of reads per cytosine is higher than a threshold
DMRcaller implements three statistical tests: (i) Fisher’s exact test, (ii) the Score test (which is a z-test; see Supplementary Section S2 for details) and (iii) Beta regression test for biological replicates. For all statistical tests, we adjust the P-values for multiple testing using Benjamini and Hochberg’s method (25) to control the false discovery. Note that first two of these tests lead to similar results, but Score test is slightly faster to compute compared to Fisher’s exact test (see below). Thus, in our analysis, when not computing DMRs on biological replicates, we used Score test to compute differential methylation.
Merging DMRs
Adjacent DMPs, cytosines or bins are merged using an iterative process, where neighbouring DMPs, cytosines or bins in the same sequence context (within a certain distance of each other) are joined only if the three conditions listed in Algorithm 1 are still met. These regions are then called DMRs.
Finally, DMRs can be filtered as follows:
Algorithm 2. Filter DMRs
Remove DMRs whose lengths are less than a minimum size
Remove DMRs with fewer cytosines than a threshold value
Additional functionality
For a set of potential DMRs (e.g. genes, transposable elements or CpG islands), DMRcaller can compute if these regions are differentially methylated by first pooling all reads in each region and then evaluating whether the three conditions in Algorithm 1 are met.
BS-seq data and processing
We used four previously published BS-seq datasets of Arabidopsis thaliana plants: (i) WT replicate 1 (GSM1242401), (ii) WT replicate 2 (GSM980986), (iii) first generation met1-3 (GSM981031) and (iv) drm1/2 cmt2/3 (ddcc) (GSM1242404) (20,22). These datasets were generated using 3-week-old leaves from A. thaliana plants in the Columbia background that were grown under continuous light. In addition, for the biological replicates analysis we used an additional previously published BS-seq dataset of A. thaliana plants: (i) WT (GSM2384978) and (ii) first generation met1-3 (GSM2384980) (26). These datasets were generated using 2-week-old seedlings from A. thaliana plants in the Columbia background that were grown under long-day conditions (16 h light, 8 h dark).
Furthermore, we analysed a bisulfite sequencing dataset from rice endosperm (GSM560563) and embryo (GSM560562) together with a list of embryo and endosperm specifically expressed genes published in (27).
Finally, we also used two BS-seq dataset in human IMR90 and H1 cell lines published in (15). In particular, we used a preprocessed version of this dataset from ListerEtAlBSseq Bioconductor metadata package (28).
Computing DMRs
The workflow to compute DMRs is listed in Figure 1. We selected DMRs that contain at least one cytosine, have a size of at least 50 bp, display a difference in methylation levels of at least 40% and this difference is statistically significant according to the Score test with an adjusted P-value ≤ 0.05. DMRs within a certain distance (equal to twice the window size) of each other were joined if all these conditions were still met. For methylKit, methylSig and methylPipe we used the set of parameters provided in (12) and only varied the window size in the case of methylKit and methylSig.
RESULTS
Considerations on the methods to detect DMRs
The neighbourhood method assumes the computation of DMCs with no prior filtering or smoothing of the data. Although this is the only implemented method able to call directly single DMCs, one of the disadvantages is that, in the majority of libraries, a consistent subset of cytosines will often have too few reads for the statistical test to be able to call those cytosines as differentially methylated. This is due to the unequal amplification of DNA produced during the library preparation (29), a problem that can be only partially compensated by a higher sequence coverage.
Supplementary Figure S2 shows the coverage of two biological replicates for a BS-seq of A. thaliana WT plants. Even for a genome as small as A. thaliana (≈130 Mb), only around 60% of the cytosines in CpG context have at least 10 reads in two different BS-seq experiments. In the case of cytosines in CpHpH context, the coverage is even lower than that. This means that DMRs might not be properly detected in a large part of the genome. In another example, a 30× coverage for BS-seq experiment in human cells lead to similar coverage as in the case of A. thaliana (60% of the cytosines had at least 10 reads) (15).
One possible solution to alleviate this problem is to smooth the data using a smoothing kernel, which could allow identification of DMRs with much lower coverage (7). The noise filter method uses a moving average to remove non-homogeneous coverage of the genome. The main assumption of the method is that neighbouring cytosines display correlated methylation levels, which seems to be valid in the case of CpG methylation in mammals (30). To determine the usefulness of the noise filter method, we need to test whether this assumption is valid for other organisms (e.g. plants) for both CpG and non-CpG methylation. For this, we considered WT A. thaliana plants and computed the Pearson’s correlation coefficient between methylation levels of cytosines in the same context that are separated by a fixed distance. Supplementary Figure S3 confirms that CpG methylation display high correlation within 1 Kb, which is similar to the case of CpG methylation in mammalian systems (30). Furthermore, methylation in CpHpG context seems to display similarly high correlation as CpG. In contrast to that, CpHpH methylation displays a low correlation which steeply drops for distances higher than 20 bp, possibly reflecting the different pathway of methylation in this cytosine context in plants (21,23). Together, these results indicate that the noise filter method can be applied on CpG methylation and potentially to CpHpG methylation, but is likely not suitable for CpHpH methylation if used with window sizes higher than 20 bp.
The last method implemented in DMRcaller is the bins method ((DMRcaller-B). This method partitions the genome in fixed size tilling bins, pooling together all reads in each bin and then performing a statistical test to determine which of the bins display different levels of methylation. Pooling reads within 100 bp bins leads to high number of reads and smaller differences become statistically significant. This is a popular method which is implemented in several other R packages (e.g. (6,9)), but one question which has not been addressed yet is how binning the reads affects the accuracy.
Comparison with other tools
In Table 1, we listed several R/Bioconductor packages that compute DMRs. In addition to DMRcaller, only methylPipe, methylKit and methylSig can handle non-CpG methylation and, thus, can be applied to plants or to study non-CpG methylation in mammals. It is worthwhile noting that whilst the majority of other packages implement only one method (tilling bins, smoothing, calling differential methylation at individual cytosines, etc.), DMRcaller implements three algorithms, thus, allowing more flexibility in applying the most appropriate detection method to call DMR for each cytosine context.
Table 1.
R/Bioconductor packages for calling DMRs on WGBS
| Package | Link | non-CpG | |
|---|---|---|---|
| DMRcaller | http://bioconductor.org/packages/DMRcaller/ | Yes | |
| methylKit | https://github.com/al2na/methylKit | Yes | (6) |
| methylSig | https://github.com/sartorlab/methylSig | Yes | (9) |
| BiSeq | http://bioconductor.org/packages/BiSeq/ | No | (8) |
| bsseq | http://bioconductor.org/packages/bsseq/ | No | (7) |
| methylPipe | http://bioconductor.org/packages/methylPipe/ | Yes | (12) |
| RnBeads | http://bioconductor.org/packages/RnBeads/ | No | (11) |
| BEAT | http://bioconductor.org/packages/BEAT/ | No | (13) |
| M3D | http://bioconductor.org/packages/M3D/ | No | (14) |
| DSS | http://bioconductor.org/packages/DSS/ | No | (10) |
In our analysis, we compared DMRcaller (bins and noise filter methods) with two of the most popular tools for detecting differential methylation: methylKit and methylSig. To test the performance of our tool to detect DMRs, we first considered the case of CpG methylation in A. thaliana. METHYLTRANSFERASE1 (MET1) is the main methyltransferase involved in the maintenance of CpG methylation in A. thaliana (26,31,32) and, in met1-3 mutant, CpG methylation is completely removed; see Supplementary Figure S4A. One parameter that needs to be selected for all four methods is the bin size (window size in the case of DMRcaller-NF) and, since all methods lead to DMRs of different size, we computed the DMRs genome coverage (total size of DMRs) for each method to compare the results of the different tools. Taking in account that CpG methylation in met1-3 mutant is absent, we assume that all DMRs that are detected in the comparison with wild type methylation are true (there is no false positive). Therefore, the method that can recover the highest genome coverage of DMRs will perform best. Figure 2 shows that all tilling bin methods lead to similar DMR genome coverage (39.76 Mb for methylKit, 39.26 Mb for methylSig and 41.84 Mb for DMRcaller-B) with the DMRcaller-B displaying a slightly higher value. This can be explained by the fact that, in DMRcaller, DMRs are joined if the initial conditions are still met, which will result in larger DMRs. Interestingly, DMRcaller-NF produces the highest DMR genome coverage (48.14 Mb), which suggests that this method is the most sensitive of the four methods considered in our analysis.
Figure 2.

Genome coverage of CpG DMRs between WT and met1-3 Arabidopsis thaliana plants as a function of window/bin size. The graph plots the total size of DMRs computed using: (i) methylKit, (ii) methylSig, (iii) DMRcaller-B and (iv) DMRcaller-NF.
To investigate the difference between DMRcaller and other tools to call DMRs, we compared regions uniquely identified by DMRcaller-NF with common regions identified by both DMRcaller and methylKit (the method that displayed highest DMR genome coverage excluding DMRcaller). We observed that DMR portions identified only by DMRcaller are mostly adjacent to common regions identified by both methods (10.1 Mb, which represents ≈82% of DMRcaller-NF specific DMRs), indicating that most of DNA regions uniquely identified by DMRcaller are extension of DMRs called by methylKit. These adjacent DMR portions are consistently less methylated in WT samples if compared to common portions (see Supplementary Figure S5B). Considering that CpG methylation in met1-3 mutant is always absent, this suggests that the smoothing function implemented in the noise filter method can efficiently call regions with a lower methylation change, using information from neighboring higher methylated genomic areas. In contrast, DMRs exclusively called by DMRcaller-NF which are not adjacent to commonly identified regions display higher methylation but are shorter in size (see Supplementary Figure S5C), indicating that smoothed data can more efficiently be used to call smaller DMRs, by increasing the amount of informative positions used in the statistical test. Consistently with this, the DMRcaller-NF specific regions also display a slightly lower coverage compared to common DMRs (see Supplementary Figure S5A).
Furthermore, we evaluated whether the statistical test used impacted the results. Supplementary Figure S6A and B shows that the there is an almost perfect overlap between the DMRs called in CpG context using the Score test or the Fisher’s exact test and this is valid for both using the noise filter method or the bins method. Nevertheless, the Score test leads to faster computing of DMRs specifically for noise filter method (where the statistical test is performed for each position in the genome); see Supplementary Figure S6C and D.
Next, to test the accuracy of DMRcaller, we considered a condition where only a few differences in CpG methylation were expected (compare to previous example). For this, we computed the DMRs in CpG context for two BS-seq dataset previously published in (15): (i) IMR90 and (ii) H1 cells. Figure 3A shows genome coverage of DMRs called by the four methods ( DMRcaller-B, DMRcaller-NF, methylKit and methylSig) on chromosome 1 of the human genome and shows that methylKit and methylSig display lower genome coverage of DMRs (32.50 and 29.39 Mb) compared to the two DMRcaller methods (35.14 and 35.85 Mb). Similar as in the case of CpG methylation in A. thaliana, there is an optimal bin/window size that leads to highest genome coverage of DMRs for each method, which corresponds to the highest sensitivity for the methods.
Figure 3.

Genome coverage of DMRs between H1 and IMR90 human cells as a function of window/bin size. The graph plots the total size of DMRs computed using: (i) methylKit, (ii) methylSig, (iii) DMRcaller-B and (iv) DMRcaller-NF. DMRs on chromosome 1 of the human genome were computed between H1 and IMR90 cells with the four methods. (A) Straight lines represent the genome coverage of DMRs on the actual methylation data and dashed lines the genome coverage of DMRs on the scrambled methylation data. (B) The lines represent the difference in genome coverage of DMRs between the actual methylation data and scrambled methylation data.
Using the Fisher’s exact test or a Score test could potentially lead to high false positive rates (33–35). Since a good algorithm to call DMRs should be able to discriminate the natural epigenetic variation at level of single cytosine (biological noise) from a consistent stretch of DNA differentially methylated, we generated a scrambled methylation dataset for estimation of false positive call, randomly swapping the methylation values between all cytosines for each methylation context. We used this scrambled dataset to compute DMRs with the same parameters as in the case of the real data for all the methods tested. Due to the fact that the scrambled dataset consists of random methylation data, DNA sequence stretches with consistent differential methylation should appear by chance at a very low rate and the calling algorithm should detect as fewer/smaller DMRs as possible (assuming that all difference found are actually false positive). Moreover, increasing the window/bin size threshold should sharply decrease the genome coverage of DMRs for the scrambled data, whilst should still allow the identification of longer DMRs in the real data.
Figure 3A shows that, for each method, there is a window/bin size that maximises the genome coverage of DMRs in the real dataset whilst displaying low values in the scrambled dataset. Whilst the DMR genome coverage was used as measurement of sensitivity in the analysis of met1-3 mutant plants, here, we used the difference of the DMR genome coverage calculated for real and scrambled data as estimation of the accuracy in the IMR90 versus H1 comparison. The difference between genome coverage of DMRs in the real dataset and the artificial dataset displays a maximum, indicating that there is a bin/window size that leads to high sensitivity (maximising the genome coverage of DMRs in the real dataset) and at the same time a high accuracy (minimizing the genome coverage of DMRs in the scrambled dataset); see Figure 3B.
Comparing the four methods, we found that DMRcaller-NF computes DMRs in CpG context with the highest accuracy among the tested tools (displaying the highest DMR genome coverage in the actual methylation dataset and lowest DMR genome coverage in the scrambled dataset). This is not surprising since CpG methylation highly correlated spatially in both humans (30) and plants (Supplementary Figure S3), and the noise filter algorithm takes advantage of this to increase the significance of the statistical test. Taking this forward, our approach of using scrambled methylation data can be used to compute the window/bin size that will display the highest sensitivity (large genome coverage of DMRs in the real dataset) and at the same time a high accuracy (low-genome coverage of DMRs in the scrambled dataset).
Next, we split the DMRs into regions where there is more methylation in H1 than in IMR90 and regions that display more methylation in IMR90 compared to H1. All methods seem to agree that there is more methylation in H1 than in IMR90 (see Supplementary Figure S7), which is consistent with previous observations (15). Our results show that the majority of CpGs are recovered by all four methods and the overlap between them is between 62 and 78% (Supplementary Figure S7A). Most importantly, between 74 and 78% of the DMRs identified by methylKit and methylSig are also identified by DMRcaller-B and DMRcaller-NF (Supplementary Figure S7A).
In our comparison, we did not include methylPipe, due to the fact that one cannot call DMRs that contain less than five differentially methylated CpGs and, thus, we could not estimate the accuracy with the scrambled data. Nevertheless, we computed the DMRs with methylPipe using the default parameters provided in (12) and compared the results to the ones of the other four methods. Supplementary Figure S7 confirms that methylPipe detects DMRs that have a smaller genome coverage (20.9 Mb with higher methylation in H1 cells compared to 35.4 Mb for DMRcaller-NF) and the overlap with the DMRs computed by the other methods is usually lower (between 38 and 65%).
Finally, we also compared the speed of these tools. DMRcaller (both DMRcaller-B and DMRcaller-NF) displays slower times compared to methylKit and methylSig, nevertheless, DMRcaller is still fast enough for this not to be an issue; see Figure 4. For example, DMRcaller computes the DMRs in less than 30 min on human chromosome 1 (≈ 247 Mb) between H1 and IMR90 cells for window sizes between of 50 and 5000 bp using 10 CPUs. Note that the joining of adjacent DMRs is the cause of the reduce in speed and computing DMRs without merging them leads to similar speeds to to methylKit and methylSig.
Figure 4.

Speed of computing DMRs. The CPU time to compute the DMRs from Figure 3 on methylation data using 10 CPUs on a Mac Pro computer with Intel Xeon E5 2.7GHz 12-core.
DMRs in non-CpG context
In plants, methylation in CpHpG context is maintained through a positive feedback loop in between KRYPTONITE (KYP; also known as SUVH4) and CHROMOMETHYLASE 3 (CMT3) or CHROMOMETHYLASE 2 (CMT2) (36–38), whilst methylation in CpHpH context is maintained by the RNA-directed DNA methylation (RdDM) pathway (21,23) . The quadruple mutant ddcc (drm1 drm2 cmt3 cmt2) leads to complete loss of both CpHpG and CpHpH methylation (22); see Supplementary Figure S4B and C.
Using a similar approach as in the case of CpG methylation, we called DMRs with the four methods for different bin/window sizes. For CpHpG methylation, all four methods lead to very similar genome coverage of DMRs (14.87 Mb for methylKit, 15.36 Mb for methylSig, 16.41 Mb for DMRcaller-B and 15.68 Mb for DMRcaller-NF); see Figure 5A. For CpHpH methylation, we found that all tilling bins methods lead to similar results, but the noise filter method is almost unable to detect DMRs; see Figure 5B. This can be explained by the fact that CpHpH, is the only context where the methylation shows very limited spatial correlation; see Supplementary Figure S3.
Figure 5.

Genome coverage of non-CpG DMRs between WT and ddcc Arabidopsis thaliana plants as a function of window/bin size. The graph plots the total size of DMRs computed using: (i) methylKit, (ii) methylSig, (iii) DMRcaller-B and (iv) DMRcaller-NF. (A) The genome coverage of DMRs in CpHpG context. (B) The genome coverage of DMRs in CpHpH context.
Taking together, these results showed that DMRCaller is able to call more DMRs compared to other existing tools designed to detect differences in non-CpG context methylation, in conditions where all methylatransferases involved in CpHpG and CpHpH methylation were mutated. However, such extreme condition is rather artificial and not representing a physiological scenario, where epigenetic changes only occur at specific functional regions. Therefore, we used the previously established functional correlation of CpHpG hypomethylation and gene expression observed in rice endosperm to test DMRcaller performances in a developmental study (27). In particular, we used the bins method to call DMRs in CpHpG context in rice endosperm compared to embryo from published dataset (27). Then we tested the level of overlap of CpHpG DMRs with a stringent group of 165 genes that displayed a strong preference for endosperm expression, compared with a control group of 153 genes that are preferentially express in the embryo (as defined in (27)). For all windows used, we always observed that DMRs tend to overlap more endosperm-preferred genes (10–22%) compared to embryo-preferred genes (1–10%), providing evidence of functional annotation (see Figure 6). Remarkably, DMRcaller outperformed methylKit and methylSig in both total number of DMRs called and proportion of endosperm-preferred genes overlapping DMRs (see Supplementary Figure S8).
Figure 6.

Overlap between CpHpG DMRs called with DMRcaller and preferential expression of genes in rice endosperm or embryo.
Computing DMRs from biological replicates
To investigate the DMRcaller performance in the detection of DMRs from biological replicates, we considered again the case of WT and met1-3 mutant A. thaliana plants and used one biological replicate from (20) and the second biological replicate from (26). In this analysis we considered DMRcaller bins (DMRcaller-B) and noise filter (DMRcaller-NF) methods and we pooled together the reads from the two replicates. We also used DMRcaller and considered explicitly the biological replicates using the implemented beta regression method in conjunction with binning the data (DMRcaller-BR) or neighbourhood method (DMRcaller-NR); see ‘Materials and Methods’ section. In addition, we used three other methods that model explicitly biological replicates: methylKit (6), methylSig (9) and DSS (10). Due to the long computational time of some of the tools included in this analysis, we detected DMRs only on chromosome 1, using various window sizes. Figure 7A shows that DMRcaller-NF detects most DMRs (9.4 Mb) (consistently with what was previously observed), whilst DSS detected least DMRs (6.8 Mb); see Figure 3. DMRcaller-BR identified a similar number of DMRs as DMRcaller-NF (9.2 Mb) and majority of these (≈80%) were detected by all methods; see Figure 7C. When comparing the DMR methylation levels for each sample, we observed that the DMRs detected by DMRcaller-NF display similar methylation levels as the ones detected by DMRcaller-B, DMRcaller-BR and DSS; see Figure 7D.
Figure 7.

Computing DMRs with biological replicates. We computed DMRs using: (i) methylKit, (ii) methylSig, (iii) DSS, (iv) DMRcaller Bins (DMRcaller-B), ($v$) DMRcaller Noise filter (DMRcaller-NF) and (vi) DMRcaller with bins and beta regression (DMRcaller-BR). DMRs between WT plants and met1-3 plants were computed on chromosome 1. (A) Genome coverage. (B) We considered the DMRs identified by DMRcaller-NF method and the DMRs identified by DSS and split the DMRs into common DMRs, DMRcaller-NF specific and DSS specific. Furthermore, the DMRs specific to each method were split based on their relative location into: adjacent to common ones (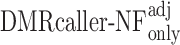 and
and 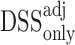 ) or far from common ones (
) or far from common ones (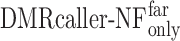 and
and 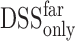 ). (C) Overlap of DMRs between different methods. (D) Methylation level in DMRs for each sample.
). (C) Overlap of DMRs between different methods. (D) Methylation level in DMRs for each sample.
Finally, we used the neighbourhood method (DMRcaller-N) to identify ∼300 000 single cytosines in CpG context that are differentially methylated on chromosome 1 of met1-3 mutant compared to WT (pooling together reads from replicates) (20,26). Over 81% of these cytosines were included in DMRs called by all methods tested (DMRcaller-NF, DSS, DMRcaller-BR, DMRcaller-NR), whilst ∼14% were not included in DMRs by any of these; see Supplementary Figure S9. Both DMRcaller-NF and DMRcaller-NR detected slightly more DMCs compared to DSS and DMRcaller-BR (85.6% compared to 81%). This suggests that, in the case tested here, independently of whether we model explicitly biological replicates or pool together reads, the detection of differentially methylated CpGs would lead to similar results.
DISCUSSION
DMRcaller is a simple to use, fast (see Figure 4), powerful and versatile R/Bioconductor package that is able to compute DMRs between two samples. The package implements three methods to compute DMRs: (i) noise filter, (ii) bins and (iii) neighbourhood. The first two methods (noise filter and bins) assume that there is spatial correlation between methylated cytosine (see Supplementary Figure S3) and aim to address low coverage in BS-seq experiments. They achieve this by either smoothing the data with a noise filter (the noise filter method) or binning the data into tilling bins (the bins method). The neighbourhood method can be applied on high coverage dataset and assumes calling individual cytosines as DMCs and joining adjacent DMCs to form longer DMRs.
Most importantly, this package is among few that can compute DMRs in non-CpG context. Non-CpG methylation is an well-established epigenetic mark in plants (19–23) and, recent work, shows that this methylation exists also in some mammalian tissues (16–18). Having the capability to compute DMRs in CpG and non-CpG contexts supports an integrative analysis of DNA methylation and can be useful in investigating the interaction between these types of DNA methylation.
Versions of some of these methods are implemented in several other packages, but, in contrast to DMRcaller, these other tools implement only one method. Comparisons of these tools indicate relative success of each method for different datasets, but it seems that none of these methods produce the best results on all datasets. This suggests that the method to detect DMRs should be selected depending on the methylation context, coverage and tissues. Our analysis revealed that, similar to mammals, CpG methylation is spatially correlated in plants and, in addition, non-CpG methylation can also display strong spatial correlation (in the case of CpHpG methylation). Due to this property, we found that the noise filter method has the highest sensitivity (can detect the largest genome coverage of DMRs) in CpG context; see Figures 2 and 3.
Furthermore, we propose a new method to estimate the window/bin size by comparing the genome coverage of DMRs on a dataset where methylation data were scrambled and on the real dataset. To increase sensitivity, we selected values that lead to higher genome coverage of DMRs on the real dataset (to avoid missing differential methylated regions) and, to increase accuracy, we selected values that lead to lower genome coverage of DMRs on the scrambled dataset. Maximizing the difference between there two values would result in a bin/window size that increases sensitivity and accuracy at the same time. Our analysis showed that, for certain window sizes (specific for each methylation context), DMRcaller computes a high number of DMRs on the real dataset and a low number of DMRs on the scrambled dataset, which suggests low false negative and false positive rates.
The scrambled dataset permuted the methylation levels to form a null distribution of the methylome profile, from which no difference is expected only if the global methylation level is the same. Thus, we applied the scrambled dataset only for comparison where global methylation levels were similar (75−85%).
Overall, we found that, for both CpG and CpHpG methylation, tilling bins methods and noise filter method will detect DMRs that will cover a large part of the genome (with either DMRcaller-NF and DMRcaller-B slightly outperforming the other methods); See Figures 2, 3 and 5. Nevertheless, DMRcaller-NF call less DMRs in the scramble dataset compared to the other methods. This means that the results of DMRcaller-NF are more reliable when the methylation levels are spatially correlated (for CpG and CpHpG context).
In contrast, in the case of CpHpH methylation, there is a reduced spatial correlation of methylation levels and DMRcaller-NF is not the appropriate method to detect DMRs. In this case, all tilling bins methods perform well and detect DMRs with high sensitivity and accuracy using bin sizes between 100 and 500 bp. Nevertheless, due to the higher variability of CpHpH methylation, biological replicates would be necessary to detect DMRs with high confidence.
Finally, we show that DMRcaller computes DMRs very fast, within similar time scales as other fast methods (methylKit and methylSig) making the package applicable to larger genomes and that there is a high overlap between biological replicates and DMRcaller is able to identify DMRs that recapitulate known biology.
Supplementary Material
ACKNOWLEDGEMENTS
Most of this work was carried out in Dr Jerzy Paszkowski lab and we would like to thank him for the support, the feedbacks and the critical read of the manuscript. We would also like to thank Robert Stojnic, Laurent Gatto and Herve Pages for useful discussions and comments on the R/Bioconductor package and Hajk Georg Drost, Tyler Gorrie-Stone and the anonymous reviewers for comments on the manuscript. We would like to acknowledge the use of the genomics cluster at School of Biological Sciences, University of Essex and thank Stuart Newman for his help with the cluster.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council [322621]; Gatsby Foundation [AT3273/GLE] and University of Essex. Funding for open access charge: University of Essex.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002; 16:6–21. [DOI] [PubMed] [Google Scholar]
- 2. Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012; 13:484–492. [DOI] [PubMed] [Google Scholar]
- 3. Sun D., Xi Y., Rodriguez B., Park H.J., Tong P., Meong M., Goodell M.A., Li W.. MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 2014; 15:R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. R Development Core Team. R:a language and environment for statistical computing. R Found.Stat. Comput. 2014; Vienna: http://www.R-project.org/. [Google Scholar]
- 5. Gentleman R., Carey V., Bates D., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Akalin A., Kormaksson M., Li S., Garrett-Bakelman F.E., Figueroa M.E., Melnick A., Mason C.E.. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012; 13:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hansen K., Langmead B., Irizarry R.. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012; 13:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hebestreit K., Dugas M., Klein H.-U.. Detection of significantly differentially methylated regions in targeted bisulfite sequencing data. Bioinformatics. 2013; 29:1647–1653. [DOI] [PubMed] [Google Scholar]
- 9. Park Y., Figueroa M.E., Rozek L.S., Sartor M.A.. MethylSig: a whole genome DNA methylation analysis pipeline. Bioinformatics. 2014; 30:2414–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feng H., Conneely K.N., Wu H.. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014; 42:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Assenov Y., Muller F., Lutsik P., Walter J., Lengauer T., Bock C.. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods. 2014; 11:1138–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kishore K., de Pretis S., Lister R., Morelli M.J., Bianchi V., Amati B., Ecker J.R., Pelizzola M.. methylPipe and compEpiTools: a suite of R packages for the integrative analysis of epigenomics data. BMC Bioinformatics. 2015; 16:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akman K., Haaf T., Gravina S., Vijg J., Tresch A.. Genome-wide quantitative analysis of DNA methylation from bisulfite sequencing data. Bioinformatics. 2014; 30:1933–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mayo T., Schweikert G., Sanguinetti G.. M3D: a kernel-based test for shape changes in methylation profiles. Bioinformatics. 2015; 31:809–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.-M. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009; 462:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lister R., Mukamel E.A., Nery J.R., Urich M., Puddifoot C.A., Johnson N.D., Lucero J., Huang Y., Dwork A.J., Schultz M.D. et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013; 341:1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schultz M.D., He Y., Whitaker J.W., Hariharan M., Mukamel E.A., Leung D., Rajagopal N., Nery J.R., Urich M.A., Chen H. et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015; 523:212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He Y., Ecker J.R.. Non-CG methylation in the human genome. Annu. Rev. Genomics Hum. Genet. 2015; 16:55–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lister R., O’Malley R.C., Tonti-Filippini J., Gregory B.D., Berry C.C., Millar A.H., Ecker J.R.. Highly Integrated Single-Base resolution maps of the epigenome in arabidopsis. Cell. 2008; 133:523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stroud H., Greenberg M., Feng S., Bernatavichute Y., Jacobsen S.. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell. 2013; 152:352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matzke M.A., Mosher R.A.. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014; 15:394–408. [DOI] [PubMed] [Google Scholar]
- 22. Stroud H., Do T., Du J., Zhong X., Feng S., Johnson L., Patel D.J., Jacobsen S.E.. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014; 21:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Du J., Johnson L.M., Jacobsen S.E., Patel D.J.. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015; 16:519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krueger F., Andrews S.R.. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011; 27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benjamini Y., Hochberg Y.. Controlling the false discovery Rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995; 57:289–300. [Google Scholar]
- 26. Catoni M., Griffiths J., Becker C., Zabet N.R., Bayon C., Dapp M., Lieberman-Lazarovich M., Weigel D., Paszkowski J.. DNA sequence properties that determine susceptibility to epiallelic switching. EMBO J. 2017; 36:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zemach A., Kim M.Y., Silva P., Rodrigues J.A., Dotson B., Brooks M.D., Zilberman D.. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:18729–18734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kishore K. ListerEtAlBSseq: BS-seq data of H1 and IMR90 cell line excerpted from Lister et al. 2009. 2015; R package version 1.4.2http://bioconductor.org/packages/ListerEtAlBSseq/. [Google Scholar]
- 29. Meyer C.A., Liu X.S.. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat. Rev. Genet. 2014; 15:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eckhardt F., Lewin J., Cortese R., Rakyan V.K., Attwood J., Burger M., Burton J., Cox T.V., Davies R., Down T.A. et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006; 38:1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kankel M.W., Ramsey D.E., Stokes T.L., Flowers S.K., Haag J.R., Jeddeloh J.A., Riddle N.C., Verbsky M.L., Richards E.J.. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics. 2003; 163:1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saze H., Scheid O.M., Paszkowski J.. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 2003; 34:65–69. [DOI] [PubMed] [Google Scholar]
- 33. Ayyala D.N., Frankhouser D.E., Ganbat J.-O., Marcucci G., Bundschuh R., Yan P., Lin S.. Statistical methods for detecting differentially methylated regions based on MethylCap-seq data. Brief. Bioinform. 2016; 17:926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y., Baheti S., Sun Z.. Statistical method evaluation for differentially methylated CpGs in base resolution next-generation DNA sequencing data. Brief. Bioinform. 2016; 19:374–386. [DOI] [PubMed] [Google Scholar]
- 35. Shafi A., Mitrea C., Nguyen T., Draghici S.. A survey of the approaches for identifying differential methylation using bisulfite sequencing data. Brief. Bioinform. 2017; bbx013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Du J., Zhong X., Bernatavichute Y., Stroud H., Feng S., Caro E., Vashisht A., Terragni J., Chin H., Tu A. et al. Dual binding of chromomethylase domains to H3K9me2-Containing nucleosomes directs {DNA} methylation in plants. Cell. 2012; 151:167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Du J., Johnson L., Groth M., Feng S., Hale C., Li S., Vashisht A., Gallego-Bartolome J., Wohlschlegel J., Patel D. et al. Mechanism of {DNA} methylation-directed histone methylation by {KRYPTONITE}. Mol. Cell. 2014; 55:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zabet N.R., Catoni M., Prischi F., Paszkowski J.. Cytosine methylation at CpCpG sites triggers accumulation of non-CpG methylation in gene bodies. Nucleic Acids Res. 2017; 45:3777–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.