


Abstract
Epigenetic alterations, a well-recognized cancer hallmark, are driven by chromatin regulators (CRs). However, little is known about the extent of CR deregulation in cancer, and less is known about their common and specialized roles across various cancers. Here, we performed genome-wide analyses and constructed molecular signatures and network profiles of functional CRs in over 10 000 tumors across 33 cancer types. By integration of DNA mutation, genome-wide methylation, transcriptional/post-transcriptional regulation, and protein interaction networks with clinical outcomes, we identified CRs associated with cancer subtypes and clinical prognosis as potential oncogenic drivers. Comparative network analysis revealed principles of CR regulatory specificity and functionality. In addition, we identified common and specific CRs by assessing their prevalence across cancer types. Common CRs tend to be histone modifiers and chromatin remodelers with fundamental roles, whereas specialized CRs are involved in context-dependent functions. Finally, we have made a user-friendly web interface-FACER (Functional Atlas of Chromatin Epigenetic Regulators) available for exploring clinically relevant CRs for the development of CR biomarkers and therapeutic targets. Our integrative analysis reveals specific determinants of CRs across cancer types and presents a resource for investigating disease-associated CRs.
INTRODUCTION
Epigenetics, the study of stable, heritable traits that are not attributable to changes in the DNA sequence, has emerged as a means of elucidating critical regulation in cancer. Chromatin regulators (CRs) are indispensable upstream regulatory factors of epigenetics. According to regulatory roles in epigenetics, CRs are usually grouped into three major categories: DNA methylators, histone modifiers, and chromatin remodelers (1–3). DNA methylators and histone modifiers can code and decode various modifications on cytosine and histone residues and are usually further divided into readers, writers, and erasers (4,5). Readers usually contain specific domains that can recognize specific cytosine or histone residues and determine the modification type and state (1,2,5). Writers and erasers usually play roles in adding and removing certain modifications and from specific cytosine or histone residues, as in methylation and demethylation (1,2,5). Chromatin remodelers are a special type of CRs that can disrupt the contact between nucleosomes and DNA, shuffle nucleosomes around, replace them or remove them from the chromatin, and cause abnormal epigenetic modifications (1,2,5).
The alteration of epigenetic marks is a prevalent feature in cancer (6). An increasing number of studies have found that dysfunction of CRs can occur at different molecular levels. For example, it is widely accepted that mutations can perturb CR functions. Yan et al. have found that genetic alteration of DNMT3A (a DNA methylation transferase) can induce genome-wide alterations of DNA methylation and gene expression. Moreover, patients with DNMT3A mutations have poor prognosis compared with those without such mutations (7). In addition, a number of CRs have been found to be dysregulated in gene expression across cancer types. Damaschke et al. have observed that CHD8 and CTCF (two chromatin remodelers) are up-regulated and down-regulated, respectively, in prostate tumor samples. They found that dysregulation of these CRs results in structural abnormalities in chromatins and epigenetic alterations of numerous cancer-associated genes, which finally lead to increased tumor volume, extracapsular extension, and metastases in prostate cancer patients (8). These studies demonstrate that CRs hold crucial roles in epigenetics and that various molecular alterations can cause functional perturbation of CRs.
With the development of high-throughput sequencing, although hundreds of CRs have been identified, distinguishing critical CRs in cancer remains a major challenge. Currently, functional aberrations of individual CRs have been discovered in multiple cancer types. However, it is often difficult to determine if a CR can exhibit the same role or unique roles in different tumor contexts. For instance, the up-regulated expression of EZH2 (a lysine methyltransferase) was found to promote tumor cell proliferation by increasing the promoter occupancy of trimethylation of H3K27 in various types of cancer (9–12). These analyses of multiple cancers for EZH2 expression revealed its common role in carcinogenesis. In another study, Gonzalez-Perez et al. found that several functional CRs have varied mutation frequencies that depend on the tumor type. In addition, they found that CRs in the same multi-protein complexes show mutually exclusive alterations in each cancer lineage (2). Taken together, these previous studies primarily identified dysfunctional CRs in a specific cancer or characterized the dysregulation of a given CR in multiple cancer types. These results suggest that there is widespread perturbation of CRs on multiple molecular levels. Integrative analysis of omics datasets of CRs is needed to evaluate their activities and discover potential oncogenic CRs across cancer types.
Here, we performed an integrative analysis of CRs in 10,969 tumors from patients across 33 cancer types. A computational method was proposed to prioritize functional CRs in cancer; this method integrates genetic mutations, gene expression, miRNA regulation, protein–protein interaction networks (PPIN), and regulation of epigenetic modifications. In-depth analysis was performed to explore the functional features of common and specific CRs in cancer. Moreover, we found that functional CRs can contribute to individual cancer type and pan-cancer reclassification. To better delineate the alterations and effects of CRs in cancer, we have developed and presented FACER (http://bio-bigdata.hrbmu.edu.cn/FACER/), a convenient and friendly resource for biomedical researchers to further investigate CRs. Systematic analyses of multi-omics data across cancer types could help identify functional CRs in cancer and further elucidate the commonalities and differences in mechanisms across cancer types.
MATERIALS AND METHODS
Manually curation of chromatin regulators
A list of CRs was collected from the literatures by manual curation (13–18). Information on biological categories, functions, and involved protein complexes was also collected from these publications. We ultimately collected 870 CRs and classified them into three major categories and seven subcategories by considering the regulatory patterns described in the literatures (Supplementary Figure S1 and Supplementary Table S1).
Somatic mutations of CRs in cancer
Somatic mutations of CRs sequenced by whole-exome sequencing were collected from The Cancer Genome Atlas (TCGA) project. All sequencing platforms were considered, and silent mutations were removed from our analyses. We calculated the mutation frequency of a CR in specific cancer types, defined as the percentage of patients with at least one somatic mutation on this CR.
mRNA and miRNA transcriptome across cancer types
Both mRNA and miRNA expression profiles for all cancer types were also obtained from TCGA (Supplementary Figure S2 and Supplementary Table S2). For mRNA expression data, gene expression was measured through mapping RNA-Seq by Expectation Maximization (RSEM) for all types of cancer. For glioblastomas (GBM), the miRNA expression profile was measured by microarray, whereas miRNA-Seq datasets were used for the remaining cancer types. The expression of miRNAs was measured as reads per million (RPM). We first removed genes (or miRNAs) with RSEM (or RPM) expression values of 0 in all samples and then log2-transformed the expression levels. Considering the limited number of normal samples, we combined all normal samples together as the control for further analysis (19).
Transcriptional regulation of CRs across cancer types
TF–gene interactions assayed by chromatin immunoprecipitation followed by sequencing (ChIP-seq) were downloaded from the ChIPBase database (20). To identify the TF-CR interactions in specific cancer types, we used a linear regression model to evaluate the regulatory activity between the TF and CR on the basis of gene expression data. All candidate TF-CR pairs with adjusted P<0.01 were identified as the TF-CR interactions in the cancer.
miRNA regulation of CRs across cancer types
miRNA-gene interactions were collected from the combination of all experimentally validated pairs from databases, including miRecords (21), miRTarBase (22) and TarBase (23). Similar as transcriptional regulation, miRNA-CR regulation in the context of cancer was identified on the basis of their expression correlation measured by Pearson correlation coefficient. Candidate miRNA–CR interactions with adjusted P < 0.05 were retained for further analysis.
Protein–protein interactions of CRs
A proteome-scale interaction network was generated by high-throughput affinity-purification mass spectrometry (24). We extracted the CR-related interactions, which consist of 2915 interactions among 554 CRs and directly interacting partners.
Regulation of DNA methylation by CRs across cancer types
DNA methylation datasets detected by Illumina Infinium HumanMethylation450 BeadChip array were also downloaded from TCGA data portal. After selecting the promoter CpG islands (CGIs) and open sea regional clusters on the genome, we calculated the aberrant hypermethylation (over CGI probes) and hypomethylation (over open sea probes) values for each tumor sample compared with normal samples (Supplementary Methods). To evaluate the global regulatory effect of a given CR on the DNA hypermethylation (or hypomethylation) in a specific cancer type, we computed the significance of Pearson correlation (P value) between CR expression and aberrant hypermethylation (or hypomethylation) of tumor samples (25).
Clinical information of tumor samples
We obtained clinical characteristics of the patients from TCGA, including survival state, survival time, disease stage and histologic grade.
Prioritization of functional CRs based on multi-omics data in cancer
We proposed a computational method to prioritize functional CRs in each cancer type by integrating multi-omics features of CRs, which involved three steps (Figure 1 and Supplementary Figure S3). First, seven functional features that characterize the activity, regulation by differentially expressed TFs and miRNAs, central roles in PPIN, and aberrant regulation of epigenetic modifications of CRs were calculated. The activity levels of CRs were assessed on the basis of their mutation frequency and the extent of their differential expression in cancer. Moreover, we evaluated the aberrant regulatory activity of CRs by assessing their DNA hypermethylation and hypomethylation levels. All the CRs were ranked on the basis of the seven features in each cancer type. Second, we constructed classifiers to prioritize functional CRs for each cancer type on the basis of the aggregated ranks, which integrated the seven separate ranks using the robust rank aggregation method (26). The aggregated ranks represent the global functional impact of CRs. Using the cancer hallmark–related CRs as the gold standard data set, we trained the classifiers using all possible feature combinations for individual cancer types. Third, we selected the most effective combined feature (MECF) and obtained the corresponding functional CRs in each cancer type.
Figure 1.

The workflow for prioritizing the cancer-related functional CRs. The processes for prioritizing functional CRs for each cancer type involved three steps. First, seven functional features were defined, and CRs were ranked based on the features. Second, the individual ranks were integrated into aggregated ranks, and multiple classifiers were constructed on the basis of the aggregated ranks. Third, the classifiers were evaluated and the prioritized CRs obtained in each cancer type. ‘RRA’ represents the robust rank aggregation method. The cancer type abbreviations are those used by TCGA.
Defining the functional features of CRs in cancer
We defined seven features to evaluate the functional impact of each candidate CR in cancer (Table 1). Fmu is the mutation frequency of the CR. Fde is the value of –log(P), where P is the P value from the Student t-test used to evaluate the significance of differential expression of CRs in cancer. Ftf is the proportion of differentially expressed TFs (adjusted P < 0.01 by t-test and |log2(fc)| > 1) to the overall TF set that regulates this CR. Fmi is the proportion of differentially expressed miRNAs (adjusted P < 0.01 by t-test and |log2(fc)| > 1) to the overall miRNAs that regulate the CR. Fppi is the number of interacting partners of the CR in the PPIN. Fhyperm and Fhypom are negatively logarithmic P values of correlation between CR expression and DNA hypermethylation and hypomethylation scores, respectively. Each CR was ranked by these features in descending order. That is, in a specific cancer type, a CR with a higher value for a feature was considered to have greater importance with regard to that feature in that cancer.
Table 1.
The definition and calculation of seven features
| Feature | Feature value (for each CR in each cancer) |
|---|---|
| F mu | Num. of mutated samples / Num. of all samples |
| F de | –log10(P), P represents the significance of CR differential expression calculated by t-test |
| F tf | Num. of diff-expressed TFs/number of regulatory TFs |
| F mi | Num. of diff-expressed miRNAs/number of regulatory miRNAs |
| F ppi | Num. of CR neighbors in PPIN |
| F hyperm | –log10(P), where P represents the significance of CR regulation to sample hypermethylation |
| F hypom | –log10(P), where P represents the significance of CR regulation to sample hypomethylation |
Evaluating the functional impact of CRs by aggregated ranks
We first constructed gold standard positive and negative control sets. The true positives were defined as known cancer hallmark–related CRs. Cancer hallmark–related genes were obtained from MSigDB V4.0 (27). Compiling a list of negative controls is currently difficult or even impossible. Thus, we randomly selected the same number of non–hallmark-related CRs as negative controls. This process was repeated 100 times.
To identify the functional CRs in each cancer type, classifiers were developed on the basis of all possible combinations of the seven features. In total, there are 127 feature combinations. For each feature combination, multiple ranks of each CR were aggregated to a final rank based on the robust rank aggregation method (26). For each feature combination, the robust rank aggregation method comprised two steps based on multiple feature ranks (one to seven feature ranks) of 870 CRs as follows. Let g be the number of CRs and f be the number of features. First, we obtained normalized ranks with the maximal rank value of 1 by dividing ranks by the maximal rank value g. For each CR, we got the corresponding rank vector r = (r1, …, rj…, rf), where rj denotes the normalized rank of this CR in the jth feature. Second, a scoring method that produces comparable scores for CRs was performed. The scoring method was based on the assumption that all normalized feature ranks come from a uniform distribution; the method determined how probable it is to obtain 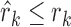 when the rank vector
when the rank vector 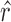 is generated by the null model. The probability that
is generated by the null model. The probability that 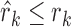 was denoted by βk,f(
was denoted by βk,f(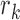 ) and calculated as follows:
) and calculated as follows:
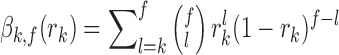 |
Then we defined minβ as the minimum of βk,f(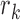 ):
):
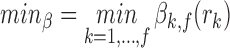 |
and obtained the final score for the rank vector r s(r), which was defined as the adjusted P value of minβ by Bonferroni correction. For each feature combination, according to the final score of each CR, we obtained the aggregated rank. Using the gold standard data set and negative control set mentioned above, we validated the prediction efficiency of each classifier by calculating the area under the receiver operating characteristics curve (AUC). With the use of 100 randomly selected negative controls, the AUC of each classifier was represented by the mean value of 100 AUCs.
Prioritizing functional CRs
For a given cancer type, the classifier with the highest AUC was selected as the optimal classifier. In addition, the corresponding feature combination of this classifier was considered the MECF. We then identified top-ranked 100 CRs as the functional CRs in the cancer.
Survival analysis
We performed survival analysis to identify the association of functional CRs with patients’ overall survival. For given CR sets, we divided the tumor samples into different numbers of subtypes based on those CRs’ expression levels. Kaplan-Meier survival plots and log-rank tests were used to evaluate the survival differences between groups of patients. This process was performed using the R package ‘survival’ (28).
Construction of pan-cancer and cancer-specific CR networks
A pan-cancer–associated CR network was constructed by integrating CR-related interactions from three aspects, including protein interaction, TF regulation and miRNA regulation. We first obtained direct and indirect interaction partners for common CRs from PPIN. For TF and miRNA regulation, we ranked each regulation by the frequency of cancer types. The top 10% of regulations, with the highest frequency, were added to the CR-related network. For cancer-specific networks, the TF/miRNA-CR interactions observed in the specific cancer were selected. In these CR-related networks, three types of nodes (common/specific CR, TF, miRNA) and three types of edges (CR-CR, TF-CR and miRNA-CR) were included.
Reclassification of tumor samples based on CR-related transcriptome data
We developed an integrated subtype classification system based on transcriptome data for functional CRs, and for TFs and miRNAs that target the functional CRs. In addition, the TFs and miRNAs need to be differentially expressed in cancer. Next, we followed a two-step approach called COCA (cluster-of-cluster assignment) to identify the cancer subtypes (29). First, consensus clustering for the CR, TF, and miRNA expression profiles was performed separately for each. Second, the results of single-level clustering were processed into a Boolean matrix and then taken as the input of second-level consensus clustering for tumor samples. As a result, we obtained the high-order subtypes for 33 cancer types. We also performed reclassification for all of tumor samples from all 33 cancer types following the COCA approach by integrating the expression profiles of cancer-specific CRs, and of TFs and miRNAs that target the specific CRs. Only TFs and miRNAs that showed expression perturbation in at least one cancer type were considered.
RESULTS
Functional characterization and prioritization of CRs across 33 cancer types
Multi-level deregulation of CRs was observed in cancer; thus we proposed an integrative method to identify cancer-related CRs by integrating multiple functional features of CRs. We first collected a relatively comprehensive list of CRs by manual curation (2–5,25,30–32), which involved 870 CRs. These CRs were further classified into three categories and seven subcategories (Supplementary Figure S1 and Supplementary Table S1). Next, we integrated multi-omics data from tumor patients representing 33 cancer types in TCGA (Supplementary Figure S2 and Supplementary Tables S2 and S3) and proposed feature classifiers to prioritize functional CRs. In these classifiers, we defined seven features to evaluate the functional impact of each CR in a given cancer (Figure 1). The seven features involved two indices that measure CR mutation frequency and the extent of differential expression; three indices that evaluate the targeting of CR at the transcription, post-transcription, and protein interaction levels; and two indices that assess the regulation of CR on DNA hypermethylation and hypomethylation. We next ranked all CRs by these features in descending order and compared the relative ranks of cancer hallmark–related CRs and other CRs. We found that hallmark-related CRs had significantly lower ranks than other CRs across the seven features we defined (Supplementary Figure S4). These results indicate that these features can be used to identify functional CRs in cancer. Thus, we performed robust rank aggregation (26) to prioritize functional CRs by considering the seven features synthetically. Considering the AUC of the classifiers constructed using different combinations of features, we selected the classifier that gave the highest AUC. The MECFs for each cancer were also identified. We found that the AUCs for classifiers that combined multi-dimensional features were significantly higher than those based on individual features (Supplementary Figure S5). These results suggest that it is necessary to combine multiple features for the prioritization of functional CRs. On the basis of the classifier with the highest AUC, we identified the 100 top-ranked CRs in each cancer. Finally, 640 CRs were totally identified as functional CRs across the 33 cancer types (Supplementary Table S4).
Next, we investigated the potential dysregulation mechanism of the prioritized CRs. Cancer-related functional CRs tend to show widespread perturbation across cancer types compared with other CRs (Figure 2A). For example, functional CRs in colon adenocarcinoma (COAD) exhibit perturbations in six aspects, including higher mutation frequency, differential expression, stronger regulation by differentially expressed TFs and miRNAs, higher centrality in PPIN, and stricter regulation of genome hypermethylation (Wilcoxon rank-sum test, Figure 2B–G). On the other hand, the perturbations of functional CRs vary across cancer types (Figure 2A), even for cancers with similar tissue origins. For instance, for uterine corpus endometrial carcinoma (UCEC) and uterine carcinosarcoma (UCS), which both originate from the uterus, the perturbations of CRs were different despite their significant number of shared functional CRs (hypergeometric P = 2.31e–07). Although both of the functional CRs in UCEC and UCS are located in the center of PPIN, the functional CRs in UCEC are likely to show expression perturbation and play critical roles in the regulation of hypomethylation. In the contrast, the functional CRs in UCS exhibit higher mutation frequency compared with other CRs.
Figure 2.

The global features of functional CRs across cancer types. (A) A global view of the seven features of functional CRs across cancer types. The columns represent the significance of the tests of whether the feature values for functional CRs were higher compared with those of other CRs. The values are colored based on the adjacent color map. (B–G) The comparison results of functional CRs and other CRs in COAD for the seven features, including CR mutation frequency (B), negatively logarithmic P values of CR differential expression (C), proportion of differentially expressed TFs to the overall TF set that regulates this CR (D), proportion of differentially expressed miRNAs to the overall miRNA set that regulates this CR (E), CR degree in PPIN (F) and negatively logarithmic P values of correlation between CR expression and DNA hypermethylation and hypomethylation (G). (H) The summary MECFs for 33 cancer types. The right bars show the number of cancers with a certain number of features in their MECFs. Each point set represents the MECF of the cancer indicated on the right. The node size indicates the frequency of features across cancers, and different colors correspond to different features. (I) The frequency of the seven features in MECFs across 33 cancer types. ‘Fun-CR’ represents functional CRs and ‘O_CR’ represents other CRs.
To investigate the contribution of each feature in prioritizing the functional CRs in cancer, we next identified the MECFs in each cancer type. Interestingly, we found that MECFs were different across 33 cancer types. In particular, the MECFs in 30 cancer types consisted of more than two features, and the MECFs in six cancers included four features (Figure 2H). These observations suggest multi-level functional perturbation of CRs in cancer. However, it is not clear to what extent each feature contributes to functional CR prioritization. Thus, we calculated the number of cancer types each feature was involved in. Mutation frequency and centrality in PPIN were found to be the top two recurrent features (Figure 2I), which were involved in 25 and 29 cancer types, respectively. These results suggest that genes with high mutation frequency or with high centrality in PPIN are likely to be involved in cancer development. For example, IDH1, a DNA methylation eraser, was found to be the functional CR in three cancer types with high mutation frequency (78%, 13.9% and 10.7% in brain lower grade glioma (LGG), cholangiocarcinoma (CHOL), and acute myeloid leukemia (LAML), respectively). Another example is a histone deacetylase (HDAC1), which was found to be a functional CR in 16 cancer types. This CR exhibits a higher centrality (in top 11%) in PPIN. Both of these CRs (IDH1 and HDAC1) are well-known targets of epigenetic cancer drugs, such as IDH inhibitors or HDAC inhibitors (33). Moreover, we found that as the expression of IDH1 increased, the DNA methylation on open sea clusters decreased in LGG patients (Supplementary Figure S6). Taken together, these results suggest multiple-level perturbation of CRs across cancer. Integration of omics data can help prioritize functional CRs in cancer.
Histone modifiers and chromatin remodelers are more likely functional CRs
In accordance with epigenetic regulation patterns, we grouped the functional CRs that we identified into three major categories: DNA methylators, histone modifiers and chromatin remodelers. In addition, we further divided the DNA methylators and histone modifiers that we identified into readers, writers and erasers (Supplementary Figure S7). We next calculated the number of functional CRs in different categories and found that the majority of these CRs were histone modifiers across various cancer types. Moreover, we found that DNA methylation readers and histone modification writers were the most common functional CRs across all 33 cancer types (Figure 3A). Next, we explored whether this result is biased according to the numbers of CRs in different categories. We calculated the odds ratios for different categories and subcategories of cancer-context functional CRs and found that histone modifiers and chromatin remodelers were universally prioritized as functional CRs while DNA methylators were rewired across cancer types (Figure 3B, top). Moreover, histone modification readers and writers as well as chromatin remodelers played important roles across cancer types compared with DNA methylation erasers and histone modification erasers (Figure 3B, bottom). Interestingly, we found that specific categories of CRs were likely to occur in several cancer lineages. For instance, DNA methylation readers tended to be related to gastrointestinal and gynecologic cancers, while DNA methylation writers tended to play roles in cerebral, kidney, and hematologic diseases. However, we found that the CRs are different, although the specific CR category was involved in some cancer types. For example, among DNA methylation writers, DNMT3B was identified in GBM, while DNMT3A was likely to be critical in LGG.
Figure 3.

Epigenetic category of functional CRs across cancer types. (A) Pie charts show the numbers of CRs in different categories, and bar charts show the numbers of CRs in different categories across cancer types. The cancers in bar charts are ordered as in Figure 2A. ‘M’ represents DNA methylator, ‘H’ represents histone modifier, ‘Cr’ represents chromatin remodeler, ‘Two’ represents two types, ‘a’ means ambiguous, ‘R’ represents reader, ‘W’ represents writer and ‘E’ represents eraser. (B) The odds ratios for different categories of CRs across cancer types. The cancer type abbreviations are those used by TCGA. (C) The CGI methylation in KIRP tumor samples is proportional to the expression of DNMT1 and DNMT3B. Darker color represents higher expression. (D) Two groups of KIRP samples distinguished by the expression of DNMT1 and DNMT3B show significantly different CGI methylation and different survival rates.
Potential functions of the prioritized CRs in cancer
As cancer-related hallmarks provide a framework for understanding remarkable biological processes in cancer, we next focused on different categories of CRs in the context of cancer hallmarks (34). Functional enrichment analysis showed that DNA methylators, histone modifiers and chromatin remodelers were all enriched in at least one cancer hallmark, especially in the hallmark ‘genome instability and mutation’, highlighting the extent of genome alternations in cancer (Supplementary Figure S8 and Supplementary Table S5). Histone modifiers were enriched in seven hallmarks, indicating their widespread roles in cancers. Functional histone modifiers in almost all cancer types were enriched in the functions ‘evading apoptosis’, ‘genome instability and mutation’, ‘insensitivity to antigrowth signals’, and ‘self-sufficiency in growth signals’.
A number of studies have proposed that epigenetics-related phenotypes, such as the CpG island methylator phenotype (CIMP), define distinct subgroups of cancer (35). As CRs could regulate the genome-wide methylation pattern, we next explored to what extent the functional CRs contributed to the CIMP phenotype. We found that two DNA methyltransferases, DNMT1 and DNMT3B, were prioritized as kidney renal papillary cell carcinoma (KIRP) related CRs, ranked at 14 and 19, respectively. These two CRs showed expression perturbation in KIRP. We next investigated the association between the expressions of these two DNA methylators with hypermethylation of KIRP tumor samples. We found that the patients highly expressing two DNA methylators have higher hypermethylation on genome CGIs (P = 7.15e–04, Figure 3C). Moreover, the patients showed a significant difference in survival (log-rank P = 1.98e–04, Figure 3D). Next, we systematically analyzed the regulation of genome-wide DNA methylation levels from all prioritized DNA methylators in the corresponding cancers. We found that 90% of prioritized DNA methylators significantly regulated DNA hypermethylation and hypomethylation (P < 0.05, Supplementary Figure S9 and Supplementary Table S6). For example, IDH2, an eraser of DNA methylation, was found to significantly decrease the genome-wide DNA methylation levels of LGG samples, including CGI (R = –0.31, P = 1.85e–13) and open sea regions (R = 0.19, P = 1.79e–05). These results suggest that the perturbed expression of DNA methylators contributes to the CIMP phenotype and that DNA methylators influence the prognosis of cancer patients by regulating genome methylation.
We also revealed the functions of histone modifiers in histone modifications. For example, we found that PHF19, a writer for H3K36me3 as a component of polycomb repressive complex 2 (PRC2), was prioritized as a breast invasive carcinoma (BRCA) related CR in our analyses. In addition, PHF19 was up-regulated in patients with BRCA tumors. In two other independent breast cancer cell line data sets that we analyzed previously (36), the expression of PHF19 was also up-regulated compared with that in a normal breast cell line. Moreover, the genome-wide H3K36me3 marks showed an obvious increase in cancer (36). These results suggest that the up-regulated expression of PHF19 contributes to the high H3K36me3 marks and further induces the development of breast cancer.
Common and specific CRs play different roles in cancer
Plenty of studies have described the shared molecular characteristics among different cancer types as well as the specific traits of individual cancers (34,37). Thus, we aimed to explore the similarity and specificity of various cancer types based on the similarity of prioritized functional CRs. We calculated the Simpson index between pairs of cancers on the basis of the prioritized CRs and found that CRs were generally shared across different cancer types. Specifically, 471 of 528 cancer pairs significantly shared the prioritized chromatin regulators (P < 0.05, Supplementary Figure S10). This phenomenon was more obvious in pairs of cancers with similar tissue origins, such as COAD and rectum adenocarcinoma (READ) (56% shared), ovarian serous cystadenocarcinoma (OV) and UCS (65% shared), and lung squamous cell carcinoma (LUSC) and head and neck squamous cell carcinoma (HNSC) (50% shared). Furthermore, we found that the overlap between LAML and other cancer types was smaller than the overlaps between the solid cancer types, which suggests a difference between blood cancers and solid tumors. Next, we calculated the number of related cancer types for each CR. The results showed that as the number of cancer types increased, the number of CRs related to those cancer types decreased. In addition, we further grouped functional CRs according to the number of cancer types they occurred in. Specifically, 32 CRs that occurred in more than half of the cancer types (≥17 cancers) were defined as common CRs, whereas 193 CRs exclusively to one specific cancer type were defined as specific CRs, and the remaining CRs were defined as mixed CRs (Figure 4A). We found that the proportions of the three groups of CRs across the 33 cancer types varied from 7% to 30% of common CRs and from 0% to 23% of specific CRs (Figure 4B). Next, we analyzed the differential expression of common chromatin regulators across cancer types. A total of 15 CRs were found to be consistently differentially expressed (false discovery rate<0.05) across more than half of the 33 cancer types. Among these CRs, 14 were up-regulated in cancer, and 1 was down-regulated (Supplementary Figure S11A). Interestingly, 11 of these 15 CRs showed consistent deregulation across cancer types compared with the corresponding adjacent normal samples (Supplementary Figure S11B). These results indicate that common CRs tend to show the same direction of expression perturbation across cancer types. Moreover, it was observed that the functional CRs in LAML involved relatively fewer common and relatively more specific CRs compared with other cancers (8% and 19%, respectively), which may be a consequence of the difference between hematologic diseases and solid tumors.
Figure 4.

Common and specific CRs in cancer. (A) The definitions of common, specific, and mixed CRs in cancer. Bars show the number of functional CRs with certain numbers across cancer types. (B) The frequencies of the types of CRs across cancer types. (C) The biological categories and ranks of the three types of CRs. The colors in the heat map show the ranks of CRs. The cancers are ordered as in Figure 4B.
We next analyzed the distribution of the common, specific and mixed CRs in cancer in different epigenetic categories. Overall, all of these types of CRs included DNA methylators, histone modifiers, and chromatin remodelers (Figure 4C and Supplementary Table S7). The common CRs in cancer were found to be enriched mostly in readers and writers for histone modification and chromatin remodelers (19%, 56% and 19%, Supplementary Figure S12). Among these CRs, we identified some well-known oncogenes, such as KMT2C, SMARCD1 and SMARCA4 (1,33,38). For cancer-specific CRs, we found that these CRs included 36% of the DNA methylation erasers and 35% of the histone modification erasers. The specific CRs have previously been found to play specific roles in specific cancer types. For example, the lung adenocarcinoma (LUAD) specific DNA methylation eraser APOBEC3F has been found to play important roles in the immune response in non–small cell lung cancer (39). Another LUAD–specific DNA methylation eraser, APOBEC3C, has been reported to be significantly associated with a decreased risk of lung cancer (40). Taken together, these results suggest the commonalities and specificities of CR sets among multiple cancer types.
Distinct functional features contribute to common and specific CRs in cancer
To uncover the potential perturbation mechanism of CRs in cancer, we next investigated the seven original functional features of common and specific CRs. Tracing the original feature ranks of 32 common CRs, we found that their aberrations varied across cancer types, even in cancers with similar tissue origins (Figure 5A and Supplementary Figure S13). For instance, KMT2C was prioritized as a functional CR of both kidney chromophobe (KICH) and KIRP; however, its dysfunction mechanism differed between these two cancer types. KMT2C showed a higher mutation frequency in KIRP patients, whereas the aberrant regulation of miRNAs and its regulation of DNA methylation over open sea regions were additional features in KICH patients. On the other hand, it is apparent that functional CRs in the same cancer type play diverse roles (Figure 5A), suggesting the advantage of integrating multiple features to identify functional CRs. Next, we compared the functional features of specific CRs between their related cancers and other cancer types. For example, the GBM-specific CRs showed a higher mutation frequency and more dysregulated expression in GBM patients than in other tumor samples (Figure 5B). Moreover, both regulation density from differentially expressed TFs/miRNAs and the aberrant regulation of DNA methylation of specific CRs were found to be stronger in related cancer types (Supplementary Figure S14A and B).
Figure 5.

Portrayal of common and specific CRs in cancer. (A) Circos plot displays 32 common and 193 specific CRs. The top right sector shows the mean ranks of common CRs at seven individual features and the aggregated feature levels in corresponding cancer types. The adjacent heat map indicates the related cancer types for each common CR. The remaining sectors each indicate a unique cancer type with related specific CRs. Only cancers with at least one specific CR are shown. The pie charts inside show the categories of common and specific CRs. (B) The features of GBM-specific CRs in GBM and other cancer types, including mutation frequency and the extent of differential expression. (C) Feature values of common and specific CRs in 33 cancers and in their related cancer types are shown at the top and bottom, respectively. Features are defined as in Table 1.
Next, we globally compared the seven features in all cancer types by focusing on common and specific CRs. We found that common CRs in cancer had higher values for all seven features compared with specific CRs, especially in four functional features (all P < 0.05, Wilcoxon rank-sum test), including mutation frequency, differential expression, degree in PPIN, and regulation of genome hypermethylation (Figure 5C, top, and Supplementary Table S8). However, limiting the CRs to the related cancer types brought different observations. The significantly higher mutation frequency and greater differential expression of the common CRs were diminished considerably, and specific CRs showed significantly higher regulation of hypermethylation (P = 1.93e–03, Figure 5C, bottom). Moreover, specific CRs tend to be targeted by more differentially expressed TFs and miRNAs (P = 5.95e–04, P = 2.71e–04, respectively) and showed a tendency toward greater regulation of DNA hypomethylation (P = 0.06, Figure 5C, bottom). These results suggest that common CRs have extensive perturbation mechanisms across cancers while specific CRs have intensive aberrant mechanisms primarily in their unique related cancer types.
Cancer is rarely a consequence of perturbation in a single gene but, rather, reflects the rewiring of a network (41). We next constructed pan-cancer and cancer-specific associated CR networks (as described in Materials and Methods). Network analysis revealed that common CRs tended to form a tightly connected network compared with specific CRs, with more internal interactions and targeting by more common TFs and miRNAs (Supplementary Figure S14C). In total, 79.6% of miRNAs (39 of 49) in the pan-cancer network have been reported to be involved in multiple cancer types (13–18). Function enrichment analysis for common and specific CRs further revealed their different biological functions. Common CRs in cancer were highly enriched for basic cellular processes in multiple cancer types, while specific CRs were related to the cancer-specific processes (Supplementary Figure S14D). In summary, distinct functional features contributed to common and specific CR prioritization in cancer. Common CRs play basic and extensive roles while specific CRs hold special and intensive responsibilities in carcinogenesis.
Functional CRs contribute to cancer subtyping with clinical relevance
It is well known that cancers exhibit multiple subtypes with different molecular perturbations and clinical outcomes (42). The identification of clinically meaningful subtypes based on these molecular profiles is helpful for personalizing therapy in cancer. We therefore sought to explore the contribution of CRs to cancer subtyping. Based on cancer-context functional CR–associated transcriptome data, we performed subtype classification for the 33 cancer types. As a result, tumor samples were clustered into 3–6 subtypes across cancer types. The patients in different cancer subtypes showed significantly different survival for 21 of 33 cancer types (log-rank P < 0.05, Supplementary Figure S15). These observations suggest the validity and effectiveness of molecular classification based on cancer-context functional CR–associated multi-omics data.
Next, we analyzed the molecular patterns and clinical differences across different subtypes for each cancer type. For instance, we classified adrenocortical carcinoma (ACC) tumor samples into four subtypes (Figure 6A, Supplementary Figure S16, and Supplementary Table S9). The classification result was highly consistent with that of a previous study (43). Subtype 4, which was seen in only one sample, was excluded from further analyses. Comparing the clinical and pathological features across the three remaining subtypes of ACC, we found that patients in subtype 1 had the most invasive and malignant tumors. We found that several CRs, TFs, and miRNAs that contribute to the subtype identification were associated with cancer. RB1 is an example, which was found to be associated with aggressive ACC previously (44). Moreover, the differences between these three subtypes were also demonstrated by other independent CR features, including different mutation frequencies and the different DNA methylation patterns. Patients in subtype 1 of ACC showed the highest mutation frequency of TP53 and CGI methylation, while patients in subtype 3 showed high mutation of KDM6B and the lowest open sea methylation. In addition, we identified several subtypes for LGG and bladder urothelial carcinoma (BLCA) tumor samples. Differential molecular patterns across the subtypes were demonstrated by the CR-associated multi-omics features and showed different clinical behaviors for different subtypes (Figure 6B and C and Supplementary Table S9). In particular, we found that TP53, which is a common CR related to 25 cancer types, showed different somatic mutation frequencies across different subtypes of ACC, LGG and BLCA and was overexpressed in the LGG subtype 2. These results indicate that functional CRs not only characterize the tumor samples but also contribute to cancer subtyping.
Figure 6.

Cancer subtypes based on functional CRs. (A) Molecular patterns and clinical behaviors of ACC subtypes. The top panel represents the COCA results. Comparison section indicates the consistency of the subtype classification with previous studies. The Clinical section shows the difference in clinical factors between the subtypes. The Mutation section shows the differential mutation of the ACC-related functional CRs. The Methylation section denotes the hypermethylation and hypomethylation of subtypes. The CR, TF, and miRNA sections represent the differentially expressed CRs, TFs, and miRNAs. The bottom panel shows the overall survival (OS) outcomes for subtypes. (B and C) Molecular subtypes of LGG and BLCA. Detailed legends are shown in Supplementary Figure S16.
Specific CRs contribute to pan-cancer reclassified types
Exploration of the shared molecular alterations across cancer types spanning multiple tissues of origin has been meaningful and challenging, as these findings can bring novel approaches for personalizing therapy (45). In the above analyses, we divided single cancer types into subtypes with differential molecular patterns and clinical behaviors. Next, we extended the analysis in another direction to seek the possible convergence of different cancer types that share similar molecular patterns. Here we used the transcriptome datasets of 193 cancer specific CRs to reclassify all patients using the COCA method (described in Materials and Methods). Finally, we mainly obtained 37 reclassified types, which included >1% of pan-cancer samples or >20% samples of any cancer type. We found that the pan-cancer reclassified types showed a considerably consistent correspondence with the tissue-of-origin types (Figure 7A). Kaplan–Meier analysis was performed to investigate the prognosis of patients from 37 types. We found that the patients in different subtypes significantly differed in survival (Supplementary Figure S17).
Figure 7.

The reclassified molecular subtypes of pan-cancer patients. (A) Comparison between the COCA reclassified types and the tissue-of-origin types for pan-cancer samples. The colors of COCA classes are explained in Supplementary Figure S16. (B) The top 12 genes mutated in the tumor types, which are converted into class 6. Mutations shared across tumor entities are depicted by colored boxes. Functional CRs are marked with colored borders. (C) The differential molecular patterns and clinical behaviors between KIRP samples in class 15 and those in class 16. KIRP-specific CRs and related TFs/miRNAs are marked. (D) The clinical outcomes for KIRP, LUSC, BLCA, and MESO samples in different COCA classes. Detailed legends are shown in Supplementary Figure S16.
In the COCA results, some tumor samples from similar tissues were clustered into one class, such as the GBM and LGG samples in class 1 as well as the COAD and READ samples in class 4. Moreover, tumor samples that derived from different tissues could be converged owing to their similar molecular mechanisms, as in the tumor samples in classes 6, 13, 20, and 21. Focusing on the top 12 genes with mutations in the different cancer types in these classes, we found that these genes were greatly shared across different tumor entities (Supplementary Figure S18). For example, for patients in class 6, which mainly spanned five cancer types (esophageal carcinoma (ESCA), BLCA, cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), HNSC, and LUSC) and involved 455 squamous-like tumor samples, 65% of mutated genes such as TP53 and KMT2C were shared across these five cancer types (Figure 7B).
We next focused on the cancer types that were classified into multiple classes. Differential expression of the cancer-related CRs, TFs, and miRNAs was observed among different classes for a given cancer type (Supplementary Figure S19). These molecular differences suggest different molecular mechanisms in different subtypes. For instance, KIRP samples were clustered into two independent classes (class 15 and class 16). RNA expression analysis showed that not only the KIRP-specific CRs, TFs, and miRNAs that were used to do the classification but also other CRs, TFs, and miRNAs exhibited differential expression between the classes, indicating a significant difference between these two classes (Figure 7C and Supplementary Table S10). Moreover, other independent omics data also showed a difference between the two classes, as the KIRP tumor samples in class 15 showed significantly higher hypermethylation and hypomethylation (P = 5.10e–6 and P = 2.86e–3, respectively) compared with the KIRP samples in class 16. In addition, comparing clinical behaviors between class 15 and class 16, we found that class 15 was enriched for malignant type 2 samples and showed higher mortality with worse clinical outcomes (Figure 7C). Moreover, survival analyses of sub-cohorts showed significantly different outcomes in several cancer types, including LUSC, BLCA and mesothelioma (MESO) (Figure 7D). Taking these results together, we propose an integrated molecular taxonomy for pan-cancer patients based on cancer-specific CR molecular datasets, which present molecularly defined cancer types, unlike prior tissue classification systems.
DISCUSSION
CRs play various roles in tumorigenesis. In this study, we proposed feature-based classifiers and prioritized functional CRs on a genome-wide scale for each individual cancer type. Analysis of the functional features revealed recurrent multi-omics effects of functional CRs across 33 cancer types. Comparing the recurrence levels of seven features, we found that the mutation and interaction network features were the most contributory features, which were defined as the elements of most effective combined features in 25 and 29 cancer types. Analysis of functional CRs indicated that histone modifiers and chromatin remodelers are ubiquitously prioritized, while DNA methylators are rewired across cancer types. Moreover, readers and writers for histone modification as well as chromatin remodelers have common roles across cancer types. Furthermore, we classified the functional CRs depending on the number of cancer types they occurred in, and we found that common CRs are associated with basic cellular processes and play general roles in multiple cancer types, while specific CRs are enriched in cancer-related biological processes and play intensive roles in specific cancer types. By comparison between common, specific and mixed CRs as well as DNA methylators, histone modifiers and chromatin remodelers, we found that DNA methylation readers as well as histone modification readers and writers were with more common CRs, suggesting that these three categories of CRs tend to be aberrant across cancer types. We also found that DNA methylation erasers and histone modification erasers tend to be dysregulated in specific cancer type. Moreover, molecular reclassification of pan-cancer samples based on specific CRs provides independent prognostic information beyond tumor stage and tissue of origin. These observations suggest that functional CRs could act as biomarkers that contribute to classification of cancer subtypes and pan-cancer samples.
Increasing studies have showed that processes of DNA methylation, histone modification and chromatin remodeling in tumors are not independent, but closely related (46,47). For instance, histone modifiers can modulate the genome methylation pattern by mediating the modifications on histones (46). Besides, histone modifiers and chromatin remodelers have been found to be able to influence genome methylation by altering the activity of DNA methylators by adding or removing histone modification marks on DNA methylators or by changing the state of the chromatin. Thus, we investigated the regulation by all the CRs of DNA hypermethylation and hypomethylation following the approaches of a previous study to identify the functional CRs (25). Analyzing the specific regulation of functional CRs on epigenetics, 90% of the prioritized CRs were found to significantly regulate the DNA methylation. Moreover, we found that DNA methylators and histone modifiers can induce the development of cancer by regulation of DNA methylation and histone modifications, such as DNMT1, DNMT3A and PHF19. We also identified several CRs that can regulate genome-wide DNA methylation. For instance, histone deacetylases (HDAC1, HDAC2, HDAC3, HDAC4, HDAC5, HDAC7 and HDAC11) showed significant regulation of the sample methylation in multiple cancer types. All these clues demonstrated the complex relationship between DNA methylation, histone modification, and chromatin remodeling. As additional data become available, further investigations of the regulation by CRs of histone modification and chromatin remodeling will extend the integrative analysis.
With the development of high-throughput sequencing technology, datasets are increasingly being generated to investigate TF–gene regulation. We integrated additional transcriptional regulatory information from ChIPBase v2.0 (20), TRRUST (48) and data from Boyle et al. (49). In total, 99 497 transcriptional regulatory pairs were added (Supplementary Figure S20), which was threefold the size of the original dataset. Based on these integrated data, we re-calculated the Ftf features for each CR. We found that the new Ftf values of CRs significantly correlated with the original values across cancer types (Supplementary Figure S21). Moreover, we re-trained the classifiers on the basis of the new features and further prioritized the CRs in cancer. We found that the results were the same for 22 out of 33 cancer types. For the other cancer types, the prioritized CRs significantly overlapped with those prioritized in our original analyses (Supplementary Table S11). All these results indicate that the method is robust to differences in transcriptional regulatory data.
In addition, a free, web-accessible database called FACER (http://bio-bigdata.hrbmu.edu.cn/FACER/) was presented (Supplementary Figure S22). The web site offers three layers of information for 640 cancer-related functional CRs, including basic information (biological function and involved protein complex), epigenetic category, related cancer types, and details of seven aberrant features of functional CRs. The common and specific roles of functional CRs are also stored in FACER. Users can obtain this detailed information for CRs of interest, cancer types of interest, or both. We believe that this database will serve as a valuable public resource for further investigations of CRs. Furthermore, we review the number of publications in PubMed database for each cancer-related functional CR, which is listed in Supplementary Table S12. This result will help researchers understand the studies of CRs at a holistic level.
In summary, we developed feature-based classifiers to systematically identify cancer-context functional CRs for 33 cancer types and revealed the molecular perturbations of CRs in different categories as well as common and specific CRs. The global dissection of functional CRs across various cancer types will serve as a valuable resource for further investigating the function of epigenetics in tumorigenesis.
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully thank the TCGA Research Network for providing data for this work. We thank the Department of Scientific Publications at MD Anderson for editorial assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Program on Key Basic Research Project (973 Program) [2014CB910504]; funds for Creative Research Groups of the National Natural Science Foundation of China [81421063]; National High Technology Research and Development Program of China (863 Program) [2014AA021102]; the National Natural Science Foundation of China [61473106, 31571331, 61502126, 61603116]; Cancer Prevention and Research Institute of Texas (CPRIT) New Investigator Grant [RR160021]; AASLD Foundation Pinnacle Research Award in Liver Disease, China Postdoctoral Science Foundation [2016T90309, 2015M571436, 2016M591544, LBH-Z14134 LBH-Z16142]; Natural Science Foundation of Heilongjiang Province [QC2015020]; Funds for the Graduate Innovation Fund of Heilongjiang Province [YJSCX2015-6HYD]; Weihan Yu Youth Science Fund Project of Harbin Medical University, University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province Youth Science and technology innovation personnel training project [UNPYSCT-2016189 and UNPYSCT-2017060]; Harbin Special Funds for Innovative Talents in Science and Technology Research Project [2015RAQXJ091, 2017RAQXJ164, 2017RAGXJ004]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Plass C., Pfister S.M., Lindroth A.M., Bogatyrova O., Claus R., Lichter P.. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013; 14:765–780. [DOI] [PubMed] [Google Scholar]
- 2. Gonzalez-Perez A., Jene-Sanz A., Lopez-Bigas N.. The mutational landscape of chromatin regulatory factors across 4,623 tumor samples. Genome Biol. 2013; 14:r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Medvedeva Y.A., Lennartsson A., Ehsani R., Kulakovskiy I.V., Vorontsov I.E., Panahandeh P., Khimulya G., Kasukawa T., Consortium F., Drablos F.. EpiFactors: a comprehensive database of human epigenetic factors and complexes. Database. 2015; 2015:bav067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ram O., Goren A., Amit I., Shoresh N., Yosef N., Ernst J., Kellis M., Gymrek M., Issner R., Coyne M. et al. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. 2011; 147:1628–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Q., Huang J., Sun H., Liu J., Wang J., Wang Q., Qin Q., Mei S., Zhao C., Yang X. et al. CR Cistrome: a ChIP-Seq database for chromatin regulators and histone modification linkages in human and mouse. Nucleic Acids Res. 2014; 42:D450–D458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brien G.L., Valerio D.G., Armstrong S.A.. Exploiting the epigenome to control Cancer-Promoting Gene-Expression programs. Cancer Cell. 2016; 29:464–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan X.J., Xu J., Gu Z.H., Pan C.M., Lu G., Shen Y., Shi J.Y., Zhu Y.M., Tang L., Zhang X.W. et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011; 43:309–315. [DOI] [PubMed] [Google Scholar]
- 8. Damaschke N.A., Yang B., Blute M.L. Jr., Lin C.P., Huang W., Jarrard D.F.. Frequent disruption of chromodomain helicase DNA-binding protein 8 (CHD8) and functionally associated chromatin regulators in prostate cancer. Neoplasia. 2014; 16:1018–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bachmann I.M., Halvorsen O.J., Collett K., Stefansson I.M., Straume O., Haukaas S.A., Salvesen H.B., Otte A.P., Akslen L.A.. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006; 24:268–273. [DOI] [PubMed] [Google Scholar]
- 10. Visser H.P., Gunster M.J., Kluin-Nelemans H.C., Manders E.M., Raaphorst F.M., Meijer C.J., Willemze R., Otte A.P.. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br. J. Haematol. 2001; 112:950–958. [DOI] [PubMed] [Google Scholar]
- 11. Kleer C.G., Cao Q., Varambally S., Shen R., Ota I., Tomlins S.A., Ghosh D., Sewalt R.G., Otte A.P., Hayes D.F. et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. PNAS. 2003; 100:11606–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Varambally S., Cao Q., Mani R.S., Shankar S., Wang X., Ateeq B., Laxman B., Cao X., Jing X., Ramnarayanan K. et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008; 322:1695–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y., Qiu C., Tu J., Geng B., Yang J., Jiang T., Cui Q.. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014; 42:D1070–D1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang Q., Wang Y., Hao Y., Juan L., Teng M., Zhang X., Li M., Wang G., Liu Y.. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009; 37:D98–D104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang Q., Qiu C., Yang J., Wu Q., Cui Q.. miREnvironment database: providing a bridge for microRNAs, environmental factors and phenotypes. Bioinformatics. 2011; 27:3329–3330. [DOI] [PubMed] [Google Scholar]
- 16. Wang D., Gu J., Wang T., Ding Z.. OncomiRDB: a database for the experimentally verified oncogenic and tumor-suppressive microRNAs. Bioinformatics. 2014; 30:2237–2238. [DOI] [PubMed] [Google Scholar]
- 17. Ruepp A., Kowarsch A., Theis F.. PhenomiR: microRNAs in human diseases and biological processes. Methods Mol. Biol. 2012; 822:249–260. [DOI] [PubMed] [Google Scholar]
- 18. Liu X., Wang S., Meng F., Wang J., Zhang Y., Dai E., Yu X., Li X., Jiang W.. SM2miR: a database of the experimentally validated small molecules' effects on microRNA expression. Bioinformatics. 2013; 29:409–411. [DOI] [PubMed] [Google Scholar]
- 19. Jiang P., Freedman M.L., Liu J.S., Liu X.S.. Inference of transcriptional regulation in cancers. PNAS. 2015; 112:7731–7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou K.R., Liu S., Sun W.J., Zheng L.L., Zhou H., Yang J.H., Qu L.H.. ChIPBase v2.0: decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 2017; 45:D43–D50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang J.H., Li J.H., Jiang S., Zhou H., Qu L.H.. ChIPBase: a database for decoding the transcriptional regulation of long non-coding RNA and microRNA genes from ChIP-Seq data. Nucleic Acids Res. 2013; 41:D177–D187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hsu S.D., Tseng Y.T., Shrestha S., Lin Y.L., Khaleel A., Chou C.H., Chu C.F., Huang H.Y., Lin C.M., Ho S.Y. et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014; 42:D78–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vergoulis T., Vlachos I.S., Alexiou P., Georgakilas G., Maragkakis M., Reczko M., Gerangelos S., Koziris N., Dalamagas T., Hatzigeorgiou A.G.. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res. 2012; 40:D222–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huttlin E.L., Ting L., Bruckner R.J., Gebreab F., Gygi M.P., Szpyt J., Tam S., Zarraga G., Colby G., Baltier K. et al. The BioPlex network: a systematic exploration of the human interactome. Cell. 2015; 162:425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang Z., Jones A., Widschwendter M., Teschendorff A.E.. An integrative pan-cancer-wide analysis of epigenetic enzymes reveals universal patterns of epigenomic deregulation in cancer. Genome Biol. 2015; 16:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kolde R., Laur S., Adler P., Vilo J.. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012; 28:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. T T. A Package for Survival Analysis in S. version 2.38. 2015; https://CRAN.R-project.org/package=survival.
- 29. Cancer Genome Atlas, N Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tirosh I., Reikhav S., Sigal N., Assia Y., Barkai N.. Chromatin regulators as capacitors of interspecies variations in gene expression. Mol. Syst. Biol. 2010; 6:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saxena A., Carninci P.. Long non-coding RNA modifies chromatin: epigenetic silencing by long non-coding RNAs. BioEssays. 2011; 33:830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang H.T., Kathrein K.L., Barton A., Gitlin Z., Huang Y.H., Ward T.P., Hofmann O., Dibiase A., Song A., Tyekucheva S. et al. A network of epigenetic regulators guides developmental haematopoiesis in vivo. Nat. Cell Biol. 2013; 15:1516–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fratta E., Montico B., Rizzo A., Colizzi F., Sigalotti L., Dolcetti R.. Epimutational profile of hematologic malignancies as attractive target for new epigenetic therapies. Oncotarget. 2016; 7:57327–57350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu J., Li Y., Lu J., Pan T., Ding N., Wang Z., Shao T., Zhang J., Wang L., Li X.. The mRNA related ceRNA-ceRNA landscape and significance across 20 major cancer types. Nucleic Acids Res. 2015; 43:8169–8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Suzuki M., Wada H., Yoshino M., Tian L., Shigematsu H., Suzuki H., Alaa M., Tamura H., Fujiwara T., Nagato K. et al. Molecular characterization of chronic obstructive pulmonary disease-related non-small cell lung cancer through aberrant methylation and alterations of EGFR signaling. Ann. Surg. Oncol. 2010; 17:878–888. [DOI] [PubMed] [Google Scholar]
- 36. Li Y., Li S., Chen J., Shao T., Jiang C., Wang Y., Chen H., Xu J., Li X.. Comparative epigenetic analyses reveal distinct patterns of oncogenic pathways activation in breast cancer subtypes. Hum. Mol. Genet. 2014; 23:5378–5393. [DOI] [PubMed] [Google Scholar]
- 37. Kleppe A., Albregtsen F., Vlatkovic L., Pradhan M., Nielsen B., Hveem T.S., Askautrud H.A., Kristensen G.B., Nesbakken A., Trovik J. et al. Chromatin organisation and cancer prognosis: a pan-cancer study. Lancet. Oncol. 2018; 19:356–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang W., Ernst P.. Distinct functions of histone H3, lysine 4 methyltransferases in normal and malignant hematopoiesis. Curr. Opin. Hematol. 2017; 24:322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Araujo J.M., Prado A., Cardenas N.K., Zaharia M., Dyer R., Doimi F., Bravo L., Pinillos L., Morante Z., Aguilar A. et al. Repeated observation of immune gene sets enrichment in women with non-small cell lung cancer. Oncotarget. 2016; 7:20282–20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu M., Wang Y., Wang C., Shen W., Liu J., Geng L., Cheng Y., Dai J., Jin G., Ma H. et al. The eQTL-missense polymorphisms of APOBEC3H are associated with lung cancer risk in a Han Chinese population. Sci. Rep. 2015; 5:14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yi S., Lin S., Li Y., Zhao W., Mills G.B., Sahni N.. Functional variomics and network perturbation: connecting genotype to phenotype in cancer. Nat. Rev. Genet. 2017; 18:395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hofree M., Shen J.P., Carter H., Gross A., Ideker T.. Network-based stratification of tumor mutations. Nat. Methods. 2013; 10:1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zheng S., Cherniack A.D., Dewal N., Moffitt R.A., Danilova L., Murray B.A., Lerario A.M., Else T., Knijnenburg T.A., Ciriello G. et al. Comprehensive Pan-Genomic characterization of adrenocortical carcinoma. Cancer Cell. 2016; 30:363. [DOI] [PubMed] [Google Scholar]
- 44. Ragazzon B., Libe R., Assie G., Tissier F., Barreau O., Houdayer C., Perlemoine K., Audebourg A., Clauser E., Rene-Corail F. et al. Mass-array screening of frequent mutations in cancers reveals RB1 alterations in aggressive adrenocortical carcinomas. Eur. J. Endocrinol. 2014; 170:385–391. [DOI] [PubMed] [Google Scholar]
- 45. Hoadley K.A., Yau C., Wolf D.M., Cherniack A.D., Tamborero D., Ng S., Leiserson M.D.M., Niu B., McLellan M.D., Uzunangelov V. et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014; 158:929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cervoni N., Szyf M.. Demethylase activity is directed by histone acetylation. J. Biol. Chem. 2001; 276:40778–40787. [DOI] [PubMed] [Google Scholar]
- 47. Lehnertz B., Ueda Y., Derijck A.A., Braunschweig U., Perez-Burgos L., Kubicek S., Chen T., Li E., Jenuwein T., Peters A.H.. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol.: CB. 2003; 13:1192–1200. [DOI] [PubMed] [Google Scholar]
- 48. Han H., Cho J.W., Lee S., Yun A., Kim H., Bae D., Yang S., Kim C.Y., Lee M., Kim E. et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018; 46:D380–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boyle A.P., Araya C.L., Brdlik C., Cayting P., Cheng C., Cheng Y., Gardner K., Hillier L.W., Janette J., Jiang L. et al. Comparative analysis of regulatory information and circuits across distant species. Nature. 2014; 512:453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.